

Abstract
Cysteine residues are susceptible to oxidation to form S-sulfinyl (R-SO2H) and S-sulfonyl (R-SO3H) post-translational modifications. Here we present a simple bioconjugation strategy to label S-sulfinated proteins using reporter-linked maleimides. After alkylation of free thiols with iodoacetamide, S-sulfinated cysteines react with maleimide to form a sulfone Michael adduct that remains stable under acidic conditions. Using this sequential alkylation strategy, we demonstrate differential S-sulfination across mouse tissue homogenates, as well as enhanced S-sulfination following pharmacological induction of ER stress, lipopolysaccharide stimulation, and inhibitors of the electron transport chain. Overall, this study reveals a broadened profile of maleimide reactivity across cysteine modifications, and outlines a simple method for profiling the physiological role of cysteine S-sulfination in disease.
Keywords: protein modifications, Michael addition, sulfur, redox chemistry, cysteine
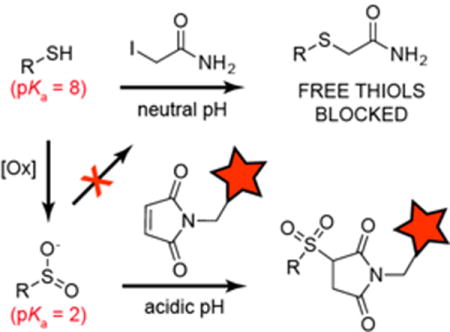
Graphical abstract
Maleimide, but not iodoacetamide, reacts with aryl and alkyl sulfinic acid standards and S-sulfinated proteins. The resulting sulfonyl-succinimide adduct is stable under acidic conditions, providing a sequential alkylation strategy for selective sulfinic acid labelling in biological samples. This study reveals a broadened profile of maleimide reactivity across cysteine modifications in proteins.
In select cellular environments and physiological states, certain redox-active cysteine residues are susceptible to rapid oxidation to S-sulfenylcysteine (Cys-SOH). This transient modification typically reacts with a second thiol to form a disulfide[1], but when inaccessible to cellular reductants or if the oxidative load in the local environment is too high, cysteine can undergo further oxidation to a sulfinic acid (Cys-SO2H)[2]. This intermediate oxidation state exists as a kinetically long-lived species, as further oxidation proceeds 25 – 50 times slower than thiol and sulfenic acid oxidation[3]. Sulfinic acid lifetimes can be further extended by the local protein environment. Indeed, sulfinic acids are estimated to occupy 5% of soluble protein thiols[4], establishing a major contribution to basal oxidative damage and enzyme inactivation across the proteome[5].
Despite the predicted prevalence of S-sulfination, there have been no robust methods for direct analysis or enrichment. Two methods were recently reported leveraging nitroso group reactivity for direct covalent labeling of endogenous S-sulfinated proteins (Scheme 1). The first strategy begins by sulfinic acid addition to an aryl-nitroso linked probe, followed by attack by the transient oxyanion towards an ortho-ester to form a stable benzioxazolone ring[6]. The second strategy uses S-nitrosothiol-linked probes to form a thiosulfonate with S-sulfinylated proteins[5]. Mammalian cell lysates were labeled with biotin-conjugate of S-nitrosoglutathione (GSNO-biotin), enriched on streptavidin beads, and analyzed by mass spectrometry. Hundreds of S-sulfinated proteins were identified, including peroxiredoxins, DJ-1, and many metabolic enzymes. This method is not ideal, since the GSNO-biotin probe degrades down within a few hours. Importantly, both strategies require complete alkylation of cellular thiols to prevent non-specific reactions. Furthermore, S-sulfonated cysteines (R-SO3H) are chemically inert, and cannot cross react with any electrophilic probes targeting S-sulfination.
Scheme 1. Nitroso-directed methods for labeling endogenous S-sulfinated cysteine residues in proteins.
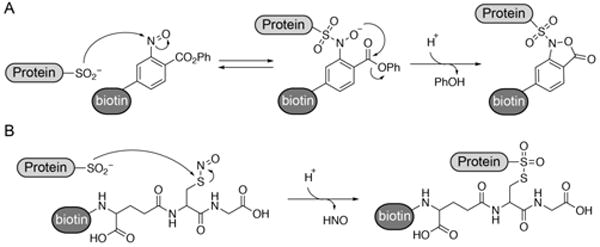
(A) Aryl-nitroso probes react with protein sulfinic acids to form a stable benzioxazolone ring. (B) S-nitrosoglutathione probes react with protein sulfinic acids to form a thiosulfonate linkage.
We previously reported that sulfinic acids do not react with 2-iodoacetamide (IAM) in aqueous buffers[5], allowing orthogonal alkylation to reporter-linked S-nitrosothiols. Sulfinic acids are reported to participate in Michael additions, yet the reaction has not been translated to biological systems[7]. Building from these studies, we find that N-ethyl maleimide (NEM) reacts with both aryl (sodium phenylsulfinate) and alkyl (sodium 3-methoxy-3-oxopropane-1-sulfinate; SMOPS) sulfinic acids in aqueous buffer (Figure 1 and S1). When incubated at 10:1 (maleimide:SMOPS) ratio, we observed ~90% conversion of SMOPS to the corresponding sulfonyl-succinimide. Since IAM only reacts with thiols, we reasoned IAM and maleimide could be used sequentially for selective sulfinic acid detection. Furthermore, the pKa of cysteine sulfinate (pKa = ~2) is 6 pH units below the pKa of cysteine thiol (pKa = 8.3)[2]. Accordingly, we devised a sequential labeling strategy for proteome-wide analysis of S-sulfination. Reduced cysteines are first labeled with IAM at neutral pH, and then switched to acidic pH for orthogonal cysteine sulfinic acid labeling by NEM (Figure 2A).
Figure 1. N-ethyl maleimide reacts with NEM but not IAM.
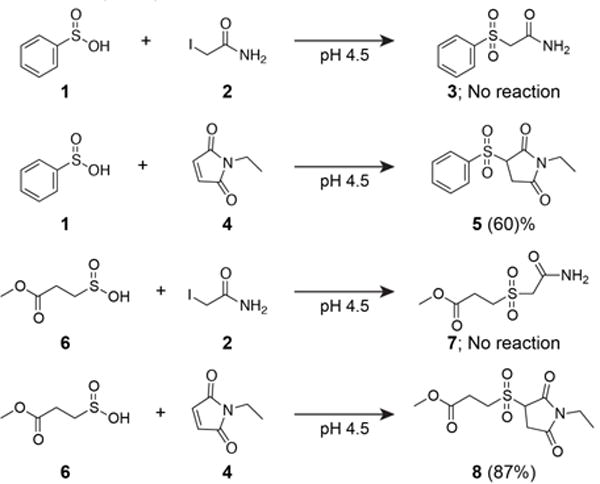
Reactions were carried out in 10:1 (electrophile : nucleophile) in degassed 6 M urea / citrate-phosphate buffer (pH 4.5) for 2 hours and assayed by high resolution LC-MS. Similar yields were achieved at pH 7.4 in phosphate buffer. Values represent conversion efficiency of the reaction mixture measured by LC-MS. Supporting LC-MS extracted ion chromatographs are reported in Figure S2.
Figure 2. Sequential alkylation strategy for detection endogenous S-sulfination.
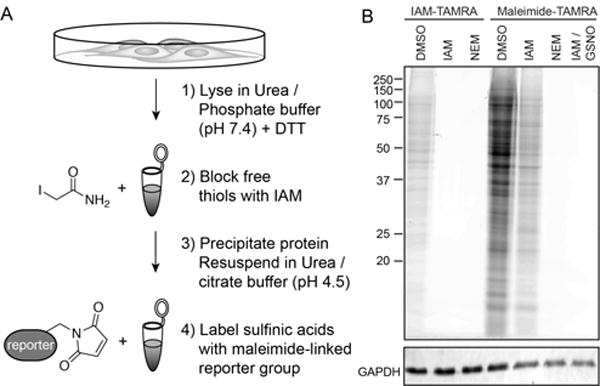
(A) Schematic of sequential alkylation strategy for labeling endogenous S-sulfinated proteins. Proteins are denatured in 6 M urea / PBS buffer (pH 7.4) for IAM labeling, then precipitated and resuspended in 6 M urea / citrate-phosphate buffer (pH = 4.5) for maleimide labeling. (B) IAM-TAMRA and Maleimide-TAMRA (50 μM) labeling of HEK-293T cell lysates after pre-treatment (30 min) with various electrophiles. IAM-TAMRA (50 μM), IAM (50 mM), NEM (50 mM), and GSNO (200 μM).
For maleimide-linked probes to succeed as a useful tool for S-sulfination profiling, the sulfonyl succinimide conjugate must be stable and irreversible in biocompatible buffers. To explore the product stability, N-ethyl-3-(phenylsulfonyl)succinimide was incubated across a panel of buffers at different pH values (Figure S3). When buffered below pH 6, there was no significant decomposition of the sulfonyl succinimide product (5), demonstrating sufficient stability for biochemical analysis.
We then examined if the conjugate can undergo a retro-Michael displacement of phenylsulfinate[8]. If the sulfonyl-succinimide conjugate is reversible, the released phenylsulfinate could react with excess maleimide to form a scrambled species. After two hours at pH 4.5, we observed no NEM-maleimide scrambling, confirming that under these conditions, the sulfonyl-succinimide is stable and not reversible (Figure S1). Based on these findings, orthogonal maleimide conjugation occurs at low pH, preventing lysine cross-reactivity while minimizing succinimide hydrolysis.
Next, the sequential labeling protocol was adapted for analysis of sulfinic acids in mammalian cell lysates (Figure 2A). Fresh HEK-293T cells were lysed in 6 M urea to denature proteins and incubated with dithiothreitol (DTT) to reduce disulfides, S-nitrosothiols, persulfides, or any other reversible cysteine modification. Importantly, pre-alkylation of cysteine with either IAM or maleimide in phosphate-buffered saline (PBS, pH 7.4) blocked all iodoacetamide-tetramethylrhodamine (IAM-TAMRA) labeling (Figure 2B). Once the lysate is pre-blocked with IAM, addition of maleimide-TAMRA at pH 4.5 led to a broad profile of labeled proteins. Importantly, pre-incubation with IAM alkylates any cellular thiols, including free cysteine and glutathione. In combination with the additional protein precipitation step, there is little chance for thiol interference with the sulfonyl succinimide product. We also find that 4′,4′-dithiopyridine (DPS; aldrithiol) reacts efficiently with both phenylsulfinate and SMOPS (93%) (Figure S4). Moreover, GSNO addition blocked all maleimide labeling, either by transnitrosation of unreacted thiols or by thiosulfonate formation with sulfinic acids[5]. Thus, even after denaturation, reduction, and alkylation with IAM, a persistent profile of proteins react with maleimide at pH 4.5, consistent with conjugation to sulfinic acids.
The Parkinson’s disease protein 7 (DJ-1/PARK7) contains a network of hydrogen bonds that stabilize sulfinic acid modification at Cys106, presumably inactivating its deglycase activity[9]. DJ-1 has 3 conserved cysteines (Cys46, Cys53, and Cys106), but only Cys106 forms a stable sulfinic acid[5, 10] (Figure 3A and 3B). Interestingly, oxidized DJ-1 (Cys106-SO2H) is reported to impart additional antioxidant properties[10]. Thus, we prepared purified, recombinant DJ-1 protein to validate the chemical identity of the IAM-resistant maleimide protein conjugate. DJ-1 protein was treated with increasing concentrations of hydrogen peroxide, followed by sequential IAM and NEM treatment. In the absence of peroxide, DJ-1 Cys106 is primarily reduced and alkylated by IAM, leaving only residual NEM labeling by in-gel fluorescence analysis (Figure 3C). Incubation with 5 or 100 equivalents of hydrogen peroxide increased maleimide labeling, indicative of increased sulfinic acid formation.
Figure 3. Analysis of sulfonyl succinimide formation on recombinant human DJ-1.
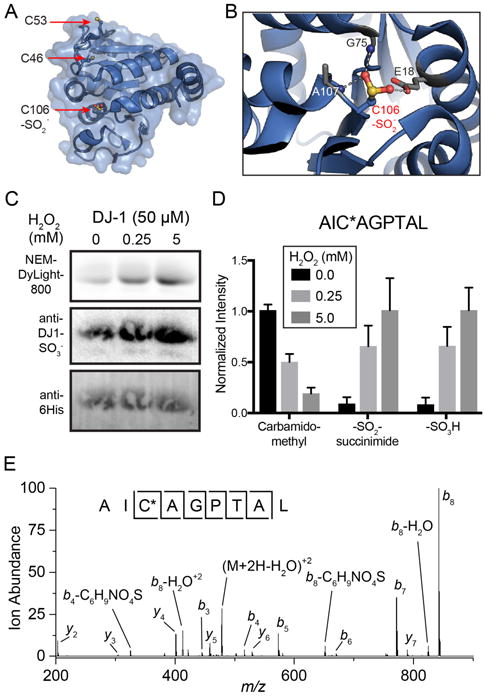
(A) Structure of DJ-1 (PDB: 1SOA) highlighting the three encoded cysteines. Cys46 and Cys53 are surface exposed, while Cys106 is buried in the putative active site. (B) DJ-1 Cys106 forms a stabilized sulfinic acid through a network of hydrogen bonds. (C) Profiling S-sulfination of DJ-1 by sequential alkylation with IAM and maleimide-DyLight 800. Recombinant DJ-1 was pretreated hydrogen peroxide, followed by DTT, IAM, and maleimide-DyLight 800 to detect sulfinic acids. (D) Label-free mass spectrometry quantitation of DJ-1 Cys106 oxidation summed and normalized from the pepsin digested peptides AICAGPTAL AND AICAGPTALL. Error bars represent standard deviations from 4 biological replicates. (E) Collision-induced dissociation MS/MS spectra of the DJ-1 N-ethyl-3-(sulfonyl-Cys106)succinimide conjugated peptide (AIC*AGPTAL). (* = sulfonyl-succinimide).
Since the sulfonyl-succinimide is only stable in acidic buffers, the peroxide treated DJ-1 samples were digested with pepsin at pH 1.5 and analyzed by high resolution mass spectrometry. Similar to the gel-based analysis, we observed robust peroxide-dependent sulfonyl-succinimide labeling exclusively on Cys106 (Figure 3D). Furthermore, there were no succinimide or sulfonyl-succinimide adducts detected on either Cys46 or Cys53, which lack the stabilizing hydrogen bond network of Cys106. More detailed analysis of the MS/MS spectra confirmed accurate annotation of the sulfonyl succinimide conjugate, as well as a minor, but reproducible neutral loss corresponding to release of succinimide sulfinic acid (Figure 3E). Based on these findings, the sequential labeling strategy captures endogenous sulfinic acids as sulfonyl succinimide conjugates, which are sufficiently stable for site-specific mass spectrometry analysis.
Next, we profiled pharmacological induction of redox stress in RAW 264.7 cells. The mitochondrial electron transport chain inhibitors oligmoycin, rotenone, and paraquat all induced minor increases in maleimide-TAMRA labeling (Figure 4A). Interestingly, potassium cyanide induced more significant labeling, suggesting it is a strong inducer of sulfinic acid formation. Alternatively, antimycin A reduced labeling, which could signify further oxidation of sulfinic acids to unreactive sulfonic acids. Lipopolysaccharide (LPS) and tunicamycin stimulated more labeling, suggesting sulfinic acids may be more readily induced outside of the mitochondria, either through LPS-mediated induction of NADPH oxidases or through tunicamycin-stimulation of endoplasmic reticulum (ER) stress. Interestingly, a single ~75 kD protein dominated maleimide-TAMRA labeling across the proteome, suggesting at least one major S-sulfinated protein in macrophage cells. Collectively, stimulus-dependent maleimide labeling confirms detection of an IAM-resistant population of modified thiols, further supporting direct conjugation to sites of S-sulfination.
Figure 4. Profiling S-sulfination in stimulated cells and tissues.
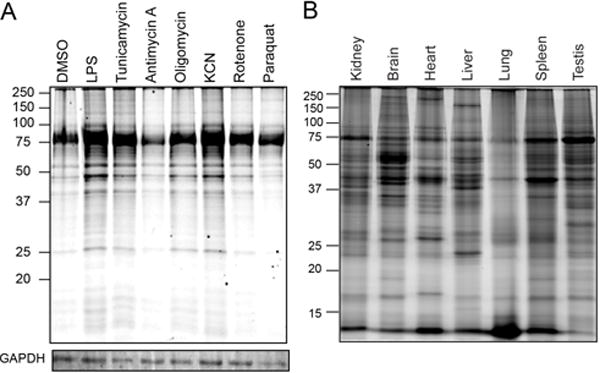
(A) Stimulus-dependent induction of protein S-sulfination in RAW 264.7 cells (24 hr); LPS (100 ng / mL), tunicamycin (10 μM), antimycin A (50 μM), oligomycin (5 μMds), KCN (1 mM), rotenone (1 uM), and paraquat (40 μM). (B) S-sulfination profile across different mouse tissues.
Similar to previous results with GSNO-biotin, maleimide-biotin labeled a distinct profile of S-sulfinated proteins across different mouse tissues[5]. While a similar 75 kD protein is present across all tissues, there are clear differences that could reflect differential protein expression or redox states. Despite continuous exposure to high concentrations of oxygen, lung homogenates show the lowest overall labeling. Indeed, the low molecular weight <15 kD band, presumably thioredoxin, is labeled more efficiently in lung than other tissues. Clearly, site-specific mass spectrometry-based proteomic annotation of these tissue-specific oxidation events will greatly enhance our understanding of cellular redox regulation and how cysteine can reach higher oxidation states in proteins. As compared to thiol-succinimide conjugates, sulfonyl-succinimide labeled peptides have different retention times and mass due to the two additional oxygens (+32 D), providing clear orthogonality for mass spectrometry annotation.
Cysteine thiols are both nucleophilic and sensitive to the cellular redox environment. While cysteines typically form disulfide bonds when exposed to oxidants, certain thiols are further oxidized to sulfinic acids and sulfonic acids. Here we demonstrate maleimide reacts with cysteine sulfinic acids in proteins. Using a sequential alkylation strategy, maleimide probes can selectively label protein sulfinic acids on chemical standards, recombinant protein, and in cell homogenates from stimulated cells. The resulting sulfonylcysteine-succinimide adduct is stable in acidic buffers, providing a simple strategy to profile S-sulfination in biological samples.
While this approach simplifies the direct detection of protein S-sulfination, it also demonstrates potential challenges in profiling such a transient, intermediate posttranslational modification. While increased labeling can be attributed to enhanced S-sulfination, decreased labeling could represent either lower S-sulfination or further conversion to S-sulfonation (R-SO3H). Thus, changes in maleimide labeling cannot be used alone to assess redox status, and will require more detailed analysis. Furthermore, mass spectrometry profiling studies using this approach will require site-specific peptide analysis to avoid detection thiols that may have escaped complete iodoacetamide alkylation. Acid stable proteases, such as pepsin or Glu-C, may be necessary to stabilize the modification during proteolysis. With these points in mind, future studies will explore site-specific S-sulfination across distinct physiological states, while providing a simple and accessible method for direct S-sulfination analysis.
Experimental Section
Synthetic methods, characterization, and mass spectrometry methods are provided in the Supporting Information.
HPLC assays of sulfonyl succinimide formation and reversibility
Stock solutions of benzene sulfinic acid (20 mM), N-ethylmaleimide (200 mM), and iodoacetamide (200 mM) were freshly prepared in DMSO for each experiment. 50 μL of benzene sulfinic acid and 50 μL of either N-ethylmaleimide or iodoacetamide were added to 900 μL of 6 M urea in citrate-phosphate buffer at pH 4.5 for 1 hr. After 60 minutes, the reaction mixture was analyzed by HPLC. Stock solutions of benzene sulfinic acid (10 mM), maleimide (100 mM), and 1-ethyl-3-(phenylsulfonyl)succinimide (100 mM) were prepared in DMSO. 50 μL of benzene sulfinic acid and 50 μL of maleimide solutions were added to 900 μL of 6 M urea in citrate-phosphate buffer at pH 4.5. After 30 minutes, the reaction mixture was analyzed by HPLC. Next, 50 μL of 1-ethyl-3-(phenylsulfonyl)succinimide and 50 μL of maleimide stock solution were added to 900 μL of 6 M urea in citrate-phosphate buffer at pH 4.5. After 2 hours, the reaction mixture was analyzed by HPLC.
Gel-based profiling of protein S-sulfination
Mouse RAW 264.7 cells and human HEK-293T cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum (JR Scientific) and 1% (v/v) penicillin / streptomycin / glutamine (Gibco). Cells were harvested at 80% confluence with a cell scraper in 6 M urea / PBS buffer supplemented with 1 mM IAM (pH 7.4 and lysed by sonication (4 °C, 10% duty cycle, 15 seconds). Protein concentrations were measured using the BioRad DC assay, and then diluted 2 mg / mL. Next, lysates were incubated with 2.5 mM dithiothreitol for 30 minutes, followed by 50 mM iodoacetamide with 10 mM EDTA and 2% (w/v) SDS for 1 hr in the dark. The reaction was quenched by chloroform/methanol precipitation, and re-solubilized in 6 M urea in citrate-phosphate buffer (pH 4.5). Next, lysates were incubated with 50 μM maleimide-TAMRA for 30 minutes. For tissue homogenate studies, tissues were collected from an 18 month-old C57/B6 mice and frozen immediately in liquid nitrogen, and later dounce homogenized in aqueous 6 M urea / PBS (pH 7.4) supplemented with 1 mM IAM. The homogenate was cleared by centrifugation at 3000× g for 5 minutes, followed by 5000× g for 5 minutes, and the supernatant was processed as described above. Protein samples were resolved by SDS-PAGE (10% or 12% gels) and images using a Typhoon flatbed fluorescence gel scanner. Next, the resolved proteins were transferred to the Immobilon-FL membrane (Millipore) and blocked with 5% bovine serum albumin (BSA, Fisher) in Tris-buffered Saline-Tween 20 (TBS-T, pH 7.4) for 1 hr at room temperature, followed by washing with TBST (3 × 5 minutes). After blocking with 5% BSA, immunoblotting was performed with the primary and secondary antibodies, and detected using an Azure Biosystems c600 imager. For GAPDH detection, blots were probed with the anti-GAPDH mouse monoclonal antibody (mAb 6C5, Calbiochem, 1 μg / mL, 2.5% BSA, 0.02% sodium azide, TBS-T, pH 7.0), washed and probed with a secondary Alexa Fluor 488 nm donkey-anti-mouse antibody conjugate (IgG H +L, Life Technologies, 2 μg / mL antibody, 0.06% sodium azide, TBS-T) for 1 hr at room temperature. For 6x-His Tag detection, blots were probed with the anti-6x-His Tag monoclonal antibody (mAb IgG2b, Thermo Fisher, 1 μg / mL, 2.5% BSA, 0.02% sodium azide, TBS-T, pH 7.0), washed and probed with a secondary Alexa Fluor 488 nm donkey-anti-mouse antibody for 1 hr at room temperature. Mouse studies were approved by the University of Michigan Institutional Animal Care and Use Committee (PRO0005707).
Supplementary Material
Acknowledgments
We would like to thank John E. Crellin, Hashim F. Motiwala, Rachel Pricer (U. Michigan) for help with chemical synthesis, analysis, and figure preparation. Financial support was provided by the National Institutes of Health DP2 GM114848, T32 CA140044 Proteome Informatics of Cancer Training Grant (A.M.K), T32 GM008597 Chemical Biology Interfaces Training Grant (S.E.H.), S10 OD021619 (Orbitrap Fusion Lumos Tribrid Mass Spectrometer), a National Science Foundation Graduate Research Fellowship (A.M.K.), the American Heart Association 14POST20420040 (J.D.M.), and the University of Michigan.
Footnotes
Supporting information for this article can be found at XXX.
References
- 1.Paulsen CE, Carroll KS. Chem Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lo Conte M, Carroll KS. J Biol Chem. 2013;288:26480–26488. doi: 10.1074/jbc.R113.467738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chauvin JR, Pratt DA. Angew Chem, Int Ed. 2016 [Google Scholar]
- 4.Hamann M, Zhang T, Hendrich S, Thomas JA. Methods Enzymol. 2002;348:146–156. doi: 10.1016/s0076-6879(02)48634-x. [DOI] [PubMed] [Google Scholar]
- 5.Majmudar JD, Konopko AM, Labby KJ, Tom CT, Crellin JE, Prakash A, Martin BR. J Am Chem Soc. 2016;138:1852–1859. doi: 10.1021/jacs.5b06806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lo Conte M, Carroll KS. Angew Chem, Int Ed. 2012;51:6502–6505. doi: 10.1002/anie.201201812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Lu G-P, Cai C, Chen F, Ye R-L, Zhou B-J. ACS Sustainable Chem Eng. 2016;4:1804–1809. [Google Scholar]; b) Matsuda I, Akiyama K, Toyoshima T, Kato S, Mizuta M. Bull Chem Soc Jpn. 1975;48:3675–3677. [Google Scholar]; c) Trost BM, Kallander LS. J Org Chem. 1999;64:5427–5435. doi: 10.1021/jo990195x. [DOI] [PubMed] [Google Scholar]
- 8.Baldwin AD, Kiick KL. Bioconjugate Chem. 2011;22:1946–1953. doi: 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richarme G, Liu C, Mihoub M, Abdallah J, Leger T, Joly N, Liebart JC, Jurkunas UV, Nadal M, Bouloc P, Dairou J, Lamouri A. Science. 2017 doi: 10.1126/science.aag1095. [DOI] [PubMed] [Google Scholar]
- 10.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. Proc Natl Acad Sci U S A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.