

Abstract
Here we report a ratiometric fluorescent probe for chemoselective conjugation to sulfenic acids in living cells. Our approach couples an α-fluoro-substituted dimedone to an aminonaphthalene fluorophore (F-DiNap), which upon sulfenic acid conjugation is locked as the 1,3-diketone, changing the fluorophore excitation. F-DiNap reacts with S-sulfenylated proteins at equivalent rates to current probes, but the α-fluorine substitution blocks side-reactions with biological aldehydes.
Graphical Abstract
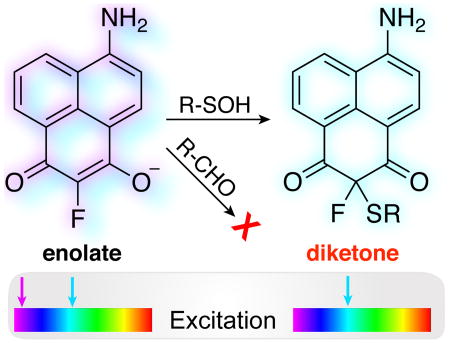
When exposed to reactive oxygen species, the cysteine thiol group can be modified to induce temporary and sometimes permanent protein damage. Cysteine thiols are initially oxidized to sulfenic acids, which quickly react with a second thiol to form disulfides1, 2. In the absence of an impendent thiol, sulfenic acids can be long-lived, establishing S-sulfenylation as a reversible protein post-translational modification.3 In several examples, S-sulfenylation has been shown to act as a redox regulatory switch, coordinating complex cellular cues as part of conserved redox-dependent signaling cascades1-3.
Active methylene containing 1,3-diketones such as dimedone are well established as the primary tools to detect sulfenic acids in biological systems4-9. When added to proteins, the nucleophilic α-carbon reacts with S-sulfenylated thiols to form a stable thioether, covalently bridging the probe to the site of S-sulfenylation. This covalent adduct can then be detected either by anti-dimedone antibodies10 or by conjugation to a reporter group4, 5, 7, 8, 11. Strained cyclooctynes, benzoxaborazoles, and boronic acids have also been explored as sulfenic acid labeling reagents11, 12. Despite this expanded toolset, each approach relies on tethered fluorophores or secondary conjugation to a reporter group13, 14, which complicate imaging S-sulfenylation in living cells by increasing background or requiring excessive washing.
Dimedone exists primarily as an enolate at physiological pH (pKa = 4.3)15. The electron-rich carbon reacts with a sulfenic acid to eliminate water, forming a transient diketone that quickly deprotonates to form the enolate product (Scheme S1)16. Accordingly, only one α-hydrogen is necessary to react with a sulfenic acid. Therefore, substitution of a single α-hydrogen with a small blocking group should have no effect on sulfenic acid conjugation, but would preclude subsequent tautomerization and trap the product in the diketone form. Thus, after reaction with a sulfenic acid, the enolate-to-diketone transition might switch the chemical and electronic properties of a conjugated dye to provide a direct approach for live-cell imaging of this dynamic redox post-translational modification.
Aminonaphthalene fluorophores, such as Lucifer Yellow, are typically linked to biomolecules at the imide nitrogen for use in biological imaging17. We hypothesized that replacement of the imide nitrogen with a methylene carbon would create a sulfenic acid-reactive fluorophore, potentially spectroscopically responsive to protein conjugation. A series of dimedone-naphthalene (DiNap) probes were synthesized in two steps (Schemes S2-S4). In the enolate form, H-DiNap (3a) has a single α-hydrogen, while the substituted Me-DiNap (3b) and F-DiNap (3c) have none (Scheme 1). Next, each DiNap probe was evaluated for labelling efficiency using the mutant E.coli peroxiredoxin AhpC (C166S), which lacks the resolving thiol and forms a stable sulfenic acid1. H-DiNap and F-DiNap labeled AhpC (C166S) (Figure 1a), but there was essentially no labeling by Me-DiNap. Importantly, F-DiNap yields a similar profile of labeled proteins as the alkyne-linked dimedone derivative Dyn-218 (Figure S1), labels AhpC (C166S) in an oxidation-dependent manner (Figure S2), and reacts to completion with a sulfenamide standard (Figure S3). Furthermore, sulfenylation with small-molecule sulfenamides yielded a >50 ppm 19F-NMR shift (Figure S4), indicating a new distinct spectroscopic reporter group for sulfenic acid detection.
Scheme 1.
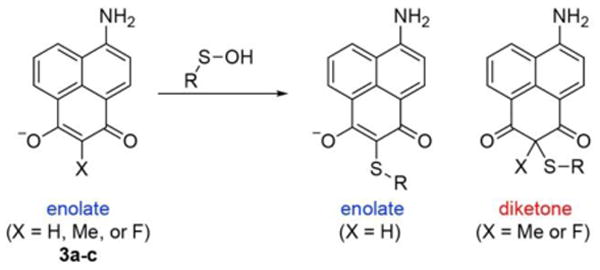
DiNap probes react with S-sulfenylated proteins. After sulfenic acid conjugation, unsubstituted probes undergo a second deprotonation to return to the enolate form. However, α-substituted Me- and F-DiNap probes are locked in the diketone form after conjugation.
Fig. 1.
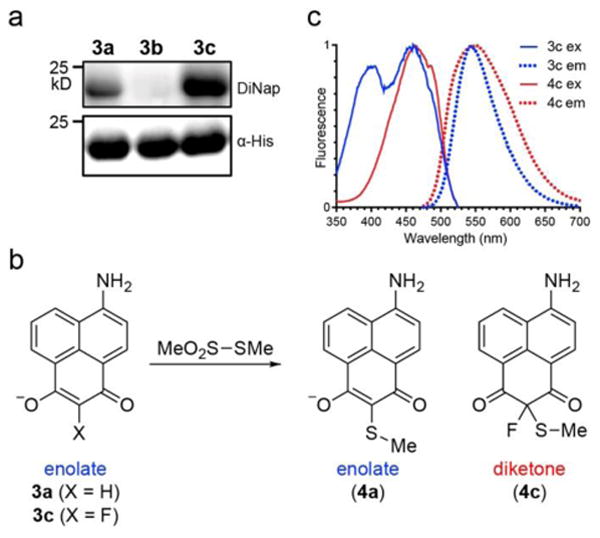
F-DiNap is an excitation ratiometic probe for protein S-sulfenylation. (a) Comparison between 3a-c conjugation to AhpC (C166S) by in-gel fluorescence (Ex. 488 nm, Em: 555/20 nm). (b) Methyl methane thiosulfonate (MeO2S-SMe) reacts with 3a and 3c to yield conjugated standards in aqueous buffer. (c) F-DiNap fluorescent excitation and emission spectra before and after conjugation to MeO2S-SMe (S-methyl methanethiosulfonate, MMTS).
In these probes, the aminonaphthalene fluorophore is directly conjugated and sensitive to the electronics of the sulfenic acid reactive 1,3-diketone. Both H- and F-DiNap probes exhibit two major excitation peaks at 400 nm and 460 nm, and one emission peak centered near 540 nm. When protonated, the 400 nm excitation peak is slightly reduced in H-DiNap (3a), but completely absent in F-DiNap (3c) (Figure S5). Using these pH-sensitive shifts, the pKa of H-DiNap (pKa = 5.3) was measured to be slightly higher than F-DiNap (pKa = 4.8) (Figure S6), confirming both probes are predominantly present in their reactive enolate form. Based on these findings, we hypothesize the reactive enolate primarily contributes to the 400 nm excitation peak, which is eliminated by protonation or conversion to the keto form by reaction with a sulfenic acid. Since small-molecule sulfenic acids are unstable (t1/2 <1 min)19, and the F-DiNap reaction product has poor solubility, making it challenging to accurately measure the reaction rate and response with small molecule standards. To address this technical problem, we found that S-methyl methane thiosulfonate (MMTS) can act as simple surrogate to provide a direct route to the sulfenylated standards 4a and 4c (Figure 1b and Scheme S5). While the reaction with MMTS is slow in aqueous buffer, it was accelerated in organic solvent under basic conditions. Both 3a and 3c have similar spectral properties (Table S1), although 3a is nearly three times brighter than 3c. Following conjugation to MMTS, the 400 nm excitation peak is reduced in 4a and completely absent in 4c (Figure 1c and Figure S7). In addition, 3a is nearly 7-times brighter than 4a, which largely cancels out any effective excitation ratio change (Table S1). Conversely, 3c and 4c have the same effective brightness, presenting a potential excitation ratiometric sensor for imaging protein S-sulfenylation, in a similar manner as the popular calcium sensor Fura-220, 21. For F-DiNap, sequential 400 nm and 488 nm excitation result in different emission intensities at a fixed wavelength (550 nm), establishing a ratiometric measure of sulfenic acid conjugation (Figure S8).
In live cells, F-DiNap must compete with a range of reactive thiols and redox detoxifying enzymes to efficiently react with sulfenic acids. For example, S-sulfenylated albumin forms a disulfide with glutathione 100-fold faster than the corresponding reaction with dimedone. Therefore, fast reaction rates increase the likelihood of capturing the transient S-sulfenylated species22. A recent analysis of α-fluoro dimedone derivatives reports at least a 100-fold slower reaction rate than dimedone using a cyclic sulfenamide standard23. Using in-gel fluorescence analysis, here we find that F-DiNap labels AhpC (C166S) at an equivalent rate as dimedone, while H-DiNap is slightly slower (Figures S9 – S10, Table 1). Based on this analysis, F-DiNap probes do not compromise the rate of reaction with a common protein within a complex proteome.
Current 1,3-dione-based probes have the potential to participate in condensation reactions with cellular aldehydes24, 25, which like sulfenic acids, are formed under oxidative stress. Since the Knoevenagel condensation requires the presence of two α-protons, we predicted that α-fluorine on 3c will prevent any reaction with biological aldehydes (Figure 2a). After a brief incubation of dimedone with pyridoxal or glyceraldehyde in Dulbecco's phosphate-buffered saline (DPBS), we observed complete conversion to the condensation products (Figures S11 and S12) at a rate of 0.10 ± 0.01 M-1 s-1 (Table 1 and Figure S13), as well as secondary conjugate addition to the resulting electrophilic unsaturated ketone. Since aldehyde condensation occurs at near the same rate as sulfenic acid conjugation, current 1,3-dione-based probes likely interrupt a variety of metabolic pathways. For these metabolic aldehydes, F-DiNap avoids aldehyde cross-reactivity, introducing a new chemo-selective approach for detecting biological sulfenic acids.
Fig. 2.
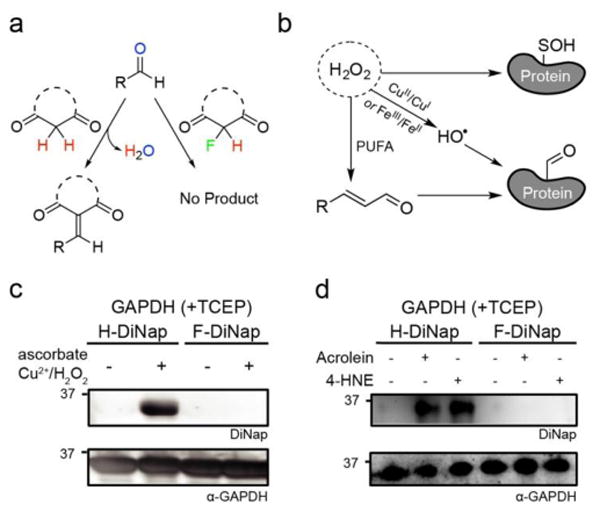
F-DiNap is does not react with protein-linked aldehydes. (a) α-Fluoro substituted probes do not form condensation products with aldehydes. (b) Reactive oxygen species promote the formation of cysteine S-sulfenylation, but also generate protein-linked aldehydes and lipid peroxidation ene-al products. (b) GAPDH oxidized under Fenton conditions reacts with H-DiNap, but not F-DiNap. (c) Addition of acrolein (1 mM) or 4-hydroxynonenal (HNE) (1 mM) to GAPDH increases labelling by H-DiNap, but not F-DiNap. In-gel fluorescence ex. 488 nm, em. 555/20 nm.
Under basal conditions, aldehydes are generally not present in proteins. However, aldehydes can be formed under conditions of oxidative stress, in similar conditions as found to promote sulfenic acid formation (Figure 2b). In a process analogous to the Fenton reaction, Cu+1/Cu+2 or Fe+2/Fe+3, ascorbate, and hydrogen peroxide react to release hydroxyl radicals, which modify nearby proteins by direct oxidation of several amino acid side chains or the peptide backbone to form protein-linked aldehydes at diffusion-limited rates26. To examine the reactivity of DiNap derivatives under these conditions, recombinant glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was incubated with CuSO4, ascorbate, and peroxide. Incidentally, these conditions mimic the common copper-catalyzed azide-alkyne cycloaddition (CuAAC) conditions used for alkynyl dimedone labeling of peroxide treated samples. Next, tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was added to reduce any sulfenic acids, leaving aldehydes as the sole electrophiles. Under these Fenton conditions, GAPDH was efficiently labeled with H-DiNap, but there was no detectable labeling by F-DiNap (Figure 2b).
Peroxidation of long-chain polyunsaturated fatty acids (PUFAs) releases a series of α,β-unsaturated aldehydes including acrolein and 4-hydroxynonenal (4-HNE). These ene-als undergo conjugate addition primarily with cysteine, lysine, and histidine to tether a reactive aldehyde to proteins. To explore any cross-reactivity, dimedone, acrolein and N-acetyl cysteine were combined together and allowed to react in DPBS. HPLC analysis revealed a series of aldehyde condensation products, while F-DiNap was completely unreactive under these conditions (Figure S14). Next, the biological ene-als acrolein and 4-HNE were added to AhpC (C166S) or GAPDH-overexpressing bacterial lysates. H-DiNap labeling of AhpC (C166S) was greatly enhanced by each ene-al, while F-DiNap labeling remained constant (Figure S15). Similarly, reduction with TCEP after ene-al treatment eliminated all subsequent sulfenic acid labeling by F-DiNap, confirming that α-fluoro substitution of active methylene compounds prevents condensation with lipid-derived aldehyde byproducts on proteins (Figure 2c). This selectivity over peroxide-generated aldehydes that are formed concurrently with sulfenic acids is a key improvement over previous methods.
When added to cultured mammalian cells, F-DiNap is cell permeant, allowing live cell labeling and gel-based fluorescence profiling of S-sulfenylation. In different cancer cell lines, we corroborated previous reports demonstrating higher levels of protein S-sulfenylation in MDA-MD-231 cells10 (Figure S16). Next, CH27 B-cells were incubated with H- or F-DiNap probes and imaged by total internal reflection (TIRF) microscopy, alternating excitation between 405 and 488 nm lasers, to selectively fluorescence image fluorophores at the cell membrane near the glass surface. Over the time course of 15 minutes, H-DiNap labeled cells showed no change in excitation ratio. In contrast, F-DiNap labeled cells demonstrated a robust excitation ratio change, revealing distinct hotspots at the cell membrane (Figure 3a and Figure S17) with no signs of cell blebbing or death. In some F-DiNap labeled cells, the rapid F-DiNap reaction rates enabled detection of spatially restricted excitation ratio changes within 6 seconds of hydrogen peroxide stimulation, revealing sites of rapid membrane-localized redox stress (Figure 3b and Figure S18).
Fig. 3.
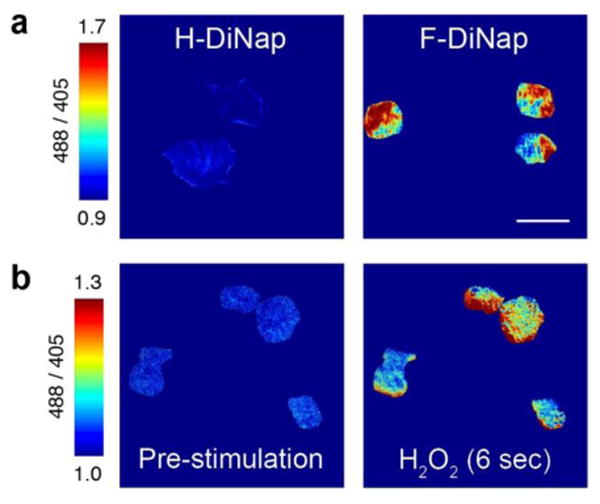
Live cell excitation ratiometric imaging of sulfenic acids in CH27 B-cells with DiNap probes. (a) Images were acquired by TIRF microscopy 15 minutes after probe addition, showing spatially resolved ratio changes with F-DiNap. The ratios were calculated after background subtraction of regions outside of cells, and dividing the emission intensities from each separate excitation. Scale bars are displayed to highlight scale and range of ratios displayed. (b) Hydrogen peroxide (1 mM) induces rapid F-DiNap ratio changes within a few seconds. Data is normalized to excitation ratios measured after 9 minutes of probe labeling. Images are representative of 3 biological replicates. Scale bar = 20 μm.
Next we examined if F-DiNap could detect physiological changes in cellular redox regulation and protein oxidation. The transcription factor Snail (SNAI1) is elevated in metastatic cancers, and plays a key role in reprogramming cells to undergo epithelial-to-mesenchymal transition (EMT) through the loss of the cellular adhesion protein E-cadherin27. Quantitative mass spectrometry profiling of Snail-expressing cells recently demonstrated a >3-fold elevation of many antioxidant enzymes, including peroxiredoxins, superoxide dismutase, and thioredoxin28. Based on these data, we examined if Snail overexpression affects redox homeostasis to influence protein S-sulfenylation levels in live cells. Confocal excitation ratiometric F-DiNap imaging in live MCF10A cells confirmed reduced S-sulfenylation in Snail-expressing cells (Figure 4a), particularly outside of the nucleus. When cells were lysed and analyzed by in-gel fluorescence, Snail over-expression dramatically reduced F-DiNap labeling (Figure 4b). Snail over-expression also enhanced cell viability following exposure to excess hydrogen peroxide, confirming induction of an antioxidant response that functionally represses cellular S-sulfenylation (Figure 4c).
Fig. 4.
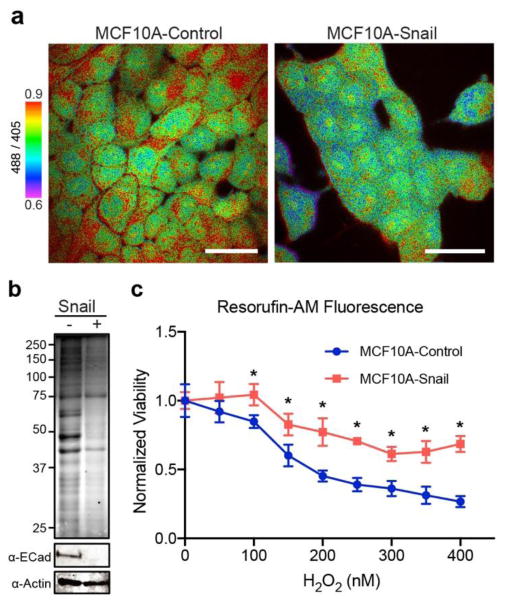
Snail expression attenuates F-DiNap labeling in MCF10A cells. (a) Confocal ratiometric imaging of F-DiNap in Snail-expressing cells. Images are representative of 3 technical replicates. Scale bar = 50 μm. (b) In-gel F-DiNap fluorescence of attenuated S-sulfenylation in Snail-expressing MCF10A cells. (c) Snail over-expression protects MCF10A cells from peroxide-dependent reduction in cell viability. Cell viability was measured after 24 hours by resorufin-AM fluorescence. Data is representative of 3 biological replicates each with 6 technical replicates.
In summary, we present a simple probe for gel-based profiling of protein S-sulfenylation. α-Fluorination of 1,3-dione probes significantly improves the selectivity towards sulfenic acids over biological aldehydes while not sacrificing rate of reaction. F-DiNap is compatible with live-cell excitation ratiometric fluorescence imaging of S-sulfenylation, allowing direct assessment of cellular redox states. By extension, this design should be transferrable to brighter dye scaffolds, faster 1,3-diketone scaffolds, chemoselective affinity probes for mass spectrometry profiling, and 19F-NMR probes for multimodal analysis of protein S-sulfenylation.
Supplementary Material
Footnotes
Electronic Supplementary Information (ESI) available: Synthetic methods, chemical characterization, experimental methods, supporting figures. See DOI: 10.1039/x0xx00000x.
Notes and references
- 1.Ellis HR, Poole LB. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 2.Paulsen CE, Carroll KS. Chem Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Devarie-Baez NO, Silva Lopez EI, Furdui CM. Free Radical Res. 2016;50:172–194. doi: 10.3109/10715762.2015.1090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poole LB, Zeng BB, Knaggs SA, Yakubu M, King SB. Bioconjug Chem. 2005;16:1624–1628. doi: 10.1021/bc050257s. [DOI] [PubMed] [Google Scholar]
- 5.Reddie KG, Seo YH, Muse Iii WB, Leonard SE, Carroll KS. Mol Biosyst. 2008;4:521–531. doi: 10.1039/b719986d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benitez LV, Allison WS. J Biol Chem. 1974;249:6234–6243. [PubMed] [Google Scholar]
- 7.Qian J, Wani R, Klomsiri C, Poole LB, Tsang AW, Furdui CM. Chem Commun. 2012;48:4091–4093. doi: 10.1039/c2cc17868k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qian J, Klomsiri C, Wright MW, King SB, Tsang AW, Poole LB, Furdui CM. Chem Commun. 2011;47:9203–9205. doi: 10.1039/c1cc12127h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, Gupta V, Carroll KS, Liebler DC. Nat Commun. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seo YH, Carroll KS. Proc Natl Acad Sci U S A. 2009;106:16163–16168. doi: 10.1073/pnas.0903015106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu CT, Benkovic SJ. J Am Chem Soc. 2013;135:14544–14547. doi: 10.1021/ja407628a. [DOI] [PubMed] [Google Scholar]
- 12.Poole TH, Reisz JA, Zhao W, Poole LB, Furdui CM, King SB. J Amer Chem Soc. 2014;136:6167–6170. doi: 10.1021/ja500364r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGarry DJ, Shchepinova MM, Lilla S, Hartley RC, Olson MF. ACS Chem Biol. 2016;11:3300–3304. doi: 10.1021/acschembio.6b00742. [DOI] [PubMed] [Google Scholar]
- 14.Yin Q, Huang C, Zhang C, Zhu W, Xu Y, Qian X, Yang Y. Org Biomol Chem. 2013;11:7566–7573. doi: 10.1039/c3ob41434e. [DOI] [PubMed] [Google Scholar]
- 15.Qian J, Klomsiri C, Wright MW, King SB, Tsang AW, Poole LB, Furdui CM. Chem Commun. 2011;47:9203–9205. doi: 10.1039/c1cc12127h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagner G. Tetrahedron. 2013;69:7243–7252. [Google Scholar]
- 17.Stewart WW. Cell. 1978;14:741–759. doi: 10.1016/0092-8674(78)90256-8. [DOI] [PubMed] [Google Scholar]
- 18.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Nat Chem Biol. 2011;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta V, Carroll KS. Biochim Biophys Acta. 2014;1840:847–875. doi: 10.1016/j.bbagen.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grynkiewicz G, Poenie M, Tsien RY. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 21.Tsien RY, Rink TJ, Poenie M. Cell Calcium. 1985;6:145–157. doi: 10.1016/0143-4160(85)90041-7. [DOI] [PubMed] [Google Scholar]
- 22.Turell L, Botti H, Carballal S, Ferrer-Sueta G, Souza JM, Duran R, Freeman BA, Radi R, Alvarez B. Biochemistry. 2008;47:358–367. doi: 10.1021/bi701520y. [DOI] [PubMed] [Google Scholar]
- 23.Gupta V, Paritala H, Carroll KS. Bioconjug Chem. 2016;27:1411–1418. doi: 10.1021/acs.bioconjchem.6b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deb ML, Bhuyan PJ. Tetrahedron Lett. 2005;46:6453–6456. [Google Scholar]
- 25.Kudirka R, Barfield RM, McFarland J, Albers AE, de Hart GW, Drake PM, Holder PG, Banas S, Jones LC, Garofalo AW, Rabuka D. Chem Biol. 2015;22:293–298. doi: 10.1016/j.chembiol.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 26.Requena JR, Chao CC, Levine RL, Stadtman ER. Proc Natl Acad Sci U S A. 2001;98:69–74. doi: 10.1073/pnas.011526698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohkubo T, Ozawa M. J Cell Sci. 2004;117:1675–1685. doi: 10.1242/jcs.01004. [DOI] [PubMed] [Google Scholar]
- 28.Hernandez JL, Davda D, Majmudar JD, Won SJ, Prakash A, Choi AI, Martin BR. Mol Biosyst. 2016;12:1799–1808. doi: 10.1039/c6mb00019c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.