

Abstract
Colorectal cancer results from the malignant transformation of colonic epithelial cells. Stromal fibroblasts are the main component of the tumour microenvironment, and play an important role in the progression of this and other neoplasias. Wnt/β‐catenin signalling is essential for colon homeostasis, but aberrant, constitutive activation of this pathway is a hallmark of colorectal cancer. Here we present the first transcriptomic study on the effect of a Wnt factor on human colonic myofibroblasts. Wnt3A regulates the expression of 1,136 genes, of which 662 are upregulated and 474 are downregulated in CCD‐18Co cells. A set of genes encoding inhibitors of the Wnt/β‐catenin pathway stand out among those induced by Wnt3A, which suggests that there is a feedback inhibitory mechanism. We also show that the PKP2 gene encoding the desmosomal protein Plakophilin‐2 is a novel direct transcriptional target of Wnt/β‐catenin in normal and colon cancer‐associated fibroblasts. PKP2 is induced by β‐catenin/TCF through three binding sites in the gene promoter and one additional binding site located in an enhancer 20 kb upstream from the transcription start site. Moreover, Plakophilin‐2 antagonizes Wnt/β‐catenin transcriptional activity in HEK‐293T cells, which suggests that it may act as an intracellular inhibitor of the Wnt/β‐catenin pathway. Our results demonstrate that stromal fibroblasts respond to canonical Wnt signalling and that Plakophilin‐2 plays a role in the feedback control of this effect suggesting that the response to Wnt factors in the stroma may modulate Wnt activity in the tumour cells.
Keywords: colon cancer, normal and cancer‐associated fibroblasts, Wnt/β‐catenin signalling, PKP2/Plakophilin‐2, gene regulation
Short abstract
What's new?
Cancer‐associated fibroblasts in the tumour microenvironment play a critical role in colorectal cancer (CRC) progression. In this transcriptomic study of human colon fibroblasts, profound changes in fibroblast gene expression profile were linked to Wnt3A signalling. Wnt3A signalling was associated particularly with the induction of inhibitors of the Wnt/β‐catenin pathway, indicative of an inhibitory feedback mechanism. Among the genes most strongly upregulated was PKP2, a novel transcriptional target of Wnt/β‐catenin signalling in both normal and colon cancer‐associated fibroblasts. Plakophilin‐2 protein, previously known to be widely expressed in carcinomas, was further found to antagonize Wnt/β‐catenin transcriptional activity.
Abbreviations
- CAF
cancer‐associated fibroblasts
- CMS
consensus molecular subtype
- CRC
colorectal cancer
- ECM
extracellular matrix
- EMT
epithelial‐mesenchymal transition
- NF
normal fibroblasts
- PCF
pericryptal fibroblasts
- S.D.
standard deviation
- TSS
transcription start site
Colorectal cancer (CRC) is one of the leading causes of cancer‐related death in Western countries.1, 2 CRC results from the malignant transformation of epithelial cells lining the luminal surface of the colonic crypt, a process driven by cumulative mutations in both oncogenes and tumour suppressor genes.3, 4 Aberrant activation of the Wnt/β‐catenin pathway is a hallmark of colorectal carcinoma and plays a key role in initiation and progression of the tumour.5 Nevertheless, it is now well‐accepted that the surrounding tumour stroma plays an essential role in CRC progression.6, 7, 8, 9 Therefore, understanding the reciprocal interaction between colon carcinoma cells and cancer‐associated fibroblasts (CAF) and other cell types in the tumour stroma is of key importance in the design of new, more efficient strategies against this neoplasia.
Tumour stroma is composed of extracellular matrix (ECM) proteins and a variety of cell types including endothelial cells, pericytes, infiltrating immune cells, adipocytes and CAF. A recent collaborative study has classified CRC into four consensus molecular subtypes (CMS). CMS4, the type with the worst prognosis, is characterized by strong transforming growth factor (TGF)‐β activation and high stromal component.8 Another study has revealed that the CMS4 subtype displays high expression of immune signatures with overexpression of markers of lymphocytes and cells of monocytic origin with strong angiogenic and inflammatory components.9 Moreover, most genes upregulated by TGF‐β in stem‐like/mesenchymal, poor prognosis CRC are expressed by stromal cells: CAF, leukocytes or endothelial cells.10, 11 These data highlight the relevance of tumour stroma for CRC progression.
Pericryptal fibroblasts (PCF) in healthy colon stroma secrete a variety of molecules, including Wnt factors, that modulate adjacent epithelial cell growth and differentiation.7 Secreted Wnt ligands bind their receptors located at the plasma membrane of stem cells and proliferative progenitors at the bottom of the colon crypt, and trigger a signalling cascade that regulates gene expression in the nucleus. The main effector of this pathway is β‐catenin. Wnt binding to Frizzled and low density lipoprotein (LDL) receptor‐related protein (LRP) 5/6 receptor heterodimers leads to phosphorylation of the cytoplasmic tail of LRP and to subsequent inhibition of a multiprotein complex that is responsible for β‐catenin degradation.5 The tumour suppressors and scaffold proteins APC (encoded by the adenomatous polyposis coli gene) and Axin, as well as the kinases Casein Kinase 1 (CK1) and Glycogen Synthase Kinase 3β (GSK3β) are the main components of this β‐catenin destruction complex. In the absence of Wnt ligands, CK1 and GSK3β catalyse β‐catenin N‐terminal phosphorylation which triggers β‐catenin ubiquitination and subsequent degradation by the proteasome. Inhibition of the β‐catenin destruction complex in response to Wnt signalling results in β‐catenin accumulation in the cytoplasm and its translocation into the nucleus, where it behaves as a transcriptional co‐activator for LEF/TCF transcription factors. The LEF/TCF family is composed of four members (TCF‐1 to −4) that bind to β‐catenin/TCF binding sites in promoters and enhancers of target genes and regulate their expression.5 Aberrant activation of the Wnt/β‐catenin pathway is thought to be the initial event and a driving force of colorectal tumorigenesis, and most human CRC carry mutations in genes that encode intracellular members of this pathway (including APC, CTNNB1/β‐catenin and AXIN1/2 genes).5
In spite of the abundant literature on Wnt/β‐catenin signalling in CRC and colon epithelial cells, studies are lacking on how Wnt signalling affects colon PCF. It is highly likely that Wnt factors secreted by PCF and possibly other crypt cell types bind not only Wnt receptors in crypt epithelial cells, but also those in PCF themselves, triggering a Wnt signalling cascade. Therefore, we studied the transcriptomic response to Wnt3A in established human normal colonic myofibroblasts (CCD‐18Co). To our knowledge, this is the first study that explores the transcriptomic effect of Wnt proteins on human colon myofibroblasts. Our analysis rendered a total of 1,136 regulated genes, of which 662 were upregulated and 474 were downregulated. The gene encoding the desmosomal protein Plakophilin‐2 (PKP2) was upregulated by Wnt3A in CCD‐18Co as well as in primary cultures of normal fibroblasts (NF) and CAF derived from CRC patients. Like β‐catenin, plakophilins are members of the armadillo family that are located both in the cytoplasm and in the nucleus.12 Here we show that PKP2 is a β‐catenin/TCF target gene whose expression is regulated through several β‐catenin/TCF binding sites present in its promoter and in an enhancer sequence located 20 kb upstream from its transcription start site (TSS). Moreover, our data suggest that Plakophilin‐2 may act as an antagonist of β‐catenin/TCF complexes on Wnt‐activated promoters.
Material and Methods
Cells and cell culture
CCD‐18Co (ATCC CRL‐1459) human colon myofibroblasts were purchased from the ATCC and cultured in Minimum Essential Medium (MEM, Life Technologies, Carlsbad, CA). IMR‐90 fibroblasts (ATCC CCL‐186), human embryonic kidney (HEK)‐293T cells, HeLa cells, and MCF7 breast cancer cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Life Technologies). All media were supplemented with 10% Foetal Bovine Serum (FBS, Life Technologies) and MEM also with l‐glutamine and sodium pyruvate (both from Life Technologies). Cell lines were periodically authenticated with the GenePrint 10 System (Promega, Fitchburg, WI) and the results were sent for comparison against the ATCC cell line database (Manassas, VA). Cells were treated with 200 µg/ml recombinant human Wnt3A (rhWnt3A, R&D Systems, Minneapolis, MN) in 0.1% phosphate buffer saline–bovine serum albumin (PBS‐BSA) to a final concentration of 100 ng/ml, or with the corresponding vehicle (PBS‐BSA) concentration.
Primary cultures of human colon NF and CAF were obtained following the explant outgrowth technique13, 14 from fresh surgical specimens of colon primary tumour and morphologically normal colonic mucosa (at least 5 cm from the surgical margin) of the same patient resected from CRC patients subjected to surgery at the Hospital Universitario La Paz (Madrid, Spain). Samples were provided by the IdiPAZ Biobank (PT13/0010/0003: Plataforma de Apoyo a la Investigación en Ciencias y Tecnologías de la Salud en la Red de Biobancos 2013), integrated into the Spanish Biobank Network (www.redbiobancos.es). Briefly, tissue samples were incubated for 30 min with PBS containing 0.5 mg/ml Primocin (Invivogen, San Diego, CA), 0.1 mg/ml gentamicin and 0.5 µg/ml amphotericin‐B (both from Life Technologies). Then, tissue samples were cut into small pieces of approximately 3 mm3 in size and seeded in cell culture flasks in FBS with 0.25 mg/ml Primocin. After 1 week and to facilitate fibroblast growth, FBS was replaced by Fibroblast Growth Medium‐2 (FGM‐2, Lonza, Basel, Switzerland). Fibroblasts grew around the explants for approximately 3 weeks. Then, tissue fragments were removed and fibroblasts were routinely subcultured at 1:2 ratio in FGM‐2. Experiments were performed with primary fibroblasts at the seventh passage at most. This study was approved by the Ethics Committee for Clinical Research of the Hospital Universitario La Paz (HULP‐PI‐1425), and all patients gave written informed consent.
RNA sequencing and analysis of gene expression
CCD‐18Co cells were stimulated with rhWnt3A or vehicle, and three biological replicates per sample were collected and sequenced on the Illumina HiSeq 2000 platform (Spanish National Cancer Research Centre, CNIO). Image analysis, per‐cycle basecalling and quality score assignment was performed with Illumina Real‐Time Analysis software. The Illumina2bam tool (Wellcome Trust Sanger Institute—NPG) was used to convert BCL files to bam format. To align fastq reads, we employed TopHat15 version 2.0.14 against hg19/GRCh37.75 assembly of the human reference genome,16 using standard parameters. Feature alignments were quantified using HTSeq‐counts from HTSeq framework version 0.6.0,17 using the GRCh37.75 GTF gene model. We used the edgeR package version 3.12.118 for R statistical computing software to normalize the count data, and statistical analysis to identify differentially expressed genes.
RNA isolation and RT‐qPCR
Total RNA was extracted from cells with the RNeasy mini kit (Qiagen, Hilden, Germany). Complementary DNA was synthesized from 0.5 μg of total RNA using the ImProm‐II Reverse Transcription System (Promega). Then, the qPCR reaction was performed in a CFX384 Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA), using TaqMan Universal Master Mix II without UNG (Applied Biosystems, Foster City, CA). Thermal cycling of the qPCR reaction was initiated with a denaturation step at 95°C for 10 min, and consisted of 40 cycles (denaturation at 95°C 15 sec, annealing and elongation at 60°C for 30 sec). We used TaqMan probes for PKP2 (Hs00428040_m1), PTGER3 (Hs00168755_m1), APCDD1 (Hs00537787_m1), DKK2 (Hs00205294_m1), NKD1 (Hs00263894_m1), NKD2 (Hs01108239_m1), WNT16 (Hs00365138_m1), TRPA1 (Hs00175798_m1), ODZ3 (Hs01111780_m1), ILDR2 (Hs01025498_m1), AMIGO2 (Hs00827141_g1), PKP1 (Hs00240873_m1), PKP3 (Hs00170887_m1), PKP4 (Hs00269305_m1), VIM (Hs00958111_m1) and ACTA2 (Hs00426835_g1) (Applied Biosystems). The expression levels were normalized to the levels of human ribosomal protein RPLPO (Taqman probe set 4310879E). All experiments were performed in triplicate.
Western blotting
Whole‐cell extracts were prepared by cell lysis with RIPA buffer plus protease‐ and phosphatase‐inhibitors for 20 min on ice, followed by centrifugation at 13,200 rpm for 15 min at 4°C. Proteins were separated by SDS‐PAGE, transferred to PVDF membranes and incubated with antibodies against Plakophilin‐2 (sc‐136039, Santa Cruz Biotechnology, Santa Cruz, CA; 651101, Progen, Heidelberg, Germany), β‐catenin (610154, BD Transduction Laboratories, San José, CA), GSK3β (610201, BD Transduction laboratories), Vimentin (M7020, clone V9, Dako), α‐SMA (A5228, clone 1A4, Sigma‐Aldrich), GAPDH (G9545, Sigma‐Aldrich) and β‐actin (sc‐1616, Santa Cruz Biotechnology), and then with HRP‐conjugated secondary antibodies. Antibody binding was visualized using the ECL detection system (GE Healthcare, Chalfont St. Gills, UK). Films were scanned with a HP Scanjet G2710 (Palo Alto, CA) and images were processed using Adobe Photoshop CS6 (San Jose, CA). Quantification was done by densitometry, using ImageJ (National Institutes of Health, Bethesda, MD).
PKP2 promoter and enhancer constructs
Genomic DNA, obtained from CCD‐18Co human normal colon myofibroblasts, was used to amplify by PCR two upstream sequences of the PKP2 promoter. PCR was performed using the Certamp Kit for complex amplifications (Biotools, Madrid, Spain) and primers 5′‐CATCTCAGCATCATGGTTGG‐3′ and 5′‐GAGAAACTCCAACAAGAGGC‐3′ for the −1509/–735 fragment, containing the putative β‐catenin/TCF binding sites, and 5′‐TTAATCAACGTTAGCAGGGC‐3′ and 5′‐ACAGGATGGATTTCCGCTCG‐3′ for the control −817/–299 fragment. The resulting PCR products were cloned into the pGEM‐T Easy vector (Promega) and sequenced. Subsequently, a second PCR was performed to generate suitable restriction sites using primers: 5′‐TTGGTACCTTCTATTGGGGTGGGAGC‐3′ and 5′‐ATAAAGCTTGAGAAACTCCAACAAGAGGC‐3′ for the −1509/–735 fragment, and 5′‐ ATGGTACCTTAATCAACGTTAGCAGGGC‐3′ and 5′‐TACAAGCTTACAGGATGGATTTCCGCTCG‐3′ for the −817/–299 fragment. The PCR products were digested with KpnI and HindIII (New England Biolabs, Ipswich, MA), cloned into the pGL4.23 luciferase reporter plasmid (Promega) and sequenced. Site‐directed mutagenesis of β‐catenin/TCF binding sites on the −1509/–735 PKP2 promoter construct was performed using the Agilent QuikChange II XL Site‐Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA), and the wild‐type 5′‐CCTTTG(A/T)(A/T)‐3′ sites were changed into 5′‐CCTTTGGC‐3′. Primers used for the seven single‐site mutations are shown in Supporting Information Table I. The triple mutant −1509/–735‐m347 construct was generated by three rounds of mutation on the wild‐type construct. All constructs were sequenced.
The −20666/–20091 PKP2 enhancer region was amplified by PCR using the same template and kit as above, and primers 5′‐CGGTTTTACTCAAGTAAGC‐3′ and 5′‐GCAATCTTCTTGATGAGTAG‐3′. The resulting PCR product was cloned into the pGEM‐T Easy vector and sequenced. A second PCR was performed to generate KpnI restriction sites using primers: 5′‐TTGGTACCCGGTTTTACTCAAGTAAGC‐3′ and 5′‐ATGGTACCGCAATCTTCTTGATGAGTAG‐3′. The PCR product was digested with KpnI and cloned into the pGL4.23 luciferase reporter plasmid. Constructs harbouring the PKP2 enhancer in both forward (–20666/–20091) and reverse (–20091/–20666) orientations were selected and sequenced. Site‐directed mutagenesis of β‐catenin/TCF binding sites on the −20091/–20666 PKP2 enhancer construct was performed as above and the primers used for each single‐site mutation are shown in Supporting Information Table I.
Transfections, plasmids and reporter assays
Subconfluent cells cultures were transfected in triplicate using the jetPEI transfection reagent (PolyPlus Transfection, Illkirch, France), following the manufacturer's guidelines. The reporter plasmids pTOPFLASH, pFOPFLASH, 4xwtCBF1Luc, 4xmtCBF1Luc, NF3, and that harbouring the −2235/+112 region of the DKK1 promoter upstream of the Firefly luciferase gene have been previously described.19, 20, 21, 22 The reporter plasmids harbouring wild‐type and mutant PKP2 promoter and enhancer sequences are described above. The expression plasmid pFLAG‐CMV.5.1‐Plakophilin2a‐flag encoding Plakophilin‐2 was purchased from Addgene (Cambridge, MA). The expression plasmids pMT23‐β‐catenin, pCDNA3‐TCF4‐VP16 and pCDNA3‐p65 have been described elsewhere.22, 23, 24 The expression plasmid pCDNA3‐ICND was a generous gift from Keith R. Brennan (University of Manchester, UK). A Renilla luciferase plasmid (pRL‐TK) was used in all experiments as an internal control. Firefly and Renilla luciferase activities were measured separately using the Dual Luciferase reagent kit (Promega) and a GloMax 96 microplate luminometer (Promega).
Statistical analysis
The significance of differences between two groups was assessed by the Student's t test. The differences between more than two group means were analysed with ANOVA, and considered statistically significant when the reported p values were below 0.05. A Tukey post‐hoc test was applied after a significant ANOVA to compare differences between pairs of groups.
Results
Analysis of Wnt3A transcriptome in CCD‐18Co human colon myofibroblasts
To investigate the effect of Wnt signalling on human colon myofibroblasts, we studied the transcriptional response of this cell type to Wnt3A stimulation. For this purpose, we selected the colon subepithelial myofibroblast cell line CCD‐18Co25 as a model system. CCD‐18Co cells express Frizzled receptors 1 through 7 (Supporting Information Fig. S1A; data not shown) and respond to Wnt3A treatment with a substantial increase in cytoplasmic levels of β‐catenin (Supporting Information Fig. S1B and C). We performed RNA sequencing on three independent biological replicates of CCD‐18Co cells treated with either Wnt3A (100 ng/ml) or vehicle for 24 hr, as described in the Material and Methods section. An analysis of the data rendered a total of 1,136 differentially expressed genes (FDR < 0.05), of which 662 were upregulated and 474 were downregulated in response to Wnt3A (Supporting Information Tables II and III). These genes were involved in a wide variety of cellular functions. The complete list of genes was deposited in the GEO database (accession no. GSE89124). To validate our analysis, we selected some of the upregulated (PTGER3, ODZ3, ILDR2 and AMIGO2) and downregulated (WNT16, TRPA1) genes in our study and performed RT‐qPCR on a new set of RNA samples. All these genes were confirmed as being regulated by Wnt3A (Fig. 1 a).
Figure 1.
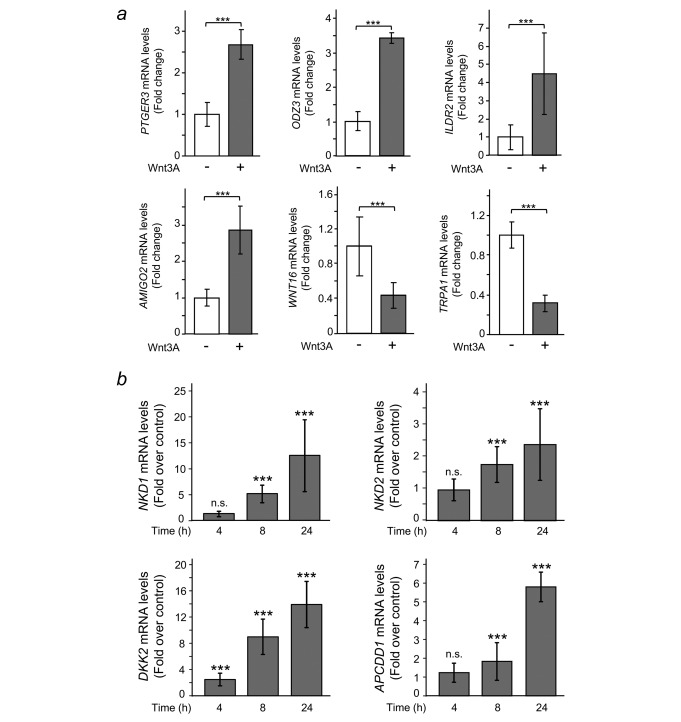
Wnt3A regulates the expression of multiple genes in CCD‐18Co human colon myofibroblasts. (a) Validation using RT‐qPCR of a representative sample of six genes (PTGER3, ODZ3, ILDR2, AMIGO2, WNT16 and TRPA1) that were up‐ or downregulated by Wnt3A in RNA‐seq experiments. Cells were treated with either Wnt3A (+) or vehicle (–) for 24 hr. The results shown are means ± S.D. of three independent biological samples, each one determined in triplicate (***p ≤ 0.001). (b) Wnt3A triggers the expression of a set of target genes encoding inhibitors of the Wnt/β‐catenin pathway. Cells were stimulated with either Wnt3A or vehicle for the indicated times and NKD1, NKD2, DKK2, and APCDD1 expression was analysed by RT‐qPCR. Results are shown as fold over control at the indicated times and are means ± S.D. of three independent biological samples, each one determined in triplicate (n.s., non‐significant; *** p ≤ 0.001).
In support of our study, a gene set enrichment analysis (GSEA) showed that the hallmark gene set that most closely correlated with our list of genes regulated by Wnt3A in CCD‐18Co cells was that of genes upregulated by activation of Wnt signalling through accumulation of CTNNB1/β‐catenin (FDR q‐val = 0.001; FWER p‐val = 0.001). Interestingly, the GSEA analysis showed a significant inverse correlation between our list of genes and genes defining epithelial‐mesenchymal transition (EMT), as in wound healing, fibrosis and metastasis (FDR q‐val = 0.022; FWER p‐val = 0.019). Consequently, genes upregulated in EMT appear to be downregulated by Wnt3A in colon myofibroblasts. In contrast, activation of the Wnt/β‐catenin pathway is known to promote EMT in epithelial cancer cells.26 Therefore, some genetic programs induced by Wnt3A in myofibroblasts differ from those induced in other cell types. This confirms previous knowledge that Wnt target genes are cell‐type specific. In agreement, a list of genes upregulated by the EWS‐FLI1 oncogenic fusion protein in Ewing's sarcoma cells showed a direct correlation with genes upregulated by Wnt3A in CCD‐18Co cells (FDR q‐val < 0.001; FWER p‐val < 0.001), although we and others have demonstrated mutual antagonism between EWS‐FLI1 and Wnt/β‐catenin signalling in Ewing's sarcoma23, 27 (Supporting Information Fig. S2).
Wnt3A induces the expression of Wnt inhibitors in human fibroblasts
Among the genes upregulated by Wnt3A in CCD‐18Co cells, we detected a set that encodes Wnt/β‐catenin pathway inhibitors, such as the extracellular antagonist Dickkopf‐2 (DKK2), the membrane‐bound inhibitor Adenomatous Polyposis Coli Downregulated 1 protein (APCDD1), and the cytoplasmic inhibitors Naked‐1 (NKD1), Naked‐2 (NKD2), and Axin 2 (AXIN2). Time‐course experiments confirmed that DKK2 was already upregulated 4 hr after treatment with Wnt3A, while NKD1, NKD2 and APCDD1 exhibited significant increases at 8 hr post‐treatment (Fig. 1 b). All these genes were also upregulated by Wnt3A in IMR‐90 human lung fibroblasts, which suggests that they may be common Wnt3A targets in fibroblasts (Supporting Information Fig. S3). Altogether, these data suggest that Wnt3A induces the expression of a variety of Wnt inhibitors that, in turn, may be responsible for feedback inhibition of the pathway in fibroblasts.
Wnt3A induces PKP2 expression in normal and colon cancer‐associated fibroblasts
For a detailed study, we chose the PKP2 gene, which is consistently upregulated by Wnt3A in CCD‐18Co cells and encodes the protein Plakophilin‐2, whose effect on Wnt signalling and relation to cancer is controversial (see the Discussion section). Plakophilin‐2 belongs to the armadillo‐repeat protein family, along with β‐catenin, plakoglobin, p120‐catenin, and other plakophilins. The PKP2 mRNA level augmented 4‐fold in response to Wnt3A (Fig. 2 a), which led to a 2‐fold increase in protein level 24 hr after treatment (Fig. 2 b). This was due to activation of the Wnt/β‐catenin signalling pathway, since pre‐treatment with the pathway inhibitor XAV‐939 impaired Plakophilin‐2 accumulation (Fig. 2 c). Moreover, Wnt5A, a noncanonical Wnt that cannot trigger β‐catenin‐induced signalling, could not upregulate PKP2 expression (Fig. 2 d). Other PKP subfamily members, including PKP3 and PKP4/p0071, were not regulated by Wnt3A in CCD‐18Co cells, which suggests that the effect on PKP2 is specific (Fig. 2 e).
Figure 2.
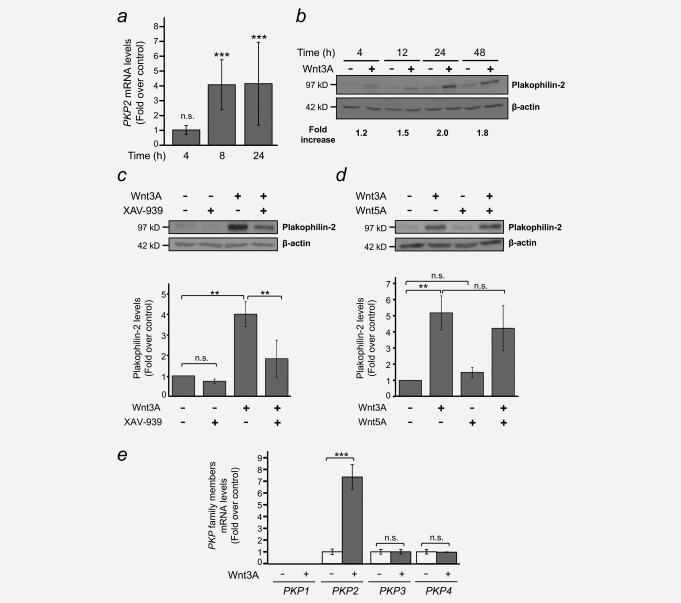
Wnt3A induces the expression of Plakophilin‐2 in CCD‐18Co human colon myofibroblasts. (a) RT‐qPCR analysis showing the upregulation of PKP2 mRNA by Wnt3A. Results are shown as fold over control at the indicated times and are means ± S.D. of three independent biological samples, each one determined in triplicate (n.s., non‐significant; *** p ≤ 0.001). (b) Western blot analysis of Plakophilin‐2 upregulation by Wnt3A in a time course experiment. β‐Actin was used as a loading control. Fold increases over vehicle‐treated controls are indicated. A representative experiment is shown. (c) The Wnt/β‐catenin pathway/tankyrase inhibitor XAV‐939 reduces Wnt3A‐dependent Plakophilin‐2 upregulation. Cells were pre‐treated for 4 hr with either 1 µM XAV‐939 (+) or vehicle (–) and then stimulated with Wnt3A (+) for 24 hr. β‐Actin was used as a loading control. A representative experiment is shown. Bar plot represents means ± S.D. of four independent experiments (n.s., non‐significant; ** p ≤ 0.01). (d) Non‐canonical Wnt5A cannot induce Plakophilin‐2 accumulation. Myofibroblasts were stimulated with either Wnt3A, Wnt5A or both for 24 hr, and Plakophilin‐2 expression was analysed by Western blot. β‐Actin was used as a loading control. A representative experiment is shown. Bar plot represents means ± S.D. of four independent experiments (n.s., non‐significant; ** p ≤ 0.01). (e) RT‐qPCR analysis of the expression of PKP family members in response to Wnt3A stimulation. CCD‐18Co human colon myofibroblasts were treated with either Wnt3A (+) or vehicle (–) for 24 hr. Results are shown as fold over control and are means ± S.D. of three independent biological samples, each one determined in triplicate (n.s., non‐significant; *** p ≤ 0.001).
To address whether PKP2 upregulation by Wnt3A also takes place in primary colonic fibroblasts we established primary cultures of NF and CAF from fresh tumour tissue, and paired adjacent normal colon mucosa obtained from biopsies of CRC patients using the explant outgrowth technique.13, 14 Both NF and CAF are vimentin and α‐SMA positive, and the expression of α‐SMA is frequently higher in CAF than in their paired NF, indicating a more myofibroblastic phenotype of CAF (Supporting Information Fig. S4A and B). Wnt3A consistently increased the expression of PKP2 in both CAF and NF, although with differences among patients (Fig. 3 a and b).
Figure 3.
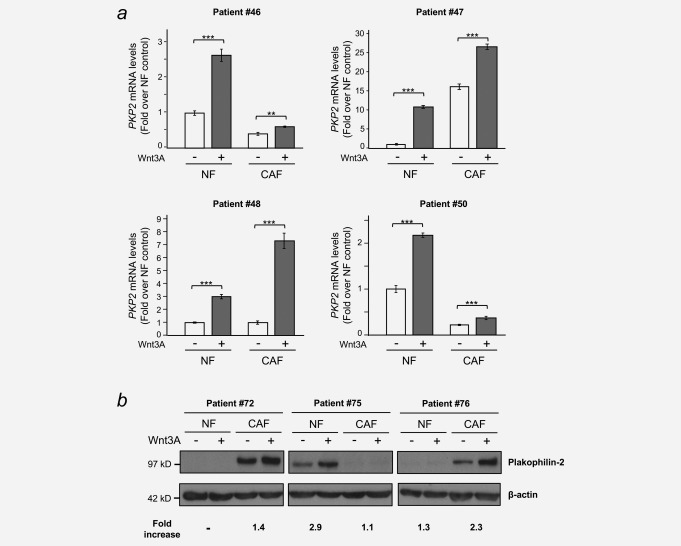
Wnt3A induces PKP2/Plakophilin‐2 expression in normal fibroblasts (NF) and in cancer‐associated fibroblasts (CAF). NF and CAF were obtained from CRC patients as described in the Materials and Methods and treated with either Wnt3A (+) or vehicle (–) for 24 hr. Total RNA and protein was purified and PKP2/Plakophilin‐2 mRNA and protein levels were analysed by RT‐qPCR (a) and western blot (b), respectively. Results in (a) are shown as fold over the NF control and are means ± S.D. of three independent biological samples, each one determined in triplicate (**p ≤ 0.01; *** p ≤ 0.001). β‐Actin was used as a loading control in (b) and fold increases over vehicle‐treated controls are indicated.
Wnt3A induces PKP2 gene transcription through β‐catenin/TCF binding sites
To get further insight into the mechanism of PKP2 gene regulation by Wnt3A, we treated CCD‐18Co cells with actinomycin D (ACTD), an inhibitor of transcription, before stimulation with Wnt3A. ACTD completely blocked the upregulation of PKP2 triggered by Wnt3A (Fig. 4 a), which argues in favour of a transcriptional mechanism.
Figure 4.
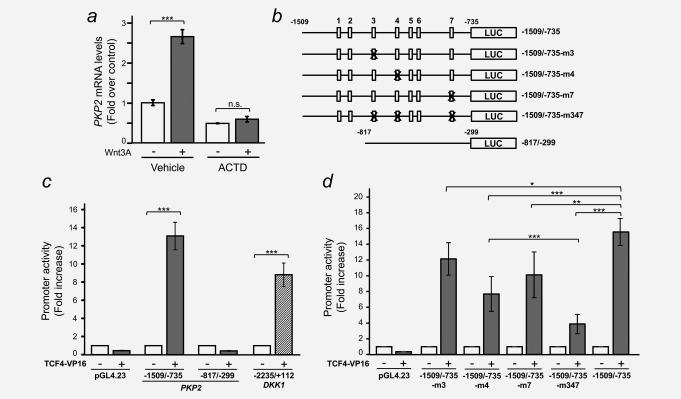
The human PKP2 gene promoter is activated by β‐catenin/TCF. (a) Actinomycin D (ACTD) blocks Wnt3A‐induced accumulation of PKP2 mRNA. CCD‐18Co cells were pre‐treated for 1 hr with either 5 µg/ml ACTD or vehicle and then stimulated with Wnt3A for 8 hr. Total RNA was purified and PKP2 mRNA levels were analysed by RT‐qPCR. Results are shown as fold over control and are means ± S.D. of three independent biological samples, each one determined in triplicate (n.s., nonsignificant; *** p ≤ 0.001). (b) Diagram of the PKP2 promoter constructs used in this study showing the location of the putative wild‐type (5′‐CTTTG[A/T][A/T]‐3′, white boxes) or mutant (5′‐CTTTGGC‐3′, X‐crossed white boxes) β‐catenin/TCF binding sites. (c) HEK‐293T cells were co‐transfected with the indicated PKP2 promoter constructs together with TCF4‐VP16 (+; a constitutively active form of TCF‐4) or an empty vector (–). The β‐catenin/TCF responsive DKK1 promoter was used as a positive control. A Renilla luciferase plasmid (pRL‐TK) was used as an internal control. Cells were lysed 48 hr after transfection and Firefly and Renilla luciferase activities were measured. Values were normalized to those of Renilla. Error bars represent S.D. (***p ≤ 0.001). (d) HEK‐293T cells were co‐transfected with the indicated PKP2 promoter wild‐type or mutant constructs together with TCF4‐VP16 (+) or an empty vector (–) and the experiment was performed as in (c). Error bars represent S.D. (*p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001).
We next made use of the UCSC Genome Browser28 and data from the ENCODE Project29 to study the regulatory regions of the human PKP2 gene. Available ChIP‐seq data for transcription factor TCF‐4 (TCF7L2 gene) in different cell types suggested two putative binding regions for this factor: a promoter region around the transcription start site (TSS) of the human PKP2 gene, and an enhancer region located about 20 kb upstream from the TSS (Supporting Information Fig. S5A). In silico analysis of both regions revealed up to 7 putative β‐catenin/TCF binding sites in the promoter (Supporting Information Fig. S5B) and up to 3 putative β‐catenin/TCF binding sites in the enhancer (Supporting Information Fig. S5C).
We PCR‐amplified and cloned both relevant sequences into the pGL4.23 plasmid upstream from the luciferase reporter gene and a minimal promoter (Figs. 4 b and 5 a). The transcriptional activity of the −1509/–735 PKP2 promoter construct containing all 7 putative β‐catenin/TCF binding sites was consistently stimulated by TCF4‐VP16, a constitutively active version of TCF‐4, in HEK‐293T, HeLa and MCF7 cells (Fig. 4 c and Supporting Information Fig. S6). Its induction was comparable to that of the DKK1 gene promoter, a well‐known β‐catenin/TCF target gene22, 30, 31 (Fig. 4 c). In contrast, a construct containing the −817/–299 promoter region that does not contain any putative β‐catenin/TCF binding sites was not activated by TCF4‐VP16 (Fig. 4 c and Supporting Information Fig. S6). Single‐site point mutations showed that only three putative β‐catenin/TCF binding sites contributed significantly to TCF4‐VP16 transcriptional activity (sites #3, #4 and #7; Fig. 4 b and d), whereas mutation of the remaining motifs (sites #1, #2, #5 and #6) had no effect (data not shown). Finally, a triple mutant targeting these three sites revealed an additive inhibitory effect on TCF4‐VP16‐mediated transcription, and some lingering activity that suggests the possibility of additional regulatory elements or residual activity of mutated sites (Fig. 4 b and d).
Figure 5.
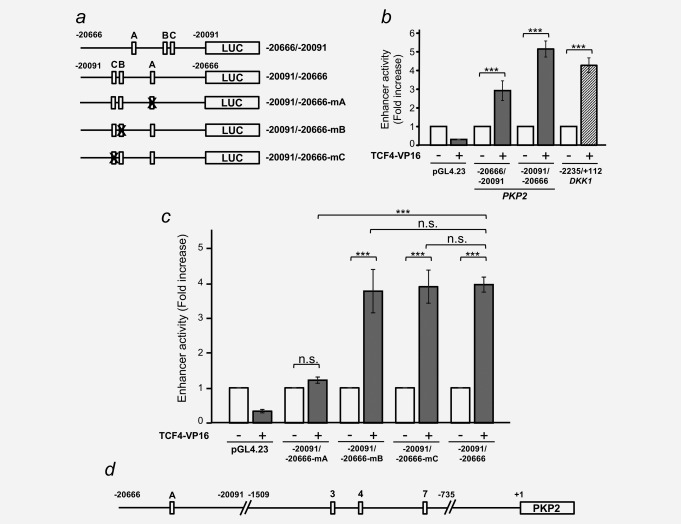
An enhancer sequence located 20 kb upstream from the PKP2 transcription start site harbours a functional β‐catenin/TCF responsive element. (a) Diagram of the PKP2 enhancer constructs used in this study showing the location of the putative wild‐type (5′‐CTTTG[A/T][A/T]‐3′, white boxes) or mutant (5′‐CTTTGGC‐3′, X‐crossed white boxes) β‐catenin/TCF binding sites. (b) HEK‐293T cells were co‐transfected with either the forward (–20666/–20091) or reverse (–20091/–20666) 575 bp PKP2 enhancer construct together with TCF4‐VP16 (+) or an empty vector (–). The DKK1 promoter was used as a positive control and pRL‐TK as an internal control. Cells were lysed 48 hr after transfection and Firefly and Renilla luciferase activities were measured. Values were normalized to those of Renilla. Error bars represent S.D. (***p ≤ 0.001). (c) HEK‐293T cells were co‐transfected with the indicated PKP2 enhancer wild‐type or mutant constructs together with TCF4‐VP16 (+) or an empty vector (–) and the experiment was performed as in (b). Error bars represent S.D. (n.s., nonsignificant; *** p ≤ 0.001). (d) A diagram summarizing the human PKP2 gene regulatory sequences found in this study. White boxes represent functional β‐catenin/TCF binding sites.
During transcription, enhancer sequences are brought into close proximity to the TSS, so that transcription factors binding to these regions can interact with the basal transcriptional machinery. We cloned the enhancer sequence described above into pGL4.23 in both forward and reverse orientations (Fig. 5 a), and showed that TCF4‐VP16 could stimulate luciferase activity from both constructs with a potency comparable to that of the DKK1 promoter, and even slightly stronger in the reverse orientation (Fig. 5 b). Therefore, we generated single‐point mutations in each of the three putative β‐catenin/TCF binding sites of the reverse‐orientated enhancer, and showed that mutations in site A completely abolished TCF4‐VP16‐mediated transcription, while mutations in sites B and C were totally innocuous (Fig. 5 c). In summary, our data suggest that β‐catenin/TCF complexes induce transcription of the PKP2 gene through at least 3 binding sites in the promoter region and one binding site in an enhancer region located 20 kb upstream from the TSS (Fig. 5 d).
Plakophilin‐2 interferes with β‐catenin/TCF transcriptional activity
Next, we aimed to address the functional effects that an increase in PKP2 gene expression triggered by Wnt3A in NF and CAF might have on β‐catenin/TCF transcriptional activity. To this end, we used the synthetic pTOPFLASH and pFOPFLASH plasmids, which contain three copies of either a wild‐type or a mutant β‐catenin/TCF binding consensus sequence upstream of a minimal c‐FOS promoter and the Firefly luciferase gene, which have been extensively used to study β‐catenin/TCF transcriptional activity.19 Transient expression of Plakophilin‐2 in HEK‐293T cells impaired activation of the pTOPFLASH reporter by wild‐type β‐catenin (Fig. 6 a). This effect was specific, since transcriptional activation of the Notch pathway reporter 4xwtCBF1Luc by ICND was unaffected by Plakophilin‐2 (Fig. 6 b), and activation of the NF‐κB reporter NF3 by p65 was potentiated, rather than inhibited, by Plakophilin‐2 (Fig. 6 c), as previously described.32 Remarkably, the inhibitory effect of Plakophilin‐2 on β‐catenin/TCF‐mediated transcription was also confirmed using the DKK1 promoter construct (Fig. 6 d). This effect was not due to a decrease in β‐catenin protein level, since this remained unaffected by Plakophin‐2 expression (Fig. 6 e). Altogether, these data show an inhibitory effect of Plakophilin‐2 on β‐catenin‐responsive promoters.
Figure 6.
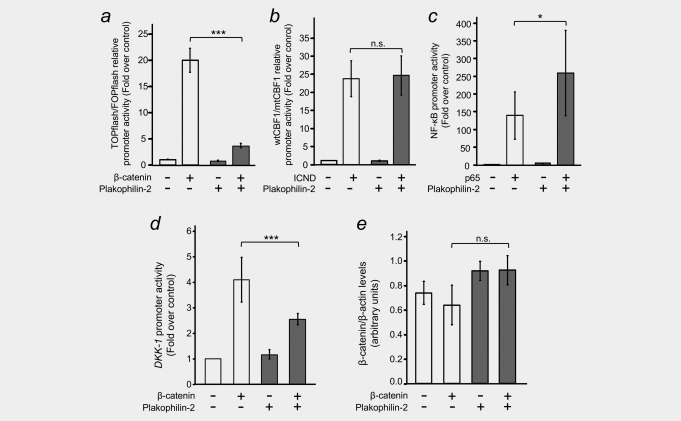
Plakophilin‐2 antagonizes β‐catenin/TCF transcriptional activity. (a) HEK‐293T cells were transfected with reporter plasmids (pTOPFLASH or pFOPFLASH) to evaluate the transcriptional activity of the Wnt/β‐catenin pathway, together with an expression vector for β‐catenin (+) and in the presence or absence of an expression plasmid for Plakophilin‐2. pRL‐TK was used as an internal control. Cells were lysed 48 hr after transfection and Firefly and Renilla luciferase activities were measured. Values were normalized to those of Renilla, related to those of the mutant pFOPFLASH construct, and represented as fold over control. Error bars represent S.D. (***p ≤ 0.001). (b) HEK‐293T cells were transfected with reporter plasmids (4xwtCBF1Luc or 4xmtCBF1Luc) to evaluate the transcriptional activity of the Notch pathway, together with an expression vector for IntraCellular Notch Domain (ICND, +) and in the presence or absence of an expression plasmid for Plakophilin‐2. The experiment was performed as in (a). (c) HEK‐293T cells were transfected with a reporter plasmid (NF3) to evaluate the transcriptional activity of the NF‐κB pathway, together with an expression vector for p65 (+) and in the presence or absence of an expression plasmid for Plakophilin‐2. Samples were processed as in (a), values were normalized to those of Renilla, and represented as fold over control. Error bars represent S.D. (*p ≤ 0.05). (d) HEK‐293T cells were transfected with the β‐catenin/TCF responsive DKK1 promoter, together with an expression vector for β‐catenin (+) and in the presence or absence of an expression plasmid for Plakophilin‐2. The experiment was performed as in (a) and values are represented as fold over control. Error bars represent S.D. (***p ≤ 0.001). (e) β‐catenin protein levels are not affected by Plakophilin‐2 expression. Extracts from the experiment in (d) were analysed by Western blot to rule out the possibility that the effects of Plakophilin‐2 on β‐catenin/TCF‐dependent transcription were due to changes in β‐catenin levels as a consequence of Plakofilin‐2 expression.
Discussion
Pericryptal stromal cells are essential for the maintenance of colon tissue homeostasis, and also play an important role in CRC progression. Wnt/β‐catenin signalling is essential in both these processes. Although the source and distribution of Wnt proteins in the intestine is still a matter of debate, stromal fibroblasts are thought to be a major source of Wnts.33, 34 Here we show that human colon myofibroblasts are highly responsive to Wnt3A, which causes a profound change in their gene expression profile leading to the up and downregulation of a large set of genes. Therefore, Wnt factors can act on epithelial cells at the bottom of the colon crypt but also on pericryptal fibroblasts that harbour functional Wnt receptors and possibly other stromal cell types. The fact that inhibitors of the Wnt/β‐catenin pathway such as DKK2, NKD1 and 2, or APCDD1 are among the strongest Wnt3A‐induced genes in colon myofibroblasts could unveil a mechanism to limit the actions of Wnt signalling on these cells or perhaps to protect them from the effects of these factors that are required for stem and proliferating cells at the bottom of the crypt, but could be deleterious for the surrounding fibroblasts. We cannot rule out the effect that secreted Wnt proteins may have on other stromal cell types in healthy and tumoural tissue, which warrants further investigation.
We compared the list of genes regulated by Wnt3A in human colon myofibroblasts with the gene sets representing the biological functions enriched in the expression patterns associated to each of the four consensus molecular subtypes in colorectal cancer (CMS 1–4).8 The pattern of expression of Wnt3A‐treated myofibroblasts clusterises closest to the molecular subtype CMS2 (Supporting Information Fig. S7A), which is characterized by WNT and MYC pathway activation.8 Therefore, although myofibroblasts are stromal cells, their molecular signature in response to Wnt3A is closest to that of CMS2 instead of CMS4, despite the latter is characterized by a high stromal component. We also found a significant enrichment of TCF7L2/TCF‐4 binding sites identified in LS174T colon cancer cells35 in genes up and downregulated by Wnt3A in CCD‐18Co myofibroblasts (Supporting Information Fig. S7B), which suggests that in spite of their different linages there is a set of genes regulated by Wnt proteins in both cell types. GSEA analysis also showed an enrichment (almost significant) of genes upregulated by Wnt3A in NIH3T336 (FDR q‐val = 0.08; FWER p‐val = 0.10; Supporting Information Fig. S7C) and dermal fibroblasts37 (FDR q‐val = 0.08; FWER p‐val = 0.18; Supporting Information Fig. S7D) among genes induced by Wnt3A in CCD‐18Co cells. Likewise, genes downregulated by Wnt3A in CCD‐18Co myofibroblasts were significantly enriched in genes repressed by Wnt3A in NIH3T336 (FDR q‐val = 0.003; FWER p‐val = 0.002; Supporting Information Fig. S7E) which suggests some conservation among genes regulated by Wnt3A in fibroblasts.
One of the genes that were most strongly upregulated by Wnt3A was PKP2, which encodes the desmosomal protein Plakophilin‐2. Although this protein is best‐known as a component of epithelial cell desmosomes, it is also involved in other types of adhesion structures, including composite junctions (area compositae) in cardiomyocytes and a very specific class of Plakophilin‐2‐containing adherens junctions (coniunctiones adhaerentes) present in mesenchymally derived cells with high proliferative activity.38 Moreover, Plakophilin‐2 seems to have additional functions that are unrelated to adhesion, since it has been consistently found inside the cell nucleus.39, 40 We show here that Plakophilin‐2 expression in HEK‐293T cells interferes with β‐catenin/TCF transcriptional activity, which implies that it might also be classified as a Wnt inhibitor. Unfortunately, CCD‐18Co cells are extremely resistant to both transient transfection and lentiviral infection, so this has precluded Plakophilin‐2 gain‐ and loss‐of‐function studies in these cells.
Our results contrast with those of Chen and colleagues who showed that Plakophilin‐2 can bind β‐catenin, and that expression of Plakophilin‐2 in SW480 colon carcinoma cells had a rather small but positive effect on β‐catenin/TCF activity.41 This discrepancy suggests that the effect of Plakophilin‐2 on β‐catenin‐mediated transcription might be cell‐type specific or, perhaps, dependent on the status of the Wnt/β‐catenin pathway, since SW480 cells harbour a mutant APC protein and have constitutively active Wnt signalling. In this regard, multipotent mesenchymal stromal cells and iPS cells from a patient carrying a PKP2 gene mutation exhibited lower β‐catenin/TCF transcriptional activity than those of a healthy control.42 Interestingly, another plakophilin family member, Plakophilin‐1, has been recently shown to be a critical mediator of Wnt signalling in tooth development and ameloblast differentiation.43 This hints at a close link between the PKP family and Wnt/β‐catenin signalling.
Our data show that PKP2 is regulated by β‐catenin through several response elements in the promoter and enhancer regions of the gene. Sequence analysis of the putative β‐catenin/TCF binding sites showed that the integrity of the core sequence 5′‐CCTTTG(A/T)(A/T)‐3′ was essential for functionality. Alterations in the first 5′ cytidine (sites #3 and #7 in the promoter) still rendered a partially functional binding site, but changes affecting one of the 3′ adenosines/thymidines (sites #1, #2, #5 and #6 in the promoter and sites B and C in the enhancer region) rendered non‐functional elements. ChIP‐seq data obtained from UCSC Genome Browser confirmed that these sites are functional in vivo, and therefore that the PKP2 gene is a bona fide Wnt target gene.
There is growing evidence of the involvement of Plakophilin‐2 and other plakophilins and desmosomal proteins in cancer.44 Plakophilin‐2 is widely expressed in carcinomas45, 46 and in some of them its expression is associated with invasive and metastatic behaviour.47, 48 Recently, Plakophilin‐2 has been shown to promote tumour development by binding the epidermal growth factor receptor (EGFR) and enhancing its ligand‐dependent and independent activity.32 In spite of these data, further research is necessary to clarify the role of Plakophilin‐2 in different types of cancer. Classically, colorectal carcinoma cells have been thought to be refractory to extracellular Wnt factors, since most of them carry intracellular mutations (APC, CTNNB1/β‐catenin and AXIN1) that result in constitutive activation of the pathway. However, it has been proposed recently that they can respond to exogenous and autocrine Wnt ligands that further activate the Wnt/β‐catenin signalling pathway.49 In addition, Wnt factors may exert some of their effects on CRC by targeting CAF. Nevertheless, we found that PKP2 expression is upregulated by Wnt3A in both normal and cancer‐associated fibroblasts of different origins. This suggests that the gene is not specifically induced in CAF, but is a general Wnt target in fibroblasts.
Plakophilin‐2 is the only plakophilin expressed in the heart and is essential for cell‐cell adhesion in cardiomyocytes.50, 51 Actually, PKP2 mutations are the leading cause of arrhythmogenic right ventricular cardiomyopathy (ARVC):52, 53 a major cause of sudden cardiac death. Therefore, further studies should determine whether PKP2 is also a Wnt target in the heart and, if so, evaluate the relevance of its Wnt‐regulated expression.
In summary, we show here that the PKP2 gene encoding Plakophilin‐2 is a novel Wnt/β‐catenin target that could function as a feedback inhibitor of β‐catenin/TCF transcriptional activity in colonic fibroblasts, and perhaps in other cell types. This is highly relevant, due to increasing evidence suggesting the involvement of Plakophilin‐2 in cancer and other pathologies, and the key role of Wnt/β‐catenin signalling in colorectal carcinogenesis.
Supporting information
Supporting Information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Figure 5
Supporting Information Figure 6
Supporting Information Figure 7
Supporting Information Table 1
Supporting Information Table 2
Supporting Information Table 3
Acknowledgements
We thank Keith Brennan and Manuel Fresno for reagents and Lucille Banham for her valuable assistance in the preparation of the English manuscript. This work was supported by the Spanish Ministerio de Economía y Competitividad and the European Regional Development Fund (Fondos FEDER) (SAF2013–43468‐R, and SAF2014–53819‐R), the Agencia Estatal de Investigación‐Fondos FEDER (SAF2016–76377‐R, AEI/FEDER, UE), the Comunidad de Madrid (S2010/BMD‐2344 Colomics2), the Instituto de Salud Carlos III–Fondos FEDER (RD12/0036/0021, and CIBERONC‐CB16/12/00273) and the Nuclear Receptors Network (Nurcamein, SAF2015–71878‐REDT).
REFERENCES
- 1. Torre LA, Bray F, Siegel RL, et al. Global cancer statistics. CA Cancer J Clin 2015; 65:87–108. [DOI] [PubMed] [Google Scholar]
- 2. Torre LA, Siegel RL, Ward EM, et al. Global cancer incidence and mortality rates and trends–an update. Cancer Epidemiol Biomarkers Prev 2016; 25:16–27. [DOI] [PubMed] [Google Scholar]
- 3. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61:759–67. [DOI] [PubMed] [Google Scholar]
- 4. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol 2011;6:479–507. [DOI] [PubMed] [Google Scholar]
- 5. Clevers H, Nusse R. Wnt/β‐catenin signaling and disease. Cell 2012; 149:1192–205. [DOI] [PubMed] [Google Scholar]
- 6. Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res 2010;316:1324–31. [DOI] [PubMed] [Google Scholar]
- 7. Medema JP, Vermeulen L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 2011; 474:318–26. [DOI] [PubMed] [Google Scholar]
- 8. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015; 21:1350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Becht E, de Reyniès A, Giraldo NA, et al. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin Cancer Res 2016; 22:4057–66. [DOI] [PubMed] [Google Scholar]
- 10. Isella C, Terrasi A, Bellomo SE, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet 2015; 47:312–9. [DOI] [PubMed] [Google Scholar]
- 11. Calon A, Lonardo E, Berenguer‐Llergo A, et al. Stromal gene expression defines poor‐prognosis subtypes in colorectal cancer. Nat Genet 2015; 47:320–9. [DOI] [PubMed] [Google Scholar]
- 12. Bass‐Zubek AE, Godsel LM, Delmar M, et al. Plakophilins: multifunctional scaffolds for adhesion and signaling. Curr Opin Cell Biol 2009; 21:708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herrera M, Islam AB, Herrera A, et al. Functional heterogeneity of cancer‐associated fibroblasts from human colon tumors shows specific prognostic gene expression signature. Clin Cancer Res 2013; 19:5914–26. [DOI] [PubMed] [Google Scholar]
- 14. Ferrer‐Mayorga G, Gómez‐López G, Barbáchano A, Fernández‐Barral A, Peña C, Pisano DG, Cantero R, Rojo F, Muñoz A, Larriba MJ. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 2016; gutjnl‐2015–310977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14:R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cunningham F, Amode MR, Barrell D, et al. Ensembl 2015. Nucleic Acids Res 2015; 43:D662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics 2015; 31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robinson MD, McCarthy DJ, Smyth GK. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Korinek V, Barker N, Morin PJ, et al. Constitutive transcriptional activation by a β‐catenin‐Tcf complex in APC‐/‐ colon carcinoma. Science 1997; 275:1784–7. [DOI] [PubMed] [Google Scholar]
- 20. Hsieh JJ, Henkel T, Salmon P, et al. Truncated mammalian Notch1 activates CBF1/RBPJk‐represed genes by a mechanism resembling that of Epstein‐Barrvirus EBNA2. Mol Cell Biol 1996; 16:952–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Solanas G, Porta‐de‐la‐Riva M, Agustí C, et al. E‐cadherin controls beta‐catenin and NF‐kappaB transcriptional activity in mesenchymal gene expression. J Cell Sci 2008; 121:2224–34. [DOI] [PubMed] [Google Scholar]
- 22. González‐Sancho JM, Aguilera O, García JM, et al. The Wnt antagonist DICKKOPF‐1 gene is a downstream target of β‐catenin/TCF and is downregulated in human colon cancer. Oncogene 2005; 24:1098–103. [DOI] [PubMed] [Google Scholar]
- 23. Navarro D, Agra N, Pestaña A, et al. The EWS/FLI1 oncogenic protein inhibits expression of the Wnt inhibitor DICKKOPF‐1 gene and antagonizes beta‐catenin/TCF‐mediated transcription. Carcinogenesis 2010; 31:394–401. [DOI] [PubMed] [Google Scholar]
- 24. Coiras M, López‐Huertas MR, Mateos E, et al. Caspase‐3‐mediated cleavage of p65/RelA results in a carboxy‐terminal fragment that inhibits IκBα and enhances HIV‐1 replication in human T lymphocytes. Retrovirology 2008; 5:109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Valentich JD, Popov V, Saada JI, Powell DW. Phenotypic characterization of an intestinal subepithelial myofibroblast cell line. Am J Physiol 1997; 272:C1513–24. [DOI] [PubMed] [Google Scholar]
- 26. Lyndsey S, Langhans SA. Crosstalk of oncogenic signaling pathways during epithelial‐mesenchymal transition. Front Oncol 2014; 4:358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pedersen EA, Menon R, Bailey KM, et al. Activation of Wnt/beta‐catenin in Ewing sarcoma cells antagonizes EWS/ETS function and promotes phenotypic transition to more metastatic cell states. Cancer Res 2016; 76:5040–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kent WJ, Sugnet CW, Furey TS, et al. The human Genome Browser at UCSC. Genome Res 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niida A, Hiroko T, Kasai M, et al. DKK1, a negative regulator of Wnt signaling, is a target of the beta‐catenin/TCF pathway. Oncogene 2004; 23:8520–6. [DOI] [PubMed] [Google Scholar]
- 31. Chamorro M, Schwartz DR, Vonica A, et al. FGF‐20 and DKK1 are transcriptional targets of beta‐catenin and FGF‐20 is implicated in cancer and development. Embo J 2005; 24:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arimoto K, Burkart C, Yan M, et al. Plakophilin‐2 promotes tumor development by enhancing ligand‐dependent and ‐independent epidermal growth factor receptor dimerization and activation. Mol Cell Biol 2014; 34:3843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kabiri Z, Greicius G, Madan B, et al. Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Development 2014; 141:2206–15. [DOI] [PubMed] [Google Scholar]
- 34. Gregorieff A, Wrana JL. Seeing is believing: Wnt3 localization in the gut epithelium. Cell Res 2016; 26:515–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hatzis P, van der Flier LG, van Driel MA, et al. Genome‐wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol Cell Biol 2008; 28:2732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen S, McLean S, Carter DE, et al. The gene expression profile induced by Wnt 3a in NIH 3T3 fibroblasts. J Cell Commun Signal 2007; 1:175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sobel K, Tham M, Stark HJ, et al. Wnt‐3a‐activated human fibroblasts promote human keratinocyte proliferation and matrix destruction. Int J Cancer 2015; 136:2786–98. [DOI] [PubMed] [Google Scholar]
- 38. Franke WW, Rickelt S, Barth M, et al. The junctions that don't fit the scheme: special symmetrical cell‐cell junctions of their own kind. Cell Tissue Res 2009; 338:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mertens C, Kuhn C, Franke WW. Plakophilins 2a and 2b: constitutive proteins of dual location in the karyoplasm and the desmosomal plaque. J Cell Biol 1996; 135:1009–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mertens C, Hofmann I, Wang Z, et al. Nuclear particles containing RNA polymerase III complexes associated with the junctional plaque protein plakophilin 2. Proc Natl Acad Sci USA 2001;98:7795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen X, Bonne S, Hatzfeld M, et al. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and β‐catenin signaling. J Biol Chem 2002; 277:10512–22. [DOI] [PubMed] [Google Scholar]
- 42. Khudiakov AA, Kostina DA, Kostareva AA, et al. The effect of plakophilin‐2 gene mutations on the activity of canonical Wnt signaling. Cell Tiss Biol 2016; 10:106–13. [PubMed] [Google Scholar]
- 43. Miyazaki K, Yoshizaki K, Arai C, et al. Plakophilin‐1, a novel Wnt signaling regulator, is critical for tooth development and ameloblast differentiation. PLOS One 2016; 11:e0152206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huber O, Petersen I. 150th Anniversary Series: Desmosomes and the Hallmarks of Cancer. Cell Commun Adhes 2015; 22:15–28. [DOI] [PubMed] [Google Scholar]
- 45. Mertens C, Kuhn C, Moll R, et al. Desmosomal plakophilin 2 as a differentiation marker in normal and malignant tissues. Differentiation 1999; 64:277–90. [DOI] [PubMed] [Google Scholar]
- 46. Schwarz J, Ayim A, Schmidt A, et al. Differential expression of desmosomal plakophilins in various types of carcinomas: correlation with cell type and differentiation. Hum Pathol 2006; 37:613–22. [DOI] [PubMed] [Google Scholar]
- 47. Papagerakis S, Shabana AH, Depondt J, et al. Immunohistochemical localization of plakophilins (PKP1, PKP2, PKP3, and p0071) in primary oropharyngeal tumors: correlation with clinical parameters. Hum Pathol 2003; 34:565–72. [DOI] [PubMed] [Google Scholar]
- 48. Takahashi H, Nakatsuji H, Takahashi M, et al. Up‐regulation of plakophilin‐2 and down‐regulation of plakophilin‐3 are correlated with invasiveness in bladder cancer. Urology 2012; 79:240.e1–8. [DOI] [PubMed] [Google Scholar]
- 49. Voloshanenko O, Erdmann G, Dubash TD, et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Comms 2013;4:2610,. doi: 10.1038/ncomms3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grossmann KS, Grund C, Huelsken J, et al. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol 2004; 167:149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pieperhoff S, Schumacher H, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. V. The importance of plakophilin‐2 demonstrated by small interference RNA‐mediated knockdown in cultured rat cardiomyocytes. Eur J Cell Biol 2008; 87:399–411. [DOI] [PubMed] [Google Scholar]
- 52. Gerull B, Heuser A, Wichter T, et al. Mutations in the desmosomal protein plakophilin‐2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 2004; 36:1162–4. [DOI] [PubMed] [Google Scholar]
- 53. Van Tintelen JP, Entius MM, Bhuiyan ZA, et al. Plakophilin‐2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2006; 113:1650–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Figure 5
Supporting Information Figure 6
Supporting Information Figure 7
Supporting Information Table 1
Supporting Information Table 2
Supporting Information Table 3