

Abstract
The importance of the Wnt‐signaling pathway on the regulation and maintenance of the intestinal stem cell (ISC) population is well recognized. However, our current knowledge base is founded on models using systems of gross deregulation of the Wnt‐signaling pathway. Given the importance of this signaling pathway on intestinal homeostasis, there is a need to explore the role of more subtle alterations in Wnt‐signaling levels within this tissue. Herein, we have used a model of Apc2 loss to meet this aim. Apc2 is a homolog of Apc which can also form a destruction complex capable of binding β‐catenin, albeit less efficiently than Apc. We show that systemic loss of Apc2 results in an increase in the number of cells displaying nuclear β‐catenin at the base of the intestinal crypt. This subsequently impacts the expression levels of several ISC markers and the fitness of ISCs as assessed by organoid formation efficiency. This work provides the first evidence that the function and fitness of ISCs can be altered by even minor misregulation of the Wnt‐signaling pathway. Our data highlights the importance of correct maintenance of this crucial signaling pathway in the maintenance and function of the ISC population. Stem Cells 2018;36:114–122
Keywords: Stem cells, Cell signaling, Animal models, Gene expression, Differentiation
Significance Statement.
To address the importance of subtle Wnt‐signaling deregulation on intestinal stem cell (ISC) function, this population of cells was analyzed within an Apc2−/− mouse model. It was shown that the ISC population is highly sensitive to changes in Wnt‐signaling levels even when such changes are below the threshold that affects the maintenance of intestinal homeostasis.
Introduction
The rapid turn‐over of cells within the intestinal epithelium is sustained by the presence of intestinal stem cells (ISCs) at the base of the intestinal crypt. The ISC population is tightly regulated in order to maintain intestinal homeostasis, and much of this regulation is controlled by the canonical Wnt‐signaling pathway. The canonical Wnt‐signaling pathway functions through β‐catenin, which builds up within the nucleus when a Wnt ligand is bound. Upon entering the nucleus, β‐catenin acts as a transcriptional co‐activator for Transcription Factor 4 (Tcf4), resulting in expression of a range of genes. In the absence of a Wnt ligand, a β‐catenin destruction complex consisting of GSK3β, Axin and adenomatous polyposis coli (APC), targets β‐catenin for phosphorylation, thereby depleting intracellular levels and preventing nuclear accumulation 1.
The role of Wnt signaling in the maintenance of the ISC compartment has been well described. Previous studies have shown that deficiency of Tcf4 (which effectively switches off Wnt signaling) results in a lack of ISCs during development 2. Alternatively, loss of the Wnt‐repressor Apc (which results in increased accumulation of nuclear β‐catenin and consequent activation of the Wnt‐signaling pathway) was shown to result in a gross expansion of undifferentiated cell types within the small intestine 3.
A homolog of Apc, Apc2, was first identified in drosophila and the structural similarities between the two proteins have led to the hypothesis that there may be some functional redundancy in the system 4. This is supported by the fact that Apc2 is evolutionarily conserved throughout a range of species from drosophila, mice, and humans 4.
Of the structural similarities between APC2 and APC, the presence of several 20‐amino acid repeat motifs is the most striking. It is these regions within Apc and Apc2 which are capable of binding β‐catenin within the destruction complex and thereby regulate Wnt signaling 5. However, Apc contains a 15‐amino acid repeat motif, lacking in Apc2, which binds β‐catenin at a higher affinity 6. The drosophila homolog of APC2 has been shown to have Wnt‐regulating capabilities, through its ability to rescue mis‐expression of Wnt target‐genes following loss of Apc 7. APC2 has also been shown to be capable of depleting the intracellular levels of β‐catenin in a human colorectal cancer (CRC) cell line through the formation of an APC‐independent β‐catenin destruction complex 8. However, in this context, APC2 depleted intracellular β‐catenin less efficiently than APC. In contrast, in APC deficient CRC cells, transient expression of APC2 resulted in a reduced readout in a TCF‐Topflash assay equivalent to that seen when APC is transiently expressed 4.
Activation of aberant Wnt signaling within the intestine is recognized to drive the development of CRC. Indeed, mutations within APC are common in this disease, being seen in more than 80% of all cases 9, whereas mutations in APC2 are rarer (22 out of 619, <4%, cBioPortal 10, 11, 12). Despite this rarity, loss of gene function is also possible through other mechanisms. One such mechanism, epigenetic regulation of gene expression through the process of DNA methylation, is a common means for controlling gene expression. Hypermethylation of the promoter regions of tumor suppressor genes is commonly observed in a number of cancer types, and is a mechanism by which many non‐commonly mutated genes are downregulated 13, 14, 15. APC2 has been shown to be hypermethylated in more than 90% of CRC tumors, indicating that downregulation of expression of APC2 is likely to be an important step in colorectal tumorigenesis 16.
It is widely believed that the ISC is not only crucial in the homeostatic maintenance of normal intestinal physiology, but this important population of cells also plays a critical role in intestinal malfunctioning, such as the development of CRC. Thus, understanding the factors that regulate the number and function of ISCs is vitally important.
Although the effect of gross Wnt‐misregulation on the ISC population has been widely studied, less is known about the effect of more subtle Wnt‐regulation on this cell compartment. In order to assess the consequences of subtle deregulation of the Wnt pathway on the ISC compartment, here we have used a mouse model carrying a mutation for constitutive loss of Apc2.
Materials and Methods
Animal Models
Unlike knockout of its homolog Apc 17, a constitutive knockout of Apc2 does not result in embryonic lethality. The Apc2−/− mouse was produced by Professor Hans Clevers’ laboratory and gifted to us for analysis of the intestinal phenotype. The Apc2−/− mouse was produced by the insertion of a neomycin cassette including a stop codon into the open reading frame of exon 14, resulting in the production of an interrupted protein, lacking several β‐catenin, and axin binding sites 18, 19, 20.
Quantitative Reverse Transcriptase Polymerase Chain Reaction (PCR) Analysis
Upon intestinal dissection, the first 10 cm of the proximal small intestine was taken for epithelial cell extraction. This was performed using the method outlined by Weiser, to produce an epithelial cell enriched population 21, which was then divided into three samples and stored at −80°C until required. Total RNA was extracted using the Trizol method. Complimentary DNA was transcribed from 5 µg of RNA using Superscript III (Invitrogen UK) and random hexamer primers (Promega).
Relative quantification was carried out using either the Fast Sybr Green master mix system (Promega), or the Taqman Universal mastermix system (Applied Biosystems) for genes with lower levels of expression. All samples were run in triplicate on the StepOne Plus PCR machine and the threshold cycle values of each gene were normalized to expression of β‐actin. Primer details can be found in the Supporting Information Methods.
Sample Preparation and Immunohistochemistry
After dissection of the 10 cm of small intestine required for epithelial cell extraction, the remaining small intestine and colon was flushed with cold water, opened longitudinally and rolled from the proximal to the distal end. The resulting “swiss roll” was secured with a pin and placed in 10% formalin for 24 hours at 4°C, and processed into paraffin blocks. Five micrometer sections were cut and rehydrated into water. The sections were then either stained with haematoxylin and eosin for counting to enable histological analysis of apoptotic and mitotic bodies, or immunohistochemistry (IHC) was performed for β‐catenin expression. IHC was performed using Mouse Envision+ kit according to manufacturer's instructions with mouse mAB anti‐β‐catenin antibody 1:200 (BD, U.K.).
Apoptotic bodies were identified by their distinctive morphology of detachment from neighbors combined with highly condensed chromatin using H&E sections, as described by Potten et al. 22. Apoptotic index was calculated by average number of apoptotic bodies per half crypt divided by the average number of cells per half crypt for each individual mouse, counting >50 crypts per mouse. For cell position analysis, position 0 was counted at the bottom of a half crypt, and the cells were numbered sequentially up to the isthmus which marks the top of the crypt.
In Situ Hybridization
In situ hyrbidization for Olfm4 was performed on formalin‐fixed paraffin‐embedded tissue prepared as for IHC. Protocol used was as outlined by Parry et al. 23.
Intestinal Organoid Culture
Intestinal organoid culture was performed from small intestinal crypt preparations as previously described 24. Organoid formation efficiency was calculated as number of grown organoids (over 150 µm in length) after 10 days in culture as a percentage of the number of crypts seeded at day 0, as measured using GelCount software (see Supporting Information Methods).
At day 10, organoids were triturated to single crypts and re‐seeded as before. Self‐renewal was measured as the number of organoids which had grown after 10 days post passage as a percentage of the number which were seeded.
Mitochondrial Activity Assay
Prestoblue (Invitrogen) was incubated 1:10 in the organoid media (and negative controls) for 2 hours at 37°C, then removed and placed into a 96 well flat‐bottomed plate for reading levels of fluorescence.
Fluorescence‐activated cell sorting (FACs) of ISC
Intestinal epithelia of both Apc2+/+ and Apc2−/− mice carrying the Lgr5Cre‐GFP 25, were disassociated to single cell level as described by Sato et al. 24. Cells were then incubated with 1:1,000 DAPI (4′,6‐diamidino‐2‐phenylindole) and sorted in a FACs medium of 1% Bovine Serum Albumin (BSA) and 0.1 mg/ml DNAseI in Dulbecco's Modified Eagle's medium (DMEM) F12 for expression of Green Fluorescent Protein (GFP) from the live cell population.
Statistics
All data analysis was carried out using SPSS statistical software (IBM). A two‐tailed Student's t test was performed for all normally distributed data, while a nonparametric Mann‐Whitney test was used for all other data sets.
Results
Apc2 Regulates Wnt Signaling Within the Murine Small Intestine
In order to assess the impact of Apc2 loss on Wnt signaling transduction within the intestine, immunohistochemical analysis of β‐catenin localization was performed on intestines from Apc2−/− and Apc2+/+ litermates. This analysis uncovered a significant increase in the number of cells possessing nuclear β‐catenin in an Apc2 deficient environment (Fig. 1A, 1B). The increase in nuclear β‐catenin as a result of Apc2 deficiency is more subtle than that observed as a result of Apc loss, and is restricted to the base of the crypts 3. However, this finding is consistent with the notion that Apc2 contributes to the formation of a β‐catenin destruction complex within the normal intestinal crypt cells. It has been shown that increased Wnt‐signaling as a result of increase nuclear β‐catenin levels does not always result in a change in expression of all Wnt target genes, which are also influenced by Bone morphogenetic protein (BMP) and Fibroblast growth factor (FGF) signaling 26, however, in the Apc2 model, increased incidence of nuclear β‐catenin translated into a small but significant upregulation of transcription levels of some Wnt target genes within the intestinal epithelia. Quantitative reverse transcriptase PCR (QRT‐PCR) using RNA extracted from isolated intestinal epithelium demonstrates subtle, yet significant over‐expression of Axin2, EphB3, and Cmyc, following the loss of Apc2, with no significant changes to Cyclin D1, EphB2, and Cd44 (Fig. 1C).
Figure 1.
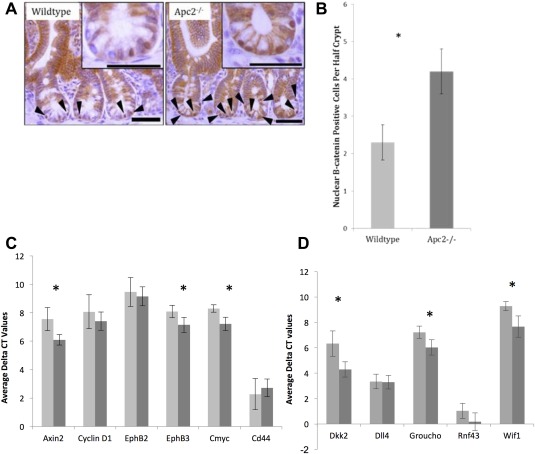
Apc2 deficiency subtly regulates Wnt signaling. (A): Immunohistochemistry for β‐catenin shows an increase in nuclear localization of β‐catenin. (B): The increase in nuclear β‐catenin is shown to be statistically significant p < .03 (n = 4). (C): Average delta CT values of Wnt target genes show a significant increase in expression of Axin2, EphB3, and Cmyc in intestinal epithelial tissue as a result of loss of Apc2 (n = 6). Light gray bars represent wildtype tissue, dark gray bars represent Apc2−/− tissue. Asterisk denotes statistical significance. (D): Average delta CT values of Wnt repressor genes show a significant increase in expression of Dkk2, Groucho, and Wif1 in intestinal epithelial tissue as a result of loss of Apc2 (n = 5). Light gray bars represent wildtype tissue, dark gray bars represent Apc2−/− tissue. Asterisk denotes statistical significance. Abbreviation: CT, cycle threshold.
Thus, these data support previous publications which have shown that Apc2 is capable of regulating Wnt signaling, and indicates that the absence of Apc2 partially compromises the destruction of β‐catenin, resulting in an increase in total Wnt signaling within the intestinal epithelium.
Interestingly, the Wnt‐inhibitors Dkk2, Groucho (GprK21), and Wif1 are also significantly upregulated in response to Apc2 deficiency, indicating that the normal regulatory feedback loops are stimulated in response to the subtly elevated levels of Wnt signaling (Fig. 1D). As Wnt‐target gene expression levels are dependent on a number of different factors including levels of Wnt‐ligands, intracellular regulatory proteins such as those involved in the destruction complex as well as expression levels of Wnt‐inhibitors, there is not a direct relationship between levels of Wnt‐inhibitors levels and expression levels of Wnt‐target genes.
Loss of Apc2 Results in Increased Apoptosis at the Base of the Intestinal Crypt
A detailed analysis of the intestines from the Apc2+/+ intestine with Apc2−/− intestine from aged matched littermates was performed in order to assess the effect that subtly upregulated Wnt signaling has on intestinal homeostasis. Overall, the intestine showed no signs of gross abnormality in the absence of Apc2 at 8 months old, with no change in numbers of the differentiated cell types, paneth cells, and goblet cells (Supporting Information Fig. 1). However, detailed analysis of the number of cells and location of apoptotic cells revealed subtle changes in intestinal homeostasis. Other than cell shedding which occurs at the tip of the villus, most apoptosis within the small intestine occurs at the crypt base 22. The total number of cells within the crypt was reduced in the Apc2−/− mice (Fig. 2A), and a trend for increased apoptosis within the crypt was observed (Fig. 2B). Although this increase was not significant, positional analysis of the apoptotic bodies indicated that while the location of apoptotic bodies in the intestinal epithelia of wildtype mice was comparable to the previously reported data with most of the apoptotic bodies within cell positions 0–10 22, apoptosis in the crypts of Apc2−/− mice occurred closer to the base. Furthermore, the level of apoptosis in the crypt base, positions 0–6, was significantly higher in the Apc2−/− (Fig. 2C, 2D). The crypt base positions 0–6 are thought to contain both of the proposed populations of ISCs; the crypt base columnar (CBC) cells interspersed between the paneth cells, and the +4 stem cell population. Thus, loss of Apc2 results in a significant increase in cell death at the base of the intestinal crypt concomitant with the position of the ISCs and their niche, without impacting on the paneth cell number, implying increased apoptosis among the ISC populations. This trend for increased total apoptosis as a result of Apc2 deficiency was replicated through the use of IHC for the apoptotic marker Caspase 3. A strong trend for increased apoptosis within the 0–6 cell position was also observed in the Caspase 3 IHC (Supporting Information Fig. 2).
Figure 2.
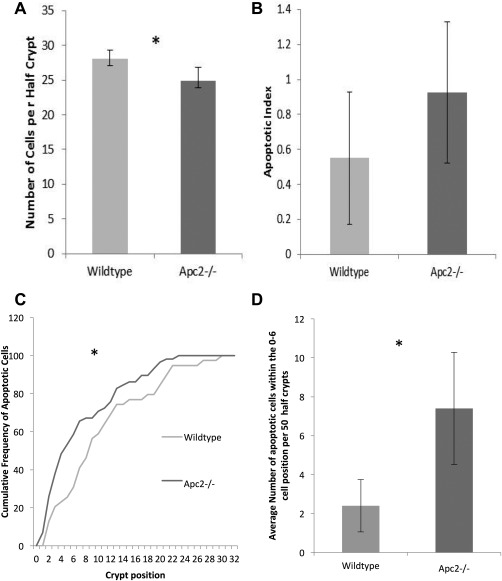
Intestinal homeostasis is regulated by Apc2. (A): Loss of Apc2 results in a decrease in number of cells per half‐crypt (n = 6). (B): Apc2 deficiency results in no significant change in number of apoptotic bodies as a percentage of crypt cells (n = 6). (C): The location of apoptotic bodies is significantly altered in absence of Apc2, with a higher proportion of them occurring at the base of the crypt. Asterisk denotes p value <.05. (D): Apc2 deficiency results in increased apoptosis at the base of the crypt in the 0–6 cell position (n = 6).
Apc has a known role in the maintenance of genomic stability and loss of Apc can drive cell death through a number of mechanisms, not solely because of its role in regulating Wnt signaling. Due to the potential functional redundancy between the Apc proteins, DNA damage markers were assessed as a proxy for genomic instability within the small intestine. Apc2 deficiency had no apparent effect on levels of genomic instability, indicating that the increased cell death observed at the base of the crypt is not due to a potential role of Apc2 in regulating DNA damage (Supporting Information Fig. 3).
Apc2 Deficiency Results in Increased Levels of Expression of the Stem Cell Markers Lgr5 and Ascl2, But Not an Increase in Stem Cell Number
Controversy surrounding the utility of proposed genes as markers of the ISCs is further compounded by the proposition that the ISC compartment comprises of two distinct populations of cells; the rapidly cycling CBC cells responsible for homeostasis, and the quiescent “+4” population of cells thought to be responsible for intestinal repair after damage 27, 28. Because of this controversy, expression levels of proposed markers for both stem cell types were analyzed, with Lgr5, Ascl2, Olfm4, and Msi1 expression representing the CBC population and Bmi1 expression representing the +4 population. Interestingly, only Lgr5 and Ascl2 were significantly upregulated in response to loss of Apc2 (Fig. 3), both of which are markers for the CBC cell population and recognized as Wnt target genes. However, the expression of the other markers associated with the CBC population were not elevated, and notably, Msi1 expression levels were significantly downregulated in the Apc2 deficient intestinal epithelium. Msi1 is a Wnt target‐gene that was originally thought to be expressed by the CBC cell 29; however, it has now been shown that Msi1 expression is not specifically limited to this compartment 30.
Figure 3.
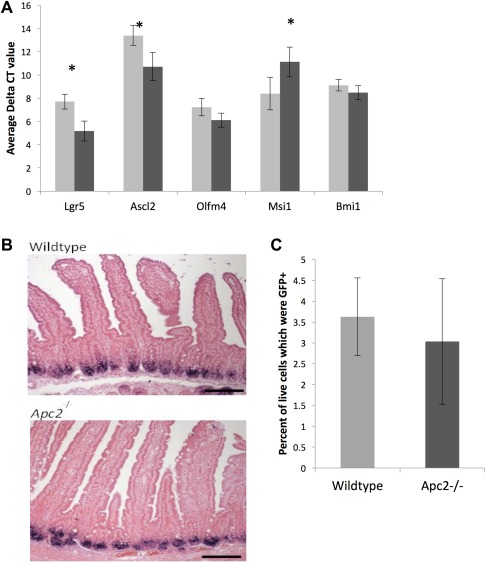
Apc2 regulation of the intestinal stem cell (ISC) markers. (A): Loss of Apc2 results in a change of expression levels of some ISC markers in the intestinal epithelium (n > 4). Light gray bars represent Wildtype tissue, dark gray bars represent Apc2−/− intestinal tissue. Asterisk indicates a p value <.05. (B): Apc2 deficiency results in no mislocation of expression of Olfm4 mRNA as determined by in situ hybridization. (C): Fluorescence‐activated cell sorting (FACs) sorting of Lgr5+ cells reveal no significant change in number of Lgr5+ cells in Apc2−/− intestinal epithelium (n = 4).
In situ hybridization analysis of the ISC marker Olfm4 (described as co‐localizing with Ascl2 31) showed a normal expression pattern in the Apc2 deficient intestine (Fig. 3B).
The Lgr5Cre‐GFP transgene created by Barker et al. enables the direct comparison of the number of Lgr5+ cells within the crypt through FACs sorting for GFP expression 25. Interestingly, despite an increase in expression of Lgr5, Apc2 deficiency resulted no change in the percentage of intestinal epithelial cells of crypt origin which were Lgr5Cre‐GFP + when sorted for GFP (Fig. 3C). Thus, overall it appears that Apc2−/− mice survive and maintain intestinal homeostasis despite a potential aberration in ISCs, such that the higher levels of Lgr5 expression does not correlate with an increase in Lgr5+ stem cell number. Furthermore, the increase of apoptosis at the base of the crypt and the reduced expression of Msi1, corroborate the notion of a potentially reduced ISC fitness.
Loss of Apc2 Results in Compromised ISC Function
A potential caveat of the FACs analysis is associated with the known mosaicsm of the Lgr5Cre‐GFP transgene. It is also possible that the deficiency for Apc2 results in a stronger silencing of the transgene rather than a reduction in the number of Lgr5 positive cells. To answer this conundrum, while also addressing the contradictory evidence between the levels of Lgr5 expression and the number of Lgr5 expressing cells, a stem cell functionality assay was utilized.
It has previously been shown that ISCs are capable of growing organoids in culture, and so organoid forming efficiency of disassociated intestinal crypts was used as a stem cell function assay. Disassociated intestinal crypts from wildtype and Apc2−/− mice were seeded at the same density, and the percentage of crypts which had formed organoids after 11 days post seeding was used as a measure of stem cell functionality. Crypts were used as an alternative to Lgr5+ single cells to avoid pre‐selection of an ISC population while ensuring a high enough organoid formation efficiency high enough to identify subtle changes.
Interestingly, an increase in expression of Lgr5 was associated with a decrease in ISC functionality, with many fewer Apc2−/− crypts capable of forming organoids than their wildtype counterparts, which is in‐line with the observed increase of apoptosis at the base of the crypt (Fig. 4A).
Figure 4.
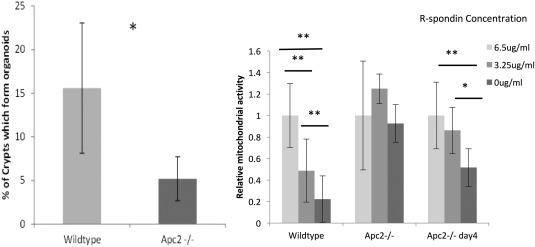
(A): Apc2 deficiency significantly reduces organoid formation efficiency of intestinal crypts n ≥ 5 (Wildtype 15.6% ± 7.5% vs. Apc2−/− 5.2% ± 2.5%, two‐tailed t test, p = .014). (B): Mitochondrial activity of Apc2−/− organoids is higher than their wildtype counterparts after 3 and 4 days of culture with reduced R‐spondin availability as assessed using PrestoBlue viability assay. * indicates p value of <.05. ** indicates p value of <.01.
Of the organoids that did grow, however, there was no difference in the growth rate (Supporting Information Fig. 4) or size of the organoids, indicating that the organoids which did grow were normal. IHC on formalin fixed, paraffin embedded organoids, and immunofluorescence on wholemount organoids revealed that Apc2 deficient organoids are also capable of developing differentiated cell types such as goblet and paneth cells (Fig. 5A, 5B). This is supported by an alternative stem cell function assay, the self‐renewal assay. In this assay, wildtype and Apc2−/− organoids were grown, and the passage efficiency was measured. Both cultures were capable of being passaged more than three times and there were no differences in the self‐renewal efficiencies between cultures (Fig. 5C). As organoid cultures have been shown to be genomically stable, we would not anticipate this passage efficiency to alter longer‐term 32, although this was not explored.
Figure 5.
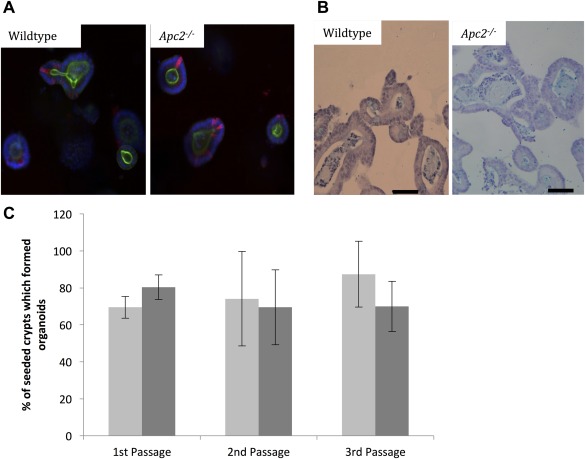
(A): Wholemount Immunofluorescence (IF) of intestinal organoids reveal no overt changes in number of paneth cells as a result of Apc2 deficiency, Red = Lysozyme, Green= phalloidin. (B): Immunohistochemistry for Alcian blue shows no overt change in number of goblet cells as a result of Apc2 deficiency. Scale bars represent 50 μm. (C): Passage efficiency was unchanged as a result of Apc2 deficiency, Light gray bars represent wildtype intestinal organoids and dark gray bars represent Apc2−/− intestinal organoids, n > 10 wells.
Wildtype organoids cultures are only capable of growth in the presence of R‐spondin within the culture medium for cell survival and organoid growth, whereas organoids grown from Wnt‐activated stem cells such as those derived from Apcflox/flox mice are not 33. In order to assess the levels of Wnt activation within the ISC compartment as a result of Apc2 deficiency, intestinal crypts from both wildtype and Apc2−/− mice were cultured in 6.5 μg/ml, 3.25 μg/ml, and 0 μg/ml R‐spondin. Wildtype crypts die in culture in the absence of R‐spondin by day 2–3, but interestingly Apc2−/− organoids survived until day 3 and only began to die by day 4 (Fig. 4B). In order to quantify organoid survival at varying concentrations of R‐spondin within the media, the Prestoblue assay, a mitochondrial activity assay was used. As day 3 appeared to be the critical day for survival, mitochondrial activity was assessed on days 3 and 4 post seeding 34. This assay showed that compared to wildtype, mitochondrial activity of Apc2−/− organoids is still high in the absence of R‐spondin at day 3, but has dropped to wildtype levels by day 4. This indicates that despite an apparent decrease in the number of functional stem cells (cells capable of forming organoids in culture), the ISCs which have overcome the apoptotic effect of increased Wnt signaling are less dependent of R‐spondin, indicating that they are more highly Wnt‐activated than wildtype.
Discussion
It is widely accepted that Wnt signaling levels are tightly regulated within the intestine in order to maintain homeostasis, with changes in Wnt signaling having potentially disastrous effects on the structural integrity of the tissue. Although the relationship between Wnt signaling and the maintenance of the ISC population has been investigated when Wnt‐signaling levels are grossly altered, the effect of subtly altered Wnt signaling on the ISC compartment has so far remained unexplored.
A constitutive Apc2 knockout proved to be a suitable model to represent subtly altered levels of Wnt signaling within the intestine. In order to analyze the effect of these altered Wnt levels on the ISC compartment, a wide range of techniques were used, which showed that loss of Apc2 resulted in an increased expression of the ISC markers Lgr5 and Ascl2 but no change in the percentage of Lgr5+ cells. Functionality of the ISCs was explored through the use of organoid culture techniques, and showed that loss of Apc2 leaves intestinal crypts less efficient at forming organoids in culture, a capability which is associated with functionality of the ISC compartment. Of the Apc2−/− organoids which did grow however there was no difference in self‐renewal efficiency compared to wildtype. These observations lead to the hypothesis that within an Apc2−/− murine intestine there is a heterogeneous population of ISCs, a proportion of which can behave normally and form organoids which grow in the same manner to wildtype organoids, and others which are more highly Wnt activated are more likely to die, (as observed by an increase in apoptosis within the first four cell positions within the crypt) and are less fit as ISCs (as observed by the overall reduced intestinal organoid formation efficiency).
Given that the levels of ISC markers did not correlate with intestinal organoid formation efficiency, it must be assumed that either levels of expression of these markers do not directly impact ISC fitness, or that organoid formation is a poor readout of ISC activity levels. The one marker which did show a correlation was expression of Msi1. Recently, it has been proposed that Msi protein levels, although not a good indication of the CBC population, play an important role in the activation of the quiescent ISC population into the cell cycle in response to injury 35. Intestinal organoid formation as described here is essentially an assay of damage response after crypt isolation. The subtle Wnt deregulation caused by Apc2 loss, and the associated reduced Msi1 expression, may well impact the ability of intestinal crypts to activate their quiescent stem cell population in response to trauma. This could explain why intestinal homeostasis was largely unaffected by Apc2 deficiency, but had an impact on ISC fitness.
Conclusion
This hypothesis is supported by the idea that levels of Wnt signaling within the intestinal epithelium must be tightly regulated 36. The Wnt “just‐right” hypothesis presents the idea that changes in Wnt signaling can affect ISC survival. In a number of tumor models, it has been shown that an increase in Wnt signaling can result in an increase in apoptosis, thereby reducing the tumor burden 37. Thus, we show for the first time that even subtle misregulation of Wnt signaling can impact on the function and fitness of the ISC population.
Author Contributions
M.A.Y.: designed the research, managed the mouse intercrosses, performed organoid culture and associated imaging, performed IHC, performed qRT‐PCR, analyzed data, manuscript drafting; C.S.D.: designed the research, performed IHC; K.R.R.: designed the research; A.R.C.: designed the research; E.T.: managed the mouse intercrosses; R.J. performed qRT‐PCR, performed western blotting; All authors: critically reviewed the manuscript, read and approved the final manuscript. The now late Professor Alan Clarke was aware of the results of the study and had approved of the manuscript plan.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Note Added in Proof
This article was published online on 13 October 2017. Minor edits have been made that do not affect data. This notice is included in the online and print versions to indicate that both have been corrected 29 December 2017.
Supporting information
Supplementary Figures
Acknowledgments
This project was funded by Cancer Research UK (ARC Program Grant C1295/A15937 and ARC project Grant C1295/A12417), supporting M.A.Y., K.R.R., and E.T. Biotechnology and Biomedical Sciences Research Council (Ph.D. Studentship to C.D.), MRC (Ph.D. Studentship to M.A.Y.). We would like to thank Johan H. Van Es and Hans Clevers for the kind provision of the Apc2 null mouse model. We thank Matthew Zverev for technical support and genotyping and Derek Scarborough and Marc Isaacs for their assistance in histology.
References
- 1. Barker N. The canonical Wnt/beta‐catenin signalling pathway. Methods Mol Biol 2008;468:5–15. [DOI] [PubMed] [Google Scholar]
- 2. Korinek V, Barker N, Morin PJ et al. Constitutive transcriptional activation by a beta‐catenin‐Tcf complex in APC‐/‐ colon carcinoma. Science 1997;275:1784–1787. [DOI] [PubMed] [Google Scholar]
- 3. Sansom OJ, Reed KR, Hayes AJ et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev 2004;18:1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Es JH, Kirkpatrick C, Van de Wetering M et al. Identification of APC2, a homologue of the adenomatous polyposis coli tumour suppressor. Curr Biol 1999;9:105–108. [DOI] [PubMed] [Google Scholar]
- 5. Rubinfeld B, Albert I, Porfiri E et al. Loss of beta‐catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res 1997;57:4624–4630. [PubMed] [Google Scholar]
- 6. Schneikert J, Vijaya Chandra SH, Ruppert JG et al. Functional comparison of human adenomatous polyposis coli (APC) and APC‐like in targeting beta‐catenin for degradation. PLoS One 2013;8:e68072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kunttas‐Tatli E, Zhou M‐N, Zimmerman S et al. Destruction complex function in the Wnt signaling pathway of drosophila requires multiple interactions between Adenomatous polyposis coli 2 and armadillo. Genetics 2012;190:1059–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakagawa H, Murata Y, Koyama K et al. Identification of a brain‐specific APC homologue, APCL, and its interaction with β‐catenin. Cancer Res 1998;58:5176. [PubMed] [Google Scholar]
- 9. Powell SM, Zilz N, Beazer‐Barclay Y et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992;359:235–237. [DOI] [PubMed] [Google Scholar]
- 10. Cerami E, Gao J, Dogrusoz U et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao J, Aksoy BA, Dogrusoz U et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Giannakis M, Mu XJ, Shukla SA et al. Genomic correlates of immune‐cell infiltrates in colorectal carcinoma. Cell Rep 2016;17:1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ehrlich M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002;21:5400–5413. [DOI] [PubMed] [Google Scholar]
- 14. Das PM, Singal R. DNA methylation and cancer. J Clin Oncol 2004;22:4632–4642. [DOI] [PubMed] [Google Scholar]
- 15. Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol 2005;2:S4–S11. [DOI] [PubMed] [Google Scholar]
- 16. Mokarram P, Kumar K, Brim H et al. Distinct high‐profile methylated genes in colorectal cancer. PLoS One 2009;4:e7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moser AR, Luongo C, Gould KA et al. ApcMin: A mouse model for intestinal and mammary tumorigenesis. Eur J Cancer 1995;31:1061–1064. [DOI] [PubMed] [Google Scholar]
- 18. Van der Meer M, Baumans V, Olivier B et al. Behavioral and physiological effects of biotechnology procedures used for gene targeting in mice. Physiol Behav 2001;73:719–730. [DOI] [PubMed] [Google Scholar]
- 19. Daly C. The roles of the Apc proteins in homeostasis and tumourigenesis [Ph.D. thesis]. Cardiff, U.K.: Cardiff University; 2013.
- 20. Daly CS, Shaw P, Ordonez LD et al. Functional redundancy between Apc and Apc2 regulates tissue homeostasis and prevents tumorigenesis in murine mammary epithelium. Oncogene 2016;36:1793–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weiser MM. Intestinal epithelial cell surface membrane glycoprotein synthesis. I. An indicator of cellular differentiation. J Biol Chem 1973;248:2536–2541. [PubMed] [Google Scholar]
- 22. Potten CS, Wilson JW Booth C. Regulation and significance of apoptosis in the stem cells of the gastrointestinal epithelium. Stem Cells 1997;15:82–93. [DOI] [PubMed] [Google Scholar]
- 23. Parry L, Young M, El Marjou F et al. Protocols for analyzing the role of paneth cells in regenerating the murine intestine using conditional cre‐lox mouse models. J Vis Exp 2015;105:53429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sato T, Vries RG, Snippert HJ et al. Single Lgr5 stem cells build crypt villus structures in vitro without a mesenchymal niche. Nature 2009;459:262–265. [DOI] [PubMed] [Google Scholar]
- 25. Barker N, Van Es JH, Kuipers J et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449:1003–1007. [DOI] [PubMed] [Google Scholar]
- 26. Nakamura Y, de Paiva Alves E, Veenstra GJ et al. Tissue‐ and stage‐specific Wnt target gene expression is controlled subsequent to β‐catenin recruitment to cis‐regulatory modules. Development 2016;143:1914–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tian H, Biehs B, Warming S et al. A reserve stem cell population in small intestine renders Lgr5‐positive cells dispensable. Nature 2011;478:255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yan KS, Chia LA, Li X et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci USA 2012;109:466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Potten CS, Booth C, Tudor GL et al. Identification of a putative intestinal stem cell and early lineage marker; musashi‐1. Differentiation 2003;71:28–41. [DOI] [PubMed] [Google Scholar]
- 30. Itzkovitz S, Lyubimova A, Blat IC et al. Single‐molecule transcript counting of stem‐cell markers in the mouse intestine. Nat Cell Biol 2011;14:106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van der Flier LG, van Gijn ME, Hatzis P et al. Transcription factor achaete scute‐like 2 controls intestinal stem cell fate. Cell 2009;136:903–912. [DOI] [PubMed] [Google Scholar]
- 32. Jung P, Sato T, Merlos‐Suárez A et al. Isolation and in vitro expansion of human colonic stem cells. Nat Med 2011;17:1225–1227. [DOI] [PubMed] [Google Scholar]
- 33. Matano M, Date S, Shimokawa M et al. Modeling colorectal cancer using CRISPR‐Cas9‐mediated engineering of human intestinal organoids. Nat Med 2015;21:256–262. [DOI] [PubMed] [Google Scholar]
- 34. Young M. The effects of aberrant Wnt signalling on the murine intestinal stem cell compartment [Ph.D. thesis]. Cardiff, U.K.: Cardiff University; 2013.
- 35. Yousefi M, Li N, Nakauka‐Ddamba A et al. Msi RNA‐binding proteins control reserve intestinal stem cell quiescence. J Cell Biol 2016;215:401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Albuquerque C, Breukel C, Van Der Luijt R et al. The ‘just‐right' signaling model: APC somatic mutations are selected based on a specific level of activation of the β‐catenin signaling cascade. Hum Mol Genet 2002;11:1549–1560. [DOI] [PubMed] [Google Scholar]
- 37. Méniel V, Song F, Phesse T et al. Cited1 deficiency suppresses intestinal tumorigenesis. PLoS Genet 2013;9:e1003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures