

Abstract
Objective
SB5 is a biosimilar agent for adalimumab (ADA). The aim of this study was to evaluate the efficacy, pharmacokinetics (PK), safety, and immunogenicity of SB5 in comparison with reference ADA in patients with rheumatoid arthritis (RA).
Methods
In this phase III, randomized, double‐blind, parallel‐group study, patients with moderately to severely active RA despite treatment with methotrexate were randomized 1:1 to receive SB5 or reference ADA at a dosage of 40 mg subcutaneously every other week. The primary efficacy end point was the response rate based on the American College of Rheumatology 20% improvement criteria (ACR20) at week 24 in the per‐protocol set (completer analysis). Additional end points included efficacy, PK, safety, and immunogenicity assessments.
Results
Of the 544 patients randomized to receive a study drug, the full analysis set comprised 542 patients (269 in the SB5 group, 273 in the reference ADA group) and the per‐protocol set comprised 476 patients (239 receiving SB5, 237 receiving reference ADA). The ACR20 response rate at week 24 in the per‐protocol set was equivalent between those receiving SB5 and those receiving reference ADA (72.4% and 72.2%, respectively); the difference in the ACR20 response rate (0.1%, [95% confidence interval −7.83%, 8.13%]) was within the predefined equivalence margin (±15%). Similar results were seen in the full analysis set (missing data being considered a nonresponse). The SB5 and reference ADA treatment groups were comparable across other end points, including the ACR 50% and ACR 70% improvement response rates, Disease Activity Score in 28 joints based on the erythrocyte sedimentation rate, PK data, incidence of treatment‐emergent adverse events, and the antidrug antibody response. Subgroup analyses showed that the efficacy and safety of SB5 and reference ADA were comparable regardless of antidrug antibody status.
Conclusion
The ACR20 response rate at week 24 was equivalent between patients treated with the biosimilar agent SB5 and those treated with reference ADA. SB5 and reference ADA were both well tolerated, with comparable safety profiles, in patients with RA.
Several biologic disease‐modifying antirheumatic drugs (DMARDs) targeted against tumor necrosis factor (TNF), such as adalimumab (ADA), certolizumab pegol, etanercept, golimumab, and infliximab 1, have been developed and approved for use worldwide in patients with rheumatoid arthritis (RA) and have yielded very positive clinical outcomes. The use of TNF inhibitors in combination therapy for the treatment of RA is recommended by the European League Against Rheumatism (EULAR) and the American College of Rheumatology (ACR) 1, 2.
Although biologic DMARDs, such as TNF inhibitors, have been used successfully for the treatment of RA, they are frequently associated with relatively high costs and substantial financial burden to patients and health care payers 3. The introduction of biosimilars offers the potential to reduce costs associated with biologic treatment and increase patient access to such therapies 3, 4, which should improve the sustainability of health care in RA.
SB5 (brand name Imraldi; Samsung Bioepis) has been approved by the European Commission as a biosimilar agent for ADA (Humira; AbbVie) (hereafter referred to as reference ADA) 5 for the treatment of RA, juvenile idiopathic arthritis, axial spondyloarthritis, psoriatic arthritis, psoriasis, pediatric plaque psoriasis, adult and adolescent hidradenitis suppurativa, Crohn's disease, pediatric Crohn's disease, ulcerative colitis, and uveitis. SB5 and the reference ADA have an identical amino acid sequence and similar physicochemical and in vitro functional properties 6. A phase I study of the pharmacokinetics (PK) of SB5 in 189 healthy individuals showed that the PK profile of the biosimilar product was equivalent to that of the reference ADA. Moreover, SB5 was well tolerated in the phase I study, with a safety profile similar to that of the reference ADA 6.
The objective of the current study was to analyze the efficacy, PK, safety, and immunogenicity of SB5 in comparison with reference ADA following 24 weeks of therapy in patients with RA whose disease remained moderately to severely active despite treatment with methotrexate (MTX).
Patients and Methods
Patients. Patients ages 18–75 years who had been diagnosed as having RA according to the ACR 1987 revised classification criteria 7, who had a disease duration of at least 6 months up to 15 years, and who had been treated with MTX for ≥6 months and had been receiving a stable dosage of MTX (10–25 mg/week) for ≥4 weeks before screening were eligible for the study. Additional requirements included the presence of active disease, defined as ≥6 swollen joints and ≥6 tender joints (from 66 and 68 joints evaluated, respectively) and either an erythrocyte sedimentation rate (ESR) of ≥28 mm/hour or serum C‐reactive protein (CRP) level of ≥1.0 mg/dl. Patients who were treated with biologic agents previously, had any known hypersensitivity to human immunoglobulin proteins or SB5 components, had an active or latent tuberculosis infection at the time of screening, had a serious infection, or had been treated with intravenous antibiotics within 8 weeks before randomization were excluded from the study. Additional eligibility criteria and exclusion criteria are listed in Supplementary Table 1 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40336/abstract). A complete list of all principal study investigators is shown in Appendix A.
Study design. This phase III multicenter, randomized, double‐blind, parallel‐group study was conducted in 51 sites in 7 countries (Bosnia and Herzegovina, Bulgaria, Czech Republic, Lithuania, Poland, Republic of Korea, and Ukraine) to evaluate the efficacy, PK, safety, and immunogenicity of SB5 in comparison with reference ADA in patients with moderately or severely active RA. This was a 52‐week study with a design that includes a single transition from the reference ADA to SB5 at week 24. Patients were initially randomized 1:1 to receive SB5 or reference ADA (40 mg subcutaneously every other week) (see Supplementary Figure 1 for details on the study design, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40336/abstract). At week 24, patients receiving reference ADA were randomized again in a 1:1 ratio to continue treatment with the reference ADA or to switch to the biosimilar agent SB5 up to week 52; patients receiving SB5 continued with SB5 for the 52 weeks of the study. Results up to 24 weeks are presented herein.
An interactive web response system (IWRS) was used to register patients, all of whom were assigned a unique patient number at the time of screening, and randomization was performed using a computer‐generated randomization scheme. Patients were randomized at the level of the study center, and patient codes generated by the IWRS were used for study drug allocation. Study drugs were prepackaged and labeled in a double‐blind manner, with identical appearance, packaging, and labeling.
As prespecified in the protocol, for the 24‐week interim report, the biostatistician, medical monitor, medical writer, pharmacologist, and safety monitoring personnel (all employees of Samsung Bioepis) were unblinded so that they could perform a formal analysis of the primary efficacy data; patients, investigators, joint assessors, and other study personnel remained blinded throughout the study. The overall randomization code was broken only for reporting purposes for this interim analysis, once all appropriate clinical data had been entered into the database, all data queries had been resolved, and the assignment of patients to the analysis sets had been completed.
The study was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation Guidelines for Good Clinical Practice. The study protocols were reviewed and approved by an independent ethics committee or institutional review board for each study center. Written informed consent was obtained from all patients before study entry.
Study end points. The primary end point of the study was the response rate at week 24 based on the ACR 20% improvement response criteria (ACR20) 8. Secondary end points at week 24 included the ACR 50% and ACR 70% improvement response rates, Disease Activity Score in 28 joints based on the ESR (DAS28‐ESR) 9, and EULAR response (good, moderate, or no response) 10. In addition, post hoc analyses were performed to determine the Clinical Disease Activity Index (CDAI) 11, Simplified Disease Activity Index (SDAI) 12, remission rates based on a DAS28‐ESR of <2.6, a SDAI score of ≤3.3, or a CDAI score of ≤2.8, and the proportion of patients with low disease activity based on a DAS28‐ESR of ≤3.2, a SDAI score of 3.3–11.0, or a CDAI score of 2.8–10.0. PK assessments, including determination of the serum trough concentration (Ctrough) up to 24 weeks, were performed in a subset of patients using blood samples collected from the PK population at multiple time points (0, 4, 8, 12, 16, and 24 weeks). The serum concentrations of the reference ADA or the biosimilar ADA were measured in a central laboratory (Covance Laboratories, North Yorkshire, UK) using an enzyme‐linked immunosorbent assay specific for ADA.
Safety assessments included monitoring for vital sign abnormalities, clinical laboratory abnormalities, adverse events (AEs; graded as mild, moderate, or severe), serious AEs, and treatment‐emergent AEs (TEAEs).
Immunogenicity assessments included monitoring for the development of antidrug antibodies and neutralizing antibodies. Immunogenicity was analyzed as either an “emergent” response (≥1 positive antidrug antibody result up to week 24 among patients negative for antidrug antibodies at baseline) or a “boosted” response (antidrug antibodies with increased titers at any time compared with baseline). Antidrug antibodies were detected using Meso Scale Discovery electrochemiluminescence bridging, in which an SB5 single‐tagged immunoassay was utilized. Antidrug antibody levels were measured at 0, 4, 8, 16, and 24 weeks.
Subgroup analyses of the 2 treatment groups were performed to further examine the ACR responses (ACR20, ACR50, and ACR70) and PK profiles by antidrug antibody status. In addition, the ACR20 response and TEAEs were summarized by age group (<65 years versus ≥65 years).
Statistical analysis. The equivalence margin in the current study was determined on the basis of findings in previous studies of ADA 13, 14 and regulatory guidelines 15, 16. A meta‐analysis of 2 previous studies 13, 14 estimated a risk difference of 0.424 (95% confidence interval [95% CI] 0.239, 0.609) with a random‐effects model, and a risk difference of 0.383 (95% CI 0.306, 0.461) with a fixed‐effects model, using the Mantel‐Haenszel weight. To preserve 50% of the effect of ADA over placebo, one‐half of the lower limit of the 95% CI for the treatment difference between ADA and placebo was calculated to be 0.12 (0.5 × 0.239) from a random‐effects model, and 0.15 (0.5 × 0.306) from a fixed‐effects model. A sample size of ≥245 patients per treatment group was required to achieve a significance level of 5% and a power of 80% for the primary analysis. Sample size was determined based on an equivalence margin of ±15%, an expected response rate of 63%, and a 12% dropout rate. The primary end point was analyzed for the difference between treatment groups, with 95% CIs, using the nonparametric covariance method, with geographic region used as stratification factor and baseline CRP level used as a covariate.
The efficacy outcomes of the ACR20, ACR50, and ACR70 response rates were analyzed in the full analysis set (FAS) and in the per‐protocol set (PPS). The PPS was the primary analysis set and included patients who completed the week 24 visit, had adherence in the range of 80–120% of the expected number of study drug and MTX doses, and were without any major protocol deviations that could affect the efficacy assessment. The FAS comprised all randomized patients (intent‐to‐treat principle); patients who were inadvertently randomized were excluded from this analysis, provided that they did not receive study drug. Supportive analysis was performed using an exponential time‐response model 17 with a prespecified equivalence margin to compare the ACR20 response between those receiving SB5 and those receiving reference ADA over the time course of the study.
Demographic and baseline characteristics were compared between the SB5 and reference ADA treatment groups using the chi‐square test for categorical variables or analysis of variance for continuous variables. Change in the DAS28‐ESR score from baseline to week 24 was deemed equivalent in the 2 treatment groups if the 2‐sided 95% CI of the treatment difference was contained within the equivalence margin of ±0.6. Descriptive statistics were used for the PK, safety, and immunogenicity analyses. Statistical analyses were performed using SAS version 9.2 or higher (SAS Institute).
Results
Patient characteristics and duration of exposure. The study was initiated on May 12, 2014, and the last patient visit for the 24‐week report was on May 8, 2015. The 24‐week results are presented herein. The mean duration of exposure at the 24‐week cutoff date was comparable between the SB5 and reference ADA groups (150.7 days versus 148.7 days). Of the 747 patients who were screened, 544 were randomized to receive either SB5 (n = 271) or reference ADA treatment (n = 273). Patient demographics and baseline disease characteristics were comparable between the SB5 and reference ADA groups (Table 1) with the exception of age, for which there was a statistically significant difference between the 2 groups (mean age 49.8 years in the SB5 group versus 52.5 years in the reference ADA group; P = 0.01). Differences in patient characteristics between the age group categories (<65 years versus ≥65 years), however, were not significant (P = 0.166). A total of 254 patients (93.7%) in the SB5 group and 254 patients (93.0%) in the reference ADA group completed 24 weeks of the study. The disposition of patients in each treatment group is depicted in Figure 1. The FAS population included 542 patients: 269 in the SB5 group (2 patients excluded on the basis of the definition of FAS) and 273 in the reference ADA group. The PPS population comprised 476 patients: 239 in the SB5 group and 237 in the reference ADA group.
Table 1.
Demographic and baseline clinical characteristics of the patients with moderate‐to‐severe rheumatoid arthritis in each treatment groupa
| Characteristic | SB5 (n = 271) | ADA (n = 273) |
|---|---|---|
| Mean ± SD age, years | 49.8 ± 12.67 | 52.5 ± 11.91 |
| Sex, no. (%) | ||
| Female | 217 (80.1) | 224 (82.1) |
| Male | 54 (19.9) | 49 (17.9) |
| Race, no. (%) | ||
| White | 271 (100) | 269 (98.5) |
| Asian | 0 (0.0) | 4 (1.5) |
| BMI, mean ± SD kg/m2 | 26.2 ± 4.76 | 27.0 ± 5.10 |
| Rheumatoid factor positive, no. (%) | 203 (74.9) | 185 (67.8) |
| Disease duration, mean ± SD years | 5.4 ± 4.4 | 5.5 ± 4.3 |
| Weekly methotrexate dose, mean ± SD mg | 15.13 ± 4.62 | 15.35 ± 4.41 |
| Swollen joint count, mean ± SD | 15.8 ± 8.03 | 15.5 ± 7.54 |
| Tender joint count, mean ± SD | 23.9 ± 11.69 | 24.1 ± 10.82 |
| CRP, mean ± SD mg/ml | 11.5 ± 19.04 | 12.6 ± 18.99 |
| ESR, mean ± SD mm/hour | 39.6 ± 13.27 | 39.6 ± 13.86 |
| HAQ DI score, mean ± SD | 1.3 ± 0.61 | 1.4 ± 0.64 |
| VAS score, mean ± SD mm | ||
| Pain | 59.2 ± 20.70 | 60.8 ± 19.71 |
| Patient's global assessment | 58.5 ± 20.29 | 59.4 ± 18.65 |
| Physician's global assessment | 59.8 ± 16.87 | 60.6 ± 15.38 |
| DAS28‐ESR, mean ± SD | 6.5 ± 0.74 | 6.5 ± 0.71 |
ADA = reference adalimumab; BMI = body mass index; CRP = C‐reactive protein; ESR = erythrocyte sedimentation rate; HAQ DI = Health Assessment Questionnaire disability index; VAS = visual analog scale; DAS28‐ESR = Disease Activity Score in 28 joints based on the ESR.
Figure 1.
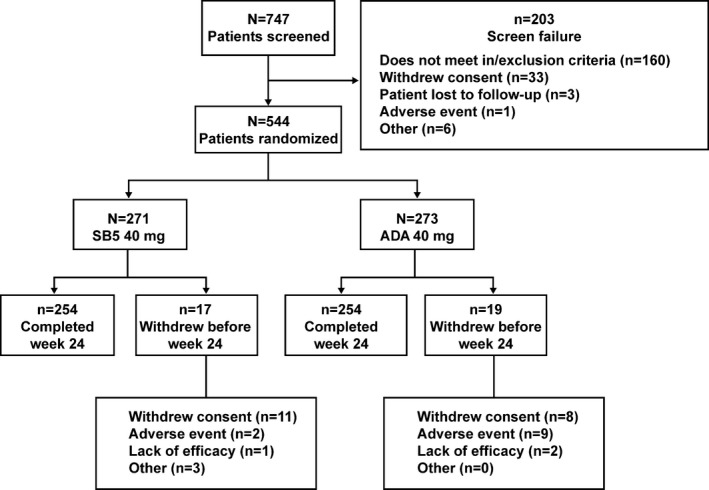
Disposition of the patients in each treatment group. ADA = reference adalimumab.
Efficacy assessments. The primary efficacy end point (the ACR20 response rate at week 24 in the PPS) was 72.4% (173 of 239 patients) in the SB5 group and 72.2% (171 of 237 patients) in the reference ADA group (Figure 2A). Similar responses were observed in the FAS population, with an ACR20 response rate of 68.0% (183 of 269 patients) in the SB5 group and 67.4% (184 of 273 patients) in the reference ADA group. The adjusted difference in the ACR20 response rate between the SB5 group and reference ADA group was 0.1% (95% CI −7.83%, 8.13%) in the PPS, and 0.8% (95% CI −7.03%, 8.56%) in the FAS, both of which were within the predefined equivalence margin (−15% to 15%). The secondary end points of the ACR50 and ACR70 response rates at week 24 in the PPS and FAS were also equivalent between the SB5 and reference ADA groups (Figures 2B and C).
Figure 2.
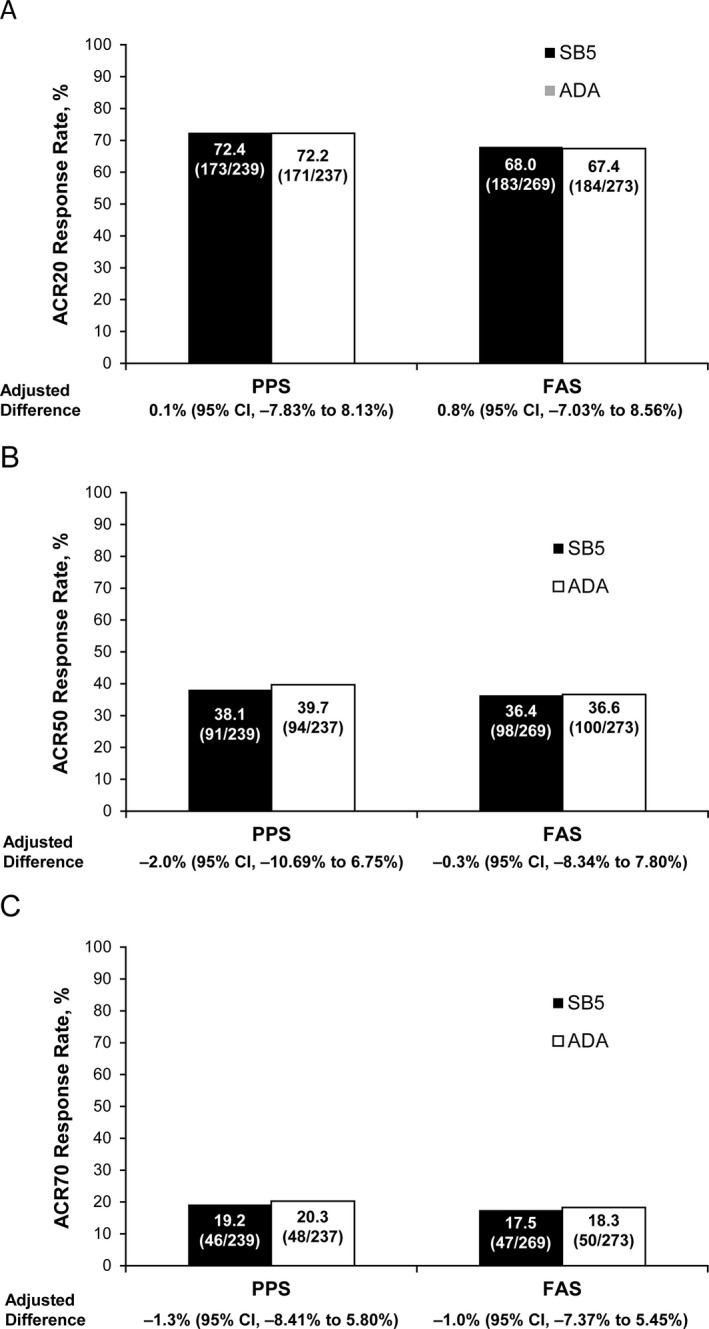
Response rates based on the American College of Rheumatology (ACR) improvement response criteria at week 24 in the per‐protocol set (PPS) and full analysis set (FAS). Patients receiving the biosimilar agent SB5 and those receiving the adalimumab (ADA) reference product were assessed for response rates and adjusted differences in response rates (with 95% confidence intervals [95% CIs]) according to the ACR criteria for 20% improvement (ACR20) (A), 50% improvement (B), and 70% improvement (C). Values shown are the percentage (no./total no.) of patients.
The time‐response curves for the ACR20 response were equivalent between the SB5 and reference ADA groups and support the robustness of the primary analysis (Figure 3). The between‐group difference in the ACR20 time‐response was 8.07 (95% CI −11.884, 28.022); the upper limit of the CI was lower than the prespecified equivalence margin of 64.31.
Figure 3.
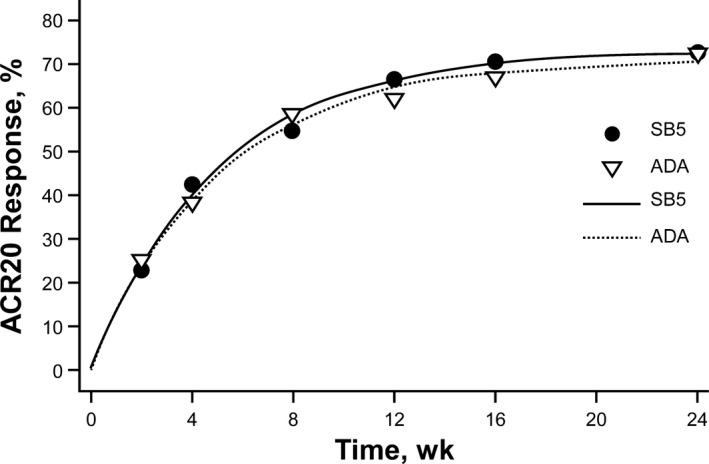
Time‐response curves of the American College of Rheumatology 20% improvement response (ACR20) in the per‐protocol set of patients treated with SB5 or reference adalimumab (ADA). The symbols in each curve represent the actual ACR20 responses in each treatment group at each visit, and the curve is fitted by nonlinear mixed models using an exponential time‐response model.
The mean change from baseline to week 24 in the DAS28‐ESR was comparable between the SB5 and reference ADA groups (−2.74 versus −2.68) (results in Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40336/abstract). The least squares mean for the treatment difference (SB5 versus reference ADA) in the DAS28‐ESR at week 24 was −0.04 (95% CI −0.26, 0.17), which was contained within the predefined equivalence margin (−0.6 to 0.6).
At week 24, a comparable proportion of patients in the SB5 and reference ADA groups had a good EULAR response (34.1% versus 34.6%) or moderate EULAR response (59.2% versus 58.8%). The mean change in disease activity scores from baseline to week 24 was comparable between the SB5 and reference ADA groups, both for the SDAI (−25.98 versus −25.00) and the CDAI (−25.39 versus −24.33) (see Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40336/abstract). Rates of remission and proportions of patients with low disease activity were also comparable between the SB5 and reference ADA groups, regardless of whether these rates were based on the DAS28‐ESR, the SDAI, or the CDAI (see Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40336/abstract).
Subgroup analyses based on the antidrug antibody status showed that the ACR responses were comparable between the SB5 and reference ADA subgroups of antidrug antibody–positive patients and antidrug antibody–negative patients (results in Supplementary Figures 2A–D, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40336/abstract). ACR improvement responses at week 24 were lower in antidrug antibody–positive patients treated with SB5 compared with antidrug antibody–negative patients treated with SB5, whereas there was a weaker correlation between the presence of antidrug antibodies and clinical efficacy in the reference ADA group. The ACR20 response rate at week 24 was comparable within each of the treatment groups when analyzed by age group category (<65 years versus ≥65 years), being 72.9% (156 of 214 patients) and 68.0% (17 of 25 patients), respectively, in the SB5 group and 72.3% (146 of 202 patients) and 71.4% (25 of 35 patients) in the reference ADA group.
Pharmacokinetic results. Analyses of the PK profiles in 356 patients (178 patients in each treatment group) showed that the mean Ctrough values up to week 24 were comparable between the SB5 and reference ADA treatment groups (Figure 4A). Subgroup analyses based on the antidrug antibody status showed lower Ctrough levels in the antidrug antibody–positive subgroups compared with the antidrug antibody–negative subgroups, regardless of treatment group assignment (Figure 4B).
Figure 4.
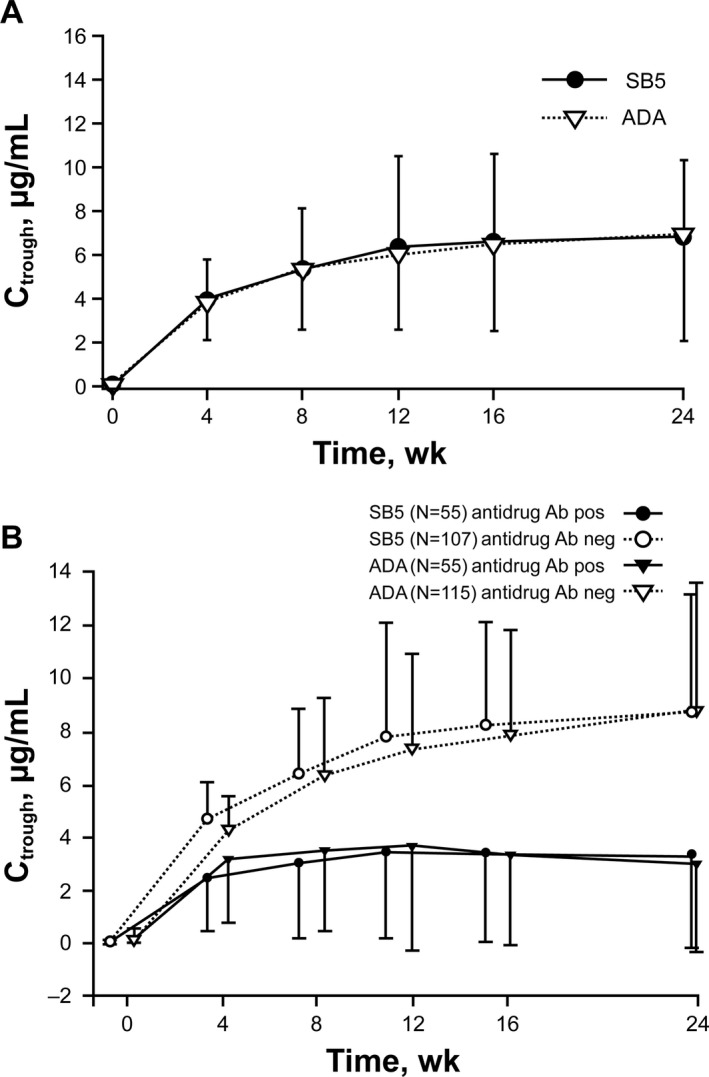
Serum trough concentration (Ctrough) of SB5 and reference adalimumab (ADA) at week 24 among patients in each treatment group (A) and among subsets of antidrug antibody–positive patients and antidrug antibody–negative patients within each treatment group (B). Bars show the mean ± SD at each time point.
Safety results. The TEAEs reported in the study were as expected for the population and drug class, and the majority were mild to moderate in severity. Overall, 35.8% of patients in the SB5 group and 40.7% in the reference ADA group experienced TEAEs up to week 24 (Table 2). The most common TEAEs were nasopharyngitis, headache, bronchitis, and an increase in alanine aminotransferase level. TEAEs considered related to the study drug were reported in 10.1% of SB5‐treated patients and 11.7% of reference ADA–treated patients. Serious TEAEs were reported in 3 patients (1.1%) in the SB5 group and 8 patients (2.9%) in the reference ADA group. None of the patients developed active tuberculosis. Other serious infections were reported in 1 patient (0.4%) in the SB5 group (Escherichia urinary tract infection) and 2 patients (0.7%) in the reference ADA group (bronchopneumonia and staphylococcal sepsis). The proportion of patients reporting injection site reactions was comparable between the SB5 and reference ADA groups (3.0% versus 2.9%). Up to week 24, the proportion of patients experiencing TEAEs leading to treatment discontinuation was lower in the SB5 group than in the reference ADA group (0.7% versus 3.7%). Two patients in the reference ADA group experienced malignancy (lymphoma and metastases to the spine in 1 patient, papillary thyroid cancer in 1 patient). There were 2 deaths that occurred up to week 24 (attributable to cardiac arrest and pulmonary embolism); both were reported to occur in patients in the reference ADA group and were not considered to be related to the study drug.
Table 2.
Safety findings in the study populationa
| Variable | SB5 (n = 268) | ADA (n = 273) |
|---|---|---|
| Patients with ≥1 TEAE | 96 (35.8) | 111 (40.7) |
| TEAEs reported in ≥2% of patients | ||
| Nasopharyngitis | 13 (4.9) | 25 (9.2) |
| Headache | 9 (3.4) | 7 (2.6) |
| Bronchitis | 7 (2.6) | 7 (2.6) |
| Increased ALT levels | 6 (2.2) | 8 (2.9) |
| Spinal pain | 6 (2.2) | 7 (2.6) |
| Nausea | 5 (1.9) | 6 (2.2) |
| Patients with ≥1 serious TEAE | 3 (1.1) | 8 (2.9) |
| Serious infections | 1 (0.4) | 2 (0.7) |
| Active tuberculosis | 0 (0.0) | 0 (0.0) |
| Injection site reactionsb | 8 (3.0) | 8 (2.9) |
| Malignancyc | 0 (0.0) | 2 (0.7) |
| Deathd | 0 (0.0) | 2 (0.7) |
Values are the number (%) of patients. ADA = reference adalimumab; TEAE = treatment‐emergent adverse event; ALT = alanine aminotransferase.
Defined as the high‐level group term of administration site reaction.
Lymphoma and metastases to the spine related to the study drug were reported in 1 patient, and papillary thyroid cancer not related to the study drug was reported in 1 patient.
The reported deaths were attributable to cardiac arrest in 1 patient and pulmonary embolism in 1 patient; neither was considered to be related to the study drug.
TEAEs were reported by 36.0% of patients ages <65 years and 34.5% of patients ages ≥65 years in the SB5 group, and by 40.3% of patients ages <65 years and 42.5% of patients ages ≥65 years in the reference ADA group. Similarly, the proportion of patients who experienced any serious TEAEs, as well as the most commonly reported TEAEs, were comparable within each treatment group between patients ages <65 years and those ages ≥65 years. All 3 serious infections occurred in patients who were <65 years of age.
Immunogenicity. The incidence of antidrug antibodies up to week 24 was comparable between the SB5 and reference ADA groups (33.1% versus 32.0%). At the same time point, emergent antidrug antibodies were reported in 32.4% of patients (80 of 247) in the SB5 group and 31.4% of patients (82 of 261) in the reference ADA group, and boosted antidrug antibody titers were found in 42.1% of patients (8 of 19) in the SB5 group and 50% of patients (4 of 8) in the reference ADA group. Approximately one‐half of the antidrug antibodies were found to be neutralizing in both treatment groups. The percentage of patients who were negative for neutralizing antibodies at baseline but who had ≥1 neutralizing antibody–positive result up to week 24 was comparable between the SB5 and reference ADA groups (13.6% versus 14.6%).
Discussion
These results demonstrate equivalent efficacy between the biosimilar agent SB5 and the reference ADA with regard to the primary end point (ACR20 response rate) and other efficacy end points. The ACR20 response rate observed in this study is similar to that demonstrated in other historical studies of ADA 13, 14, 18, 19 and similar to the findings reported for other ADA biosimilars in development 20, 21. The ACR20 time‐response model showed equivalent efficacy over multiple time points assessed during the study. In addition, secondary end points such as the ACR50 and ACR70 response rates, the DAS28‐ESR, and the EULAR responses were comparable between the SB5 and reference ADA groups.
The safety profile of SB5 was also comparable to that of reference ADA, with similar incidence rates of TEAEs, serious TEAEs, serious infections, and injection site reactions. Fewer patients in the SB5 group (2 patients [0.7%]) than in the reference ADA group (10 patients [3.7%]) experienced TEAEs leading to treatment discontinuation. Most of these TEAEs were considered to be related to the study drug.
Comparable immunogenicity with regard to the incidence of antidrug antibodies (treatment‐boosted and treatment‐emergent antidrug antibodies), as defined by the American Association of Pharmaceutical Scientist guidelines 22, was also demonstrated. It is known that the formation of antibodies to ADA is associated with increased clearance and reduced efficacy of ADA 5, 23. ACR responses at week 24 in the SB5 group were lower in antidrug antibody–positive patients compared with antidrug antibody–negative patients, but there was a weaker correlation between the presence of antidrug antibodies and clinical efficacy in the reference ADA treatment group, owing to the small number of antidrug antibody–positive patients and a fluctuation in the ACR responses.
The analyses of PK, which are generally considered to be more sensitive than clinical efficacy end points 24, showed a clear trend toward lower Ctrough levels in antidrug antibody–positive patients than in antidrug antibody–negative patients in both the SB5 and reference ADA groups. When efficacy was compared by Ctrough levels (≥1.274 μg/ml versus <1.274 μg/ml), which is considered the optimal cutoff trough level of ADA for achievement of a good EULAR response at week 24 25, there was a clear trend in both treatment groups toward higher efficacy in patients with higher Ctrough levels (≥1.274 μg/ml) compared with patients with lower Ctrough levels (<1.274 μg/ml).
Patient age had no notable effect on the efficacy results, which is consistent with previous findings showing that the efficacy of DMARD therapy in elderly patients with RA was comparable with that in younger patients 26. We also found no notable influence of patient age on treatment safety, although according to historical data on ADA, the frequencies of serious infections and malignancies have been found to be higher among ADA‐treated patients ages ≥65 years compared with those ages <65 years 5.
Several ADA biosimilars are currently in various stages of clinical development, with varying study designs 27. The current study was conducted to demonstrate equivalence in terms of clinical efficacy in a representative study population. The study was designed to be sensitive enough to detect potential differences in efficacy between SB5 and the reference ADA in accordance with European Medicines Agency (EMA) guidelines 28, 29. The study was not focused on demonstrating efficacy per se, because the efficacy of ADA in RA has already been well established. Among the approved therapeutic indications for ADA, RA has been the most thoroughly studied and validated, and reasonably sensitive methods to determine disease activity are available; therefore, examining the efficacy of SB5 in comparison with reference ADA in RA is an appropriate choice for demonstrating similarity in efficacy between the 2 products.
Based on the guidelines of the EMA 28 and the US Food and Drug Administration 24, demonstration of the safety and efficacy of treatment for one indication can be extrapolated to other indications when biosimilar comparability has been determined by thorough physicochemical and structural analyses and by in vitro functional tests complemented with clinical data (efficacy and safety data or PK/pharmacodynamic data). SB5 and the reference ADA were demonstrated to be highly comparable in a comprehensive similarity exercise using analytic procedures and specific biologic assays. No significant differences with regard to PK have been observed across indications for the marketed ADA product. The similarity of PK characteristics between SB5 and the reference ADA was confirmed in the most sensitive population, healthy individuals 6, and results of the present study provide additional supporting evidence of the PK profile of these agents in patients with moderate‐to‐severe RA. In terms of safety, the types and frequencies of AEs observed during preapproval studies of ADA were similar across indications.
Thus, the findings from this phase III multicenter, randomized, double‐blind, parallel‐group study showed equivalent efficacy between SB5 and the ADA reference product, as demonstrated by the ACR20 response rates at week 24 and other secondary efficacy end points at week 24. SB5 was well tolerated and possessed PK, safety, and immunogenicity profiles comparable to those of the reference ADA.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Weinblatt had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Weinblatt, Cheong, Ghil.
Acquisition of data
Baranauskaite, Niebrzydowski, Dokoupilova, Zielinska, Jaworski, Racewicz, Pileckyte, Jedrychowicz‐Rosiak, Cheong, Ghil.
Analysis and interpretation of data
Weinblatt, Cheong, Ghil.
Role of the Study Sponsor
Samsung Bioepis funded the study and facilitated the study design, provided writing assistance for the manuscript, and reviewed and approved the manuscript prior to submission. The authors independently collected the data, interpreted the results, and had the final decision to submit the manuscript for publication. Samsung Bioepis also funded the medical writing assistance provided by Beena John, PhD (Complete Healthcare Communications, Chadds Ford, PA; a CHC Group company). Publication of this article was not contingent upon approval by Samsung Bioepis.
Supporting information
Supplementary Table 1
Supplementary Table 2
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
The authors thank the patients who were involved in this study and the study personnel who made this work possible.
Appendix A. Principal Study Investigators
Principal study investigators, in addition to the authors of this article, are as follows: S. Sokolovic, M. Mekic, N. Prodanovic, B. Gajic, E. Karaselimovic‐Dzambasovic, and B. Pojskic (Bosnia and Herzegovina); A. Toncheva, P. Dimitar, L. Rodina, M. Geneva‐Popova, I. Staykov, and R. Stoilov (Bulgaria); L. Podrazilova, Z. Mosterova, G. Simkova, J. Kopackova, Z. Stejfova, J. Vencovsky, Z. Urbanova, L. Janska, and D. Galatikova (Czech Republic); S. Stropuviene and I. Sniuoliene (Lithuania); K. Sitek‐Ziolkowska, M. Rell‐Bakalarska, R. Kolasa, S. Daniluk, B. Sliwowska, M. Bartosik‐Twardowska, J. Brzezicki, M. Konieczny, and S. Jeka (Poland); J. Choe, S. Bae, and Y. Kang (Republic of Korea); and L. Prystupa, Z. Vyacheslav, I. Gasanov, R. Yatsyshyn, D. Rekalov, O. Iaremenko, M. Stanislavchuk, and V. Tseluyko (Ukraine).
ClinicalTrials.gov identifier: NCT02167139.
Supported by Samsung Bioepis.
Dr. Weinblatt has received consulting fees and/or honoraria from AbbVie, Amgen, Novartis, Roche, GlaxoSmithKline, Merck, Samsung, Crescendo Bioscience, and AstraZeneca (less than $10,000 each) and Bristol‐Myers Squibb, Lilly, Pfizer, and UCB (more than $10,000 each), and has received research funding from Amgen, Bristol‐Myers Squibb, Crescendo Bioscience, Sanofi, and UCB. Dr. Baranauskaite has received consulting fees (less than $10,000) and research support from Samsung Bioepis. Drs. Niebrzydowski, Dokoupilova, Zielinska, Jaworski, Racewicz, Pileckyte, and Jedrychowicz‐Rosiak have received research support from Samsung Bioepis.
References
- 1. Smolen JS, Landewe R, Breedveld FC, Buch M, Burmester G, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2013 update. Ann Rheum Dis 2014;73:492–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singh JA, Furst DE, Bharat A, Curtis JR, Kavanaugh AF, Kremer JM, et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease‐modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken) 2012;64:625–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dorner T, Strand V, Cornes P, Goncalves J, Gulacsi L, Kay J. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis 2016;75:974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hirsch BR, Lyman GH. Biosimilars: a cure to the U.S. health care cost conundrum? Blood Rev 2014;28:263–8. [DOI] [PubMed] [Google Scholar]
- 5. Humira (adalimumab) prescribing information. North Chicago (IL): AbbVie; 2016. [Google Scholar]
- 6. Shin D, Kim Y, Kim HS, Fuhr R, Körnicke T. A phase 1 pharmacokinetic study comparing SB5, an adalimumab biosimilar, and adalimumab reference product Humira. Ann Rheum Dis 2015;74 Suppl 2:459. [Google Scholar]
- 7. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 8. Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38:727–35. [DOI] [PubMed] [Google Scholar]
- 9. Prevoo ML, van ‘t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty‐eight–joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 10. Van Gestel AM, Prevoo ML, van ‘t Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis: comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism criteria. Arthritis Rheum 1996;39:34–40. [DOI] [PubMed] [Google Scholar]
- 11. Aletaha D, Nell VP, Stamm T, Uffmann M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther 2005;7:R796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smolen JS, Breedveld FC, Schiff MH, Kalden JR, Emery P, Eberl G, et al. A Simplified Disease Activity Index for rheumatoid arthritis for use in clinical practice. Rheumatology (Oxford) 2003;42:244–57. [DOI] [PubMed] [Google Scholar]
- 13. Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti–tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo‐controlled, 52‐week trial. Arthritis Rheum 2004;50:1400–11. [DOI] [PubMed] [Google Scholar]
- 14. Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, et al. Adalimumab, a fully human anti–tumor necrosis factor α monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum 2003;48:35–45. [DOI] [PubMed] [Google Scholar]
- 15. European Medicines Agency . Guideline on the choice of the non‐inferiority margin. July 2005. URL: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003636.pdf.
- 16. US Food and Drug Administration . Guidance for industry: non‐inferiority clinical trials. November 2016. URL: http://www.fda.gov/downloads/Drugs/Guidances/UCM202140.pdf.
- 17. Emery P, Vencovsky J, Sylwestrzak A, Leszczynski P, Porawska W, Baranauskaite A, et al. A phase III randomised, double‐blind, parallel‐group study comparing SB4 with etanercept reference product in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis 2017;76:51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, van Vollenhoven R, et al. The PREMIER study: a multicenter, randomized, double‐blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 2006;54:26–37. [DOI] [PubMed] [Google Scholar]
- 19. Weinblatt ME, Keystone EC, Furst DE, Kavanaugh AF, Chartash EK, Segurado OG. Long term efficacy and safety of adalimumab plus methotrexate in patients with rheumatoid arthritis: ARMADA 4 year extended study. Ann Rheum Dis 2006;65:753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cohen S, Genovese MC, Choy E, Perez‐Ruiz F, Matsumoto A, Pavelka K, et al. Efficacy and safety of the biosimilar ABP 501 compared with adalimumab in patients with moderate to severe rheumatoid arthritis: a randomised, double‐blind, phase III equivalence study. Ann Rheum Dis 2017;76:1679–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jani RH, Gupta R, Bhatia G, Rathi G, Ashok Kumar P, Sharma R, et al. A prospective, randomized, double‐blind, multicentre, parallel‐group, active controlled study to compare efficacy and safety of biosimilar adalimumab (Exemptia; ZRC‐3197) and adalimumab (Humira) in patients with rheumatoid arthritis. Int J Rheum Dis 2016;19:1157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shankar G, Arkin S, Cocea L, Devanarayan V, Kirshner S, Kromminga A, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides‐harmonized terminology and tactical recommendations. AAPS J 2014;16:658–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long‐term follow‐up. JAMA 2011;305:1460–8. [DOI] [PubMed] [Google Scholar]
- 24. US Food and Drug Administration . Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. April 2015. URL: http://www.fda.gov/downloads/DrugsGuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf.
- 25. Chen DY, Chen YM, Tsai WC, Tseng JC, Chen YH, Hsieh CW, et al. Significant associations of antidrug antibody levels with serum drug trough levels and therapeutic response of adalimumab and etanercept treatment in rheumatoid arthritis. Ann Rheum Dis 2015;74:e16. [DOI] [PubMed] [Google Scholar]
- 26. Koller MD, Aletaha D, Funovits J, Pangan A, Baker D, Smolen JS. Response of elderly patients with rheumatoid arthritis to methotrexate or TNF inhibitors compared with younger patients. Rheumatology (Oxford) 2009;48:1575–80. [DOI] [PubMed] [Google Scholar]
- 27. Lai Z, La Noce A. Key design considerations on comparative clinical efficacy studies for biosimilars: adalimumab as an example. RMD Open 2016;2:e000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. European Medicines Agency . Guideline on similar biological medicinal products containing biotechnology‐derived proteins as active substance: non‐clinical and clinical issues. December 2014. URL: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf.
- 29. European Medicines Agency . Guideline on similar biological medicinal products containing monoclonal antibodies: non‐clinical and clinical issues. May 2012. URL: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1
Supplementary Table 2
Supplementary Figure 1
Supplementary Figure 2