

Abstract
Objective
Cardiac diseases are established risk factors for ischemic stroke incidence and severity. Conversely, there is increasing evidence that brain ischemia can cause cardiac dysfunction. The mechanisms underlying this neurogenic heart disease are incompletely understood. Although it is established that ischemic stroke is associated with cardiac arrhythmias, myocardial damage, elevated cardiac enzymes, and plasma catecholamines in the acute phase, nothing is known about the delayed consequences of ischemic stroke on cardiovascular function.
Methods
To determine the long‐term cardiac consequences of a focal cerebral ischemia, we subjected young and aged mice to a 30‐minute transient middle cerebral artery occlusion and analyzed cardiac function by serial transthoracic echocardiography and hemodynamic measurements up to week 8 after surgery. Finally, animals were treated with metoprolol to evaluate a pharmacologic treatment option to prevent the development of heart failure.
Results
Focal cerebral ischemia induced a long‐term cardiac dysfunction with a reduction in left ventricular ejection fraction and an increase in left ventricular volumes; this development was associated with higher peripheral sympathetic activity. Metoprolol treatment prevented the development of chronic cardiac dysfunction by decelerating extracellular cardiac remodeling and inhibiting sympathetic signaling relevant to chronic autonomic dysfunction.
Interpretation
Focal cerebral ischemia in mice leads to the development of chronic systolic dysfunction driven by increased sympathetic activity. If these results can be confirmed in a clinical setting, treating physicians should be attentive to clinical signs of heart failure in every patient after ischemic stroke. Therapeutically, the successful β‐blockade with metoprolol in mice could also have future clinical implications. Ann Neurol 2017;82:729–743
It is well established that cardiac diseases, such as atrial fibrillation, valvular heart disease, or congestive heart failure, are important risk factors for ischemic stroke (IS).1 In population‐based studies, about 30% of IS are caused by cardiac diseases.2, 3 However, there is accumulating evidence that this relationship is not monodirectional and cerebrovascular disorders can conversely alter cardiovascular and autonomic function.4, 5, 6 IS has been associated with changes in autonomic cardiac dynamics, cardiac arrhythmias, acute myocardial damage, elevated cardiac enzymes (troponin, brain natriuretic peptide [BNP]) and plasma catecholamines, and increased susceptibility to sudden death both in humans and in laboratory animals.7, 8, 9 Within the first 3 months after IS, about 19% of patients have a fatal or serious nonfatal cardiac event, including acute myocardial infarction or ventricular tachycardia, and this mortality cannot be entirely explained by concomitant cardiac disease.5 Rather, cerebral ischemia may directly contribute to the generation of cardiac dysfunction via an alteration of the balance of the sympathetic and parasympathetic tone.10, 11 Whereas most of the previous studies on neurogenic heart dysfunction focused on the acute or subacute stage after stroke, with a special emphasis on myocardial infarction, cardiac arrhythmias, or the occurrence of early stress cardiomyopathy (Takotsubo syndrome), very little is known about the delayed consequences of ischemic brain damage on cardiovascular function, in particular the evolvement of chronic cardiac failure.8, 12 As potential cardiac dysfunction upon brain ischemia may prompt clinicians to perform long‐lasting cardiac monitoring or even start cardioprotective medication, a detailed mechanistic characterization of the long‐term consequences of IS on cardiac function is necessary.
In this longitudinal study (Stroke‐Induced Cardiac FAILure in mice and men [SICFAIL]), we evaluated long‐term cardiac function in mice subjected to focal IS by transient middle cerebral artery occlusion (tMCAO). Finally, we assessed whether β‐blocker treatment protects from chronic cardiac dysfunction.
Materials and Methods
Animals
In this study, 6‐ to 8‐week‐old (n = 159) or 1‐year‐old (n = 18) male C57BL/6J mice were used. Animal experiments were approved by the legal state authorities and performed according to the recommendations for research in experimental stroke studies and the current Animal Research: Reporting of In Vivo Experiments guidelines.13, 14
Ischemia Model
Focal cerebral ischemia was induced by a 30‐minute tMCAO, as previously described.15 Briefly, mice were anesthetized with 2% isoflurane in O2. A servo‐controlled heating blanket was used to maintain core body temperature close to 37 °C throughout surgery. After a midline neck incision, a standardized silicone rubber–coated No. 6.0 nylon monofilament (6023910PK10; Doccol, Sharon, MA) was inserted into the right or the left common carotid artery and advanced via the internal carotid artery to occlude the origin of the middle cerebral artery. After 30 minutes, mice were reanesthetized, and the occluding filament was removed to allow reperfusion. All animals were operated on by the same experimenter to reduce infarct variability; operation time per animal did not exceed 15 minutes. Stroke volumes and right hemispheric atrophy (left hemispheric volume − right hemispheric volume) were assessed 24 hours and 8 weeks, respectively, after tMCAO, based on 2,3,5‐triphenyl tetrazolium chloride (TTC) staining. Mice were randomly assigned to the operator by an independent researcher who was not involved in data analysis. Investigators involved in the surgery and evaluation of all readout parameters were blinded to the experimental groups.
Assessment of Functional Outcome
The adhesive removal test was used to monitor neurologic function over the whole time period (8 weeks).16 For this, we placed small pieces of adhesive tape (5 × 5mm) on the forepaws of the mice, and put the animals into a test cylinder. Two time measurements were taken: (1) the time to contact the contralesional paw and the adhesive and (2) the time to remove the adhesive on the contralesional side (maximum 120 seconds). In our study, we focused on the time to removal and did not assess the time to contact. A training period of 5 days before surgery obtained an optimal level of performance and limited individual variations. After surgery, the test was performed regularly every week on the same day of the week and at the same time of day.
Pharmacologic Treatment
Metoprolol (2.5mg/kg/day; M5391, Sigma‐Aldrich, St Louis, MO) was administered orally via the drinking water.17 In pilot experiments, we assessed the daily drinking water intake of mice housed under standard conditions in our animal unit (3ml/day). Mice were weighed every week, and the concentration of metoprolol in the drinking water was adjusted accordingly. Treatment was initiated directly after the surgery and continued for 8 weeks.
Echocardiography
Cardiac function was assessed with echocardiography before tMCAO and in weeks 1, 3, and 8 after tMCAO. Echocardiography was performed and evaluated as previously described.18 Serial cardiac ultrasound analyses were performed on a VisualSonics Vevo 1100 imaging system (Fujifilm, Tokyo, Japan) with a 30MHz ultrasound probe under light anesthesia with isoflurane (1.5 vol%) and spontaneous respiration by a single researcher experienced in rodent echocardiography who was blinded to the treatment group. From 2‐dimensional short‐axis imaging, endocardial borders were traced at end systole and end diastole utilizing a prototype offline analysis system (Vevo LAB 1.7.0, Fujifilm VisualSonics). Measurements were performed at the midpapillary muscle level. The end‐systolic (smallest) and end‐diastolic (largest) cavity areas were determined. From 2‐dimensionally targeted M‐mode tracings, end‐diastolic and end‐systolic diameters were measured and fractional shortening ([(end‐diastolic diameter − end‐systolic diameter)/end‐diastolic diameter] × 100) was calculated.
Left Ventricular Catheterization
Hemodynamic measurements were performed with the open chest method in week 8 according to published protocols.19 The chest of the anesthetized (2% isoflurane in O2), fixed, and intubated animals was entered through the midriff and a small stitch was made at the left ventricular (LV) apex, leaving the pericardium as intact as possible. The 1.4F micromanometer‐tipped pressure/volume catheter (Millar Instruments, Houston, TX) was then moved carefully into the LV along the cardiac longitudinal axis under continuous monitoring of the pressure/volume waveform. With the catheter fixed in place, terminal measurements of LV performance assessed by ejection fraction, heart rate, end‐systolic and end‐diastolic volumes and pressures, and maximal pressure increase and decrease rates were performed in 1% isoflurane‐anesthetized mice.
Determination of Plasma BNP, Epinephrine, Norepinephrine, and Cortisol Levels
Blood plasma was collected from the retro‐orbital venous plexus under short isoflurane inhalation (2% in O2)20 and stored at −80 °C until BNP, catecholamine, and cortisol concentrations were determined. Plasma BNP levels were measured using a commercially available kit from Sigma‐Aldrich (RAB0386, St. Louis, MO). Plasma epinephrine, norepinephrine, and cortisol levels were measured using the 2‐CAT (A‐N) Research/Cortisol ELISA kit (BA E‐6500/MS E‐5000; Labor Diagnostika Nord, Nordhorn, Germany) following the manufacturer's protocol.
Quantitative Real‐Time Polymerase Chain Reaction
Quantitative real‐time polymerase chain reaction (PCR) analysis was performed according to standard procedures.21 The following primers were purchased from Applied Biosystems (Foster City, CA) or Thermo Fisher Scientific (Waltham, MA): BNP (Mm01255770_g1), tumor necrosis factor α (TNF‐α; Mm00443258_m1), matrix metallopeptidase‐9 (MMP‐9; Mm00442991_m1), tyrosine hydroxylase (TH; Mm00447557_m1), and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; TaqMan Predeveloped Assay Reagents for gene expression, part number: 4352339E). GAPDH was used as an endogenous control. PCR was performed with equal amounts of cyclic deoxyribonucleic acid and water control in the StepOnePlus Real‐Time PCR System (Applied Biosystems) using the TaqMan Universal 2X PCR Master Mix (Applied Biosystems). Each sample was measured in triplicate, and data points were examined for integrity by analysis of the amplification plot. The comparative cycle threshold method was used for relative quantification of gene expression as described elsewhere.22
Histology
Picrosirius red staining of paraffin‐embedded LV tissue sections (7 µm) was performed according to standard procedures.23 Tissue collagen content was analyzed using fluorescence microscopy (Z1m Imager, Carl Zeiss, Oberkochen, Germany) as previously described.24
Nissl‐stained cryoembedded brain slices (10 µm) were analyzed and acquired with a DMi8 microscope equipped with the DMC 2900 camera control unit and LAS X software (Leica, Wetzlar, Germany).
Protein Extraction and Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis Analysis
Sample preparation and high‐resolution protein gel electrophoresis for myosin heavy chain (MHC) analysis were executed as previously described.25 After 30 hours, the gels were fixed and silver‐stained. A gel documentation system (Bio‐Rad, Hercules, CA) was used to scan the stained gels.
Small Animal 18F‐Fluorodeoxyglucose Positron Emission Tomography Imaging
18F‐Fluorodeoxyglucose (18F‐FDG) was synthesized in house with a 16MeV Cyclotron (GE PETtrace 6; GE Healthcare, Milwaukee, WI) using GE FASTlab methodology according to the manufacturer's instructions. Before use, radiochemicals were analyzed by high‐performance liquid chromatography for radiochemical identity and purity. A dedicated small‐animal positron emission tomography (PET) scanner (Inveon; Siemens, Erlangen, Germany) was used to verify the location of injured area in the brain induced by tMCAO on 18F‐FDG imaging.26 All animals were maintained under anesthesia with 2% isoflurane during PET image acquisition. 18F‐FDG (30MBq) was injected intraperitoneally into the tMCAO mice (n = 10) and sham animals (n = 10) after a 16‐hour fasting period to minimize physiologic cardiac glucose uptake. One hour after tracer injection, data acquisition was initiated for a period of 7 minutes. Data were reconstructed using the ordered subset expectation maximization 2D algorithm with 16 subsets and 4 iterations. PET images were analyzed by AMIDE‐bin, version 1.0.2. Same‐size regions of interest for tMCAO and sham‐operated groups were drawn manually on images of the horizontal brain sections both ipsilateral and contralateral to the occlusion site. The mean concentration of radioactivity within the regions of interest was expressed as the percentage injected dose per tissue cubic centimeter.
Statistics
All results are expressed as mean ± standard error of the mean. The numbers of animals (n = 9) necessary to detect a standardized effect size on the fractional shortening and the ejection fraction ≥ 0.15% were determined via a priori sample size calculation with the following assumptions: α = 0.05, β = 0.2 (power 80%), mean 20% standard deviation of the mean (GraphPad Stat Mate 2.0; GraphPad Software, San Diego, CA). For statistical analysis, the GraphPad Prism 6 software package was used. Data were tested for Gaussian distribution with the D'Agostino‐Pearson omnibus normality test and then analyzed by 1‐way analysis of variance (ANOVA), or in the case of measuring the effects of 2 factors simultaneously, by 2‐way ANOVA with post hoc Bonferroni adjustment for p values. Nonparametric functional outcome scores were compared by Kruskal–Wallis test with post hoc Dunn corrections. If only 2 groups were compared, an unpaired, 2‐tailed Student t test, or in the case of nonparametric functional outcome, the Wilcoxon–Mann–Whitney test, was applied. For comparison of survival curves, the log‐rank test was used. Probability values < 0.05 were considered to indicate statistically significant results.
Results
Focal Cerebral Ischemia Leads to Chronic Systolic Dysfunction
Before analyzing cardiac function, we assessed the extent of hypoxic neurodegeneration after a 30‐minute tMCAO. For this, brain samples were stained with TTC 24 hours after the 30‐minute right hemispheric tMCAO occlusion. In line with other studies using this occlusion time, we found rather small infarcts that were, in most cases, restricted to the basal ganglia, and more importantly, also to the insular cortex (Fig 1). In comparison, there was no visible tissue damage in the brain slices of control (ctrl; ie, no interventional treatment) and sham‐operated mice. Importantly, 8 weeks after surgery, decreased 18F‐FDG uptake was visible in the infarct region compared with sham‐treated animals, reflecting tissue damage (p < 0.001). This indicates that the ischemic lesion influenced metabolic brain function over the whole follow‐up time. Accordingly, functional analysis of the animals using the adhesive removal test also indicated functional impairment of the tMCAO group versus the sham‐operated mice over the whole observation time (p < 0.05). Although the survival rate of the tMCAO‐treated animals showed a 20% reduction (80%) compared with sham‐operated and control animals (100%), mortality was only seen between days 1 and 7 after surgery, indicating a direct stroke dependence. Furthermore, 8 weeks after surgery a strong correlation (Pearson r = 0.88, p < 0.001) between the right hemispheric atrophy and the ejection fraction levels was observed by which low ejection fraction level correlated with high atrophy and further damaged tissue in the insular area.
Figure 1.
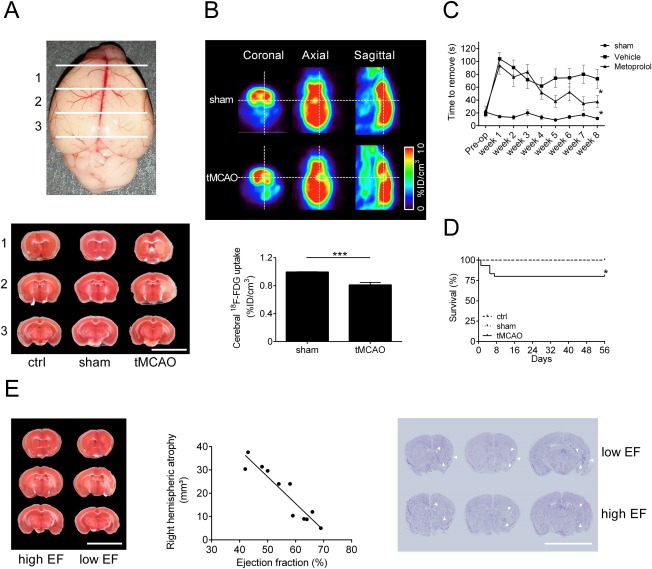
Infarct volumes and functional outcome 24 hours and 8 weeks after focal cerebral ischemia. (A) Upper panel: schematic overview of coronal brain sections. Lower panel: representative 2,3,5‐triphenyl tetrazolium chloride (TTC) stains of 3 corresponding coronal brain sections of control (ctrl), sham‐operated, and transient middle cerebral artery occlusion (tMCAO)‐treated C57BL/6J mice euthanized on day 1 after tMCAO (scale bar = 10mm). (B) Upper panel: representative coronal, horizontal, and sagittal 18F‐fluorodeoxyglucose (18F‐FDG) positron emission tomography images of sham‐operated and tMCAO‐treated mice 8 weeks after tMCAO. Lower panel: densitometric quantification of cerebral 18F‐FDG uptake (n = 10 per group; ***p < 0.001; %ID = percentage injected dose). (C) Time to remove the adhesive tape from the contralesional forepaw of sham‐operated, vehicle (tMCAO)‐treated, and metoprolol (tMCAO + β‐blocker)‐treated mice (n = 10 per group; *p < 0.05). (D) Survival rate of ctrl, sham‐operated, and tMCAO‐treated mice until day 56 after surgery (n = 15 per group; *p < 0.05). (E) Left panel: representative TTC stains of 3 corresponding coronal brain sections of 2 tMCAO‐treated C57BL/6J mouse with a low/high ejection fraction (EF) euthanized on week 8 after tMCAO (scale bar = 10mm), showing high brain atrophy in mice with low EF. Middle panel: right hemispheric atrophy quantified by TTC staining correlated with left ventricular ejection fraction (n = 11, correlation Pearson r = 0.88, p < 0.001). Right panel: representative comparison of Nissl‐stained anterior brain lesion areas (arrowheads) of tMCAO‐treated mice with high EF (no/less insular damage) and low EF (insular damage; scale bar = 10mm).
In a second step, we analyzed the impact of right and left hemispheric tMCAO on cardiac function. For this, we investigated C57BL/6J mice treated with a 30‐minute tMCAO and compared them with sham‐operated and control animals as negative controls. There were no statistically significant differences in the LV ejection fraction, heart rate, heart/lung weight to body weight ratio, volumes, pressures, or maximal LV contractility and relaxation between the 2 control groups and animals treated with a left hemispheric stroke 8 weeks after the surgery (Fig 2). In contrast, right hemispheric tMCAO‐treated mice showed a significant difference in LV ejection fraction and an increase in LV end‐systolic and end‐diastolic volumes, indicative of the development of a mild cardiac dysfunction (p < 0.001; ejection fraction: sham, p < 0.05; tMCAO [left], p < 0.01; ctrl, p < 0.001). Also at this time point, right hemispheric tMCAO‐treated animals presented a significant heart rate elevation, an increase in the heart/lung weight to body weight ratio, and an increase in the end‐diastolic pressure, whereas LV end‐systolic pressure showed no differences (p < 0.001; heart rate: sham, p < 0.05; ctrl, p < 0.01; heart weight/body weight ratio: ctrl/sham, p < 0.05; tMCAO [left], p < 0.01). Furthermore, we found a progressive decline in LV maximal pressure increase/decrease rate and fractional shortening over the 8‐week follow‐up period (p < 0.05), confirming the previous results. These findings strongly support the hypothesis that a right hemispheric tMCAO can induce a chronic cardiac dysfunction in mice.
Figure 2.
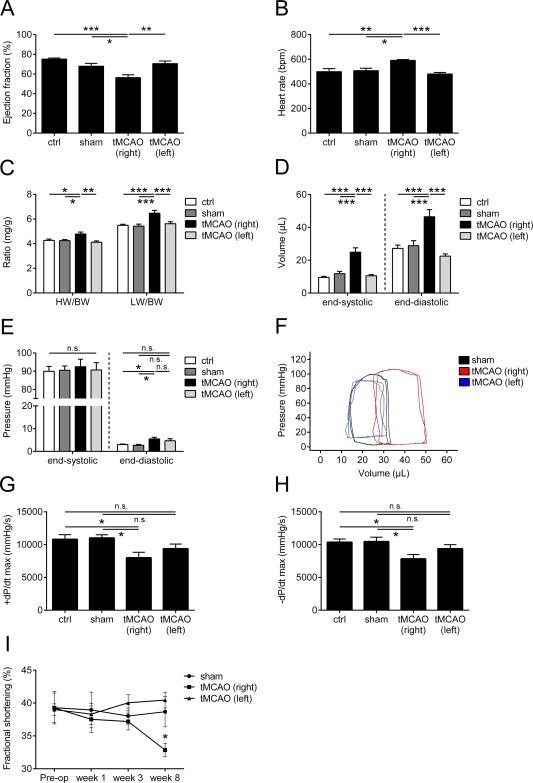
Development of chronic cardiac dysfunction after right and left transient middle cerebral artery occlusion (tMCAO). (A–H) Hemodynamic parameters (ejection fraction (A), heart rate (B), heart/lung weight (HW/LW) to body weight (BW) ratio (C), end‐systolic and end‐diastolic left ventricular (LV) volume (D), end‐systolic and end‐diastolic LV pressure (E), representative pressure–volume loops in sham (black) and right (red) and left (blue) tMCAO‐treated mice (F), and maximal LV pressure increase/decrease rate (+dP/dt, −dP/dt, respectively; G, H)) assessed 8 weeks after tMCAO. (I) Natural course of fractional shortening in sham‐operated and right and left tMCAO‐treated C57BL/6J mice (n = 10 or 11 animals per group for all experiments; *p < 0.05, **p < 0.01, ***p < 0.001; ctrl = control; n.s. = not significant).
Translation of preclinical experimental stroke data to the clinical setting is disappointing.27 One reason for this is the use of young and healthy animals for rodent studies, whereas the typical stroke patient is aged and comorbid. International guidelines on the conduction of animal stroke studies recommend the analysis of aged animals.14, 28 Therefore, to increase the translational validity, we repeated the key experiment of our study with aged animals (1‐year‐old C57BL/6J mice). After 8 weeks, right hemispheric tMCAO‐treated mice showed a significant decrease in LV ejection fraction as an indicator of the development of a mild cardiac dysfunction (Fig 3; p < 0.05). Accordingly, fractional shortening declined after week 3 of the follow‐up period (p < 0.05). Furthermore, heart rate and heart/lung weight to body weight ratio presented a significant increase in tMCAO‐treated mice compared with sham‐operated animals (p < 0.05; lung weight/body weight ratio, p < 0.01). These results indicate that the development of cardiac dysfunction after tMCAO is a common effect independent of age, which means that it is not restricted to young and healthy animals, but is also relevant in a clinically meaningful setting. All further analyses were therefore performed with tissue from animals treated with a right hemispheric stroke.
Figure 3.
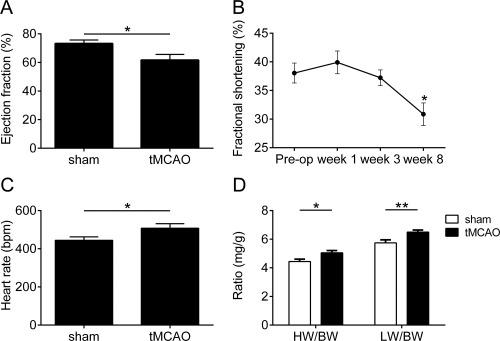
Chronic cardiac dysfunction development after transient middle cerebral artery occlusion (tMCAO) in aged mice. (A) Ejection fraction assessed by hemodynamics 8 weeks after right tMCAO. (B) Natural course of fractional shortening of old C57BL/6J mice treated with right tMCAO. (C, D) Heart rate (C) and heart weight (HW)/lung weight (LW) to body weight (BW) ratio (D) assessed by hemodynamics 8 weeks after tMCAO (n = 9 animals per group for all experiments; *p < 0.05, **p < 0.01).
Focal Cerebral Ischemia is Associated with the Increase of Biomarkers for Cardiac Dysfunction and Slow Cardiac Remodeling
To evaluate the pathologic significance of the development of cardiac dysfunction after right hemispheric tMCAO, we next analyzed LV tissue of young animals for biomarkers of cardiac dysfunction and remodeling 8 weeks after the surgery. IS led to an upregulation of BNP (p < 0.01) and TNF‐α (p < 0.05) gene expression 8 weeks after the surgery (Fig 4). Also, the sham operation led to elevated TNF‐α gene expression levels. In addition, mRNA levels of MMP‐9, a proteolytic enzyme involved in the remodeling of the extracellular matrix, were increased (p < 0.05). As further confirmation of the echocardiographic and hemodynamic findings, plasma BNP levels, as a typical biomarker for cardiac dysfunction, were upregulated in tMCAO‐treated mice compared with controls (sham, p < 0.05; ctrl, p < 0.001). We analyzed the MHC protein isoform content, because the relative proportions of α‐ and β‐MHC have been shown to be an independent indicator of the molecular response of the sarcomere to various external and internal stimuli.25 In contrast to the group without any treatment, β‐MHC was detectable in the tMCAO and sham‐operated groups (sham, p < 0.01; tMCAO, p < 0.001).29 Accumulation of β‐MHC was significantly higher in the tMCAO compared with the sham group (p < 0.01). As myocardial fibrosis plays a role in the development of chronic cardiac dysfunction, we evaluated the extracellular remodeling and measured the collagen content in the LV tissue. In marked contrast to the control groups, tMCAO‐treated animals presented a significant increase in LV collagen (p < 0.001). These data suggest that the development of chronic cardiac dysfunction after tMCAO is causally linked to the elevation of biomarkers for cardiac dysfunction and extracellular remodeling processes, such as β‐MHC expression and slow LV fibrosis.
Figure 4.
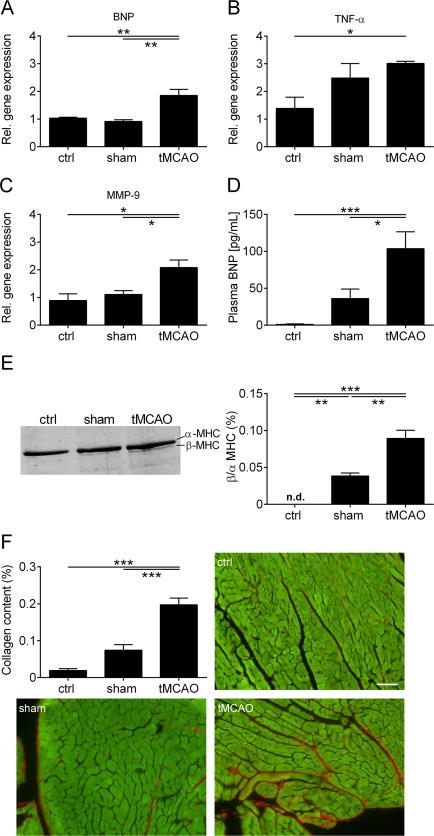
Increase of biomarkers for cardiac dysfunction and slow left ventricular (LV) remodeling after right transient middle cerebral artery occlusion (tMCAO). (A–C) Relative (Rel.) gene expression of brain natriuretic peptide (BNP) (A), tumor necrosis factor α (TNF‐α) (B), and matrix metallopeptidase‐9 (MMP‐9) (C) in the LV tissue of control (ctrl), sham‐operated, and tMCAO‐treated mice 8 weeks after surgery (n = 4 or 5 per group; *p < 0.05, **p < 0.01). (D) Plasma BNP levels from ctrl, sham‐operated, and tMCAO‐treated mice 8 weeks after tMCAO (n = 8–12 per group; *p < 0.05, ***p < 0.001). (E) Left panel: representative silver‐stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis of α‐ and β‐myosin heavy chain (MHC) distribution in the left ventricle of ctrl, sham‐operated, and tMCAO‐treated mice 8 weeks after surgery. Right panel: densitometric quantification (n = 4 per group; **p < 0.01, ***p < 0.001; n.d. = not detected). (F) Densitometric quantification and representative images of the collagen (red) content in LV tissue (green) sections (7 µm) using picrosirius red staining (n = 4 or 5 per group; scale bar = 50 µm; ***p < 0.001).
Chronic Systolic Dysfunction after Focal Cerebral Ischemia Is Associated with Increased Catecholamine and Cortisol Levels in the Periphery
Because a right insular stroke is associated with an acute and prolonged sympathetic stimulation,9, 30, 31 we next evaluated the impact of the autonomic nervous system on the development of chronic cardiac dysfunction after right hemispheric tMCAO. For this purpose, we analyzed the brain tissue mRNA levels of TH, which is the rate‐limiting enzyme in catecholamine synthesis. Eight weeks after surgery, there were no statistically significant differences in the brain cortex (p > 0.05), but in the basal ganglia (p < 0.05) and LV heart tissue (p < 0.05) differences were significant (Fig 5). These findings were corroborated by measuring plasma catecholamine levels of norepinephrine and epinephrine and cortisol levels 8 weeks after treatment. tMCAO‐treated mice presented significantly increased levels of plasma norepinephrine, epinephrine, and cortisol compared with both controls (p < 0.05; cortisol: ctrl, p < 0.01). Consequently, at this step, we can conclude that elevated catecholamine and cortisol levels are associated with development of cardiac dysfunction.
Figure 5.
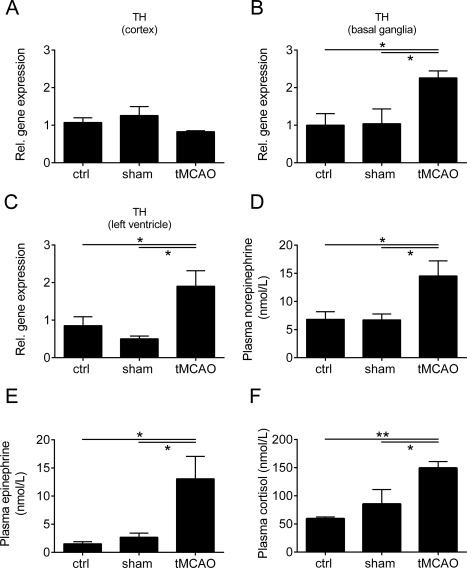
Chronic systolic dysfunction is associated with increased sympathetic activity. (A–C) Relative (Rel.) gene expression of tyrosine hydroxylase (TH) in the brain cortex (A), basal ganglia (B), and left ventricular heart tissue (C) of control (ctrl), sham‐operated, and transient middle cerebral artery occlusion (tMCAO)‐treated mice 8 weeks after surgery (n = 5 or 6 per group; *p < 0.05). (D–F) Plasma norepinephrine (D), epinephrine (E), and cortisol (F) levels from ctrl, sham‐operated, and tMCAO‐treated mice 8 weeks after tMCAO (n = 7–11 per group; *p < 0.05, **p < 0.01).
Therapeutic Intervention with Metoprolol Prevents the Development of Systolic Dysfunction after Focal Cerebral Ischemia
Given the increased sympathetic activity in association with cardiac dysfunction, we tested the efficacy of β‐blockade using metoprolol, a selective β1‐blocker that is approved and well known from treatment of human heart failure.32 Due to the mode of action of a β‐blockade, we observed a significant decrease in heart rate compared with vehicle‐treated animals (right hemispheric tMCAO treatment + normal drinking water; Fig 6; p < 0.05). Furthermore, metoprolol treatment protected right tMCAO‐treated mice from increased volume load, impairment of cardiac pump performance, and heart/lung weight to body weight ratio elevation (p < 0.05; end‐systolic volume, p < 0.01; end‐diastolic volume, p < 0.001). In contrast, LV pressure did not differ between metoprolol‐ and vehicle‐treated animals (p > 0.05). Also, the progressive decline in fractional shortening over the 8‐week follow‐up period was significantly reduced (p < 0.05). Functional analysis of the animals using the adhesive removal test also indicated neurologic improvement of the metoprolol group versus vehicle‐treated mice beginning at 4 weeks of follow‐up (see Fig 1C; p < 0.05). These results strongly support the hypothesis that development of chronic cardiac dysfunction can be prevented by β‐blocker treatment.
Figure 6.
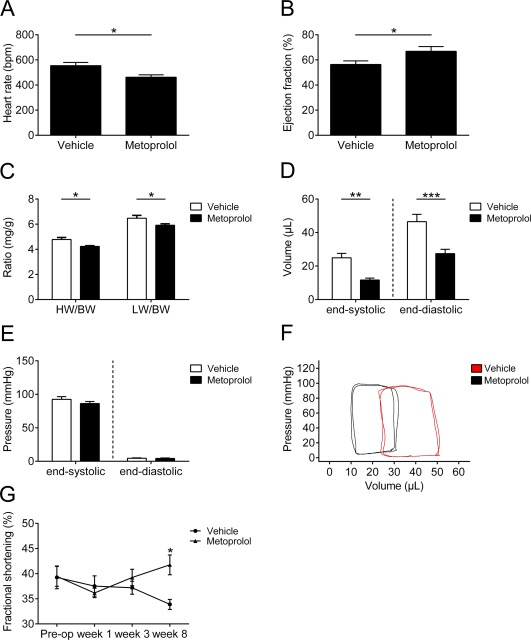
Therapeutic treatment with metoprolol after right transient middle cerebral artery occlusion (tMCAO) prevents development of systolic dysfunction. (A–F) Hemodynamic parameters of vehicle‐ or metoprolol‐treated mice assessed 8 weeks after tMCAO: heart rate (A), ejection fraction (B), heart weight (HW)/lung weight (LW) to body weight (BW) ratio (C), end‐systolic and end‐diastolic left ventricular (LV) volume (D), end‐systolic and end‐diastolic LV pressure (E), and representative pressure–volume loops in vehicle (red) and metoprolol‐treated mice (black; F). (G) Time course of fractional shortening of vehicle‐ or metoprolol‐treated C57BL/6J mice after tMCAO (n = 9–11 animals per group for all experiments; *p < 0.05, **p < 0.01, ***p < 0.001).
Metoprolol Influences Cardiac Biomarker Profile and Cardiac Remodeling
In a next step, we sought to determine whether the cardioprotective effects observed after metoprolol treatment also influence profiles of biomarkers in the LV that are important for the pathophysiology of cardiac dysfunction, and cardiac remodeling 8 weeks after right hemispheric tMCAO. Treatment with metoprolol led to a significant decrease in BNP (p < 0.01), TNF‐α (p < 0.05), and MMP‐9 (p < 0.05) gene expression compared with vehicle (Fig 7). Plasma BNP measurements showed a significantly lower level in the metoprolol‐treated group compared with vehicle treatment (p < 0.01). Similar to these effects, a significant decrease in β/α‐MHC relation and collagen content in the LV of metoprolol‐treated mice was detected (p < 0.05; collagen content, p < 0.01). In summary, pharmacologic treatment with metoprolol influences the profile of cardiac biomarkers that are relevant for the development of cardiac dysfunction. In addition, β‐blockade seems to decelerate extracellular cardiac remodeling, as shown by collagen content reduction in heart tissue after treatment.
Figure 7.
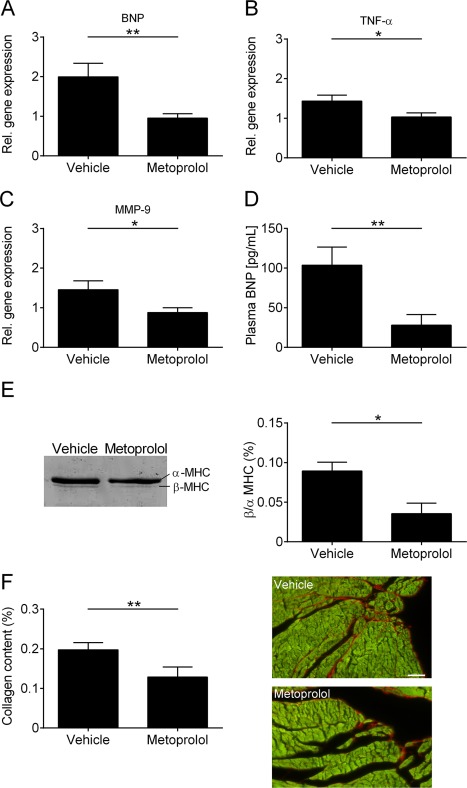
Influence of metoprolol on the cardiac biomarker profile and left ventricular (LV) remodeling. (A–C) Relative (Rel.) gene expression of brain natriuretic peptide (BNP) (A), tumor necrosis factor α (TNF‐α) (B), and matrix metallopeptidase‐9 (MMP‐9) (C) in the LV tissue of vehicle‐ and metoprolol‐treated mice 8 weeks after surgery (n = 5 per group; *p < 0.05, **p < 0.01). (D) Plasma BNP levels from vehicle‐ and metoprolol‐treated mice 8 weeks after transient middle cerebral artery occlusion (n = 8–11 per group; **p < 0.01). (E) Left panel: representative silver‐stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis of α‐ and β‐myosin heavy chain (MHC) distribution in the left ventricle of vehicle‐ and metoprolol‐treated mice 8 weeks after surgery. Right panel: densitometric quantification (n = 4 per group; *p < 0.05). (F) Densitometric quantification (left panel) and representative images (right panel) of the collagen (red) content in LV tissue (green) sections (7 µm) using picrosirius red staining (n = 4 or 5 animals per group; scale bar = 50 µm; **p < 0.01).
Pharmacologic β‐Blockade Inhibits Sympathetic Signaling Relevant for Chronic Autonomic Dysfunction
To prove whether metoprolol treatment also has an impact on the chronic sympathetic overstimulation after IS, we determined the gene expression levels of TH in the brain and the LV heart tissue of metoprolol‐treated mice and their respective controls 8 weeks after right hemispheric tMCAO. We could not find statistically significant differences in the brain cortex (Fig 8; p > 0.05). In contrast, gene expression of TH was decreased in the basal ganglia and LV tissue of metoprolol‐treated mice compared with vehicle‐treated animals (p < 0.05). In addition, metoprolol reduced catecholamine levels of norepinephrine (p < 0.01) and epinephrine (p < 0.05) and cortisol levels (p < 0.05) in the blood plasma compared with vehicle‐treated mice. These results, together with the increase in sympathetic neurotransmitters in the plasma of tMCAO animals compared with control mice (see Fig 5), indicate a causal effect of chronic cardiac dysfunction driven by a marked increase in sympathetic tone after focal cerebral ischemia. Furthermore, our data suggest that this detrimental effect can be prevented or ameliorated by β‐blockade.
Figure 8.
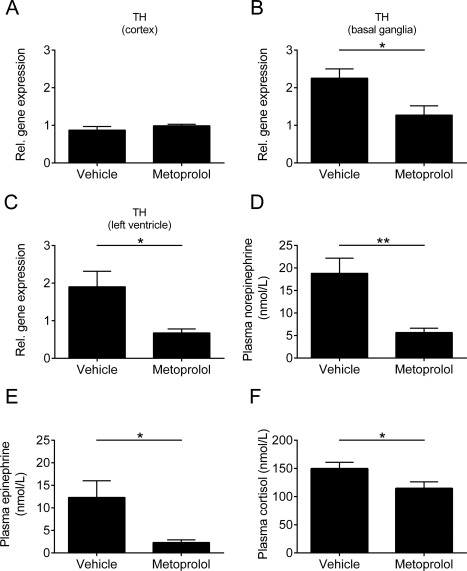
Pharmacologic treatment with metoprolol inhibits signaling relevant for chronic autonomic dysfunction. Relative (Rel.) gene expression of tyrosine hydroxylase (TH) in the brain cortex (A), basal ganglia (B), and left ventricular heart tissue (C) of vehicle‐ and metoprolol‐treated mice 8 weeks after surgery (n = 5 or 6 per group; *p < 0.05). (D–F) Plasma norepinephrine (D), epinephrine (E), and cortisol (F) levels from vehicle‐ and metoprolol‐treated mice 8 weeks after transient middle cerebral artery occlusion (n = 9–11 per group; *p < 0.05, **p < 0.01).
Discussion
Here we show, for the first time, that right hemispheric IS can induce a chronic systolic dysfunction in mice. This effect was observed in both young and aged animals, representing a clinically meaningful setting. An increased sympathetic activity in terms of upregulated catecholamine and cortisol levels and increased heart rate could be identified as the underlying mechanism. Finally, we demonstrate that an antisympathetic treatment approach using the β‐blocker metoprolol stopped the autonomic dysregulation and prevented development of chronic cardiac dysfunction.
Several clinical and animal studies have observed associations between IS and cardiac diseases,8, 11 such as cardiac arrhythmias,33 stress‐induced cardiomyopathy (Takotsubo syndrome),34 myocardial infarction,35 paroxysmal arterial hypertension,36 or autonomic dysfunction.9 Thereby, autonomic dysfunction is underlying the impaired physiologic regulation of heart rate and blood pressure as well as the increased secretion of catecholamines and cortisol.6, 37, 38, 39 The insular cortex has been identified as a crucial area within the central nervous system relevant for autonomic regulation.11, 30, 40, 41, 42 Furthermore, there is increased evidence that the lateralization of the insular area is associated with autonomic responses, where the right side focuses on sympathetic and the left side on parasympathetic function.9 In the current study, 30‐minute tMCAO had an impact on autonomic regulation (see Fig 5), due to a direct lesion of the right insular cortex area (see Fig 1), or—in cases without macroscopic lesion detection—alternatively due to ischemic disruption of the autonomic pathways downstream of the insula that led to cardiac dysfunction development in comparison to left‐sided insular cortex lesions (see Fig 2).44 As sympathetic overactivity is associated with myocardial injury and cardiac dysfunction up to heart failure development,40, 41, 43 it could be the missing link between IS and chronic cardiac dysfunction. In contrast to reports showing acute cardiac dysfunction after stroke,7 until now, no study has specifically investigated chronic development of systolic dysfunction after IS. Therefore, the present study adds to our understanding that, in mice, stroke‐induced sympathetic overactivity can be observed over a long time and leads to chronic systolic dysfunction accompanied by morphologic cardiac changes. The balance between sympathetic and vagal activity is probably critical to the ultimate effect on the heart. Former studies observed a direct vagal innervation of the left ventricle.45 Thus, further investigations are required to unravel how crucial the impact of the vagal tone is in the context of stroke‐induced chronic systolic dysfunction development. Our data also showed an increase in β‐MHC in sham‐operated animals. This finding is surprising at first glance, but is in accordance with previous reports on LV remodeling in the heart after myocardial infarction.29 Furthermore, the raised levels of collagen and marker gene expression of BNP and MMP‐9 fit very well with observations of other heart failure research studies.46, 47, 48, 49
The improved cardiac outcome after metoprolol treatment confirms our hypothesis of stroke‐driven chronic cardiac dysfunction by increased sympathetic activity.32 Despite metoprolol being a selective postsynaptic β1‐adrenoceptor inhibitor acting mainly on cardiac receptors, it has been shown that it also lowers plasma norepinephrine levels and release, by inhibiting presynaptic facilitating adrenoceptors in rats.50 This fits very well with our observation of reduced catecholamine levels after chronic metoprolol treatment in stroke animals.
Another important finding of our study was the functional improvement of metoprolol‐treated animals in the adhesive removal test (see Fig 1C). One explanation could be that β‐blocker treatment calms down the animals, leading to a better grip of the tape. Another study indicated that rats treated with selective β1 adrenoceptor antagonists showed neuroprotective effects after transient focal ischemia.51 Further investigations are required to unravel the main mechanism for the improved neurological recovery after β‐blocker therapy.
There are several limitations of our study that must be considered. First, the poststroke animals with proven echocardiographic dysfunction had no phenotype of heart failure. Therefore, the medical relevance is unknown. Second, despite using aged mice, the translational validity is not totally clear, as there might be differences regarding anatomical and pathophysiologic features in the context of chronic heart failure development. Third, even if our stroke model showed the detrimental cardiac effect, further analysis with other stroke models still needs to be done to clarify the dependence of cardiac dysfunction development on lesion localization. Finally, for animal welfare reasons, we had to restrict our observation to 8 weeks and therefore cannot predict development of cardiac function beyond that observation period. Therefore, we can exclude neither spontaneous recovery nor further deterioration of cardiac function after the 8‐week follow‐up period.
A strength of the SICFAIL study is the bitranslational concept. In parallel with the animal study, we are conducting a prospective clinical trial. In this trial, cardiac function of IS patients is being measured using serial echocardiographic examinations at different time points after stroke onset. The results of the clinical part of the study are expected at the end of 2017. Should the observation of chronic cardiac dysfunction be reproduced in human stroke patients, this could change routine diagnostic and therapeutic strategies. Besides echocardiography in the acute phase of stroke, subsequent examinations could be necessary to identify later deterioration in cardiac function and to treat patients who might develop chronic cardiac dysfunction. Further clinical studies would be necessary to delineate the best treatment strategy in that clinical setting. At this point, it is worth noting that stroke patients usually suffer from pre‐existing cardiovascular comorbidities. This makes it hard to distinguish to what extent the ischemic stroke or the comorbidities contribute to subsequent chronic cardiac dysfunction.
In conclusion, tMCAO in mice leads to the development of chronic systolic dysfunction driven by increased sympathetic activity. If this result can be confirmed in a clinical setting, treating physicians should be attentive to clinical signs of heart failure in every patient after IS. In mice, the detrimental effect of autonomic imbalance leading to cardiac dysfunction can be prevented by β‐blockade with metoprolol. If these results can be translated to a clinical setting, then the implications for practice could be significant.
Author Contributions
M.B., U.H., P.U.H., S.F., O.R., P.K., and C.K. contributed to the conception and design of the study. M.B., R.A.W., E.T., T.H., K.S., and P.K. contributed to the acquisition and analysis of data. M.B., R.A.W., U.H., T.H., K.S., P.U.H., S.F., O.R., P.K., and C.K. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Acknowledgment
This work was supported by grants from the Bundesministerium für Bildung und Forschung (BMBF01EO1504) through the Comprehensive Heart Failure Center Würzburg.
We thank L. Frieß, S. Braunschweig, S. Hellmig, S. Umbenhauer, H. Wagner, and M. Abeßer for excellent technical assistance.
Contributor Information
Peter Kraft, Email: kraft_p1@ukw.de.
Christoph Kleinschnitz, Email: christoph.kleinschnitz@uk-essen.de.
References
- 1. Endres M, Heuschmann PU, Laufs U, Hakim AM. Primary prevention of stroke: blood pressure, lipids, and heart failure. Eur Heart J 2011;32:545–552. [DOI] [PubMed] [Google Scholar]
- 2. Kolominsky‐Rabas PL, Weber M, Gefeller O, et al. Epidemiology of ischemic stroke subtypes according to TOAST criteria: incidence, recurrence, and long‐term survival in ischemic stroke subtypes: a population‐based study. Stroke 2001;32:2735–2740. [DOI] [PubMed] [Google Scholar]
- 3. Hajat C, Heuschmann PU, Coshall C, et al. Incidence of aetiological subtypes of stroke in a multi‐ethnic population based study: the South London Stroke Register. J Neurol Neurosurg Psychiatry 2011;82:527–533. [DOI] [PubMed] [Google Scholar]
- 4. Samuels MA. The brain‐heart connection. Circulation 2007;116:77–84. [DOI] [PubMed] [Google Scholar]
- 5. Kumar S, Selim MH, Caplan LR. Medical complications after stroke. Lancet Neurol 2010;9:105–118. [DOI] [PubMed] [Google Scholar]
- 6. Soros P, Hachinski V. Cardiovascular and neurological causes of sudden death after ischaemic stroke. Lancet Neurol 2012;11:179–188. [DOI] [PubMed] [Google Scholar]
- 7. Min J, Farooq MU, Greenberg E, et al. Cardiac dysfunction after left permanent cerebral focal ischemia: the brain and heart connection. Stroke 2009;40:2560–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Finsterer J, Wahbi K. CNS‐disease affecting the heart: brain‐heart disorders. J Neurol Sci 2014;345:8–14. [DOI] [PubMed] [Google Scholar]
- 9. Xiong L, Leung HH, Chen XY, et al. Comprehensive assessment for autonomic dysfunction in different phases after ischemic stroke. Int J Stroke 2013;8:645–651. [DOI] [PubMed] [Google Scholar]
- 10. Colivicchi F, Bassi A, Santini M, Caltagirone C. Cardiac autonomic derangement and arrhythmias in right‐sided stroke with insular involvement. Stroke 2004;35:2094–2098. [DOI] [PubMed] [Google Scholar]
- 11. Vargas ER, Soros P, Shoemaker JK, Hachinski V. Human cerebral circuitry related to cardiac control: a neuroimaging meta‐analysis. Ann Neurol 2016;79:709–716. [DOI] [PubMed] [Google Scholar]
- 12. Laowattana S, Zeger SL, Lima JA, et al. Left insular stroke is associated with adverse cardiac outcome. Neurology 2006;66:477–483; discussion 463. [DOI] [PubMed] [Google Scholar]
- 13. Dirnagl U. Bench to bedside: the quest for quality in experimental stroke research. J Cereb Blood Flow Metab 2006;26:1465–1478. [DOI] [PubMed] [Google Scholar]
- 14. Kilkenny C, Browne WJ, Cuthill IC, et al. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 2010;8:e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kleinschnitz C, Pozgajova M, Pham M, et al. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation 2007;115:2323–2330. [DOI] [PubMed] [Google Scholar]
- 16. Bouet V, Boulouard M, Toutain J, et al. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat Protoc 2009;4:1560–1564. [DOI] [PubMed] [Google Scholar]
- 17. Blain A, Greally E, Laval S, et al. Beta‐blockers, left and right ventricular function, and in‐vivo calcium influx in muscular dystrophy cardiomyopathy. PloS One 2013;8:e57260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frey A, Popp S, Post A, et al. Experimental heart failure causes depression‐like behavior together with differential regulation of inflammatory and structural genes in the brain. Front Behav Neurosci 2014;8:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pacher P, Nagayama T, Mukhopadhyay P, et al. Measurement of cardiac function using pressure‐volume conductance catheter technique in mice and rats. Nat Protoc 2008;3:1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grouzmann E, Cavadas C, Grand D, et al. Blood sampling methodology is crucial for precise measurement of plasma catecholamines concentrations in mice. Pflugers Arch 2003;447:254–258. [DOI] [PubMed] [Google Scholar]
- 21. Heydenreich N, Nolte MW, Gob E, et al. C1‐inhibitor protects from brain ischemia‐reperfusion injury by combined antiinflammatory and antithrombotic mechanisms. Stroke 2012;43:2457–2467. [DOI] [PubMed] [Google Scholar]
- 22. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 23. Puchtler H, Waldrop FS, Valentine LS. Polarization microscopic studies of connective tissue stained with picro‐sirius red FBA. Beitr Pathol 1973;150:174–187. [DOI] [PubMed] [Google Scholar]
- 24. Vogel B, Siebert H, Hofmann U, Frantz S. Determination of collagen content within picrosirius red stained paraffin‐embedded tissue sections using fluorescence microscopy. MethodsX 2015;2:124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nahrendorf M, Spindler M, Hu K, et al. Creatine kinase knockout mice show left ventricular hypertrophy and dilatation, but unaltered remodeling post‐myocardial infarction. Cardiovasc Res 2005;65:419–427. [DOI] [PubMed] [Google Scholar]
- 26. Disselhorst JA, Brom M, Laverman P, et al. Image‐quality assessment for several positron emitters using the NEMA NU 4‐2008 standards in the Siemens Inveon small‐animal PET scanner. J Nucl Med 2010;51:610–617. [DOI] [PubMed] [Google Scholar]
- 27. O'Collins VE, Macleod MR, Donnan GA, et al. 1,026 experimental treatments in acute stroke. Ann Neurol 2006;59:467–477. [DOI] [PubMed] [Google Scholar]
- 28. Albers GW, Goldstein LB, Hess DC, et al. Stroke Treatment Academic Industry Roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra‐arterial and neuroprotective therapies. Stroke 2011;42:2645–2650. [DOI] [PubMed] [Google Scholar]
- 29. Nordbeck P, Bonhof L, Hiller KH, et al. Impact of thoracic surgery on cardiac morphology and function in small animal models of heart disease: a cardiac MRI study in rats. PLoS One 2013;8:e68275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walter U, Kolbaske S, Patejdl R, et al. Insular stroke is associated with acute sympathetic hyperactivation and immunodepression. Eur J Neurol 2013;20:153–159. [DOI] [PubMed] [Google Scholar]
- 31. Sander D, Klingelhofer J. Changes of circadian blood pressure patterns after hemodynamic and thromboembolic brain infarction. Stroke 1994;25:1730–1737. [DOI] [PubMed] [Google Scholar]
- 32. Chatterjee S, Biondi‐Zoccai G, Abbate A, et al. Benefits of beta blockers in patients with heart failure and reduced ejection fraction: network meta‐analysis. BMJ 2013;346:f55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Simula S, Muuronen AT, Taina M, et al. Effect of middle cerebral artery territory ischemic stroke on QT interval. J Stroke Cerebrovasc Dis 2014;23:717–723. [DOI] [PubMed] [Google Scholar]
- 34. Yoshimura S, Toyoda K, Ohara T, et al. Takotsubo cardiomyopathy in acute ischemic stroke. Ann Neurol 2008;64:547–554. [DOI] [PubMed] [Google Scholar]
- 35. Mathias TL, Albright KC, Boehme AK, et al. The impact of myocardial infarction vs. pneumonia on outcome in acute ischemic stroke. J Cardiovasc Dis 2014;2:1–3. [PMC free article] [PubMed] [Google Scholar]
- 36. Phillips AM, Jardine DL, Parkin PJ, et al. Brain stem stroke causing baroreflex failure and paroxysmal hypertension. Stroke 2000;31:1997–2001. [DOI] [PubMed] [Google Scholar]
- 37. Feibel JH, Hardy PM, Campbell RG, et al. Prognostic value of the stress response following stroke. JAMA 1977;238:1374–1376. [PubMed] [Google Scholar]
- 38. Myers MG, Norris JW, Hachniski VC, Sole MJ. Plasma norepinephrine in stroke. Stroke 1981;12:200–204. [DOI] [PubMed] [Google Scholar]
- 39. Christensen H, Boysen G, Johannesen HH. Serum‐cortisol reflects severity and mortality in acute stroke. J Neurol Sci 2004;217:175–180. [DOI] [PubMed] [Google Scholar]
- 40. Ay H, Koroshetz WJ, Benner T, et al. Neuroanatomic correlates of stroke‐related myocardial injury. Neurology 2006;66:1325–1329. [DOI] [PubMed] [Google Scholar]
- 41. Cheshire WP Jr, Saper CB. The insular cortex and cardiac response to stroke. Neurology 2006;66:1296–1297. [DOI] [PubMed] [Google Scholar]
- 42. Oppenheimer S. The insular cortex and the pathophysiology of stroke‐induced cardiac changes. Can J Neurol Sci 1992;19:208–211. [PubMed] [Google Scholar]
- 43. Korpelainen JT, Sotaniemi KA, Myllyla VV. Autonomic nervous system disorders in stroke. Clin Auton Res 1999;9:325–333. [DOI] [PubMed] [Google Scholar]
- 44. Parati G, Esler M. The human sympathetic nervous system: its relevance in hypertension and heart failure. Eur Heart J 2012;33:1058–1066. [DOI] [PubMed] [Google Scholar]
- 45. Saper CB, Kibbe MR, Hurley KM, et al. Brain natriuretic peptide‐like immunoreactive innervation of the cardiovascular and cerebrovascular systems in the rat. Circ Res 1990;67:1345–1354. [DOI] [PubMed] [Google Scholar]
- 46. Yan AT, Yan RT, Spinale FG, et al. Plasma matrix metalloproteinase‐9 level is correlated with left ventricular volumes and ejection fraction in patients with heart failure. J Card Fail 2006;12:514–519. [DOI] [PubMed] [Google Scholar]
- 47. Feldman AM, Combes A, Wagner D, et al. The role of tumor necrosis factor in the pathophysiology of heart failure. J Am Coll Cardiol 2000;35:537–544. [DOI] [PubMed] [Google Scholar]
- 48. Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res 2005;66:22–32. [DOI] [PubMed] [Google Scholar]
- 49. Mahadavan G, Nguyen TH, Horowitz JD. Brain natriuretic peptide: a biomarker for all cardiac disease? Curr Opin Cardiol 2014;29:160–166. [DOI] [PubMed] [Google Scholar]
- 50. Berg T. β1‐Blockers lower norepinephrine release by inhibiting presynaptic, facilitating β1‐adrenoceptors in normotensive and hypertensive rats. Front Neurol 2014;5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goyagi T, Horiguchi T, Nishikawa T, Tobe Y. Post‐treatment with selective beta1 adrenoceptor antagonists provides neuroprotection against transient focal ischemia in rats. Brain Res 2010;1343:213–217. [DOI] [PubMed] [Google Scholar]