

Abstract
Unfolded protein response (UPR) has roles not only in resolving the accumulation of unfolded proteins owing to endoplasmic reticulum (ER) stress, but also in regulation of cellular physiological functions. ER stress transducers providing the branches of UPR signaling are known to localize in distal dendritic ER of neurons. These reports suggest that local activation of UPR branches may produce integrated outputs for distant communication, and allow regulation of local events in highly polarized neurons. Here, we demonstrated that synaptic activity‐ and brain‐derived neurotrophic factor (BDNF)‐dependent local activation of UPR signaling could be associated with dendritic functions through retrograde signal propagation by using murine neuroblastoma cell line, Neuro‐2A and primary cultured hippocampal neurons derived from postnatal day 0 litter C57BL/6 mice. ER stress transducer, inositol‐requiring kinase 1 (IRE1), was activated at postsynapses in response to excitatory synaptic activation. Activated dendritic IRE1 accelerated accumulation of the downstream transcription factor, x‐box‐binding protein 1 (XBP1), in the nucleus. Interestingly, excitatory synaptic activation‐dependent up‐regulation of XBP1 directly facilitated transcriptional activation of BDNF. BDNF in turn drove its own expression via IRE1‐XBP1 pathway in a protein kinase A‐dependent manner. Exogenous treatment with BDNF promoted extension and branching of dendrites through the protein kinase A‐IRE1‐XBP1 cascade. Taken together, our findings indicate novel mechanisms for communication between soma and distal sites of polarized neurons that are coordinated by local activation of IRE1‐XBP1 signaling. Synaptic activity‐ and BDNF‐dependent distinct activation of dendritic IRE1‐XBP1 cascade drives BDNF expression in cell soma and may be involved in dendritic extension.
Cover Image for this issue: doi. 10.1111/jnc.14159.
![]()
Keywords: BDNF, dendrite, endoplasmic reticulum stress, IRE1, unfolded protein response, XBP1
Abbreviations used
- ATF6
activating transcription factor 6
- BDNF
brain‐derived neurotrophic factor
- BiP
binding immunoglobulin protein
- bp
base pair
- cAMP
cyclic adenosine monophosphate
- ChIP
chromatin immunoprecipitation
- CNS
central nervous system
- CREB
cAMP response element‐binding protein
- eIF2α
α subunit of eukaryotic initiation factor 2
- ERAD
ER‐associated degradation
- ER
endoplasmic reticulum
- GFP
green fluorescence protein
- GluR
glutamate receptor
- IRE1
inositol requiring 1
- LTP
long‐term potentiation
- MAP2
microtubule‐associated protein 2
- p38 MAPK
p38 mitogen activated protein kinase
- PERK
protein kinase R‐like ER kinase
- PKA
protein kinase A
- PSD95
postsynaptic density protein 95
- ROCKII
Rho‐associated protein kinase II
- S6K1
ribosomal protein S6 kinase 1
- TrkB
tropomyosin‐related kinase B
- UPRE
unfolded protein response element
- UPR
unfolded protein response
- XBP1
x‐box‐binding protein 1
The endoplasmic reticulum (ER) is a central cellular organelle responsible for calcium ion storage, lipid metabolism, and folding and post‐translational modification of secretory and membrane proteins. Multiple environmental and intracellular abnormalities, such as disturbances in calcium ion homeostasis cause accumulation of unfolded proteins in the ER lumen, resulting in perturbed ER function. These abnormalities are known as ER stress (Kaufman 2002; Ron 2002; Rutkowski and Kaufman 2004). Eukaryotic cells transduce signals dealing with unfolded proteins, referred to as the unfolded protein response (UPR) (Kaufman 2002; Ron 2002; Ron and Walter 2007). In mammalian cells, the three major canonical branches of the UPR are the inositol‐requiring kinase 1 (IRE1) (Tirasophon et al. 1998), protein kinase R‐like ER kinase (PERK) (Harding et al. 1999), and activating transcription factor 6 (ATF6) (Yoshida et al. 2000) pathways. Under ER stress conditions, these ER‐resident ER stress transducers activate three distinct responses: (i) induction of folding capacity by expression of chaperone molecules (Yoshida et al. 1998; Li et al. 2000), (ii) attenuation of protein translation (Harding et al. 2000), and (iii) ER‐associated degradation to degrade unfolded proteins (Ng et al. 2000; Travers et al. 2000).
Of the three major ER stress transducers, IRE1 is the most evolutionarily conserved. IRE1 has endonuclease activity that is activated by its oligomerization and trans‐autophosphorylation (Tirasophon et al. 1998). Phosphorylated IRE1 triggers non‐conventional splicing of x‐box‐binding protein 1 (Xbp1) mRNA (unspliced Xbp1: Xbp1u) to produce mature Xbp1 mRNA (spliced Xbp1: Xbp1s) (Yoshida et al. 2001, 2003; Calfon et al. 2002), generating the x‐box‐binding protein 1 (XBP1s) transcription factor. XBP1s induces the expression of target genes, including molecular chaperones and ER‐associated degradation‐related genes (Wang et al. 1998; Yoshida et al. 2003). In the central nervous system (CNS), impaired XBP1s function is associated with increased neuronal damage. Deletion of Xbp1 in spinal cord injury models reduces locomotor recovery (Valenzuela et al. 2012). Another report has shown the importance of XBP1 in the survival of retinal ganglion cells following optic nerve crush (Hu et al. 2012). Infection with adenovirus expressing Xbp1s before nerve crush inhibits injury‐induced cell death. On the other hand, deletion of neuronal Xbp1 extends the lifespan of transgenic mice expressing a mutant superoxide dismutase‐1, an amyotrophic lateral sclerosis model (Hetz et al. 2009; Madeo et al. 2009). Xbp1 deficiency reduces the aggregation of superoxide dismutase‐1 and its toxic effects by activating autophagy in cultured motor neurons and the spinal cord, which may increase the clearance of aggregated proteins. Together, these previous studies suggest that the effects of Xbp1 deletion on neuronal damage might depend on the model. Pathologically, genomic screening has identified a polymorphism in the Xbp1 promoter as a risk factor for various neurodegenerative and psychiatric diseases such as Alzheimer's disease, bipolar disorder and schizophrenia (Kakiuchi et al. 2003, 2004; Liu et al. 2013). Additionally, IRE1‐XBP1 signaling has crucial roles in regulating neuronal homeostasis and physiological functions, indicating that the IRE1‐XBP1 branch can activate without the direct connection with an ER stress response induced by the accumulation of unfolded proteins. In Caenorhabditis elegans, loss‐of‐function mutations in Ire1, wy762, and wy782 cause deficiencies in the dendritic branches of sensory neurons (Wei et al. 2015). XBP1s also drives neurite outgrowth, cognitive processes, and memory formation via the expression of neuronal activity‐related genes such as brain‐derived neurotrophic factor (BDNF) (Hayashi et al. 2007, 2008; Martinez et al. 2016). The transfectants of primary cultured mouse hippocampal neurons expressing green fluorescence protein (GFP)‐tagged ER stress transducers including IRE1 show the localization of these transducers at distal dendrites (Murakami et al. 2007). Together, these reports suggest that the IRE1‐XBP1 branch of UPR signaling manages the integral functions and activities of neurons under basal conditions through the regulation of local events. However, the intrinsic mechanisms for physiological regulation by the local outputs of this pathway remain unclear.
BDNF is a member of the neurotrophin family, and was first identified as a critical factor for neuronal survival and neurite growth during early neuronal development (Barde et al. 1982). Various neuronal activities, including excitatory signals and activation of the glutamate receptor induce precursor BDNF, followed by its cleavage to generate the biologically active form (Zafra et al. 1991; Negro et al. 1994; Wetmore et al. 1994). This molecule is expressed throughout the CNS (Leibrock et al. 1989) and also in peripheral tissues including muscle, liver, pancreas, kidney and bladder (Lommatzsch et al. 1999). Extensive studies have shown that BDNF binds to the tyrosine kinase receptor, tropomyosin‐related kinase B (TrkB), leading to the activation of diverse signaling for the manipulation of neuronal morphogenesis and synaptic plasticity, as well as neuronal growth and survival (Binder and Scharfman 2004). Exogenous application of BDNF can promote synaptic transmission (Lohof et al. 1993; Lessmann et al. 1994). The depletion of endogenous BDNF impairs induction of long‐term potentiation (LTP) (Korte et al. 1995; Chen et al. 1999), suggesting that BDNF is important for the modulation and stabilization of synaptic functions and plasticity in distal neurites. However, neuronal activity‐dependent transcriptional regulation of BDNF and the molecular mechanisms enabling efficient transmission of BDNF‐induced signaling from distal sites to the soma remain unexplored.
In this study, we first uncovered physiological roles of UPR signaling activated in the distal dendrites of CNS neurons. We demonstrated that the IRE1‐XBP1 pathway was up‐regulated by excitatory synaptic activation in distal dendrites, followed by retrograde propagation over the distance for executing the induction of Bdnf. BDNF drove a positive feedback loop via the IRE1‐XBP1 pathway in a protein kinase A (PKA)‐dependent manner, ultimately orchestrating dendritic outgrowth and branching.
Materials and methods
Mice
C57BL/6j mice (Charles River Laboratories, Yokohama, Japan) (RRID: IMSR_JAX:000664) were used in this study. Animal housing conditions and all experimental procedures were approved by the Committee of Animal Experimentation, Hiroshima University.
Cell culture, treatment and lentivirus infection
Primary cultured mouse hippocampal neurons were prepared from the hippocampus of postnatal day 0 litter C57BL/6j mice, using a modified protocol (Murakami et al. 2007). Briefly, pregnant female mice housed in cages were randomly divided into individual cage, and postnatal day 0 litter mice were dissected for isolating hippocampi. The postnatal day 0 litter mice were not divided by the sex, because we could not distinguish their sex on postnatal day 0. All the litter mice were dissected to isolate the hippocampi. The hippocampi were digested with 0.05% papain (Worthington, Lakewood, NJ, USA), 0.02% bovine serum albumin and 0.5% glucose to isolate neurons. The extracted neurons were maintained for 10 days in neurobasal medium (Gibco, Rockville, MD, USA) supplemented with B27 (Gibco), 2 mM l‐glutamine (Gibco), 10 μM uridine (Sigma, St Louis, MO, USA) and 10 μM 5′‐fluoro‐2′‐deoxyuridine (Sigma) on dishes coated with poly‐l‐lysine (Trevigen, Gaithersburg, MD, USA). Neuro‐2A (RRID: CVCL_0470), a murine neuroblastoma cell line, was cultured in D‐MEM (Gibco) supplemented with 10% fetal calf serum. For cell treatments, 7.5 μM l‐glutamate (Wako, Osaka, Japan), 10 μM or 30 μM H89 (Sigma) and 100 ng/mL BDNF (PEPRO TECH, Rocky Hill, NJ, USA) were used. For Ire1 and Xbp1 knockdown, lentiviral vectors were constructed, using a BLOCK‐iT™ Pol α miR RNAi Expression Vector kit (Thermo Fisher Scientific, Waltham, MA, USA). Short hairpin RNA (shRNA) sequences targeting Ire1 and Xbp1 were designed using a BLOCK‐iT™ hairpin selection system (Thermo Fisher Scientific). After annealing, double‐stranded oligonucleotides were cloned into the pcDNA™6.2‐GW/EmGFP‐miR vector, using T4 DNA ligase. The double‐stranded oligonucleotides were then cloned into the pDONR™ 221 vector using Gateway BP Clonase II Enzyme mix (Thermo Fisher Scientific), followed by cloning into the pLenti‐CMV/TO‐puro vector, using Gateway LR Clonase II Enzyme mix (Thermo Fisher Scientific). To generate lentiviruses carrying Ire1 or Xbp1 shRNA with GFP, the pLenti‐based expression vectors and the ViraPower Packaging mix (Thermo Fisher Scientific) were cotransfected into the 293FT cell line. The virus‐containing supernatant was collected and concentrated, using Lenti‐X concentrator (Clontech, Mountain View, CA, USA) according to the manufacturer's protocol. Primary cultured hippocampal neurons were infected with the purified lentiviruses for 5 days before the cells were harvested or fixed for analyses.
RNA isolation and RT‐PCR
Total RNA was isolated from primary cultured hippocampal neurons, using ISOGEN (Wako) according to the manufacturer's protocol. First‐strand cDNA was synthesized from 1 μg RNA in a 20‐μL reaction volume, using a random primer (Takara, Kusatsu, Japan) and Moloney murine leukemia virus reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Xbp1, binding immunoglobulin protein (Bip) and β‐actin expression were analyzed by performing PCR in a 20‐μL reaction mixture containing 0.5 μM of each primer, 0.2 mM dNTPs, 3 units of Taq polymerase, and 10× PCR buffer (Agilent, Santa Clara, CA, USA). The PCR conditions were as follows: 94°C for 5 min; 24 cycles (Bip and Xbp1) or 16 cycles (β‐actin) of 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min; and 72°C for 5 min. The PCR products were resolved by electrophoresis on a 4.8% acrylamide gel. The primers used were: 5′‐ACACGCTTGGGAATGGACAC‐3′ (Xbp1 forward) and 5′‐CCATGGGAAGATGTTCTGGG‐3′ (Xbp1 reverse), 5′‐AGCCATCCCGTGGCATAA‐3′ (Bip forward) and 5′‐GCACAGCGGCACCATAGG‐3′ (Bip reverse) and 5′‐TCCTCCCTGGAGAAGAGCTAC‐3′ (β‐actin forward) and 5′‐TCCTGCTTGCTGATCCACAT‐3′ (β‐actin reverse).
Protein preparation and western blotting
Proteins were extracted from primary cultured hippocampal neurons in a cell lysis buffer containing 1% Triton X‐100, 20 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM EDTA, phosphatase inhibitor cocktail 3 (Sigma‐Aldrich), and protease inhibitor cocktail set I (Wako). The lysates were incubated on ice for 45 min. After centrifugation at 15 000 g for 15 min, the protein concentrations of the supernatants were determined, using a bicinchoninic acid assay kit (Thermo Fisher Scientific). Equal amounts of proteins (10 μg) were resolved, using sodium dodecyl sulfate–polyacrylamide gel electrophoresis. For immunoblotting, the following antibodies were used; anti‐β‐actin (1 : 5000; Sigma‐Aldrich, RRID: AB_476744), anti‐IRE1 (1 : 1000; Cell Signaling Technology, Beverly, MA, USA, RRID: AB_823545), anti‐alpha subunit of eukaryotic initiation factor 2 (eIF2α) (1 : 1000; Cell Signaling Technology, RRID: AB_10695409), anti‐phospho‐eIF2α (1 : 1000; Cell Signaling Technology, RRID: AB_2096481), anti‐XBP1s (1 : 250; Santa Cruz Biotechnology, Santa Cruz, CA, USA, RRID: AB_794171), anti‐p38 mitogen activated protein kinase (p38 MAPK) (1 : 1000; Cell Signaling Technology, RRID: AB_10998134), anti‐phospho‐p38 MAPK (1 : 1000; Cell Signaling Technology, RRID: AB_331296), anti‐cAMP response element‐binding protein (CREB) (1 : 1000; Cell Signaling Technology, RRID: AB_10691832), and anti‐phospho‐CREB (1 : 1000; Cell Signaling Technology, RRID: AB_2561044). Anti‐phospho‐IRE1α (1 : 1000) antibody was kindly gifted from Dr. Fumihiko Urano (Washington University in St. Louis). The density of each band was quantified, using CS Analyzer 4 image analysis software (ATTO, Tokyo, Japan).
Immunofluorescence staining
Primary cultured hippocampal neurons were fixed in 4% paraformaldehyde for 1 h and permeabilized in 0.1% Triton‐X 100 for 10 min, followed by treatment with 1% bovine serum albumin for 20 min. These procedures were performed at room temperature (25°C). The following antibodies were used; anti‐IRE1 (1 : 500; Cell Signaling Technology, RRID: AB_823545), anti‐microtubule‐associated protein 2 (MAP2) (1 : 1000; EMD Millipore, Billerica, MA, USA, RRID: AB_11213363), anti‐MAP2 (1 : 500; Abcam, Cambridge, UK, RRID: AB_2138153), anti‐postsynaptic density protein 95 (PSD95) (1 : 500; NeuroMab, Davis, CA, USA, RRID: AB_2307331), anti‐XBP1s (1 : 500; BioLegend, Sandiego, CA, USA, RRID: AB_2562960), and Anti‐phospho‐IRE1α (1 : 500). Cells were visualized under a FV1000D confocal microscope (Olympus, Tokyo, Japan). P‐IRE1 and MAP2 fluorescence intensities 40–90 μm from somata in randomly selected dendrites were measured, using ImageJ (National Institutes of Health, Rockville, MD, USA). The number of P‐IRE1‐positive puncta overlapping with PSD95‐positive postsynaptic sites was manually counted 40–90 μm from somata in randomly selected dendrites. The cells were randomly chosen from five independent cultures.
Luciferase assay
Neuro‐2A (RRID: CVCL_0470) cells were plated and transfected, using Screenfect A (Wako). Cells were transfected with a reporter plasmid carrying the mouse Bdnf promoter and firefly luciferase gene (BDNF‐Luc), or a Bdnf promoter mutant lacking the UPR element (UPRE) core sequence (ΔBDNF‐Luc), together with a pRL‐SV40 co‐reporter plasmid carrying the Renilla luciferase gene (Promega, Madison, WI, USA). After 36 h, dual luciferase activities were measured using a Dual‐Luciferase Reporter Assay System (Promega) and a GloMax Multi+ Detection System (Promega) according to the manufacturer's protocol. Relative activities were defined as the ratio between firefly and Renilla luciferase activities. The reporter plasmids BDNF‐Luc and ΔBDNF‐Luc were kindly gifted from Dr. Claudio Hetz (University of Chile).
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation (ChIP) assay was performed as previously described (Kondo et al. 2005). Briefly, primary cultured hippocampal neurons were cross‐linked using 1% formaldehyde for 15 min at 37°C, followed by inactivation with 0.15 M glycine for 5 min at 25°C. The cells were then lysed with sodium dodecyl sulfate lysis buffer and sonicated (20 × 10‐s sonication pulses at 1‐min intervals; Sonifier 250, Branson, Danbury, CT, USA). Equal amounts of chromatin from each sample were incubated overnight at 4°C with anti‐XBP1s (BioLegend, RRID: AB_2562960) or anti‐Histone H3 (Santa Cruz Biotechnology, RRID: AB_2115276) antibodies, or mouse IgG (Cell Signaling Technology, RRID: AB_10829607). Cross‐linking was reversed by incubating for 6 h at 65°C and the DNA was purified by phenol‐chloroform extraction and ethanol precipitation. Purified DNA was used for PCR analysis. The primers used to detect the mouse Bdnf promoter were: 5′‐TGCAATGCCCTGGAACGGAAT‐3′ (forward) and 5′‐GTGGGTGGGAGTCCACGAGA‐3′ (reverse). The density of each band was quantified, using CS Analyzer 4 image analysis software.
Morphological observation, and dendritic elongation and branching analysis
Primary cultured hippocampal neurons maintained for 10 days were fixed and stained with anti‐MAP2 antibody. The morphological expansion of staining was traced, using ImageJ. The length of the longest dendrite which was stained with anti‐MAP2 antibody in a randomly selected cell was traced and measured. Ten independent analyses were carried out, using blinded samples. For evaluating dendritic branching, the number of branched dendrites 20, 40, 60 and 80 μm from the center of the soma was manually counted at each distance. The cells were randomly chosen from five independent cultures.
Statistical analysis
Statistical comparisons were made using an unpaired Student's t‐test between two samples. The statistical significance of a difference was determined on the basis of a p < 0.05. Here, p‐values less than 0.05, 0.01 or 0.001 are presented as *p < 0.05, **p < 0.01 or ***p < 0.001, respectively. Double‐blind analyses were applied for all of the quantifications. To double‐blind the analysis, Dr. Saito collected the samples and then randomly passed the independent samples on to Dr. Cai and Dr. Matsuhisa.
Results
Glutamate stimulation activates the IRE1‐XBP1 pathway in dendrites
To explore the roles of UPR signaling in neurons, we focused on the dendritic roles of the IRE1‐XBP1 branch, because of previous studies that found activation of exogenously expressed GFP‐IRE1 at dendrites in response to ER stress, and that validated dendritic localization of Xbp1 mRNA (Murakami et al. 2007). We examined the localization of endogenous IRE1 by performing immunofluorescence staining analysis for IRE1 in primary cultured mouse hippocampal neurons (Fig. 1a). Immunoreactivity of endogenous IRE1 was observed in the neurites. The dendritic marker, MAP2 was detected in these IRE1‐positive neurites, indicating the localization of endogenous IRE1 at dendrites, consistent with those of exogenously expressed GFP‐IRE1 (Murakami et al. 2007). We next examined the activation mechanisms of IRE1 localized at dendrites. Various local events related to the regulation of neuronal activities such as excitatory synaptic activation are continuously induced in distal dendrites (Gulledge et al. 2005; Spruston 2008). Thus, we tested whether ER stress occurs in response to excitatory synaptic activation using exposure to glutamate, one of the major excitatory transmitters in the CNS (Headley and Grillner 1990; Nicholls and Attwell 1990; Citri and Malenka 2008). Primary cultured hippocampal neurons treated with glutamate showed the increase in phosphorylated IRE1 and accelerated splicing of Xbp1 mRNA (Fig. 1b and c). The levels of phosphorylated IRE1 and Xbp1s mRNA returned to basal levels within 12 h of exposure to glutamate. We also assessed the activation of the other branches of the UPR pathways. Phosphorylated eIF2α, downstream in the PERK pathway, was up‐regulated and peaked 3 h after glutamate treatment (Fig. 1b). Neurons stimulated with glutamate displayed the increase in Bip mRNA, which is one of the genes downstream of ATF6 (Fig. 1c). Bip expression peaked 6 h after glutamate stimulation. These temporal inductions were down‐regulated within 12 h, consistent with patterns of the IRE1‐XBP1 pathway. These data suggest the transient activation of canonical UPR signaling pathways in hippocampal neurons after glutamate stimulation. These findings are supported by previous reports suggesting the induction of ER stress by excitotoxities via the high dose of glutamate exposure (Yu et al. 1999; Kitao et al. 2001). We next investigated the activation sites of IRE1 after excitatory synaptic activation by performing immunofluorescence staining analysis. The immunoreactivities of IRE1 were detected in MAP2‐positive neurites and PSD95 (postsynaptic marker)‐positive punctate sites existing neighboring neurites of dendrites in hippocampal neurons (Fig. 1d). The localization and level of IRE1 at dendrites were not significantly changed by the treatment with glutamate. The phosphorylation of IRE1 at dendrites of hippocampal neurons was almost undetectable under normal conditions (Fig. 1e). The level of phosphorylated IRE1 was slightly increased in MAP2‐positive neurites in response to glutamate treatment (Fig. 1e and f). In contrast, a drastic increase in the phosphorylation was found in the punctate sites overlapped with immunoreactivity of PSD95 (Fig. 1e and g). Collectively, UPR branches including the IRE1 pathway are transiently activated by treatment of hippocampal neurons with glutamate. Phosphorylation of IRE1 is primarily increased at postsynaptic sites of dendrites following stimulation with glutamate. Dendritic IRE1 may be locally activated at postsynaptic sites after excitatory synaptic activation, which could modulate neuronal functions regulated by postsynaptic outputs.
Figure 1.
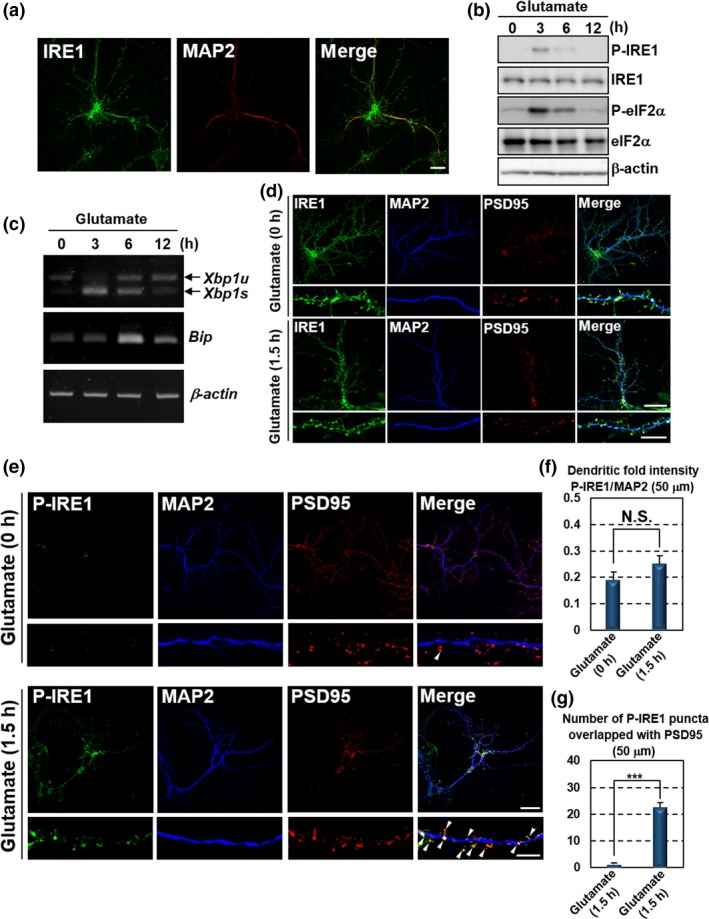
Glutamate stimulation induces local activation of the IRE1‐XBP1 pathway in distal dendrites. Primary cultured mouse hippocampal neurons maintained for 10 days were used for the analyses. (a) Immunofluorescence staining analysis of IRE1 (green) and MAP2 (dendritic marker: red). IRE1 was localized to MAP2‐positive dendrites (Bar: 20 μm). (b) Western blot analysis of phosphorylated IRE1 (P‐IRE1), IRE1, phosphorylated eIF2α (P‐eIF2α) and eIF2α. Cells were treated with 7.5 μM glutamate for the indicated times. P‐IRE1 and P‐eIF2α were transiently up‐regulated following glutamate stimulation. (c) RT‐PCR analysis of Xbp1 and Bip. Spliced Xbp1 (Xbp1s) and Bip were transiently induced by treatment with 7.5 μM glutamate for the indicated times (Xbp1u: unspliced Xbp1). (d) Immunofluorescence staining analysis of IRE1 (green), MAP2 (blue) and PSD95 (postsynaptic marker: red). IRE1 was observed at MAP2‐positive neurites and PSD95‐positive postsynaptic sites of dendrites. The localization and level were not significantly changed by the treatment with 7.5 μM glutamate for 1.5 h. The lower panels show high magnification of the dendrites shown in the upper panels. (Bar in upper panels: 20 μm, in lower panels: 5 μm). (e) Immunofluorescence staining analysis of P‐IRE1 (green), MAP2 (blue) and PSD95 (red). IRE1 was phosphorylated at PSD95‐positive postsynaptic sites of dendrites in response to 7.5 μM glutamate exposure for 1.5 h. The lower panels show high magnification of the dendrites shown in the upper panels. Arrow heads indicate immunoreactivities of P‐IRE1 overlapped with those of PSD95. (Bar in upper panels: 20 μm, in lower panels: 5 μm). (f) P‐IRE1 fluorescence intensities 40–90 μm from the somata of the dendrites in (e). The fluorescence intensities in these dendrites were not increased by glutamate treatment (mean ± SD, N = 10; number of cells prepared from five independent cultures). (g) The number of P‐IRE1‐positive puncta overlapping with PSD95‐positive postsynaptic sites 40–90 μm from the somata in (e). The number of P‐IRE1‐positive postsynaptic sites was increased by glutamate exposure (mean ± SD, N = 10; number of cells prepared from five independent cultures; ***p < 0.001).
Glutamate‐induced activation of dendritic IRE1‐XBP1 signaling facilitates Bdnf transcription in the soma
To assess the effect of glutamate‐activated dendritic IRE1 on the translocation of transcription factor XBP1s into the nucleus, hippocampal neurons were infected with lentiviruses expressing scrambled, Ire1 or Xbp1 shRNA with GFP, for 5 days prior to glutamate stimulation. GFP expression was used as a marker for lentivirus‐infected neurons. Efficient infection of hippocampal neurons (more than 90%) was confirmed by observation of GFP‐positive cells. The lentiviral infection of glutamate‐stimulated neurons substantially suppressed expression of IRE1 or XBP1s proteins (Fig. 2a and b). XBP1s expression was also inhibited in Ire1‐knockdown neurons, indicating that the production of almost all the XBP1s protein promoted by glutamate is depend on the activation of IRE1. In hippocampal neurons infected with lentivirus expressing scrambled shRNA with GFP, XBP1s accumulated into the nucleus following glutamate treatment (Fig. 2c). This accumulation was canceled in cells infected with lentiviruses expressing Ire1 or Xbp1 shRNA with GFP. Consequently, excitatory synaptic activation by glutamate treatment enhances XBP1s accumulation in the nucleus via IRE1 phosphorylation. Glutamate‐induced activation of the IRE1‐XBP1 pathway in dendrites may transduce signals over the distance to the nucleus to modulate neuronal functions and activities.
Figure 2.
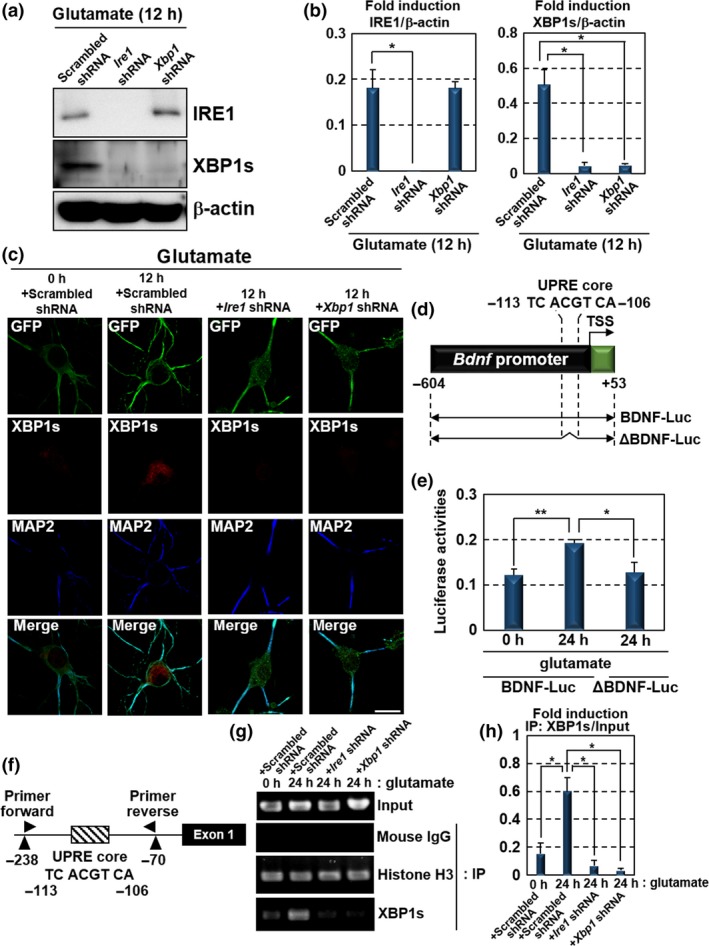
Glutamate‐induced activation of dendritic IRE1‐XBP1 signaling facilitates Bdnf transcription. (a) Western blot analysis of IRE1 and XBP1s expression in primary cultured hippocampal neurons maintained for 10 days. The cells were infected with lentiviruses expressing scrambled, Ire1 or Xbp1 shRNA, with GFP for 5 days, and treated with glutamate for 12 h. Lentiviral infection substantially suppressed the expression of IRE1 or XBP1s proteins. (b) Quantification of relative IRE1 and XBP1s protein levels in (a) (mean ± SD, N = 3; number of extracted samples prepared from independent cultures; *p < 0.05). (c) Immunofluorescence staining analysis of GFP (green), XBP1s (red) and MAP2 (blue) in hippocampal neurons maintained for 10 days. The cells were infected with lentiviruses expressing scrambled, Ire1 or Xbp1 shRNA, with GFP for 5 days and treated with 7.5 μM glutamate for 12 h. XBP1s accumulated in the nucleus in response to glutamate stimulation. This accumulation was canceled by Ire1 or Xbp1 knockdown (Bar: 20 μm). (d) Schema of the mouse Bdnf promoter region and reporter constructs. The unfolded protein response element (UPRE) core (ACGT) is located 108 bp upstream of the Bdnf transcription start site. The BDNF‐Luc construct consists of a 0.6 kbp region upstream of the Bdnf transcription start site. ΔBDNF‐Luc is a 0.6 kbp‐Luc construct lacking the UPRE core (Luc: luciferase gene, TSS: transcription start site). (e) Reporter assays using Neuro‐2A cells. Cells transfected with BDNF‐Luc or ΔBDNF‐Luc constructs in (d) were treated with 7.5 μM glutamate for 24 h. In cells transfected with ΔBDNF‐Luc constructs, reporter activities were reduced, despite treatment with glutamate (mean ± SD, N = 6; number of samples prepared from independent cultures; *p < 0.05, **p < 0.01). (f) Schema of the Bdnf promoter and the annealing sites of the primer set used in the ChIP assay. (g) ChIP assay using primary cultured hippocampal neurons maintained for 10 days. The cells were infected with lentiviruses expressing scrambled, Ire1 or Xbp1 shRNA, with GFP, for 5 days, and then treated with 7.5 μM glutamate for 24 h. Chromatin immunoprecipitation was performed, using the indicated antibodies and mouse IgG (negative control). (h) Quantitative analysis of PCR amplification after immunoprecipitation by anti‐XBP1s antibody in (g) (mean ± SD, N = 3; number of samples prepared from independent cultures; *p < 0.05).
We next investigated the roles of glutamate‐induced XBP1s in regulating neuronal functions and homeostasis. Recently, Martínez et al. have shown that XBP1 modulates synaptic plasticity, contextual memory formation and LTP through the expression of BDNF, a key component in memory consolidation (Park and Poo 2013; Lu et al. 2014; Martinez et al. 2016). However, the initial events activating XBP1 and molecular processes regulating neuronal functions in its downstream have not been understood. As shown in Figs 1 and 2(c), we found excitatory synaptic activity‐dependent activation of dendritic IRE1 and subsequent accumulation of XBP1s into the nucleus. In the CNS, the excitatory synaptic activation as a result of glutamate exposure has the potential to fine‐tune synaptic plasticity by the induction of LTP and long‐term depression (Malenka and Bear 2004; Nicoll and Schmitz 2005). Therefore, we speculated that the excitatory synaptic activation‐induced up‐regulation of IRE1‐XBP1 signaling might connect excitatory synaptic activities with modulation of synaptic plasticity and neuronal functions via the direct up‐regulation of Bdnf. A putative XBP1s consensus‐binding site, the UPRE core, is included in the Bdnf promoter region (Acosta‐Alvear et al. 2007; Martinez et al. 2016). The UPRE core is located 108 base pairs (bp) upstream of the Bdnf transcription start site and is conserved in mouse, rat and human genomes (Fig. 2d). To determine whether glutamate‐induced XBP1s binds to the Bdnf promoter and activates Bdnf transcription, we performed promoter assays using a reporter gene carrying a 0.6‐kbp promoter region of Bdnf (BDNF‐Luc). In Neuro‐2A cells (murine neuroblastoma cell line) transfected with a BDNF‐Luc construct, luciferase reporter activities were induced by the treatment with glutamate (Fig. 2e). In contrast, the induction of the reporter activities was reduced in glutamate‐stimulated Neuro‐2A cells transfected with ΔBDNF‐Luc lacking the UPRE core (Fig. 2d and e). Moreover, we carried out ChIP assays and detected robust binding of XBP1s to the endogenous Bdnf promoter in hippocampal neurons treated with glutamate (Fig. 2f–h). The high level of XBP1s binding was diminished by the knockdown of Ire1 or Xbp1. These findings suggest that glutamate‐induced IRE1 activation followed by the increase in XBP1s leads to binding of XBP1s to the UPRE core within the Bdnf promoter and facilitates Bdnf transcription in neurons. The signaling cascade may mediate the regulation of synaptic plasticity and neuronal functions modulated by excitatory synaptic activation.
BDNF activates the IRE1‐XBP1 pathway in a PKA‐dependent manner
BDNF is known to drive its own expression during the relevant processes in memory consolidation (Minichiello 2009). Our data and the previous report let us hypothesize that BDNF drives a positive feedback loop mediated by the IRE1‐XBP1 pathway to acquire the substantial effects on the regulation of neuronal functions. Thus, we examined whether BDNF activates the IRE1‐XBP1 pathway in hippocampal neurons. Treatment of hippocampal neurons with BDNF induced IRE1 phosphorylation and Xbp1 mRNA splicing, leading to the generation of Xbp1s mRNA (Fig. 3a–c). In turn, increases of phosphorylated eIF2α and Bip mRNA were not detected in these cells. These results indicate that stimulation with BDNF specifically activates the IRE1‐XBP1 pathway without the induction of the other canonical UPR branches, consistent with those of the previous report (Martinez et al. 2016). Further, we attempted to elucidate the molecular mechanisms for the induction of IRE1‐XBP1 pathway that are distinct from the general activation of UPR signaling. BDNF induces diverse signaling modules following binding to its receptor, TrkB (Sandhya et al. 2013). The downstream signaling cascades evoked by BDNF‐TrkB binding include activation of PKA signaling, which maintains synaptic plasticity (Thakker‐Varia et al. 2001). Previous studies have shown that PKA directly binds to the cytosolic site of IRE1, subsequently leading to IRE1 phosphorylation, distinct from the luminal events of ER including ER stress (Mao et al. 2011). PKA‐mediated IRE1 phosphorylation can promote Xbp1 mRNA splicing and the generation of XBP1s protein (Asada et al. 2015). Hence, we speculated that the mechanism by which BDNF up‐regulates IRE1‐XBP1 signaling, but not the other UPR branches, is via PKA activation. To test our hypothesis, we evaluated the induction of PKA signaling after the treatment of hippocampal neurons with BDNF. The phosphorylation levels of p38 MAPK and CREB, both of which are phosphorylated by PKA (Delghandi et al. 2005), were elevated in hippocampal neurons exposed to BDNF (Fig. 3a and b). Treatment of hippocampal neurons with PKA inhibitor H89 attenuated the BDNF‐induced phosphorylation of these proteins (Fig. 3d and e). BDNF‐induced IRE1 phosphorylation and Xbp1 mRNA splicing were also suppressed in the cells treated with H89 (Fig. 3d–f), indicating that BDNF up‐regulates the IRE1‐XBP1 pathway through PKA activation. These mechanisms do not affect the other canonical UPR branches, such as the PERK and ATF6 pathways.
Figure 3.
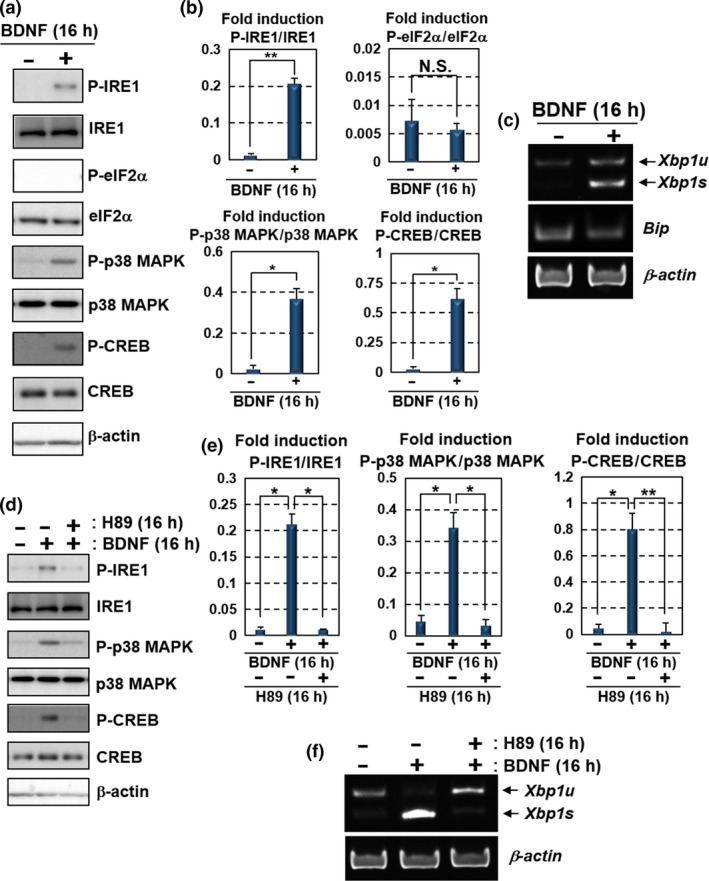
BDNF up‐regulates the IRE1‐XBP1 pathway via PKA activation. Primary cultured mouse hippocampal neurons maintained for 10 days were used for the analyses. (a) Western blot analysis of P‐IRE1, IRE1, P‐eIF2α, eIF2α, phosphorylated p38 MAPK (P‐p38 MAPK), p38 MAPK, phosphorylated CREB (P‐CREB) and CREB. Cells were treated with 100 ng/mL BDNF for 16 h. IRE1, p38 MAPK and CREB, but not eIF2α, were phosphorylated after the BDNF treatment. (b) Quantification of relative protein levels of P‐IRE1, P‐eIF2α, P‐p38 MAPK and P‐CREB in (a) (mean ± SD, N = 3; number of extracted samples prepared from independent cultures; *p < 0.05, **p < 0.01). (c) RT‐PCR analysis of Xbp1 and Bip mRNA. Xbp1s was increased by the treatment with 100 ng/mL BDNF for 16 h, but not Bip. (d) Western blot analysis of P‐IRE1, IRE1, P‐p38 MAPK, p38 MAPK, P‐CREB and CREB. The cells were treated with 100 ng/mL BDNF and 30 μM H89 (PKA inhibitor) for 16 h. The phosphorylation levels of IRE1, p38 MAPK and CREB induced by the treatment with BDNF were attenuated by treatment with H89. (e) Quantification of relative protein levels of P‐IRE1, P‐p38 MAPK and P‐CREB in (d) (mean ± SD, N = 3; number of extracted samples prepared from independent cultures; *p < 0.05, **p < 0.01). (f) RT‐PCR analysis of Xbp1 mRNA. The acceleration of Xbp1 splicing by the treatment with 100 ng/mL BDNF was inhibited by the treatment with 30 μM H89 for 16 h.
BDNF drives its own expression via activation of the PKA‐IRE1‐XBP1 cascade in dendrites
We further examined the activation of dendritic IRE1 in response to BDNF exposure by performing immunofluorescence staining analysis in hippocampal neurons. The immunoreactivities of IRE1 were observed at MAP2‐positive neurites and PSD95‐positive postsynaptic sites under normal condition (Fig. 4a). Those of the localization and the level were not significantly changed by the treatment of hippocampal neurons with BDNF or H89. Phosphorylated IRE1 were increased in dendrites of the neurons treated with BDNF compared with non‐treated cells (Fig. 4b). The signals were strongly detected in MAP2‐positive neurites (Fig. 4c). However, PSD95‐positive postsynaptic sites did not show substantial induction of phosphorylated IRE1 after the BDNF treatment, in contrast with glutamate treatment (Figs 1e, g and 4d). The different distributions of phosphorylated IRE1 found between the two treatments implicate that BDNF and glutamate have distinct mechanisms of IRE1 activation. BDNF‐induced phosphorylation of dendritic IRE1 was reduced by the H89 treatment, suggesting that BDNF promotes the phosphorylation of dendritic IRE1 via PKA activation. We tested whether BDNF enhances its own transcriptional activation through the dendritic activation of IRE1‐XBP1 signaling by performing promoter assays. Neuro‐2A cells transfected with a BDNF‐Luc construct and treated with BDNF showed the induction of luciferase reporter activities (Fig. 4e). The BDNF‐induced reporter activities were down‐regulated by treatment with H89. We found the binding of XBP1s to Bdnf promoter in hippocampal neurons treated with BDNF by performing ChIP assay (Fig. 4f and g). Binding was abolished by H89 treatment, and by Ire1 or Xbp1 knockdown. Taken together, BDNF drives the positive feedback loop via the PKA‐dependent activation of dendritic IRE1‐XBP1 signaling in hippocampal neurons. Synergistic augmentation of BDNF signaling may efficiently provide the inputs to neighboring dendrites in autocrine and paracrine manners to simultaneously regulate the functions and activities of multiple neurons.
Figure 4.
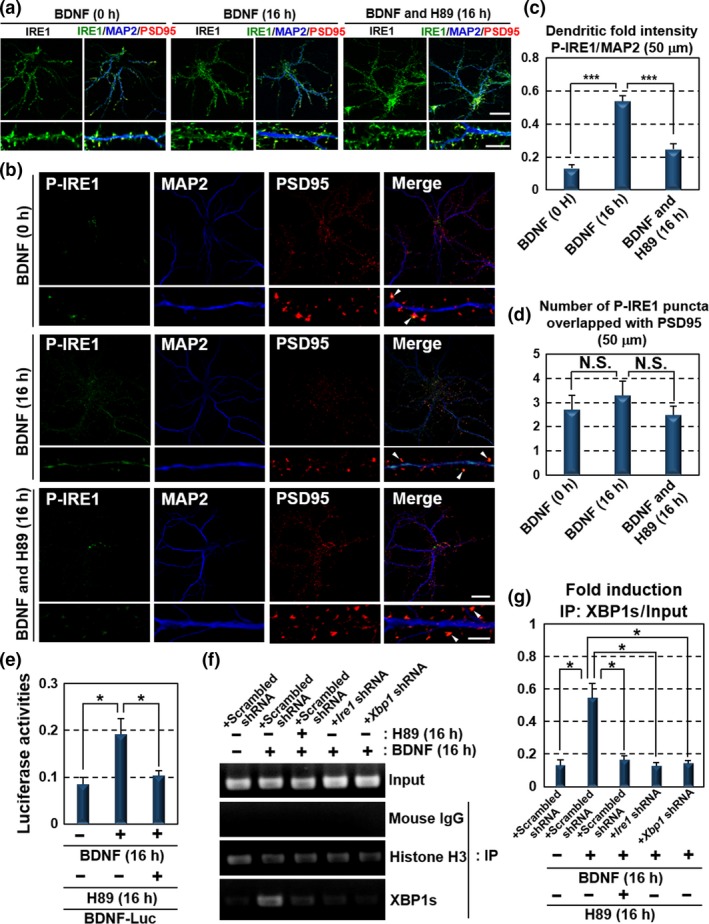
BDNF drives its own expression through dendritic activation of the PKA‐IRE1‐XBP1 pathway. (a) Immunofluorescence staining analysis of IRE1 (green), MAP2 (blue) and PSD95 (red) in primary cultured hippocampal neurons maintained for 10 days. The localization and level of IRE1 were not significantly changed by the exposure to 100 ng/mL BDNF or 30 μM H89 for 16 h. The lower panels show high magnification of the dendrites shown in the upper panels (Bar in upper panels: 20 μm, in lower panels: 5 μm). (b) Immunofluorescence staining analysis of P‐IRE1 (green), MAP2 (blue) and PSD95 (red) in primary cultured hippocampal neurons maintained for 10 days. IRE1 was phosphorylated at MAP2‐positive dendrites following exposure to 100 ng/mL BDNF for 16 h. In contrast, phosphorylation level of IRE1 in PSD95‐positive postsynaptic sites was decreased compared with those of the glutamate treatment presented in Fig. 1(e). BDNF‐induced IRE1 phosphorylation was reduced by treatment with 30 μM H89 for 16 h. The lower panels show high magnification of the dendrites shown in the upper panels. Arrow heads indicate immunoreactivities of P‐IRE1 overlapped with those of PSD95. (Bar in upper panels: 20 μm, in lower panels: 5 μm). (c) P‐IRE1 fluorescence intensities 40–90 μm from the somata of the dendrites in (b). The fluorescence intensities in these dendrites were increased by BDNF treatment (mean ± SD, N = 10; number of cells prepared from five independent cultures; ***p < 0.001). (d) The number of P‐IRE1‐positive puncta overlapping with PSD95‐positive postsynaptic sites 40–90 μm from the somata in (b). The number of P‐IRE1‐positive postsynaptic sites was not significantly increased by BDNF exposure (mean ± SD, N = 10; number of cells prepared from five independent cultures). (e) Reporter assays using Neuro‐2A cells. Cells transfected with the BDNF‐Luc constructs shown in Fig. 2(d) were treated with 100 ng/mL BDNF and 30 μM H89 for 16 h. Reporter activities induced by BDNF were inhibited by H89 treatment (mean ± SD, N = 6; number of samples prepared from independent cultures; *p < 0.05). (f) ChIP assay using primary cultured hippocampal neurons maintained for 10 days. Cells were infected with lentiviruses expressing scrambled, Ire1 or Xbp1 shRNA, with GFP for 5 days, and treated with 100 ng/mL BDNF and 30 μM H89 for 16 h. The primer set used for PCR amplification is shown in Fig. 2(f). Chromatin immunoprecipitation was performed using the indicated antibodies and mouse IgG (negative control). (g) Quantitative analysis of PCR amplification after immunoprecipitation by anti‐XBP1s antibody in (f) (mean ± SD, N = 3; number of samples prepared from independent cultures; *p < 0.05).
IRE1‐XBP1‐BDNF signaling promotes branching and expansion of dendrites
Next, we investigated how BDNF signaling, synergistically enhanced by its positive feedback loop, affects neuronal functions and homeostasis. In addition to the regulation of synaptic plasticity, BDNF modulates neurite outgrowth (Lin et al. 2006; Miyamoto et al. 2006; Hayashi et al. 2007, 2008; Martinez et al. 2016). These phenomena are essential for construction of the neural network. Moreover, previous researchers have shown the involvement of synaptic activities in neurite growth (Mazer et al. 1997; Zachor et al. 2000; Bolsover 2005). These reports suggest that BDNF signaling, initiated by the induction of synaptic activities, may regulate neurite development via the IRE1‐XBP1 signal‐dependent positive feedback loop. To verify the dendritic extension induced by BDNF and its positive feedback loop, hippocampal neurons were treated with BDNF and H89 for 4 and 3 days, respectively, and monitored the extension of dendrites by double‐blind analysis using a total of 25 neurons from five independent cultures treated with each agent (Fig. 5a and b). The sufficient expansion of dendrites stained with anti‐MAP2 antibody was observed in neurons treated with BDNF. Treatment with H89 suppressed BDNF‐induced dendritic development. The dendritic expansion was also inhibited in Ire1 and Xbp1 knockdown cells. To analyze the dendritic elongation, the length of the longest MAP2‐positive neurite in randomly selected cells was measured after the immunofluorescence staining (Fig. 5c). Dendritic extension was promoted in hippocampal neurons exposed to BDNF for 4 days. However, elongation was abrogated by treatment with H89 for 3 days. Hippocampal neurons infected with lentiviruses expressing Ire1 or Xbp1 shRNA also showed reduction in dendritic elongation following exposure to BDNF. Additionally, we counted the number of intersections between dendrites and specific distances from the soma to appraise dendritic branching (Fig. 5d and e). BDNF stimulation for 4 days encouraged dendritic branching. The dendritic branching promoted by BDNF was reduced by H89 treatment for 3 days. Hippocampal neurons infected with lentiviruses expressing Ire1 or Xbp1 shRNA also displayed fewer dendritic branches. In conclusion, the synergistic augmentation of BDNF signaling through the PKA‐IRE1‐XBP1 cascade is critical for substantial outgrowth and branching of dendrites.
Figure 5.
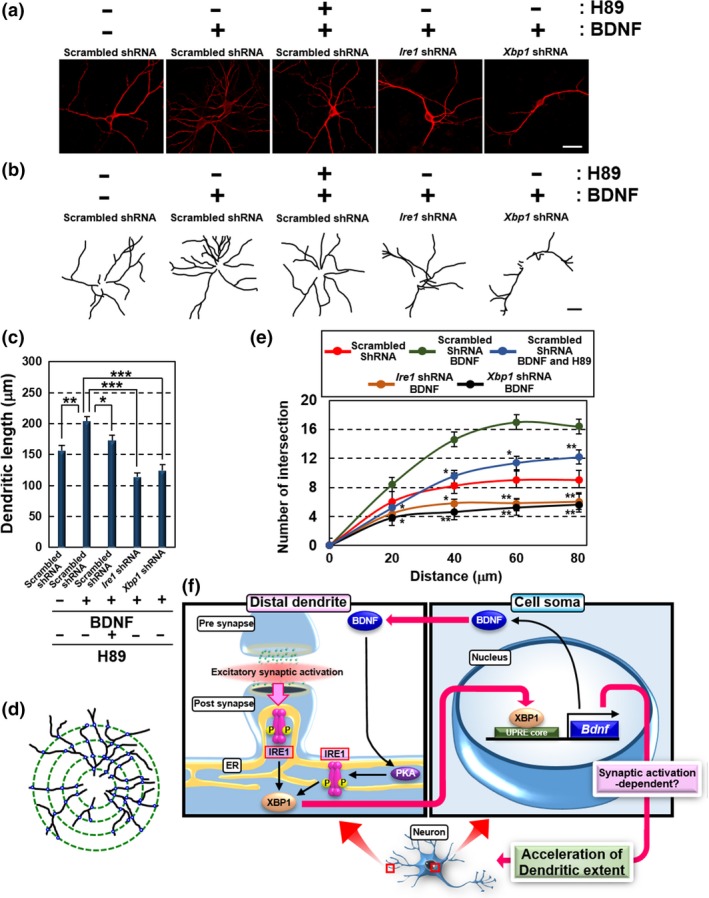
Treatment of exogenous BDNF promotes branching and elongation of dendrites via activation of PKA‐IRE1‐XBP1 cascade. (a) Immunofluorescence staining analysis of MAP2 in primary cultured hippocampal neurons maintained for 10 days. Cells were infected with lentiviruses expressing Ire1 or Xbp1 shRNA, with GFP for 5 days, and treated with either 100 ng/mL BDNF for 4 days or 10 μM H89 for 3 days (Bar: 20 μm). (b) Morphology of MAP2‐positive dendrites in (a). Dendrites were traced with black lines using ImageJ to clearly identify extension and branching (Bar: 20 μm). (c) Measurement of MAP2‐positive dendritic lengths in (a). The lengths of the longest dendrites were measured using ImageJ (mean ± SD, N = 25; number of cells prepared from five independent cultures; *p < 0.05, **p < 0.01, ***p < 0.001). (d) Schema of the method for evaluating MAP2‐positive dendritic branching in (a). Black lines indicate dendrites. Green dotted circles indicate the distance from the center of the soma (20, 40, 60 and 80 μm). Small blue circles indicate the intersections between dendrites and each distance from the soma. (e) The number of intersections between MAP2‐positive dendrites and circles on each distance from the soma in (a). The method for counting the number of intersections is shown in (d) (mean ± SD, N = 25; number of cells prepared from five independent cultures; *p < 0.05, **p < 0.01). (f) Putative model of the IRE1‐XBP1‐BDNF cascade up‐regulated by excitatory synaptic activation. Dendritic IRE1 is phosphorylated at postsynaptic sites in response to excitatory synaptic activation, followed by accumulation of XBP1s into the nucleus. XBP1s promotes transcriptional activation of Bdnf. BDNF in turn drives its own expression via PKA‐dependent activation of dendritic IRE1‐XBP1 signaling. Synaptic activity‐ and BDNF‐dependent distinct activation of the dendritic IRE1‐XBP1 cascade may comprehensively regulate dendritic extension through the expression of BDNF.
Discussion
Neurons have unique polarized morphologies, with an extensive ER network that is distributed from the soma to distal sites of neurites (Tsukita and Ishikawa 1976; Broadwell and Cataldo 1983), inferring the strong relevance among ER functions, communication over the distance, and the local regulation of neuronal activities. In terms of pathogenesis, it is conceivable that ER disruption can trigger neuronal dysfunction and enhance neurodegeneration. Indeed, a lot of previous studies have revealed that ER‐derived signaling involving UPR pathways protects neurons against abnormal events such as excitotoxicity and ischemic insults (Kitao et al. 2001; Urban et al. 2009). Disturbances to ER‐derived signaling result in neuronal death and the pathogenesis of neurodegenerative diseases (Shah et al. 2015). On the other hand, recent researches have uncovered the physiological roles of UPR in maintaining neuronal activities and functions under normal conditions (Trinh et al. 2014; Martinez et al. 2016). In this study, we demonstrated that (i) glutamate‐induced excitatory synaptic activation transiently activates UPR including the IRE1‐XBP1 branch at postsynaptic sites, (ii) XBP1s generated by glutamate stimulation translocates to the nucleus and directly facilitates the BDNF expression, (iii) BDNF in turn up‐regulates dendritic IRE1‐XBP1 signaling via PKA activation, subsequently enhancing its own expression without induction of the other UPR branches, and (iv) synergistic augmentation of BDNF signaling via a positive feedback loop through the PKA‐IRE1‐XBP1 cascade promotes dendritic elongation and branching. Our findings reveal the novel molecular mechanisms for neuronal activity‐dependent UPR activation at local sites to promote the expression of BDNF in cell soma, and for BDNF positive feedback loop through the PKA‐IRE1‐XBP1 cascade (Fig. 5f). The differential activation of dendritic IRE1 that depends on the distinct signals may provide the diversity of dendritic ER‐derived signaling to control neuronal activities. Simultaneously, the distinct activation mechanisms of local IRE1 may contribute to synergistic and comprehensive signal transduction for regulation of neuronal functions including extension of dendrites through the expression of BDNF.
We demonstrated that exogenous treatment with BDNF drives its own positive feedback loop, resulting in the acceleration of dendritic extension and branching. These results imply that synaptic activity‐dependent activation of the dendritic IRE1‐XBP1 pathway may promote dendritic extension through BDNF expression. To validate the link between excitatory synaptic activation‐induced BDNF signaling and dendritic extension, we tried to investigate the effect of glutamate on dendritic outgrowth. However, neurons must be maintained over a few days to monitor dendritic elongation. Prolonged exposure to glutamate results in neuronal cell death (Ogura et al. 1988). Therefore, we were unable to clarify whether postsynaptic activities directly affect dendritic outgrowth through the IRE1‐XBP1‐BDNF cascade by using our current experimental system. In addition to the excitotoxicity, the method of bath application of glutamate to primary cultured neurons that we adopted, is not equivalent to synaptic stimulation. The bath application of glutamate to primary cultures may cause mixed effects, because exogenous glutamate can induce glutamatergic excitation of other cells, resulting in altered neuronal activities. In future, different technical approaches for the acceleration of spontaneous excitatory synaptic activities or local stimulation of particular neuron with neurotransmitters will be required to avoid prolonged neuronal excitotoxicity and side effects. The establishment of such methods will also enable us to evaluate the effect of excitatory synaptic activation‐induced acceleration of the other UPR branches, namely the PERK and ATF6 pathways, on dendritic growth.
Dendritic IRE1 phosphorylation was up‐regulated by BDNF treatment via PKA, followed by the activation of the XBP1s as a transcription factor. Simultaneously, BDNF‐induced activation of PKA phosphorylated p38 MAPK. Phosphorylated p38 MAPK has been reported to drive the stabilization and translocation of XBP1s from the cytosol into the nucleus through phosphorylation of XBP1s (Lee et al. 2011). Therefore, phosphorylation of p38 MAPK following BDNF exposure may increase XBP1s stability and facilitate its translocation into the nucleus. BDNF‐PKA signaling may accelerate XBP1s accumulation in the nucleus via dual pathways; IRE1 phosphorylation and the increase in XBP1s, and p38 MAPK‐mediated phosphorylation and stabilization of XBP1s.
In this study, we adopted H89 as a PKA inhibitor, which has been used to block the activation of PKA in previous studies (Mao et al. 2011; Asada et al. 2015). However, a previous report suggested that H89 can also inhibit other kinase activities including ribosomal protein S6 kinase 1 (S6K1) and Rho‐associated protein kinase II (ROCKII) (Davies et al. 2000). Although expression of the constitutively active form of S6K1 (S6K1T389E) is not sufficient to induce dendritic growth, mTOR‐activated S6K1 may contribute to dendritogenesis (Malik et al. 2013; Koscielny et al. 2017). ROCKII regulates the actin cytoskeleton in the Rho‐mediated signaling pathway (Olson 2004). The treatment of mouse hippocampal neurons with ROCKII inhibitor enhances abnormal elongation of spine necks, which may have an effect on synaptic stability (Iida et al. 2007; Sfakianos et al. 2007). These previous studies suggest that treatment with H89 may influence dendritogenesis and synaptic activity through the inhibition of other kinases including S6KI and ROCKII. We could not fully exclude the possibility that the treatment of hippocampal neurons with H89 may affect dendritic outgrowth and synaptic activity by inhibiting S6K1 and ROCKII. Further studies are necessary to completely clarify the relevance of the BDNF‐induced PKA‐IRE1‐XBP1 cascade to extension of dendrites by specifically inhibiting PKA activation.
We demonstrated that IRE1‐XBP1 signaling, but not the other UPR branches, was activated by the treatment of neurons with BDNF. On contrary, another report suggests the increase in eIF2α phosphorylation, downstream of PERK, after BDNF exposure (Hayashi et al. 2007). To address this discrepancy, we reviewed the differences in cell culture conditions. Hayashi et al. reported transient induction of eIF2α phosphorylation by BDNF in hippocampal neurons cultured for 4 days. In contrast, we maintained hippocampal neurons for 10 days prior to the BDNF exposure. Because neurons exhibit exponential development during the culture period, they rapidly transit through developmental stages. Therefore, the developmental stages of the hippocampal neurons we prepared may be different from those of the neurons prepared in the previous study. The activation mechanisms and downstream cascade of UPR signaling may be different between the distinct stages of neuronal development.
We observed the increase in postsynaptic phosphorylation of IRE1 within 1.5 h after synaptic activation, indicating direct sensing of altered postsynaptic activities and rapid signal transduction. However, the activation mechanisms for UPR signaling at postsynaptic sites following postsynaptic activation remain unclear. Transient calcium influx from extracellular spaces through the ionotropic glutamate receptors up‐regulates downstream signaling cascades via phosphorylation of calcium/calmodulin dependent protein kinase α (Saneyoshi et al. 2010). Additionally, the input of the excitatory synaptic activity via metabolic glutamate receptor can promote calcium ion release from the ER lumen through the activation of the ER‐resident inositol 1, 4, 5‐phosphate receptor (Okubo et al. 2015). It is well‐known that a change in calcium ion homeostasis in the ER lumen can induce ER stress, leading to the activation of UPR signaling pathways (Kaufman 2002). Hence, depletion of calcium ions in the dendritic ER lumen caused by excitatory synaptic activation may trigger the up‐regulation of UPR signaling. We detected the activation of general UPR pathways by the application of glutamate, but not BDNF. Since BDNF cannot directly affect excitatory synaptic activities, calcium ion concentrations in the postsynaptic ER lumen may not change. Therefore, canonical UPR branches may not be activated by BDNF exposure, except for BDNF‐PKA signaling‐mediated IRE1‐XBP1 pathway. Furthermore, IRE1 phosphorylation in postsynaptic sites was barely detectable following BDNF exposure. In contrast, BDNF stimulation facilitated IRE1 phosphorylation at MAP2‐positive neurites (Fig. 4a–c), which was different from those of glutamate stimulation. These findings indicate the differences in molecular mechanisms for activating IRE1‐XBP1 signaling between treatment with glutamate and BDNF. Synaptic activity‐responsive neuronal functions may be fine‐tuned via distinct glutamate‐ or BDNF‐induced regulatory mechanisms underlying the IRE1‐XBP1 pathway.
In conclusion, we have demonstrated regulation of neuronal activities through UPR signaling over the distance. Synaptic activity‐dependent upregulation of dendritic IRE1‐XBP1 signaling is retrogradely propagated toward the soma. XBP1s translocates to the nucleus to induce transcriptional activation of Bdnf. Exogenous treatment of BDNF in turn drives its own expression via the PKA‐IRE1‐XBP1 signaling cascade, resulting in the acceleration of extension and branching of dendrites. Our present study implies that the dichotomy of dendritic IRE1 activation that depends on the synaptic activity and BDNF may induce the synergistic and comprehensive signal transduction for the regulation of dendritic extension through the expression of BDNF. These findings and prospective possibilities provide a novel insight into the machineries for communication between the neuronal soma and distal dendrites to manipulate neuronal homeostasis. Further studies should uncover the broad roles of local activation of ER‐derived UPR signaling in regulating neuronal activities and functions.
Acknowledgments and conflict of interest disclosure
This work was partly supported by grants from the Japan Society for the Promotion of Science KAKENHI (17H01424) (Japan) and The Sumitomo Electric Industries Group Corporate Social Responsibility Foundation (Japan). All authors declare no competing financial interests within this article.
All experiments were conducted in compliance with the ARRIVE guidelines.
Cover Image for this issue: doi. 10.1111/jnc.14159.
Contributor Information
Atsushi Saito, Email: saitoa@hiroshima-u.ac.jp.
Kazunori Imaizumi, Email: imaizumi@hiroshima-u.ac.jp.
References
- Acosta‐Alvear D., Zhou Y., Blais A., Tsikitis M., Lents N. H., Arias C., Lennon C. J., Kluger Y. and Dynlacht B. D. (2007) XBP1 controls diverse cell type‐ and condition‐specific transcriptional regulatory networks. Mol. Cell 27, 53–66. [DOI] [PubMed] [Google Scholar]
- Asada R., Kanemoto S., Matsuhisa K., Hino K., Cui M., Cui X., Kaneko M. and Imaizumi K. (2015) IRE1alpha‐XBP1 is a novel branch in the transcriptional regulation of Ucp1 in brown adipocytes. Sci. Rep. 5, 16580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde Y. A., Edgar D. and Thoenen H. (1982) Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder D. K. and Scharfman H. E. (2004) Brain‐derived neurotrophic factor. Growth Factors 22, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolsover S. R. (2005) Calcium signalling in growth cone migration. Cell Calcium 37, 395–402. [DOI] [PubMed] [Google Scholar]
- Broadwell R. D. and Cataldo A. M. (1983) The neuronal endoplasmic reticulum: its cytochemistry and contribution to the endomembrane system. I. Cell bodies and dendrites. J. Histochem. Cytochem. 31, 1077–1088. [DOI] [PubMed] [Google Scholar]
- Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G. and Ron D. (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP‐1 mRNA. Nature 415, 92–96. [DOI] [PubMed] [Google Scholar]
- Chen G., Kolbeck R., Barde Y. A., Bonhoeffer T. and Kossel A. (1999) Relative contribution of endogenous neurotrophins in hippocampal long‐term potentiation. J. Neurosci. 19, 7983–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A. and Malenka R. C. (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33, 18–41. [DOI] [PubMed] [Google Scholar]
- Davies S. P., Reddy H., Caivano M. and Cohen P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delghandi M. P., Johannessen M. and Moens U. (2005) The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell. Signal. 17, 1343–1351. [DOI] [PubMed] [Google Scholar]
- Gulledge A. T., Kampa B. M. and Stuart G. J. (2005) Synaptic integration in dendritic trees. J. Neurobiol. 64, 75–90. [DOI] [PubMed] [Google Scholar]
- Harding H. P., Zhang Y. and Ron D. (1999) Protein translation and folding are coupled by an endoplasmic‐reticulum‐resident kinase. Nature 397, 271–274. [DOI] [PubMed] [Google Scholar]
- Harding H. P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M. and Ron D. (2000) Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Hayashi A., Kasahara T., Iwamoto K., Ishiwata M., Kametani M., Kakiuchi C., Furuichi T. and Kato T. (2007) The role of brain‐derived neurotrophic factor (BDNF)‐induced XBP1 splicing during brain development. J. Biol. Chem. 282, 34525–34534. [DOI] [PubMed] [Google Scholar]
- Hayashi A., Kasahara T., Kametani M. and Kato T. (2008) Attenuated BDNF‐induced upregulation of GABAergic markers in neurons lacking Xbp1. Biochem. Biophys. Res. Comm. 376, 758–763. [DOI] [PubMed] [Google Scholar]
- Headley P. M. and Grillner S. (1990) Excitatory amino acids and synaptic transmission: the evidence for a physiological function. Trends Pharmacol. Sci. 11, 205–211. [DOI] [PubMed] [Google Scholar]
- Hetz C., Thielen P., Matus S. et al (2009) XBP‐1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Park K. K., Yang L. et al (2012) Differential effects of unfolded protein response pathways on axon injury‐induced death of retinal ganglion cells. Neuron 73, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida J., Ishizaki H., Okamoto‐Tanaka M. et al (2007) Synaptic scaffolding molecule alpha is a scaffold to mediate N‐methyl‐D‐aspartate receptor‐dependent RhoA activation in dendrites. Mol. Cell. Biol. 27, 4388–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakiuchi C., Iwamoto K., Ishiwata M. et al (2003) Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat. Genet. 35, 171–175. [DOI] [PubMed] [Google Scholar]
- Kakiuchi C., Ishiwata M., Umekage T., Tochigi M., Kohda K., Sasaki T. and Kato T. (2004) Association of the XBP1‐116C/G polymorphism with schizophrenia in the Japanese population. Psychiatry Clin. Neurosci. 58, 438–440. [DOI] [PubMed] [Google Scholar]
- Kaufman R. J. (2002) Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 110, 1389–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao Y., Ozawa K., Miyazaki M. et al (2001) Expression of the endoplasmic reticulum molecular chaperone (ORP150) rescues hippocampal neurons from glutamate toxicity. J. Clin. Investig. 108, 1439–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S., Murakami T., Tatsumi K., Ogata M., Kanemoto S., Otori K., Iseki K., Wanaka A. and Imaizumi K. (2005) OASIS, a CREB/ATF‐family member, modulates UPR signalling in astrocytes. Nat. Cell Biol. 7, 186–194. [DOI] [PubMed] [Google Scholar]
- Korte M., Carroll P., Wolf E., Brem G., Thoenen H. and Bonhoeffer T. (1995) Hippocampal long‐term potentiation is impaired in mice lacking brain‐derived neurotrophic factor. Proc. Natl Acad. Sci. USA 92, 8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koscielny A., Malik A. R., Liszewska E., Zmorzynska J., Tempes A., Tarkowski B. and Jaworski J. (2017) Adaptor complex 2 controls dendrite morphology via mTOR‐dependent expression of GluA2. Mol. Neurobiol. doi: 10.1007/s12035‐017‐0436‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Sun C., Zhou Y., Lee J., Gokalp D., Herrema H., Park S. W., Davis R. J. and Ozcan U. (2011) p38 MAPK‐mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 17, 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibrock J., Lottspeich F., Hohn A., Hofer M., Hengerer B., Masiakowski P., Thoenen H. and Barde Y. A. (1989) Molecular cloning and expression of brain‐derived neurotrophic actor. Nature 341, 149–152. [DOI] [PubMed] [Google Scholar]
- Lessmann V., Gottmann K. and Heumann R. (1994) BDNF and NT‐4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport 6, 21–25. [DOI] [PubMed] [Google Scholar]
- Li M., Baumeister P., Roy B., Phan T., Foti D., Luo S. and Lee A. S. (2000) ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress‐induced changes and synergistic interactions with NF‐Y and YY1. Mol. Cell. Biol. 20, 5096–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G., Bella A. J., Lue T. F. and Lin C. S. (2006) Brain‐derived neurotrophic factor (BDNF) acts primarily via the JAK/STAT pathway to promote neurite growth in the major pelvic ganglion of the rat: part 2. J. Sex. Med. 3, 821–827; discussion 828–829. [DOI] [PubMed] [Google Scholar]
- Liu S. Y., Wang W., Cai Z. Y., Yao L. F., Chen Z. W., Wang C. Y., Zhao B. and Li K. S. (2013) Polymorphism ‐116C/G of human X‐box‐binding protein 1 promoter is associated with risk of Alzheimer's disease. CNS Neurosci. Ther. 19, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohof A. M., Ip N. Y. and Poo M. M. (1993) Potentiation of developing neuromuscular synapses by the neurotrophins NT‐3 and BDNF. Nature 363, 350–353. [DOI] [PubMed] [Google Scholar]
- Lommatzsch M., Braun A., Mannsfeldt A., Botchkarev V. A., Botchkareva N. V., Paus R., Fischer A., Lewin G. R. and Renz H. (1999) Abundant production of brain‐derived neurotrophic factor by adult visceral epithelia. Implications for paracrine and target‐derived Neurotrophic functions. Am. J. Pathol. 155, 1183–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Park H. and Poo M. M. (2014) Spike‐timing‐dependent BDNF secretion and synaptic plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F., Eisenberg T. and Kroemer G. (2009) Autophagy for the avoidance of neurodegeneration. Genes Dev. 23, 2253–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka R. C. and Bear M. F. (2004) LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. [DOI] [PubMed] [Google Scholar]
- Malik A. R., Urbanska M., Gozdz A., Swiech L. J., Nagalski A., Perycz M., Blazejczyk M. and Jaworski J. (2013) Cyr61, a matricellular protein, is needed for dendritic arborization of hippocampal neurons. J. Biol. Chem. 288, 8544–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao T., Shao M., Qiu Y. et al (2011) PKA phosphorylation couples hepatic inositol‐requiring enzyme 1alpha to glucagon signaling in glucose metabolism. Proc. Natl Acad. Sci. USA 108, 15852–15857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G., Vidal R. L., Mardones P. et al (2016) Regulation of Memory Formation by the Transcription Factor XBP1. Cell Rep. 14, 1382–1394. [DOI] [PubMed] [Google Scholar]
- Mazer C., Muneyyirci J., Taheny K., Raio N., Borella A. and Whitaker‐Azmitia P. (1997) Serotonin depletion during synaptogenesis leads to decreased synaptic density and learning deficits in the adult rat: a possible model of neurodevelopmental disorders with cognitive deficits. Brain Res. 760, 68–73. [DOI] [PubMed] [Google Scholar]
- Minichiello L. (2009) TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 10, 850–860. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y., Yamauchi J., Tanoue A., Wu C. and Mobley W. C. (2006) TrkB binds and tyrosine‐phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc. Natl Acad. Sci. USA 103, 10444–10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T., Hino S. I., Saito A. and Imaizumi K. (2007) Endoplasmic reticulum stress response in dendrites of cultured primary neurons. Neuroscience 146, 1–8. [DOI] [PubMed] [Google Scholar]
- Negro A., Tavella A., Grandi C. and Skaper S. D. (1994) Production and characterization of recombinant rat brain‐derived neurotrophic factor and neurotrophin‐3 from insect cells. J. Neurochem. 62, 471–478. [DOI] [PubMed] [Google Scholar]
- Ng D. T., Spear E. D. and Walter P. (2000) The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J. Cell Biol. 150, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D. and Attwell D. (1990) The release and uptake of excitatory amino acids. Trends Pharmacol. Sci. 11, 462–468. [DOI] [PubMed] [Google Scholar]
- Nicoll R. A. and Schmitz D. (2005) Synaptic plasticity at hippocampal mossy fibre synapses. Nat. Rev. Neurosci. 6, 863–876. [DOI] [PubMed] [Google Scholar]
- Ogura A., Miyamoto M. and Kudo Y. (1988) Neuronal death in vitro: parallelism between survivability of hippocampal neurones and sustained elevation of cytosolic Ca2 + after exposure to glutamate receptor agonist. Exp. Brain Res. 73, 447–458. [DOI] [PubMed] [Google Scholar]
- Okubo Y., Suzuki J., Kanemaru K., Nakamura N., Shibata T. and Iino M. (2015) Visualization of Ca2 + Filling Mechanisms upon Synaptic Inputs in the Endoplasmic Reticulum of Cerebellar Purkinje Cells. J. Neurosci. 35, 15837–15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M. F. (2004) Contraction reaction: mechanical regulation of Rho GTPase. Trends Cell Biol. 14, 111–114. [DOI] [PubMed] [Google Scholar]
- Park H. and Poo M. M. (2013) Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23. [DOI] [PubMed] [Google Scholar]
- Ron D. (2002) Translational control in the endoplasmic reticulum stress response. J. Clin. Investig. 110, 1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. and Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Rutkowski D. T. and Kaufman R. J. (2004) A trip to the ER: coping with stress. Trends Cell Biol. 14, 20–28. [DOI] [PubMed] [Google Scholar]
- Sandhya V. K., Raju R., Verma R. et al (2013) A network map of BDNF/TRKB and BDNF/p75NTR signaling system. J. Cell Commun. Signal. 7, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T., Fortin D. A. and Soderling T. R. (2010) Regulation of spine and synapse formation by activity‐dependent intracellular signaling pathways. Curr. Opin. Neurobiol. 20, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfakianos M. K., Eisman A., Gourley S. L. et al (2007) Inhibition of Rho via Arg and p190RhoGAP in the postnatal mouse hippocampus regulates dendritic spine maturation, synapse and dendrite stability, and behavior. J. Neurosci. 27, 10982–10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S. Z., Zhao D., Khan S. H. and Yang L. (2015) Unfolded protein response pathways in neurodegenerative diseases. J. Mol. Neurosci. 57, 529–537. [DOI] [PubMed] [Google Scholar]
- Spruston N. (2008) Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci. 9, 206–221. [DOI] [PubMed] [Google Scholar]
- Thakker‐Varia S., Alder J., Crozier R. A., Plummer M. R. and Black I. B. (2001) Rab3A is required for brain‐derived neurotrophic factor‐induced synaptic plasticity: transcriptional analysis at the population and single‐cell levels. J. Neurosci. 21, 6782–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W., Welihinda A. A. and Kaufman R. J. (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers K. J., Patil C. K., Wodicka L., Lockhart D. J., Weissman J. S. and Walter P. (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER‐associated degradation. Cell 101, 249–258. [DOI] [PubMed] [Google Scholar]
- Trinh M. A., Ma T., Kaphzan H., Bhattacharya A., Antion M. D., Cavener D. R., Hoeffer C. A. and Klann E. (2014) The eIF2alpha kinase PERK limits the expression of hippocampal metabotropic glutamate receptor‐dependent long‐term depression. Learn. Mem. 21, 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S. and Ishikawa H. (1976) Three‐dimensional distribution of smooth endoplasmic reticulum in myelinated axons. J. Electron Microsc. 25, 141–149. [PubMed] [Google Scholar]
- Urban P., Pavlikova M., Sivonova M., Kaplan P., Tatarkova Z., Kaminska B. and Lehotsky J. (2009) Molecular analysis of endoplasmic reticulum stress response after global forebrain ischemia/reperfusion in rats: effect of neuroprotectant simvastatin. Cell. Mol. Neurobiol. 29, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela V., Collyer E., Armentano D., Parsons G. B., Court F. A. and Hetz C. (2012) Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death Dis. 3, e272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Z., Harding H. P., Zhang Y., Jolicoeur E. M., Kuroda M. and Ron D. (1998) Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 17, 5708–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X., Howell A. S., Dong X., Taylor C. A., Cooper R. C., Zhang J., Zou W., Sherwood D. R. and Shen K. (2015) The unfolded protein response is required for dendrite morphogenesis. eLife 4, e06963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore C., Olson L. and Bean A. J. (1994) Regulation of brain‐derived neurotrophic factor (BDNF) expression and release from hippocampal neurons is mediated by non‐NMDA type glutamate receptors. J. Neurosci. 14, 1688–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Haze K., Yanagi H., Yura T. and Mori K. (1998) Identification of the cis‐acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose‐regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M. and Mori K. (2000) ATF6 activated by proteolysis binds in the presence of NF‐Y (CBF) directly to the cis‐acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20, 6755–6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Yamamoto A., Okada T. and Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Hosokawa N., Kaufman R. J., Nagata K. and Mori K. (2003) A time‐dependent phase shift in the mammalian unfolded protein response. Dev. Cell 4, 265–271. [DOI] [PubMed] [Google Scholar]
- Yu Z., Luo H., Fu W. and Mattson M. P. (1999) The endoplasmic reticulum stress‐responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp. Neurol. 155, 302–314. [DOI] [PubMed] [Google Scholar]
- Zachor D. A., Moore J. F., Brezausek C., Theibert A. and Percy A. K. (2000) Cocaine inhibits NGF‐induced PC12 cells differentiation through D(1)‐type dopamine receptors. Brain Res. 869, 85–97. [DOI] [PubMed] [Google Scholar]
- Zafra F., Castren E., Thoenen H. and Lindholm D. (1991) Interplay between glutamate and gamma‐aminobutyric acid transmitter systems in the physiological regulation of brain‐derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc. Natl Acad. Sci. USA 88, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]