

Abstract
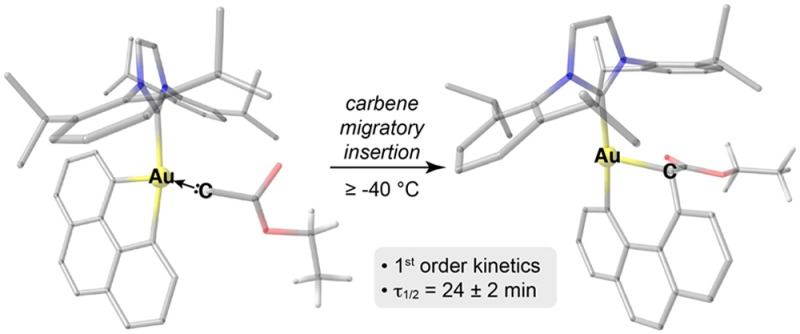
Migratory insertion of carbon-based species into transition-metal–carbon bonds is a mechanistic manifold of vast significance: it underlies the Fischer–Tropsch process, Mizoroki–Heck reaction, Ziegler–Natta and analogous late-transition-metal-catalyzed olefin polymerizations, and a number of carbonylative methods for the synthesis of ketones and esters, among others. Although this type of reactivity is well-precedented for most transition metals, gold constitutes a notable exception, with virtually no well-characterized examples known to date. Yet, the complementary reactivity of gold to numerous other transition metals would offer new synthetic opportunities for migratory insertion of carbon-based species into gold–carbon bonds. Here we report the discovery of well-defined Au(III) complexes that participate in rapid migratory insertion of carbenes derived from silyl- or carbonyl-stabilized diazoalkanes into Au–C bonds at temperatures ≥ −40 °C. Through a combined theoretical and experimental approach, key kinetic, thermodynamic, and structural details of this reaction manifold were elucidated. This study paves the way for homogeneous gold-catalyzed processes incorporating carbene migratory insertion steps.
1. Introduction
Migratory insertion of unsaturated carbon species into transition-metal carbon bonds is an elementary organometallic transformation that has proven broadly transformative.1 It features prominently in such name reactions as the Fischer–Tropsch process,2 Ziegler–Natta polymerization,3 and Mizoroki–Heck reaction.4 The former is utilized to synthesize hydrocarbon fuels and lubricants; the latter two, respectively, granted us such now-ubiquitous plastics as polyethylene and polypropylene3 and enabled the production and discovery of numerous pharmaceuticals and agrochemicals:5 their global impact on humanity has been recognized with the 1963 and 2010 Nobel Prizes in Chemistry, respectively.
Migratory insertion of unsaturated carbon-based species into metal–carbon bonds is well-established for most transition metals.1 Gold is aberrant in this regard: to our knowledge, only three examples of such reactivity have been reported to date for gold,6−9 in addition to several formal insertions10−14 that proceed via alternate mechanisms (Figure 1). Yet the ability to engage gold in this reactivity manifold is a tantalizing prospect precisely because the reactivity of organogold species is frequently complementary to that of analogous organotransition-metal complexes.9,15−23 For example, the reluctance of Au(I)-alkyl species to undergo syn-β-hydride elimination23 (a facile process for Au(III),24 however) or Au–C bond homolysis25 may enable gold-catalyzed coordination polymerization of unsaturated carbon species.
Figure 1.

Precedents and current work.
We were particularly intrigued at the outset of this venture, by the complete absence of precedent for migratory insertion of carbenes into Au–C bonds, despite numerous reports of spectroscopically observable or even isolable gold alkylidenes,26−28 insertions of electrophilic species such as SO2 into Au–C bonds,13,29,30 formal carbene insertions into Au–Cl bonds,31 and Au-catalyzed generation of carbenes from diazoalkanes and subsequent carbene transfer reactions.32,33 Concurrently with this work, an independent investigation in our laboratory into alkyl–CF3 reductive elimination serendipitously uncovered an example that was proposed to involve migratory insertion of difluorocarbene into Au–C bonds.9 With only this example known to date, migratory insertion of carbenes into Au–C bonds remains virtually unexplored.
Inspired by the well-documented polymerization of carbenes derived from α-diazocarbonyl compounds through migratory insertion at Rh(I) and Rh(III)34−40 and Pd0 and Pd(II)41−53 and oligomerization mediated by Cu0 powder54 and boranes,55 we wondered if analogous transformations could be mediated by organogold complexes. Intriguingly, more than 60 years ago, heterogeneous gold catalysts were first noted to convert diazomethane and higher diazoalkanes (though not diazocarbonyl compounds) into linear poly(alkylidene)s.56,57 Remarkably, among all the polymerization-competent heterogeneous metal catalysts, only in the case of gold was some stereoregularity observed in the produced polymers.57 (Among the homogeneous ones, only Rh(I)/Rh(III) produces highly syndiotactic poly(alkylidenes).34−36,38,39,58) More remarkably still, to date, the mechanism of this gold-catalyzed transformation remains ill-defined.59 Thus, as we set out to investigate carbene migratory insertion into Au–C bonds of well-defined homogeneous organogold species, we conjectured that our findings might also offer some insight into this long-standing question.
2. Results and Discussion
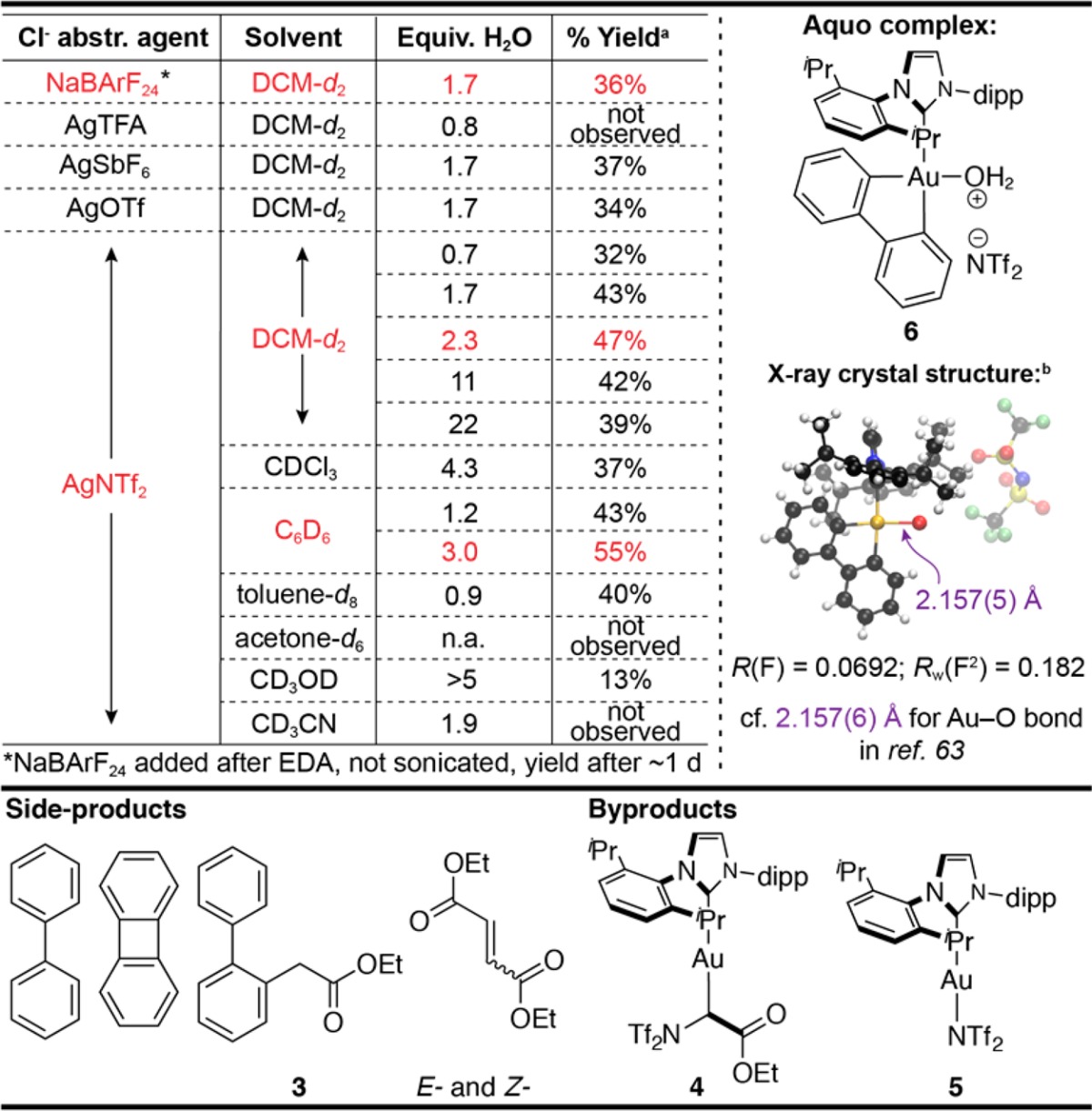
In particular, we envisioned that an organogold(III) complex with a readily available coordination site would be well-suited for diazoalkane coordination, gold-alkylidene generation, and migratory insertion of the carbene into a proximal Au–C bond.33,60 Our group recently reported that an example of such a complex derived from 1 by chloride abstraction was notable for its room-temperature persistence and Lewis acidity.61 Indeed, treatment of 1 (8.3 μM in CD2Cl2) with silver bis(trifluoromethanesulfonyl)imide (AgNTf2), followed by exposure to ethyl diazoacetate (EDA) at room temperature, led to rapid generation of fluorene derivative 2 as the major product (Table 1). We hypothesized that 2 formed via Csp2–Csp3 reductive elimination subsequent to carbene migratory insertion. Several side- and byproducts were also identified by 1H NMR spectroscopy and gas chromatography-mass spectrometry (GC-MS), including biphenylene, biphenyl, insertion/proto-deauration product 3, diethyl fumarate and maleate, and complex 4, produced via formal insertion of EDA into the Au–N bond of 5(62) (see Supplementary Figures S1 and S2).
Table 1. Initial Optimization of Reaction Conditions.

1H NMR yields.
See SI for cif file and general X-ray crystallography information.
2.1. Survey of Conditions and Substrates
Examination of multiple Ag(I) salts utilized for Cl– abstraction indicated that a weakly coordinating anion was necessary to promote the formation of 2, with –NTf2 affording the highest yield of 2. By the same token, 1 exhibited no background reactivity toward EDA (Table 1). Yet, Ag+ was not required to bring about the observed reactivity: anion metathesis with NaBArF24 in the presence of EDA similarly promoted the formation of 2, albeit more slowly (the exact time scale depended on EDA content). These observations supported the proposed mechanism that enlisted Au(III) for carbene formation from EDA, followed by migratory insertion and reductive elimination to generate 2.
The nature of the solvent was also critical to the observed transformation. The use of coordinating solvents such as methanol-d4, acetone-d6, or acetonitrile-d3 proved detrimental in this context (Table 1). On the other hand, weakly coordinating solvents such as benzene, toluene, and chlorinated alkanes supported the formation of 2. During the course of the solvent scope studies, we noticed that differences in water content in dichloromethane (DCM)-d2 (as well as other solvents) led to a variation in the yield of 2, as well as the relative content of side-products. A systematic investigation of this “water effect” revealed that about two equivalents of water (relative to 1) were optimal (Table 1). Augmented water content led to a gradual reduction in yield of 2 and generally increased formation of biphenyl and side-product 3 (Supplementary Figure S3) formed via water-promoted proto-de-auration; reduced water content below two equivalents led to a steep drop in the yield of 2 due to formation of biphenylene and 5 upon chloride abstraction (Supplementary Figure S3). Furthermore, biphenylene reductive elimination was rather rapid: in the presence of 1,3,5-trimethoxybenzene (TMB), a competitive π-donor, exchange with biphenylene was found to be approximately first-order in 1,3,5-trimethoxybenzene, which indicates that the rate-determining step occurs after reductive elimination (Supplementary Figure S4). NMR spectroscopy61 and X-ray crystallography of the cationic Au(III) intermediate formed in the presence of excess water identified it as aquo complex636 (Table 1). Exposure of 6 to EDA in DCM-d2 indeed generated 2. These results confirm the critical role of water as a ligand, which stabilizes the intermediate cationic Au(III) species from reductive elimination, but, when present in large excess, also promotes competitive proto-de-auration to generate biphenyl and 3. Notably, 3 was not formed through degradation of 2: degradation of a mass-differentiated analogue of 2 (methyl ester instead of ethyl ester) was not observed by GC-MS despite the formation of 3 (Supplementary Figure S5). Observation of 3, therefore, indicates that the product of migratory insertion can be intercepted prior to reductive elimination.
The reactivity observed for EDA extended to several other diazoalkanes, albeit with equal or diminished yield of the corresponding fluorene derivatives. Notably, 7 was not formed at all (diazoalkane hydrolysis was observed instead: see Supplementary Figure S6), and disubstituted α-diazocarbonyl compounds afforded only trace fluorene products 8 and 9, detected by GC-MS, and none of the expected products 10 and 11 (Figure 2, Supplementary Figures S7–S10). On the other hand, increased steric bulk of tert-butyl diazoacetate and the electron-withdrawing nature of the ester moiety in 2,2,2-trifluoroethyl diazoacetate were well-tolerated, leading to comparable yields (relative to EDA) of corresponding fluorene derivatives 12 and 13. Migratory insertion reactivity was also observed for representative diazoketone and silyl diazoalkane, albeit with lower yields of corresponding products (14 and 15 in Figure 2) compared to EDA. Representative azide and isocyanide—alternate nitrene and carbene surrogates—essentially did not participate in migratory insertion (see 16 and 17 in Figure 2). Hence, given its reactivity and relative structural simplicity, EDA was selected as the optimal substrate for further investigations of carbene migratory insertion at Au(III).
Figure 2.

Substrate scope. NMR yields are shown. Synthesis of pure reference samples for the observed products is described in the SI.
2.2. Investigation of Ligand Effects
With several key parameters of the transformation optimized and prevalent side-reactions identified for complex 1, the influence of the ligands in complexes 1 and 18–30 on the outcome of the transformation, e.g., the yield of products 2 or 2′, was the next critical consideration to address (Figure 3). AgNTf2 and NaBArF24 were both evaluated: the former generated the cationic Au(III) quantitatively prior to EDA addition; the latter generated it concurrently with its consumption by reaction with EDA. Saturation of the N-heterocyclic carbene (NHC) backbone (complex 18) or reduction in the NHC buried volume64−66 (complexes 19 and 20) led to a precipitous drop in the yield of 2 and an increased relative production of biphenyl and/or biphenylene, 3, and other side-products (Supplementary Figure S11). Steric shielding of the Au center evidently reduces side-reactions. Yet, despite added steric demand in the case of 21, the yield of 2 was substantially diminished compared to 1: we speculate that the phenyl groups of the ligand may irreversibly intercept the carbene intermediate prior to migratory insertion.
Figure 3.

Influence of the ligands on the yield of products 2 and 2′. When product formation was only observed by GC-MS, we designate the yield as “trace”.
Complexes with phosphorus-based ligands (22–24) with widely varying steric bulk and electronics led to low yields of 2. On the other hand, substitution of the NHC ligand in 1 (IPr) with a cyclic (alkyl)(amino)carbene (cAAC),67,68 which is both more σ-donating and simultaneously more π-accepting68 (i.e., complex 25), afforded the highest yield of 2 among the tested Au(III) complexes. However, the use of NaBArF24 was crucial to prevent premature reductive elimination of biphenylene (Supplementary Figure S12). We reasoned that the strong π-accepting ability of the cAAC promotes reductive elimination,69 leading to 2 (or biphenylene) in preference to proto-de-auration and other side-reactions. Indeed, cAAC–gold(I) complexes are known to be more resistant to oxidation compared to gold(I) complexes of imidazolin-2-ylidenes.70
We anticipated that replacement of the 2,2′-biphenyl ligand with 4,5-phenanthryl would constrain the geometry of the phenyl rings to render reductive elimination energetically unfeasible prior to migratory insertion. As expected, treatment of 26 with AgNTf2 led to no observable reductive elimination, even under rigorously anhydrous conditions. Moreover, the yield of 2′ considerably improved compared to the yield of 2 in the case of 1, with byproduct formation suppressed for both AgNTf2 and NaBArF24 (Supplementary Figure S13).
It stands to mention that multiple carbene insertions prior to reductive elimination were not observed in any of the cases examined above. Such a mechanistic scenario is desirable, as it would open the door to homogeneous gold-catalyzed carbene polymerization and offer entry to a new class of ester-laden cyclic compounds. To that end, and as captured in compounds 27–30, we explored two ligand-centric tactics to suppress reductive elimination: (1) interception of tricoordinate Au(III) via reversible intramolecular chelation and (2) stronger σ-donation and weaker π-acceptance of the l-type ligand. To address (1), we replaced each of the iPr groups on the NHC with “MeO”, capable of weakly coordinating to the cationic Au(III); indeed, slower formation of 2′ was realized, but products of multiple insertions were not observed. To address (2), we synthesized 29 and 30, which possess the most σ-donating and least π-accepting of the tested carbene ligands: a mesoionic (or abnormal) carbene66,71 (aNHC), better regarded as an ylide. Compared to 1 and 26, complexes 29 and 30 afforded significantly reduced yields of 2/2′, but also no evidence of multiple carbene insertion. Instead, in the presence of NaBArF24, both 29 and 30 led to species whose m/z and fragmentation pattern were consistent with products of BArF24/biphenyl (or phenanthryl) cross-coupling (Supplementary Figure S14). The mechanisms of these cross-coupling transformations are proposed to be analogous to the previously reported biaryl homocoupling at Au(III), which proceeds via transmetalation of an aryl group to Au(III) and extremely rapid Csp2–Csp2 reductive elimination.72 These results were instructive: ligand modification is, indeed, a powerful approach to shape the topography of the entire reaction manifold; raising or lowering one barrier may render relevant new, previously inaccessible pathways.
2.3. Experimental and Theoretical Mechanistic Studies
The results presented above are consistent with the proposed mechanism of migratory insertion at a cationic Au(III) center followed by reductive elimination.
However, to rule out alternate pathways and provide a more detailed mechanistic description, we co-opted a combination of variable-temperature (VT) NMR and computational analysis on the combination of complex 26, AgNTf2, and EDA. To begin with, we must address the nature of the Au(III) species generated by treatment with AgNTf2. In the presence of water, abstraction of chloride gives rise to AgCl(s) and an aquo complex analogous to 6. However, under rigorously anhydrous conditions, another species is exclusively formed (Figure 4) without concomitant AgCl precipitation. Evidently, instead of the expected AgCl(s) formation,73 AgNTf2 is solubilized by interaction with the Au(III) complex. 19F NMR analysis (Supplementary Figure S15) suggested that the NTf2– anion in the latter scenario is largely dissociated from Ag+: the resonance corresponding to NTf2– is 1.1 ppm upfield of AgNTf2 and only 0.3 ppm downfield of nBu4N+NTf2–.
Figure 4.

(A) Structure determination of 26·AgNTf2 in solution via variable-temperature (VT) NMR. Relevant resonances, integration, and scalar coupling constants are shown. Inset in bottom spectrum: Structure of 26·Ag+ obtained via density functional theory (DFT) (MO6-L/cc-PVDZ, Au:SDD(2f,g); vacuum, –NTf2 was omitted; color scheme: H = not shown; C = gray; N = dark blue; Cl = green; Au = yellow; and Ag = light blue. (B) Full electrospray ionization (ESI) mass spectrum of 26·AgNTf2 in CD2Cl2 (8.3 × 10–5 M) (top), simulated and measured isotope patterns (middle), and collision-induced dissociation experiment of the ion 26·Ag+ (bottom).
Furthermore, at −40 °C, the 1H NMR resonances corresponding to a portion of the 4,5-phenanthryl moiety broaden and shift upfield (Figure 4A). These observations point to the reversible coordination of Ag+ to both the phenanthrene and the Cl– anion of complex 26. Mass spectrometry analysis—m/z, isotope pattern, and MS/MS—corroborated the formation of a coordination complex between 26 and Ag+ (Figure 4B). Density functional theory (DFT) computations further supported this notion: in fact, one could regard the newly formed bimetallic complex 26·AgNTf2 as simultaneously a coordination and π-complex between AgCl and the T-shaped cationic Au(III) species (Figure 4).73,74
Addition of EDA at −80 °C led to partial conversion of 26·AgNTf2 to 31 (EDA complex and a diazonium salt) during the course of ∼2 h, with no detectable formation of 2′ (Supplementary Figure S16). However, at 233.2 K (−40 °C), consumption of 31 proceeded with first-order kinetics and a rate constant of (4.9 ± 0.4) × 10–4 s–1 (τ1/2 = 24 ± 2 min, Figure 5). The concentration-independent first-order behavior and absence of an induction period supported the proposed mechanism, wherein the carbene is generated at the Au(III) site via N2 dissociation, as opposed to alternate bimolecular pathways. Furthermore, concomitant generation of 2′ was observed, but no intermediates were detected, although side-processes were observed that accounted for the moderate yield of 2′. Hence, we can draw the following conclusions: (1) migratory insertion of carbenes into Au–C bonds is not only feasible but also rapid even at −40 °C, and (2) Csp3–Csp2 reductive elimination from Au(III) is faster than N2 loss followed by migratory insertion, but is competitive with such side-reactions as proto-de-auration.
Figure 5.

(A) Transformation monitored by 1H NMR spectroscopy. (B) 1H NMR spectra, which illustrate the concurrent consumption of 31 and formation of 2′ and 5 and, later, 4 at 233.2 K. (C) Linearized plots of VT NMR kinetics data at various initial concentrations of 31 utilized to quantify kobs and τ1/2.
DFT modeling of the reaction energy profile (Figure 6, see Computations section in the SI) provided a mechanistic picture that is consistent with the observed reactivity. Cationic Au(III) complex 32 accessible via halide abstraction coordinates water or EDA to form the corresponding adducts 6′ and 31 (ΔH = −10.7 or −6.6 kcal/mol, respectively). The latter is capable of N2 elimination to form a carbene intermediate 33 (ΔH = 16.5 kcal/mol); the transition state leading to 33 was not located, indicating that ΔH⧧ ≈ ΔH in this case (see Supplementary Figure S17). Note that similar ΔH⧧ values have been previously computed for N2 elimination from IPrCu(EDA)+ and IPrAg(EDA)+ (16.1 and 18.3 kcal/mol, respectively).75 Carbene 33 has a minute energy barrier (ΔH⧧ = 2.4 kcal/mol) and a large enthalpic driving force (ΔH = −66.2 kcal/mol) for migratory insertion to afford 34. Note that the transition state for the migratory insertion is an early one, energetically and structurally similar to carbene 33. Consequently, N2 extrusion and migratory insertion in this case are best regarded as coupled processes, which, in combination, constitute the rate-limiting step en route to 2′, with a combined ΔH⧧ ≈ 18.9 kcal/mol. This value is consistent with the observed rapid consumption of 31 even at −40 °C.
Figure 6.

Potential energy diagram for the primary reactivity of the cationic complex 32 (black) and for the second consecutive insertion of EDA (in blue) at the given level of DFT. Counterion was excluded from the computational model to reduce the complexity of the system for computational expediency. See Computations section in the SI for further details.
The fate of intermediate 34 merits careful consideration: it can be consumed through a number of competing processes, two of which have been computed and depicted in Figure 6. One is reductive elimination to yield π-complex 35 (ΔH⧧ = 3.0 kcal/mol) and ultimately 2′ and 36 after dissociation; the other is coordination of a second equivalent of EDA (37) followed by a second round of N2 dissociation (38) and migratory insertion (39) (for 37 → 39, ΔH⧧ = 21.5 kcal/mol). The latter is a desirable pathway for poly(alkylidene) synthesis, as described above. A crucial realization is that the system in consideration is in a Curtin–Hammett scenario (37 → 39 via 38/TS3 vs 34 → 35 via TS2), where 34 and 37 are likely in equilibrium, and the reaction outcome is dictated by the difference in transition state energies leading to the respective products. This difference, in this case, is 11.6 kcal/mol, which explains the absence of multiple carbene migratory insertions in this system (vide supra). Given that the electronic structure of TS3 is similar to that of carbene 38 and analogous to that of carbene 33, the π-accepting ability of the NHC ligand is expected to be a critical parameter for outcome selection. We anticipate that with further ligand design to suppress reductive elimination other decomposition pathways will enable multiple insertions at Au(III).
3. Conclusion
Thus, we have demonstrated herein the first examples of migratory insertion of carbenes derived from diazoalkanes into Au–C bonds. We identified a number of reaction pathways that occurred prior to and post-migratory insertion (e.g., reductive elimination and proto-de-auration) and optimized the conditions for the formation of products 2 and 2′.
We determined that the bulky NHC ligand in 1 and 26 (IPr) and the more π-accepting cAAC ligand in 25 and 28 lead to the highest yields of 2/2′, presumably through promotion of reductive elimination after migratory insertion in favor of other processes; furthermore, installation of the 4,5-phenanthryl ligand inhibited many of these side-processes. Mechanistic analysis revealed AgCl ligation to the T-shaped cationic Au(III) species and π-complexation with a portion of the phenanthryl ligand, which refined our conception of the chloride abstraction step with Ag+ reagents. Furthermore, N2 elimination/migratory insertion was determined to proceed with first-order kinetics and a half-life of 24 ± 2 min at −40 °C; alternative bimolecular pathways for carbene generation and transfer were ruled out. Lastly, DFT computations supported the proposed mechanism for the formation of 2′ and, by analogy, other carbene insertion products reported herein. Looking ahead, these computations point to the feasibility of EDA polymerization at a gold center; further ligand scaffold design is a promising approach to achieve this goal. Hence, these studies demonstrate for the first time the scope, viability, and mechanistic details of carbene migratory insertion into Au–C bonds, an elementary organometallic transformation with fundamental significance for gold chemistry and beyond.
Acknowledgments
We thank the National Institutes of Health (R35 GM118190) for support of this work. A.V.Z. is a Merck Fellow of the Life Sciences Research Foundation. I.J.K. is a Swiss National Science Foundation postdoctoral fellow. We thank C. Hong and C. Winslow for X-ray crystallography, E. Kreimer for elemental analysis, Z. Zhou and R. Nichiporuk for assistance with HRMS, and Prof. K. Vikse for assistance with ESI-MS of 26·AgNTf2. This work made use of the UC Berkeley Catalysis Center, as well as the College of Chemistry and QB-3 Institute NMR facilities. DFT studies were conducted at the Molecular Graphics and Computation Facility, funded by NIH grant S10OD023532.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b11435.
Author Present Address
‡ Novartis Institutes for Biomedical Research, Cambridge, Massachusetts 02139, United States.
Author Contributions
† A. V. Zhukhovitskiy and I. J. Kobylianskii contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Hartwig J. F.Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books, 2010. [Google Scholar]
- Davis B. H.; Occelli M. L.. Fischer–Tropsch Synthesis, Catalysts and Catalysis; Elsevier, 2006; Vol. 163. [Google Scholar]
- Eisch J. J. Organometallics 2012, 31, 4917–4932. 10.1021/om300349x. [DOI] [Google Scholar]
- Oestreich M.The Mizoroki-Heck Reaction; John Wiley & Sons, 2009. [Google Scholar]
- Torborg C.; Beller M. Adv. Synth. Catal. 2009, 351, 3027–3043. 10.1002/adsc.200900587. [DOI] [Google Scholar]
- Rekhroukh F.; Brousses R.; Amgoune A.; Bourissou D. Angew. Chem., Int. Ed. 2015, 54, 1266–1269. 10.1002/anie.201409604. [DOI] [PubMed] [Google Scholar]
- Rekhroukh F.; Estevez L.; Bijani C.; Miqueu K.; Amgoune A.; Bourissou D. Organometallics 2016, 35, 995–1001. 10.1021/acs.organomet.6b00040. [DOI] [PubMed] [Google Scholar]
- Rekhroukh F.; Blons C.; Estévez L.; Mallet-Ladeira S.; Miqueu K.; Amgoune A.; Bourissou D. Chem. Sci. 2017, 8, 4539–4545. 10.1039/C7SC00145B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M.; Chen T. Q.; Neubig M. E.; Hong C. M.; Theulier C. A.; Kobylianskii I. J.; Janabi M.; O’Neil J. P.; Toste F. D. Science 2017, 356, 1272–1276. 10.1126/science.aan1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C. M.; Stone F. G. A. J. Chem. Soc. D 1970, 0, 1263–1264. 10.1039/C29700001263. [DOI] [Google Scholar]
- Johnson A.; Puddephatt R. J.; Quirk J. L. J. Chem. Soc., Chem. Commun. 1972, 938b–939. 10.1039/C3972000938B. [DOI] [Google Scholar]
- Mitchell C. M.; Stone F. G. A. J. Chem. Soc., Dalton Trans. 1972, 102–107. 10.1039/dt9720000102. [DOI] [Google Scholar]
- Johnson A.; Puddephatt R. J. J. Chem. Soc., Dalton Trans. 1977, 1384–1388. 10.1039/dt9770001384. [DOI] [Google Scholar]
- Rocchigiani L.; Fernandez-Cestau J.; Agonigi G.; Chambrier I.; Budzelaar P. H. M.; Bochmann M. Angew. Chem. 2017, 129, 14049–14053. 10.1002/ange.201708640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzung M. R.; Markham J. P.; Toste F. D. J. Am. Chem. Soc. 2004, 126, 10858–10859. 10.1021/ja046248w. [DOI] [PubMed] [Google Scholar]
- Mamane V.; Gress T.; Krause H.; Fürstner A. J. Am. Chem. Soc. 2004, 126, 8654–8655. 10.1021/ja048094q. [DOI] [PubMed] [Google Scholar]
- Nieto-Oberhuber C.; López S.; Jiménez-Núñez E.; Echavarren A. M. Chem. - Eur. J. 2006, 12, 5916–5923. 10.1002/chem.200600174. [DOI] [PubMed] [Google Scholar]
- Gorin D. J.; Toste F. D. Nature 2007, 446, 395–403. 10.1038/nature05592. [DOI] [PubMed] [Google Scholar]
- Solorio-Alvarado C. R.; Wang Y.; Echavarren A. M. J. Am. Chem. Soc. 2011, 133, 11952–11955. 10.1021/ja205046h. [DOI] [PubMed] [Google Scholar]
- Mankad N. P.; Toste F. D. Chem. Sci. 2012, 3, 72–76. 10.1039/C1SC00515D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost M.; Zeineddine A.; Estévez L.; Mallet-Ladeira S.; Miqueu K.; Amgoune A.; Bourissou D. J. Am. Chem. Soc. 2014, 136, 14654–14657. 10.1021/ja506978c. [DOI] [PubMed] [Google Scholar]
- Levin M. D.; Toste F. D. Angew. Chem., Int. Ed. 2014, 53, 6211–6215. 10.1002/anie.201402924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost M.; Amgoune A.; Bourissou D. Angew. Chem., Int. Ed. 2015, 54, 15022–15045. 10.1002/anie.201506271. [DOI] [PubMed] [Google Scholar]
- Rekhroukh F.; Estevez L.; Mallet-Ladeira S.; Miqueu K.; Amgoune A.; Bourissou D. J. Am. Chem. Soc. 2016, 138, 11920–11929. 10.1021/jacs.6b07035. [DOI] [PubMed] [Google Scholar]
- Tamaki A.; Kochi J. J. Organomet. Chem. 1973, 61, 441–450. 10.1016/S0022-328X(00)86574-2. [DOI] [Google Scholar]
- Wang Y.; Muratore M. E.; Echavarren A. M. Chem. - Eur. J. 2015, 21, 7332–7339. 10.1002/chem.201406318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R.; Widenhoefer R. Chem. Soc. Rev. 2016, 45, 4533–4551. 10.1039/C6CS00171H. [DOI] [PubMed] [Google Scholar]
- Sarria Toro J. M.; García-Morales C.; Raducan M.; Smirnova E. S.; Echavarren A. M. Angew. Chem. 2017, 129, 1885–1889. 10.1002/ange.201611705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aresta M.; Vasapollo G. J. Organomet. Chem. 1973, 50, C51–C53. 10.1016/S0022-328X(00)95076-9. [DOI] [Google Scholar]
- Johnson M. W.; Bagley S. W.; Mankad N. P.; Bergman R. G.; Mascitti V.; Toste F. D. Angew. Chem., Int. Ed. 2014, 53, 4404–4407. 10.1002/anie.201400037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarria Toro J. M.; García-Morales C.; Raducan M.; Smirnova E. S.; Echavarren A. M. Angew. Chem., Int. Ed. 2017, 56, 1859–1863. 10.1002/anie.201611705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fructos M. R.; Belderrain T. R.; de Frémont P.; Scott N. M.; Nolan S. P.; Díaz-Requejo M. M.; Pérez P. J. Angew. Chem., Int. Ed. 2005, 44, 5284–5288. 10.1002/anie.200501056. [DOI] [PubMed] [Google Scholar]
- Fructos M. R.; Diaz-Requejo M. M.; Perez P. J. Chem. Commun. 2016, 52, 7326–7335. 10.1039/C6CC01958G. [DOI] [PubMed] [Google Scholar]
- Hetterscheid D. G. H.; Hendriksen C.; Dzik W. I.; Smits J. M. M.; van Eck E. R. H.; Rowan A. E.; Busico V.; Vacatello M.; Van Axel Castelli V.; Segre A.; Jellema E.; Bloemberg T. G.; de Bruin B. J. Am. Chem. Soc. 2006, 128, 9746–9752. 10.1021/ja058722j. [DOI] [PubMed] [Google Scholar]
- Jellema E.; Budzelaar P. H. M.; Reek J. N. H.; de Bruin B. J. Am. Chem. Soc. 2007, 129, 11631–11641. 10.1021/ja073897+. [DOI] [PubMed] [Google Scholar]
- Jellema E.; Jongerius A. L.; Walters A. J. C.; Smits J. M. M.; Reek J. N. H.; de Bruin B. Organometallics 2010, 29, 2823–2826. 10.1021/om100226d. [DOI] [Google Scholar]
- Franssen N. M. G.; Remerie K.; Macko T.; Reek J. N. H.; de Bruin B. Macromolecules 2012, 45, 3711–3721. 10.1021/ma300314q. [DOI] [Google Scholar]
- Walters A. J. C.; Jellema E.; Finger M.; Aarnoutse P.; Smits J. M. M.; Reek J. N. H.; de Bruin B. ACS Catal. 2012, 2, 246–260. 10.1021/cs200607s. [DOI] [Google Scholar]
- Walters A. J. C.; Troeppner O.; Ivanović-Burmazović I.; Tejel C.; del Río M. P.; Reek J. N. H.; de Bruin B. Angew. Chem., Int. Ed. 2012, 51, 5157–5161. 10.1002/anie.201200069. [DOI] [PubMed] [Google Scholar]
- Walters A. J. C.; Reek J. N. H.; de Bruin B. ACS Catal. 2014, 4, 1376–1389. 10.1021/cs500160c. [DOI] [Google Scholar]
- Ihara E.; Haida N.; Iio M.; Inoue K. Macromolecules 2003, 36, 36–41. 10.1021/ma021169v. [DOI] [Google Scholar]
- Ihara E.; Fujioka M.; Haida N.; Itoh T.; Inoue K. Macromolecules 2005, 38, 2101–2108. 10.1021/ma048293u. [DOI] [Google Scholar]
- Ihara E.; Nakada A.; Itoh T.; Inoue K. Macromolecules 2006, 39, 6440–6444. 10.1021/ma0607822. [DOI] [Google Scholar]
- Ihara E.; Kida M.; Fujioka M.; Haida N.; Itoh T.; Inoue K. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 1536–1545. 10.1002/pola.21881. [DOI] [Google Scholar]
- Ihara E.; Hiraren T.; Itoh T.; Inoue K. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 1638–1648. 10.1002/pola.22504. [DOI] [Google Scholar]
- Ihara E.; Hiraren T.; Itoh T.; Inoue K. Polym. J. 2008, 40, 1094–1098. 10.1295/polymj.PJ2008145. [DOI] [Google Scholar]
- Ihara E.; Goto Y.; Itoh T.; Inoue K. Polym. J. 2009, 41, 1117–1123. 10.1295/polymj.PJ2009065R. [DOI] [Google Scholar]
- Ihara E.; Ishiguro Y.; Yoshida N.; Hiraren T.; Itoh T.; Inoue K. Macromolecules 2009, 42, 8608–8610. 10.1021/ma901857s. [DOI] [Google Scholar]
- Franssen N. M. G.; Reek J. N. H.; de Bruin B. Polym. Chem. 2011, 2, 422–431. 10.1039/C0PY00249F. [DOI] [Google Scholar]
- Ihara E.; Takahashi H.; Akazawa M.; Itoh T.; Inoue K. Macromolecules 2011, 44, 3287–3292. 10.1021/ma200064b. [DOI] [Google Scholar]
- Ihara E.; Okada R.; Sogai T.; Asano T.; Kida M.; Inoue K.; Itoh T.; Shimomoto H.; Ishibashi Y.; Asahi T. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 1020–1023. 10.1002/pola.26480. [DOI] [Google Scholar]
- Shimomoto H.; Itoh E.; Itoh T.; Ihara E.; Hoshikawa N.; Hasegawa N. Macromolecules 2014, 47, 4169–4177. 10.1021/ma500783b. [DOI] [Google Scholar]
- Shimomoto H.; Kikuchi M.; Aoyama J.; Sakayoshi D.; Itoh T.; Ihara E. Macromolecules 2016, 49, 8459–8465. 10.1021/acs.macromol.6b01809. [DOI] [Google Scholar]
- Liu L.; Song Y.; Li H. Polym. Int. 2002, 51, 1047–1049. 10.1002/pi.1025. [DOI] [Google Scholar]
- Bai J.; Burke L. D.; Shea K. J. J. Am. Chem. Soc. 2007, 129, 4981–4991. 10.1021/ja067264r. [DOI] [PubMed] [Google Scholar]
- Saini G.; Nasini A. G. Atti della Accademia delle Scienze di Torino 1955–1956, 90, 586. [Google Scholar]
- Nasini A. G.; Trossarelli L.; Saini G. Makromol. Chem. 1961, 44, 550–569. 10.1002/macp.1961.020440144. [DOI] [Google Scholar]
- Jellema E.; Jongerius A. L.; van Ekenstein G. A.; Mookhoek S. D.; Dingemans T. J.; Reingruber E. M.; Chojnacka A.; Schoenmakers P. J.; Sprenkels R.; van Eck E. R. H.; Reek J. N. H.; de Bruin B. Macromolecules 2010, 43, 8892–8903. 10.1021/ma101792v. [DOI] [Google Scholar]
- Franssen N. M.; Walters A. J.; Reek J. N.; de Bruin B. Catal. Sci. Technol. 2011, 1, 153–165. 10.1039/c0cy00065e. [DOI] [Google Scholar]
- Liu L.; Zhang J. Chem. Soc. Rev. 2016, 45, 506–516. 10.1039/C5CS00821B. [DOI] [PubMed] [Google Scholar]
- Wu C.-Y.; Horibe T.; Jacobsen C. B.; Toste F. D. Nature 2015, 517, 449–454. 10.1038/nature14104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard L.; Gagosz F. Organometallics 2007, 26, 4704–4707. 10.1021/om7006002. [DOI] [Google Scholar]
- Komiya S.; Huffman J.; Kochi J. Inorg. Chem. 1977, 16, 2138–2140. 10.1021/ic50174a070. [DOI] [Google Scholar]
- Hillier A. C.; Sommer W. J.; Yong B. S.; Petersen J. L.; Cavallo L.; Nolan S. P. Organometallics 2003, 22, 4322–4326. 10.1021/om034016k. [DOI] [Google Scholar]
- Poater A.; Cosenza B.; Correa A.; Giudice S.; Ragone F.; Scarano V.; Cavallo L. Eur. J. Inorg. Chem. 2009, 2009, 1759–1766. 10.1002/ejic.200801160. [DOI] [Google Scholar]
- Dröge T.; Glorius F. Angew. Chem., Int. Ed. 2010, 49, 6940–6952. 10.1002/anie.201001865. [DOI] [PubMed] [Google Scholar]
- Lavallo V.; Canac Y.; Präsang C.; Donnadieu B.; Bertrand G. Angew. Chem., Int. Ed. 2005, 44, 5705–5709. 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleilhavoup M.; Bertrand G. Acc. Chem. Res. 2015, 48, 256–266. 10.1021/ar5003494. [DOI] [PubMed] [Google Scholar]
- Hegedus L. S.Transition Metals in the Synthesis of Complex Organic Molecules; University Science Books, 1999. [Google Scholar]
- Romanov A. S.; Bochmann M. Organometallics 2015, 34, 2439–2454. 10.1021/om501211p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldeco-Perez E.; Rosenthal A. J.; Donnadieu B.; Parameswaran P.; Frenking G.; Bertrand G. Science 2009, 326, 556–559. 10.1126/science.1178206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf W. J.; Winston M. S.; Toste F. D.. Nat. Chem. 2013, 6, 159. 10.1038/nchem.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniotti S.; Dalla V.; Duñach E. Angew. Chem., Int. Ed. 2010, 49, 7860–7888. 10.1002/anie.200906407. [DOI] [PubMed] [Google Scholar]
- Homs A.; Escofet I.; Echavarren A. M. Org. Lett. 2013, 15, 5782–5785. 10.1021/ol402825v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivilla I.; Sameera W. M. C.; Alvarez E.; Mar Díaz-Requejo M.; Maseras F.; Pérez P. J. Dalton Trans. 2013, 42, 4132–4138. 10.1039/c2dt32439c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.