

SUMMARY
Adipose tissue fibrosis is a hallmark of malfunction that is linked to insulin resistance and type 2 diabetes; however, what regulates this process remains unclear. Here we show that the PRDM16 transcriptional complex, a dominant activator of brown/beige adipocyte development, potently represses adipose tissue fibrosis in an UCP1-independent manner. By purifying the PRDM16 complex, we identified GTF2IRD1, a member of the TFII-I family of DNA-binding proteins, as a cold-inducible transcription factor that mediates the repressive action of the PRDM16 complex on fibrosis. Adipocyte-selective expression of GTF2IRD1 represses adipose tissue fibrosis and improves systemic glucose homeostasis independent of body-weight loss, while depleting GTF2IRD1 promotes fibrosis in a cell-autonomous manner. GTF2IRD1 represses the transcription of TGF-β-dependent pro-fibrosis genes by recruiting PRDM16 and EHMT1 onto their promoter/enhancer regions. These results suggest a mechanism by which repression of obesity-associated adipose tissue fibrosis through the PRDM16 complex leads to an improvement in systemic glucose homeostasis.
Keywords: Obesity, Diabetes, Brown adipose tissue, Beige adipocyte, Adipose tissue fibrosis, PRDM16, GTF2IRD1, UCP1-independent
eTOC Blurb
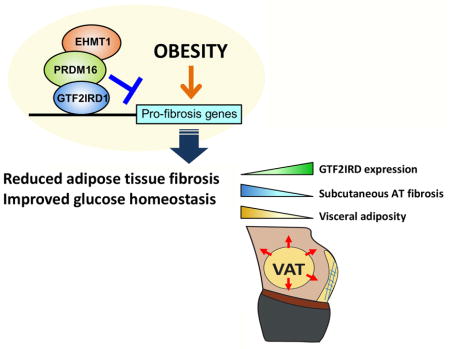
Hasegawa et al. identify GTF2IRD1 as a cold-inducible transcription factor that represses adipose tissue fibrosis through a PRDM16-EHMT1 complex. Repression of adipose tissue fibrosis by the complex improves systemic glucose homeostasis independent of UCP1-mediated thermogenesis and body-weight. In humans, GTF2IRD1 expression inversely correlates with subcutaneous WAT fibrosis and visceral adiposity.
INTRODUCTION
Dysregulation of adipose tissue homeostasis is a primary cause of obesity-related metabolic disorders, including insulin resistance, hepatic steatosis, and diabetes mellitus (Crewe et al., 2017). Mounting evidence highlights the importance of the extracellular matrix (ECM) in maintaining adipose tissue homeostasis. The accumulation of ECM proteins (e.g., collagens) during the early stages of obesity is a part of the tissue remodeling process that accompanies healthy adipose expansion; however, pathologically excessive accumulation of the ECM in adipose tissues causes fibrosis that is tightly associated with the increased infiltration of pro-inflammatory immune cells into the adipose tissues, and subsequently greater tissue inflammation (Sun et al., 2013b). Importantly, such changes to the ECM, and consequent fibrosis in human subcutaneous white adipose tissue (WAT) are strongly linked to insulin resistance and type 2 diabetes (Divoux et al., 2010; Henegar et al., 2008; Lackey et al., 2014; Muir et al., 2016; Reggio et al., 2016). For instance, collagen VI (Col6), a major ECM protein in adipose tissues, accumulates at much higher rates under obese and diabetic states (Dankel et al., 2014; Divoux et al., 2010; Khan et al., 2009; Pasarica et al., 2009; Spencer et al., 2010). In the absence of Col6, the WAT of obese mice is able to expand with less tissue fibrosis and inflammation; thus, the Col6-deficient mice display improved glucose tolerance and insulin sensitivity (Khan et al., 2009).
Two prominent pathways have been highlighted with regard to regulating adipose fibrosis: hypoxia-inducible factor 1α (HIF1α) and transforming growth factor-β (TGF-β). In obesity, adipose tissue hypoxia results in the activation of HIF1α-dependent gene transcription (Hosogai et al., 2007; Rausch et al., 2008; Ye et al., 2007). Moreover, mouse models with ectopic activation of HIF1α in adipose tissues induce fibrosis and glucose intolerance (Halberg et al., 2009), whereas treatment with the HIF1α-selective inhibitor PX-478 ameliorates adipose fibrosis, inflammation, and glucose intolerance (Sun et al., 2013a). Similarly, levels of TGF-β, a primary factor promoting tissue fibrosis, are highly elevated in both the circulation and adipose tissues of obese mice and humans (Samad et al., 1997; Yadav et al., 2011).
It is notable that TGF-β signaling exerts effects on brown/beige adipocyte biogenesis that are reciprocal to those on adipose fibrosis. For example, TGF-β treatment of adipocyte precursor cells potently inhibits beige adipocyte differentiation and expression of UCP1 (uncoupling protein 1) (Koncarevic et al., 2012; McDonald et al., 2015). By contrast, inhibiting TGF-β signaling by genetic deletion of Smad3, or by a neutralizing antibody against the activin receptor type IIB (ActRIIB), promotes brown and beige adipocyte biogenesis in vivo (Fournier et al., 2012; Koncarevic et al., 2012; Yadav et al., 2011). Furthermore, genetic ablation of myocardin-related transcription factor A (MRTFA), a G-actin-regulated transcriptional coactivator acting downstream of TGF-β signaling, has been shown to stimulate beige adipocyte differentiation (McDonald et al., 2015). The reciprocal relationship between adipose tissue fibrosis and brown/beige fat biogenesis may be simply due to body-weight loss through UCP1-mediated energy dissipation by brown and beige fat. However, our recent observations suggest that the improvements in systemic glucose homeostasis and reduced adipose tissue fibrosis, when beige adipocyte biogenesis is activated, are independent of body-weight loss and UCP1 expression. In fact, we recently found that transgenic mice expressing adipose tissue-selective PRDM16 (PR domain containing 16), which have an increased number of beige adipocytes in the subcutaneous WAT, have a remarkable improvement in glucose tolerance and reduction in their susceptibility to diet-induced adipose tissue fibrosis independent of UCP1 (Ikeda et al., 2017).
As a part of the UCP1-independent mechanisms of beige fat action in the regulation of glucose homeostasis, the present study identifies GTF2IRD1, a member of the TFII-I family of DNA-binding transcription factor, that potently represses pro-fibrosis gene expression by forming a complex with PRDM16 and EHMT1. Notably, repression of adipose tissue fibrosis by the PRDM16-EHMT1-GTF2IRD1 complex leads to a significant improvement in systemic glucose homeostasis independent of UCP1-mediated thermogenesis and body-weight loss. Our data provide an exciting avenue of improving systemic glucose homeostasis in vivo by targeting adipose tissue fibrosis.
RESULTS
Repression of Adipose Tissue Fibrosis by PRDM16 is UCP1-Independent
While transgenic mice expressing PRDM16 under control of the Fabp4 promoter/enhancer have an increased number of beige adipocytes in the subcutaneous WAT in conjunction with improved glucose tolerance, PRDM16 transgenic mice (Prdm16 Tg) fed a high-fat diet (HFD) for 10 weeks have improved systemic glucose tolerance, even when crossed onto a UCP1-deficient background (Figure 1A). This improvement was significant before any difference in body-weight emerged between the two groups (Ikeda et al., 2017). These results suggest that the mechanism underlying enhanced systemic glucose tolerance in Prdm16 Tg mice is not due to UCP1-mediated thermogenesis. Of note, we found that Prdm16 Tg and Prdm16 Tg x Ucp1−/− mice had significantly less hydroxyproline, a biochemical marker of tissue fibrosis, in the inguinal WAT and the epididymal WAT relative to their respective controls (Figure 1B), in close conjunction with the improvement in systemic glucose tolerance. Masson’s trichrome staining of WAT sections revealed that compared to corresponding littermates, Prdm16 Tg mice fed a HFD had far fewer trichrome-positive fibrotic streaks and similarly fewer crown-like structures, indicative of macrophages surrounding necrotic adipocytes (Figure 1C). Consistent with the observation of reduced hydroxyproline, the ability of adipocyte-specific Prdm16 expression to limit WAT fibrosis and macrophage accumulation was preserved in the absence of UCP1. Similarly, immunostaining of WAT sections revealed that transgenic PRDM16 expression markedly reduced the expression of endotrophin, a C-terminal cleavage product of Col6α3 that stimulates WAT fibrosis (Park and Scherer, 2012; Sun et al., 2014), and that this effect too was independent of UCP1 (Figure 1D).
Figure 1. UCP1 independent regulation of adipose fibrosis by PRDM16.

(A) Glucose tolerance test in Prdm16 Tg mice and the littermate controls (left) and in Ucp1−/−mice and Prdm16 Tg x Ucp1−/−mice (right) at 22°C. Mice were under HFD for 10 weeks. n= 7–8. * P<0.05, ** P<0.01.
(B) Hydroxyproline content in the BAT, the inguinal WAT and the epididymal WAT of mice with indicated genotypes under HFD for 14 weeks. n=6–10.
(C) Masson’s trichrome staining in the epididymal WAT of mice with indicated genotypes under HFD. Arrowheads indicate crown-like structures. Scale bars = 100 μm.
(D) Immunohistochemical staining with anti-mouse endotrophin antibody in the epididymal WAT of mice in (C). Arrowheads indicate crown-like structures. Scale bars = 100 μm.
(E) Expression profiles of pro-fibrotic genes (as indicated) in the inguinal WAT of mice with indicated genotypes under HFD for 14 weeks. The color scale shows z-scored FPKM representing the mRNA level of each gene in blue (low expression)-white-red (high expression) scheme. * P<0.05 by transgenic PRDM16 expression both in wild-type background and Ucp1−/− background.
(F) Cold-induced changes in gene expression of pro-fibrosis genes (% change relative to ambient temperature) in the inguinal WAT from wild-type (white) and Ucp1−/− mice (blue). Mice under 14 weeks of HFD were kept under ambient temperature or mild cold temperature at 16°C for 10 days. n=4–6. Data in A, B, and F are represented as mean ± SEM.
To understand the underlying mechanisms, we analyzed RNA-sequencing datasets of their inguinal WAT depots from these four genotypes. We found that a number of genes involved in connective tissue and extracellular matrix development were down-regulated in Prdm16 Tg and Prdm16 Tg x Ucp1−/− mice. Specifically, PRDM16 expression repressed genes encoding proteins comprising collagens, matrix metalloproteinases (Mmps), and Lgals3 (encoding Galectin-3) that is implicated in the development of WAT fibrosis (Martinez-Martinez et al., 2016). Repression of these pro-fibrotic genes was seen in Prdm16 Tg x Ucp1−/− mice, indicating that this repression of adipose tissue fibrosis is independent of UCP1 (Figure 1E).
Notably, WAT fibrosis was also reduced in the inguinal WAT following chronic cold exposure. We found that mild cold exposure (16°C) for 10 days significantly reduced the expression of many pro-fibrosis genes in the inguinal WAT of wild-type mice and Ucp1−/− mice (Figure 1F). Consistent with this observation, analysis of an independent microarray dataset (Xue et al., 2009) found that the expression of pro-fibrotic genes in the inguinal WAT was also down-regulated by chronic cold exposure for both 1 and 5 weeks (Figure S1A). Together, these results indicate that both PRDM16 and chronic cold exposure potently represses molecular pathways involved in adipose tissue fibrosis through an UCP1-independent mechanism.
GTF2IRD1 is a Cold-Inducible Transcription Factor that Forms a Transcriptional Complex with PRDM16 and EHMT1
Previous studies show that PRDM16 is recruited to brown/beige fat-specific genes through an interaction with DNA-binding transcription factors, such as peroxisome proliferator-activated receptors (PPARs), CCAAT-enhancer-binding proteins (C/EBPs), and Early B-Cell Factor 2 (EBF2) (Inagaki et al., 2016). However, the DNA-binding transcriptional factors that recruit the PRDM16 complex to the sites needed to regulate pro-fibrotic gene expression are unknown. Accordingly, we purified a PRDM16 transcriptional complex from the nuclear extracts of cultured beige adipocytes and subsequently characterized the subunits by liquid chromatography, coupled with tandem mass spectrometry (LC-MS/MS). We identified 413 proteins as nuclear-localized transcriptional regulators, including the previously characterized factors CtBP1 and CtBP2, EHMT1, MED1, and p107 (Inagaki et al., 2016). We then cross-referenced this list with the annotated UniProt database and found that the PRDM16 complex contains 43 DNA-binding transcriptional regulators, including C/EBPβ, PPARγ, EBF2, ZFP516, as well as uncharacterized factors (Figure 2A and Table S1).
Figure 2. GTF2IRD1 is a cold-inducible transcription factor that forms a complex with PRDM16 and EHMT1 in the BAT.

(A) Identification of nuclear-localized components in a PRDM16 transcriptional complex purified from differentiated beige adipocytes. Nuclear localized proteins and canonical DNA-binding transcription factors were listed based on the annotation database in UniProt.
(B) Gene expression profile of the identified transcriptional factors in interscapular BAT, inguinal WAT, and epididymal WAT. The color scale shows z-scored FPKM representing the mRNA level of each gene in blue (low expression)-white-red (high expression) scheme.
(C) Gene expression profile of the BAT-enriched transcriptional factors in cultured primary brown adipocytes and white adipocytes.
(D) Expression of Gtf2ird1 in BAT from mice housed at 22°C or 6°C for 3 days. n=5.
(E) Immunoblotting for GTF2IRD1 protein from mice in (D). β-actin was used as loading control.
(F) Expression of Gtf2ird1 mRNA in the indicated adipose tissues of mice treated with vehicle (saline) or CL316, 243 at a dose of 1 mg/kg for 7 days. n=5. * P<0.05, ** P<0.01, Data in D and F are represented as mean ± SEM.
(G) A EHMT1 complex was immunopurified from differentiated brown adipocytes. Endogenous GTF2IRD1 was detected by immunoblotting. Inputs are shown in lower panels.
(H) In vitro binding assay of 35S-labeled GTF2IRD1 and the indicated GST-fusion fragments of PRDM16. Coomassie brilliant blue for GST-proteins (bottom panel).
Based on our transcriptome databasets (Shinoda et al., 2015), we found that 10 transcription factors were specifically enriched in brown adipose tissue (BAT) (Figure 2B). Since adipose tissues are highly heterogeneous, we subsequently validated their expression levels in primary cultured brown adipocytes and found that the mRNA levels of 5 transcription factors, Gtf2ird1, Zfp148, Cebpb, Ebf2, and Zhx1, were enriched in primary brown adipocytes than white adipocytes (Figure 2C). In this study, we focused on Gtf2ird1 because of its clear co-enrichment with PRDM16 in mature brown adipocytes (Figure S1B–C) as well as its involvement in Williams-Beuren syndrome in which GTF2IRD1 gene is deleted in humans (Makeyev et al., 2004; Tipney et al., 2004).
Notably, exposing mice to temperatures under 6 °C for 3 days significantly increased the levels of both Gtf2ird1 mRNA (Figure 2D) and GTF2IRD1 protein (Figure 2E) in the BAT. Furthermore, daily systemic administration of a β3-adrenoceptor (β3-AR) agonist (CL316,243) to wild-type mice for 7 days significantly increased Gtf2ird1 mRNA levels in the BAT (Figure 2F). CL316,243 treatment also induced Gtf2ird1 expression in inguinal WAT and epididymal WAT, although at levels lower than that induced in BAT. Since forskolin treatment also increased Gtf2ird1 mRNA levels in cultured brown adipocytes (Figure S1D), β3-AR-dependent induction of Gtf2ird1 is mediated, at least in part, by cAMP signaling. Among eleven transcriptional variants of Gtf2ird1 in mice, variant 5 appears to be a major Gtf2ird1 transcript in BAT (Figure S1E–D).
We next investigated the interaction between GTF2RD1 and the PRDM16-EHMT1 complex; EHMT1 is a histone methyltransferase essential for PRDM16 function in brown and beige adipocyte development (Harms et al., 2014; Ohno et al., 2013). First, we detected endogenous GTF2IRD1 from the complexes that pulled down with purified EHMT1 extracted from the nuclei of differentiated adipocytes (Figure 2G). Next, in vitro binding assays using purified GTF2IRD1 and PRDM16 fragments fused to glutathione S-transferase (GST) found that GTF2IRD1 directly binds to two distinct zinc-finger domains of PRDM16 domains to which C/EBPβ and PPARγ are also known to bind (Kajimura et al., 2010) (Figure 2H). These results suggest that GTF2IRD1 is a cold-inducible and BAT-enriched transcriptional factor that interacts with both PRDM16 and EHMT1 to form a transcriptional complex.
Adipose-Selective Expression of GTF2IRD1 Represses Diet-Induced Adipose Tissue Fibrosis
To investigate the biological role of GTF2IRD1 in vivo, we generated transgenic mice in which variant 5 of Gtf2ird1 is driven by the Fabp4 promoter/enhancer in adipose tissues (Gtf2ird1 Tg). We confirmed that Gtf2ird1 mRNA and protein expression levels in the BAT and WAT of Gtf2ird1 Tg mice were significantly elevated compared to those of littermate controls (Figure S2A–B). Since a transgene driven by the Fabp4 promoter/enhancer can be detected in non-adipocytes, including macrophages (Lee et al., 2013), we first determined if macrophage activation is altered by transgenic Gtf2ird1 expression. To this end, we examined pro-inflammatory responses in the isolated macrophages from Gtf2ird1 Tg and the littermate controls by measuring pro-inflammatory cytokines (MCP1, TNF-α, and IL-6) following treatment with pro-inflammatory stimuli, such as IL-4, stearic acid (SA), palmitic acid (PA), and lipopolysaccharide (LPS). The assays found no significant differences in cytokine production and release in macrophages from Gtf2ird1 Tg and control mice (Figure 3A and Figure S2C). Thus, a specific contribution of macrophages to the differential regulation of tissue inflammatory response appears negligible in this animal model.
Figure 3. Adipose-selective expression of Gtf2ird1 represses adipose tissue fibrosis in vivo.

(A) Concentration of MCP-1, TNF-a and IL-6 secreted from the macrophages from Gtf2ird1 Tg mice and the littermate control mice (control). The isolated macrophages were stimulated with IL4, stearic acid (SA), palmitic acid (PA), or lipopolysaccharide (LPS). n=4.
(B) Immunoblotting for UCP1 in the BAT (upper panel) and the inguinal WAT (bottom panel) from Gtf2ird1 Tg mice and controls under ambient temperature. n=6–7. β-actin was used as loading control. Quantification of the UCP1 signal normalized by β-actin is shown on the right graphs. N.S., not significant.
(C) Masson’s trichrome staining in the BAT, the WAT and the epididymal WAT from Gtf2ird1 Tg and controls under 8 weeks of RD or HDF for 11, 18, and 24 weeks. Scale bars = 100 μm.
(D) Hydroxyproline content in the adipose tissues of mice in (C). * P<0.05 between Gtf2ird1 Tg mice and controls. # P<0.05, ## P<0.01 between RD and HFD. n= 5. Data in A, B, and D are represented as mean ± SEM.
(E) Hierarchical clustering and heat-map of RNA-seq transcriptome in the BAT of control and Gtf2ird1 Tg mice. The color scale shows z-scored FPKM representing the mRNA level of each gene in blue (low expression)-white-red (high expression) scheme.
(F) Repressed biological pathways in the BAT of Gtf2ird1 Tg mice and controls by Metascape.
(G) Repressed biological pathways in the inguinal WAT of Gtf2ird1 Tg mice and controls by Metascape.
(H) Ingenuity upstream analysis identified repressed signaling pathways in the BAT of Gtf2ird1 Tg mice.
(I) Expression profiles of the TGF-β regulated genes in the BAT of control and Gtf2ird1 Tg mice. Genes with red letters represent pro-fibrosis genes.
Next, we asked if Gtf2ird1 Tg mice have an altered adipose tissue thermogenesis in vivo. We found no difference in the level of UCP1 protein expressed in either the BAT or the inguinal WAT between Gtf2ird1 Tg mice and the littermate controls (Figure 3B). Gtf2ird1 Tg and control mice also displayed similar mRNA levels of the thermogenic genes in the BAT and the inguinal WAT, and whole-body heat production (Figure S2D–H). However, Gtf2ird1 Tg mice displayed far fewer developed fibrotic structures in the adipose tissue under a HFD. While trichrome staining detected very few fibrotic structures in the adipose tissue under a RD, fibrotic structures gradually developed after 11 weeks of HFD or longer in the WAT and the BAT, with the most severe fibrosis in the epididymal WAT (Figure 3C). We found significantly less fibrosis in the BAT and the inguinal WAT of Gtf2ird1 Tg mice relative to littermate controls at all the time points examined under a HFD. Fewer fibrotic structures were also found in the epididymal WAT of Gtf2ird1 Tg mice than the control mice at 11 weeks of HFD; however, the difference was not obvious following longer periods of HFD. A similar trend was found when the adipose tissues were stained with picrosirius staining (Figure S3A). In addition, quantification of hydroxyproline contents in the adipose tissue showed that the BAT and the inguinal WAT of Gtf2ird1 Tg mice were significantly less fibrotic than the controls at 11 weeks of HFD and thereafter, although no difference was seen under RD (Figure 3D). There was a trend towards reduced fibrosis in the epididymal WAT of Gtf2ird1 Tg mice at 11 weeks of HFD, but no significant difference was seen at later time points of HFD.
We next performed RNA-sequencing analysis in the adipose tissue of Gtf2ird1 Tg mice at 18 weeks of HFD. Hierarchical clustering of the RNA-seq data showed that the expression of 435 genes was significantly decreased, and that of 331 genes was significantly increased in the iBAT of Gtf2ird1 Tg vs. the littermate controls (Figure 3E). The Metascape biological pathway analysis indicates that pro-inflammatory pathways, including inflammatory responses, cytokine production, TNFα signaling, and macrophage chemotaxis, were significantly reduced in the BAT and the WAT of Gtf2ird1 Tg mice relative to control mice (Figure 3F–G, and Figure S3B). Consistent with the histological observations, biological pathways involved in adipose tissue fibrosis, including “wound healing”, “extracellular matrix”, and “HIF1-signaling”, were significantly repressed in the BAT and WAT of Gtf2ird1 Tg mice. Next, we applied Ingenuity pathway analysis to the RNA-seq dataset in order to identify signaling pathways upstream of GTF2IRD1-regulated biological processes. The analysis suggests that Gtf2ird1 Tg mice had significantly reduced expression of genes involved in pro-inflammatory cytokine-driven signaling pathways, including but not limited to those induced by IFNγ, TNFα, IL-1β, and IL-4 (Figure 3H). These data support the above observation that GTF2IRD1 represses adipose inflammation at 18 weeks of HFD in vivo. Among these signaling pathways, the TGF-β pathway was most strongly repressed in Gtf2ird1 Tg mice. We then probed GTF2IRD1-repression of TGF-β-dependent signaling more closely due to this strong repression, and also because TGF-β is a well-known inducer of fibrosis in many peripheral organs, including lung, adipose tissues, and liver (Meng et al., 2016). In fact, a large component of TGF-β-regulated genes (22/27, 81.5%), many of which (highlighted by red letters) implicated in tissue fibrosis, were repressed in Gtf2ird1 Tg mice relative to controls (Figure 3I). This change was independent of alterations in lipid metabolism, because no change was found in the expression of genes involved in de novo lipogenesis or lipolysis between the two groups (Figure S3C). Together, these data suggest that GTF2IRD1 represses HFD-induced adipose tissue fibrosis, in part, through inhibiting TGF-β-dependent pro-fibrotic signaling pathways in vivo.
A Cell-Autonomous Function of GTF2IRD1 for Inhibiting Adipocyte Fibrosis
Based on the close alignment between the reduced adipose tissue fibrosis in Gtf2ird1 Tg mice and the role of TGF-β signaling in the regulation of adipose fibrosis, we asked whether GTF2IRD1 is required, cell-autonomously, for the fibrotic responsiveness of adipocytes. We thus transduced retroviruses expressing either a scrambled control RNA (scr) or shRNAs targeting Gtf2ird1 (sh-Gtf2ird1#1 or #2) into immortalized brown adipocytes (Figure 4A). To induce fibrosis in cultured adipocytes, the transduced cells were then treated with TGF-β at doses of 0.2, 1.0, or 5.0 ng/ml under pro-adipogenic conditions. Consistent with prior work (Choy and Derynck, 2003), TGF-β treatment potently and dose-dependently impaired adipogenesis, whereas adipogenesis was impaired even at baseline when GTF2IRD1 was knocked down, and this impairment was further enhanced when the cells were treated with TGF-β (Figure 4B). GTF2IRD1 depletion also reduced the mRNA levels of BAT-specific genes, including Ucp1, Elovl3, and Cidea (Figure 4C), likely a reflection of impaired adipogenesis. In contrast, GTF2IRD1 depletion was associated with the induction of several pro-fibrotic genes, such as Col3a1, Lgals3 (encoding Galectin3), Pcolce2, Mmp2, and Fibronectin1 and this induction was further enhanced in the presence of TGF-β (Figure 4D and Figure S4A). In contrast, no significant change was found in the expression of pro-inflammatory genes by GTF2IRD1 depletion (Figure S4B).
Figure 4. GTF2IRD1 is required for the cell-autonomous capacity to regulate adipocyte fibrosis.

(A) Immunoblotting of immortalized brown adipocytes expressing shRNAs for a scrambled control (scr) or GTF2IRD1 (sh-Gtf2ird1 #1 and #2). β-actin was used as loading control.
(B) Oil-red-O staining of brown adipocytes expressing scr or sh-Gtf2ird1 cultured under an adipogenic condition medium containing TGF-β at 0.2, 1.0, or 5.0 ng ml−1. Scale bars, 200 μm.
(C) Relative mRNA expression of the BAT-related genes by qRT-PCR. * P<0.05, ** P<0.01, *** P<0.001 between scrambled control and sh-Gtf2ird1. # P<0.05, ## P<0.01, ### P<0.001 between vehicle and TGF-β. n=4.
(D) Relative mRNA expression of pro-fibrosis genes in (C).
(E) Relative mRNA expression of pro-fibrosis genes in immortalized brown adipocytes expressing GFP control or GTF2IRD1. n=4.
(F) Relative mRNA expression of pro-fibrosis genes, thermogenic genes, and pro-inflammatory genes in primary brown adipocytes derived from Gtf2ird1 Tg mice and control mice. Cells were treated with TGF-β to induce fibrosis. n=4. Data in D–F are represented as mean ± SEM.
We next employed gain-of-function approaches to ask if GTF2IRD1 represses adipocyte fibrosis in response to TGF-β signaling. First, we overexpressed a retroviral FLAG-tagged GTF2IRD1 construct in immortalized brown preadipocytes and subsequently treated with either TGF-β or vehicle in order to stimulate cellular fibrosis (Figure S4C). We found that overexpressing GTF2IRD1 potently diminished the ability of TGF-β to increase mRNA levels of the pro-fibrotic genes Lgals3 and Pcolce2 (Figure S4E), whereas no change was found in the expression of pro-inflammatory genes (Figure S4D). To recapitulate the conditions of Gtf2ird1 Tg BAT in culture, we next isolated primary SVFs from the BAT of Gtf2ird1 Tg mice and controls, and then differentiated these cells in the presence of TGF-β. Similarly, we found that primary brown adipocytes from Gtf2ird1 Tg mice expressed significantly less pro-fibrosis genes than those from the littermate control mice, whereas no significant difference was found in the expression of genes involved in thermogenesis or inflammation between the two groups (Figure 4F). Of note, we found that GTF2IRD1 also repressed pro-fibrosis genes in Ucp1−/− brown adipocytes (Figure S4E) as well as even when overexpressed in mature adipocytes by adenovirus (Figure S4F–J). Collectively, these data suggest that GTF2IRD1 negatively controls adipocyte fibrosis in a cell-autonomous manner, whereas GTF2IRD1 does not directly regulate the expression of thermogenesis genes or pro-inflammatory genes.
Regulatory Mechanisms of Pro-Fibrosis Gene Expression by GTF2IRD1 in Mice and Humans
The above results led us to hypothesize that GTF2IRD1 transcriptionally represses cellular fibrosis by recruiting the PRDM16-EHMT1 complex onto the promoter/enhancer regions of pro-fibrosis genes. To test this hypothesis, we first searched for the 8-bp core consensus binding motif sequence for GTF2IRD1 (G/A)GATT(A/G)) (Chimge et al., 2008; Thompson et al., 2007) in the regulatory regions of two representative TGF-β-regulated pro-fibrotic genes, Lgals3 and Pcolce2 (Figure 5A and Figure S5A). Among three potential sites on each gene, chromatin immunoprecipitation (ChIP) assays showed that GTF2IRD1 was significantly enriched at the regulatory regions of Lgals3 and Pcolce2 at Site A and Site D, respectively. Importantly, EHMT1 was co-enriched at these same sites (Figure 5B and Figure S5B). In addition, from a previous dataset that employed ChIP-sequencing for PRDM16 (Harms et al., 2015) we found that PRDM16 was also enriched at loci corresponding with Lgals3 (Figure 5C).
Figure 5. Regulatory mechanisms of adipose fibrosis by GTF2IRD1.

(A) Location of GTF2IRD1 binding motifs at the promoter/enhancer regions of Lgals3 (Site A–C).
(B) ChIP assays in brown adipocytes using specific antibodies against GTF2IRD1 and EHMT1. Fold enrichment at each GTF2IRD1 binding motif site in (A) was assessed by qPCR compared to IgG control. n=3. * P<0.05, ** P<0.01.
(C) Enrichment of PRDM16 on the Lgals3 gene in BAT of wild-type and Prdm16−/− (KO) mice. The dataset was obtained from the dataset in (Harms et al., 2015).
(D) Relative mRNA expression of pro-fibrosis and pro-inflammatory genes in Prdm16 KO adipocytes expressing GFP or GTF2IRD1. Cells were treated with TGF-β to induce fibrosis. n=4. Data in B and D are represented as mean ± SEM.
(E) Relative mRNA levels of pro-fibrotic genes in the abdominal subcutaneous WAT of adult human subjects (ages 25–65 years) drawn from the UCSF IDEO cohort. n=48. To ensure an ethnically mixed population of lean and obese people, the random sample included 22 Caucasian (12 obese and 10 lean) and 26 Chinese (15 obese and 11 lean) subjects. Visceral adipose tissue (VAT) mass was measured by DEXA.
(F) Correlation between GTF2IRD1 mRNA levels in the subcutaneous WAT and VAT mass. n=48.
(G) Correlation between GTF2IRD1 mRNA levels in the subcutaneous WAT and BMI.
(H) Relative mRNA levels of indicated pro-fibrotic genes comparing the groups drawn from individuals with the lowest and highest expression of GTF2IRD1 in the subcutaneous WAT. n=10 per group. Data are presented as means ± SD.
We next asked if the GTF2IRD1-mediated repression of pro-fibrosis transcription requires the PRDM16-EHMT1 complex. To this end, we overexpressed GTF2IRD1 in immortalized adipocytes from Prdm16−/− mice. We found that GTF2IRD1 failed to repress many of the pro-fibrosis genes in Prdm16−/− cells (Figure 5D). Furthermore, adipocytes lacking PRDM16 or EHMT1 exhibited a similar phenotype to GTF2IRD1-depleted adipocytes: adipocytes from Prdm16−/− mice and Ehmt1-knockdown cells expressed higher expression of pro-fibrotic genes in the presence of TGF-β (Figure S5C–D). In addition, pathway analyses of the global transcriptome datasets from inguinal WAT depots of adipocyte-specific Prdm16−/− mice (Cohen et al., 2014) and adipocyte-specific Ehmt1−/− mice (Ohno et al., 2013) showed that EMCs and pro-fibrosis genes were up-regulated in both KO mice relative to their respective controls (Figure S5E–F). These results suggest a functional requirement of the PRDM16-EHMT1-GTF2IRD1 complex to repress pro-fibrosis gene expression.
Next, we sought to determine whether the tissue-specific reciprocal relationship between Gtf2ird1 expression and that of genes involved in adipose tissue fibrosis seen in mice is mirrored in human subjects. We examined human subcutaneous WAT samples from individuals with varying degrees of obesity recruited from a multiethnic cohort of adults assembled in the San Francisco Bay Area. Examining a mixed population of Caucasian individuals and those of Chinese ethnicity revealed an overall positive correlation between increasing visceral adiposity by DEXA and increasing mRNA levels of genes indicative of WAT fibrosis, such as COL1A1, COL3A1, COL6A1, LGALS3, MMP2, and TIMP1 (Figure 5E and Figure S5G). In contrast, mRNA levels of GTF2IRD1 in the subcutaneous WAT of the same individuals showed an inverse correlation with visceral adiposity and body mass index (Figure 5F–G). In addition, PRDM16 mRNA levels in the subcutaneous WAT displayed an inverse correlation with visceral WAT mass (Figure S5H). Remarkably, placing the individuals into groups reflecting relatively high vs. low expression of GTF2IRD1 revealed that those with high GTF2IRD1 expression had lower expression of pro-fibrotic genes in the subcutaneous WAT (Figure 5H). This reduction was significant for COL1A1, COL6A1, MMP2, LGALS3, and TIMP1 and was a consistent trend for COL3A1 (P=0.08), although it did not reach statistical significance. This reciprocal relationship in humans mirrors that seen in both cultured adipocytes and adipose tissues in mice.
GTF2IRD1-Mediated Inhibition of Adipose Tissue Fibrosis Improves Systemic Glucose Homeostasis Independent of Body-Weight
Given that adipose tissue fibrosis has been associated with increased type 2 diabetes risk, our results motivated us to investigate the potential impact of reduced adipose fibrosis in the context of increased GTF2IRD1 expression on systemic glucose tolerance and insulin sensitivity in vivo. With this in mind, Gtf2ird1 Tg mice and littermate controls were fed either a RD or a HFD for up to 16 weeks. Gtf2ird1 Tg mice gained slightly less weight than controls after consuming a HFD for 15 weeks, with this phenotype persisting thereafter (Figure 6A and Figure S6A–B). Consistent with this modest reduction in body-weight gain, Gtf2ird1 Tg mice under HFD trended toward slightly higher whole-body energy expenditure (VO2) as compared to controls upon cold exposure, with no difference seen in food intake or locomotor activity (Figure S6C–E).
Figure 6. GTF2IRD1-mediated repression of adipose fibrosis is associated with improved systemic glucose homeostasis independent of body-weight.

(A) Body-weight gain of Gtf2ird1 Tg mice and the littermate controls (control) under HFD at 22°C. n=15. * P<0.05.
(B) GTT in Gtf2ird1 Tg mice and controls after 10 weeks of HFD at 22°C. n=6–7.
(C) ITT in Gtf2ird1 Tg mice and controls after 10 weeks of HFD at 22°C. n=6–7.
(D) Serum concentrations of fasting insulin in Gtf2ird1 Tg mice and controls after 11 weeks of HFD. n=6–8.
(E) Relative mRNA expression of pro-fibrosis genes in the inguinal WAT of Gtf2ird1 Tg mice and controls after 11 weeks of HFD. n=6. * P<0.05, ** P<0.01.
(F) Relative mRNA expression of pro-inflammatory genes in the inguinal WAT of Gtf2ird1 Tg mice and controls after 11 weeks of HFD. n=6.
(G) Body-weight gain of Gtf2ird1 Tg mice and controls under HFD under thermoneutrality (30°C). n=9–15.
(H) GTT in Gtf2ird1 Tg mice and controls after 10 weeks of HFD under thermoneutrality. n=6–7.
(I) Hydroxyproline content in the BAT (left) and the inguinal WAT (right) from Gtf2ird1 Tg mice and controls after 12 weeks of HFD under thermoneutrality. n=6–7. All the data are presented as means ± SEM.
Notwithstanding, Gtf2ird1 Tg mice exhibited a robust improvement in systemic glucose tolerance relative to controls when assessed after the mice consumed a HFD for 10 weeks. This improved glucose tolerance was evident before any differences in body-weight and fat mass had emerged between the two groups (Figure 6B and Figure S6A). Fasting blood glucose levels were also significantly lower in Gtf2ird1 Tg mice than in controls. Moreover, insulin tolerance was significantly better in Gtf2ird1 Tg mice than in controls after 10 weeks on a HFD (Figure 6C), and Gtf2ird1 Tg mice had reduced fasting serum insulin levels when measured at 12 weeks of HFD (Figure 6D). Importantly, inhibition of adipose tissue fibrosis was seen in the Gtf2ird1 Tg mice in conjunction with the improved systemic glucose homeostasis; at 11 weeks of HFD many of the pro-fibrosis genes were significantly reduced in the inguinal WAT and the BAT of Gtf2ird1 Tg mice relative to controls (Figure 6E, also see Figure 3C–D). In contrast, no significant change was found in the expression of pro-inflammatory genes between the two groups (Figure 6F). These results indicate that repression of adipose tissue fibrosis, rather than repression of adipose tissue inflammation, was tightly associated with an improvement in systemic glucose metabolism.
Because BAT thermogenesis in mice is active under ambient temperature at 22 °C, we asked whether the ability of GTF2IRD1 to improve systemic glucose homeostasis would be diminished in the absence of thermal stress. We thus examined Gtf2ird1 Tg and control mice that were fed a HFD at thermoneutrality (30°C). Although this abolished any differences in body-weight gain (Figure 6G) and whole-body energy expenditure (Figure S6C) between the genotypes, Gtf2ird1 Tg mice living at thermoneutrality still had better glucose tolerance compared to the littermate controls after 10 weeks of HFD (Figure 6H). Consistent with the observations, the hydroxyproline assay found significantly less adipose tissue fibrosis in the BAT and the inguinal WAT of Gtf2ird1 Tg, but not in the epididymal WAT, than controls at 10 weeks of HFD, even at 30°C (Figure 6I and Figure S6F). No difference was found in the expression of pro-inflammatory genes and thermogenic genes between the two groups (Figure S6G). Under a RD, no difference was observed in adipose tissue fibrosis and systemic glucose tolerance between the genotypes (Figure S7). These data suggest repression of adipose tissue fibrosis is a primary effect of GTF2IRD1 that alleviates diet-induced glucose tolerance and insulin resistance, even in the absence of thermal stress.
Repression of Adipose Tissue Fibrosis by GTF2IRD1 Improves Glucose Uptake and Insulin Sensitivity in BAT
To determine the tissues responsible for this improved glucose tolerance, we performed 18F-fluoro-2-deoxy-d-glucose (18F-FDG)-PET/CT scanning on Gtf2ird1 Tg and control mice under a HFD. We found that Gtf2ird1 Tg mice had significantly increased 18F-FDG uptake in the iBAT, but not in the skeletal muscle (soleus) or the liver (Figure 7A–B). This increase in glucose uptake in BAT is party through enhanced insulin sensitivity, because the BAT and the inguinal WAT from Gtf2ird1 Tg mice under HFD showed significantly higher insulin signaling based on Akt phosphorylation, whereas no difference was found in insulin signaling of either the skeletal muscle or the liver (Figure 7C–D).
Figure 7. Repression of adipose fibrosis by GTF2IRD1 improves BAT glucose uptake.

(A) 18F-fluor-deoxyglucose (FDG) uptake was measured by 18FDG-PET/CT scan under ambient temperature. Representative images of control and Gtf2ird1 Tg mice at 13 weeks of HFD are shown. n=5.
(B) Quantification of 18F-FDG uptake in the indicated organs in (A). * P<0.05.
(C) Immunoblotting for phosphorylated (S473) and total AKT in the indicated tissues from Gtf2ird1 Tg mice and the littermate controls. Mice were treated with saline or insulin before tissue harvest.
(D) Quantification of the insulin signaling assay in (C). n=4.
(E) Glucose uptake in differentiated brown adipocytes expressing scrambled control (scr) or sh-Gtf2ird1 (#2). Cells were treated with insulin (100 nM) and/or TGF-β (5 ng ml−1). * P<0.05, *** P<0.001 between scrambled control and sh-Gtf2ird1. ##P<0.01 between vehicle and TGF-β. n=6.
(F) Glucose uptake in differentiated brown adipocytes expressing GFP or GTF2IRD1. Cells were treated with insulin (100 nM) and/or TGF-β (5 ng ml−1). * P<0.05 between GFP and GTF2IRD1. ##P<0.01 between vehicle and TGF-β. n=6. Data are presented as means ± SEM.
(G) A proposed mechanism by which GTF2IRD1 controls adipose tissue fibrosis. See text for detail.
Accordingly, we asked whether GTF2IRD1 is required, cell-autonomously, for insulin-dependent glucose uptake by brown adipocytes. To this end, we measured insulin-stimulated 2-Deoxy-D-glucose (2DG) uptake in differentiated brown adipocytes that were transduced with either scr (control) or with Gtf2ird1 shRNA. We found that GTF2IRD1 deficiency significantly reduced insulin-dependent 2DG uptake relative to the control cells, a reduction that reached 71.4% when the cells were treated with TGF-β (Figure 7E). Conversely, GTF2IRD1 overexpression significantly increased insulin-dependent 2DG uptake by brown adipocytes and completely blocked the impact of TGF-β, which otherwise reduced insulin-stimulated 2DG uptake by 24% (Figure 7F). These results indicate that the mechanism by which GTF2IRD1 improves systemic glucose and insulin tolerance involves a tissue-specific enhancement of insulin sensitivity in BAT and inguinal WAT, at least in part, through blocking the inhibitory effect of TGF-β on adipose tissue fibrosis and insulin-responsive glucose uptake.
Collectively, our results point to the model illustrated in Figure 7G. In an obese state, TGF-β levels are increased in the adipose tissue and the circulation (Samad et al., 1997; Yadav et al., 2011), promoting adipose tissue fibrosis through the activation of Smad2/3 and the induction of pro-fibrotic genes, such as Lgals3 and Pcolce2. GTF2IRD1 is a BAT-enriched transcription factor that is induced by cold exposure through β3-AR-dependent signaling and that, in turn, potently inhibits TGF-β target pro-fibrosis genes by recruiting the PRDM16-EHMT1 complexes to their promoter/enhancer regions. Inhibition of adipocyte fibrosis by the PRDM16-GTF2IRD1 complex exerts a cell-autonomous effect that improves systemic glucose homeostasis independent of UCP1-mediated thermogenesis and body-weight loss.
DISCUSSION
Due to its strong association with glucose intolerance and insulin resistance in humans, adipose tissue fibrosis is emerging as a signature of unhealthy adipose tissue (Sun et al., 2013b). On the other hand, active beige fat biogenesis is often associated with improvements in glucose homeostasis and insulin sensitivity (Chondronikola et al., 2014; Hanssen et al., 2015; Sidossis and Kajimura, 2015). We demonstrate that the PRDM16 transcriptional complex not only activates brown/beige fat development, but also potently represses adipose tissue fibrosis through the direct interaction with GTF2IRD1. Notably, the time-course analysis under a HFD found that reduced adipose tissue fibrosis by GTF2IRD1 improves systemic glucose homeostasis prior to changes in adipose tissue inflammation and body-weight loss. Furthermore, the transcriptional repression of pro-fibrosis genes by PRDM16 and GTF2IRD1 occurs independent of UCP1 expression. These results support the notion that improvement of systemic glucose homeostasis, when brown/beige fat biogenesis is activated, extends beyond UCP1-dependent energy dissipation (Kajimura et al., 2015). Although the conventional consensus has been that adipose tissue fibrosis is a consequence of obesity-induced chronic inflammation and hypoxia in the adipose tissue, the present study suggests that repression of adipose tissue fibrosis through the PRDM16-GTF2IRD1 pathway is sufficient to protect animals from diet-induced glucose tolerance and insulin resistance in vivo.
To better understand adipose tissue fibrosis, it is essential to identify primary sources of ECM proteins. In general, myofibroblasts, marked by α-smooth muscle actin (α-SMA), are considered a primary cell type that synthesizes and secretes ECM components (Wynn and Ramalingam, 2012); however, it is likely that multiple cell-types contribute to ECM deposition in adipose tissues. For instance, perivascular cells expressing Nestin in the visceral WAT acquire pro-fibrotic characteristics and synthesize ECM proteins in response to PDGFRα signaling (Iwayama et al., 2015). A recent study also demonstrated that PDGFRα-positive progenitors expressing high levels of CD9 drive adipose tissue fibrosis (Marcelin et al., 2017). On the other hand, a lineage tracing study showed that the majority of ECM-producing myofibroblasts in the dermal WAT originate from Adiponectin-expressing adipocytes through a process referred to as “adipocyte–myofibroblast transition” (Marangoni et al., 2015). Additionally, inflammatory immune cells, including macrophages, appear to produce some ECM proteins (collagen I, fibronectin, and Tenascin-C) in obese WAT (Keophiphath et al., 2009). Of note, the cell-autonomous inhibitory action of GTF2IRD1 on pro-fibrosis genes is potent even when GTF2IRD1 is expressed in differentiated adipocytes; hence, the regulation of pro-fibrosis gene expression in mature adipocytes significantly contributes to ECM deposition in adipose tissues.
The mechanism of action of GTF2IRD1 is particularly intriguing because of it its relevance in Williams-Beuren syndrome (OMIM194050). This rare genetic disease caused by a heterozygous deletion of ~1.5 Mb region at chromosome 7q11.23, a region containing 26–28 genes, includes the GTF2IRD1 gene (Makeyev et al., 2004; Tipney et al., 2004). Targeted deletion of Gtf2rid1 in mice recapitulates some of the Williams-Beuren syndrome phenotypes, such as craniofacial abnormalities and diminished fear response (Enkhmandakh et al., 2009; Tassabehji et al., 2005; Young et al., 2008), although our fat-selective Gtf2rid1 transgenic mice display no developmental defects in either craniofacial or overall body-growth. Of particular relevance, there is an unusually high prevalence of glucose intolerance (up to 75%) among Williams-Beuren syndrome patients (Pober, 2010). Our data indicate that GTF2RID is required for the cell-autonomous capacity to regulate fibrosis and glucose metabolism in adipocytes. Accordingly, future studies are warranted to determine the whole-body metabolic phenotype caused by the adipocyte-specific deletion of Gtf2rid1. Beyond genetic syndromes, it is also interesting to note that the expression of GTF2IRD1 is reciprocal to that of pro-fibrotic genes within the subcutaneous WAT of human subjects drawn from an ethnically diverse population. While visceral WAT depots are considered to be the site of major fibrotic tissue in mice, subcutaneous WAT fibrosis is epidemiologically linked to metabolic disease risk in adult humans (Divoux et al., 2010; Henegar et al., 2008; Lackey et al., 2014; Muir et al., 2016; Reggio et al., 2016). In light of this study, our data in mice, which demonstrates that GTF2IRD1 represses diet-induced adipose tissue fibrosis in the BAT and the subcutaneous WAT thereby leading to an improvement in glucose tolerance, suggest a potentially important role for GTF2IRD1 in the regulation of glucose homeostasis in adult humans.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shingo Kajimura (shingo.kajimura@ucsf.edu).
Experimental Mode and Subject Details
Animals
All animal experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee for animal care and handling at the University of California, San Francisco. The Fabp4-prdm16 transgenic mouse was reported previously (Seale et al., 2011). Fabp4-Prdm16 transgenic mice in wild-type background and Ucp1−/− background were backcrossed to the Bl6 background for more than eight generations (Ikeda et al., 2017). To generate fat-selective Gtf2ird transgenic mice, the complete Gtf2ird cDNA transcript variant 5 (NM_001081465.1) was cloned under the Fabp4 gene promoter/enhancer. FVB mouse oocytes were injected with this construct by the UCSF Core Facility. Male mice were either maintained on a standard rodent chow or a 60% high-fat diet (Research Diets) at the indicated temperature under a 12hr light-dark cycle. Adipocyte tissue-specific EHMT1 KO mice (Ehmt1flox/flox) was described previously by Ohno et al. (2013). For acute cold-exposure studies, samples were obtained from 9–10 week-old male C57BL/6 mice housed at 6°C for 3 days using a rodent incubator (Power Scientific, Inc. RIS33SD). For chronic cold exposure, wild-type and Ucp1−/− mice under 12 weeks of HFD were kept under ambient temperature or 16°C for 10 days. β3 adrenergic receptor agonist CL-316,243 (Sigma) at 1 mg/kg was injected intraperitoneally into mice daily for 7 days. No animals were excluded from the analyses.
Human subjects
All subjects signed consent forms to participate in the study, which was approved by the University of California San Francisco (UCSF) Institutional Review Board. The study involved a random sample of 48 individuals (26 women, 22 men) aged 45 ±12 years (mean ±SD), drawn from the UCSF Inflammation, Diabetes, Ethnicity and Obesity (IDEO) cohort, which includes subjects recruited from clinics at the University of California San Francisco (UCSF) Medical Center and Zuckerberg San Francisco General Hospital (ZSFG). On initial enrollment, all IDEO cohort members sign consent forms including their willingness to participate in subsequent studies such as this one. To ensure an ethnically mixed population of lean and obese people, the random sample included 22 Caucasian (12 obese and 10 lean) and 26 Chinese (15 obese and 11 lean) subjects. No subjects were taking insulin, anti-inflammatory medications, glucocorticoids, or other medications likely to affect inflammation, including PPARγ agonists; they had no history of heart failure, liver failure or renal dysfunction. The subjects covered a wide range of body mass index (BMI 18.5–52 kg/m2). Individuals were excluded for smoking, not being weight-stable for the last 3 months (change >3%), having any acute or chronic inflammatory or infectious disease, cancer, or alcohol consumption >20g per day.
Anthropometric and body composition measurements
Height and weight were measured by standard procedures. Body composition was estimated by dual-energy X-ray absorptiometry (DEXA) using a Hologic Horizon/A scanner (3-minute whole body scan, <0.1 G mGy). Individuals, up to 450 lbs can be accurately measured by this device, and high-performance and “offset” scanning techniques were used to ensure complete coverage for those whose bodies were wider than the table width. Subsequent analyses used Hologic 12.4 software, following International Society for Clinical Densitometry guidelines, with precision error (1 SD) for total body fat and percent body fat of approximately 0.3 kg and 1%, respectively (calibration to correct for drifts using device-specific whole-body phantoms). Downstream analysis of these data are able to accurately estimate subcutaneous, visceral, gynoid, and android fat masses and percentages.
Subcutaneous WAT sampling
Subcutaneous WAT samples were obtained from the subjects by 2 different methods: In most cases, samples were collected by aspirational needle biopsies using a 14–16 G needle from the peri-umbilical area under local anesthesia. A minority of samples were obtained prior to either elective bariatric or other abdominal surgery. WAT samples were freed of visible connective tissue and rinsed to remove blood and clots, after which they were further washed with Krebs-Ringer bicarbonate buffer supplemented with 1% BSA. The specimens were then immediately frozen in liquid nitrogen and stored at −80°C.
Cell Isolation Procedure
Primary brown and inguinal preadipocytes were obtained from male mice by collagenase digestion following the protocol that was published previously (Liisberg Aune et al., 2013). Immortalized brown preadipocytes and inguinal white preadipocyte used in this study have been previously described (Ohno et al., 2013; Shinoda et al., 2015). Prdm16 knockout brown adipocytes, a gift from Dr. Seale, were described previously (Harms et al., 2015). Bone marrow cells were isolated and differentiated as described previously (Koliwad et al., 2010). In brief, male mice euthanized with Avertin and differentiated for 6–8 days in RPMI 1640 (Gibco) containing 10% FBS (Atlanta Biologicals), penicillin/streptomycin, and recombinant M-CSF (for BMDMs; 10 ng/mL; Peprotech).
Culture Conditions
Preadipocytes were differentiated by treating 90–95 % confluent cells in DMEM containing 10% FBS, 0.5 mM isobutylmethylxanthine, 125 nM indomethacin, 1 μM dexamethasone, 850 nM insulin, and 1 nM T3. Brown Adipocyte differentiation was induced by DMEM containing 10% FBS, 0.1 mM isobutylmethylxanthine, 25 nM indomethacin, 0.2 μM dexamethasone, 170 nM insulin, 0.2 nM T3. Two days after the induction, cells were switched to the maintenance medium containing 10% FBS, 850 nM insulin, 1 nM T3. TGF-β1 (BD Bioscience) was added from the induction at indicated concentration. To stimulate respiration, immortalized brown fat preadipocytes were incubated with 10 μM forskolin for 4 h. All chemicals for cell culture were obtained from Sigma unless otherwise indicated. After 5 days of differentiation, cells were washed with PBS and fixed with freshly prepared 4% formaldehyde for 15 min, and stained with Oil-Red-O solution for 20 min. As for the isolated macrophages, cells were washed once with PBS and starved overnight before treated for 24 hrs with 2% low-endotoxin Bovine Serum Albumin (Sigma A8806), 20 ng/mL IL-4, 200 ng/mL LPS (Sigma Aldrich L4391), 500 μM stearic acid or 500 μM palmitic acid complexed to BSA in a 2:1 molar ratio.
Method Details
Affinity Purification and Mass Spectrometry (MS)
Immortalized preadipocytes derived from mouse inguinal WAT were stably infected with retrovirus expressing FLAG-tagged PRDM16 or an empty vector. The adipocytes were grown to confluence and induced to differentiatie under pro-adipogenic conditions containing 0.5 μg/ml of rosiglitazone. Nuclear extracts from differentiated adipocytes were harvested for biochemical purification, as described in previous papers (Kajimura et al., 2009; Kajimura et al., 2008). The eluted complexes were TCA-precipitated, separated in a 4%–12% acrylamide gradient gel, and subsequently visualized by silver stain or coomassie blue dye. Gel-resolved proteins were excised, digested with trypsin, and individually analyzed by reverse-phase liquid chromatography with tandem mass spectrometry (LC-MS/MS) for peptide sequencing using a high-resolution hybrid mass spectrometer (LTQ-Orbitrap, Thermo Scientific) by employing the TOP10 method at the Taplin Biological Mass Spectrometry at Harvard Medical School. Data obtained from the LC-MS/MS was annotated using the IPI mouse database (Kersey et al., 2004). Proteins were considered significantly identified with at least two unique valid peptides, and the false discovery rate was estimated to be 0% using the target-decoy approach (Elias and Gygi, 2007).
Protein Interaction Analysis
Nuclear protein extracts were harvested from differentiated brown adipocytes expressing FLAG-PRDM16, FLAG-EHMT1, or an empty vector (control). Immunoprecipitation with FLAG-beads was carried out as previously described (Kajimura et al., 2009; Kajimura et al., 2008). Briefly, the nuclear extracts were incubated overnight at 4°C with Flag M2 agarose (SIGMA), washed, and eluted with a 3xFlag peptide. The eluted materials were analyzed by Western Blotting to detect endogenous GTF2IRD1 protein using the antibody against GTF2IRD1 (NBP1-91973, Novus). For in vitro binding assays, GST-fused PRDM16 fragments (1–223, 224–454, 455–680, 680–880, 881–1038 and 1039–1176) were described previously (Kajimura et al., 2008). 35S-labeled proteins were made with a TNT reticulocyte lysate kit (Promega). Equal amounts of GST fusion proteins (2 μg) were incubated overnight at 4°C with in vitro translated proteins in a binding buffer containing 20 mM HEPES (pH 7.7), 300 mM KCl, 2.5 mM MgCl2, 0.05% NP40, 1 mM DTT, and 10% glycerol. The sepharose beads were then washed five times with the binding buffer. Bound proteins were separated by SDS-PAGE and analyzed by autoradiography. Total protein in the gel was visualized by coomassie blue dye.
Immunoblotting
Fully differentiated adipocytes were lysed in RIPA buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton-X, 10% glycerol, and cOmplete protease inhibitors (Roche). Total protein lysates were boiled with 4× NuPage LDS loading buffer (Invitrogen) that contained 100 mM DTT, loaded on a 4–15% SDS-PAGE, and subsequently transferred onto PVDF membranes. The PVDF membrane blots were blocked in 5% milk in Tris-buffered saline with Tween 20 (TBS-T) and incubated overnight with rabbit anti-GTF2IRD1 (NBP1-91973, Novus), rabbit anti-UCP-1 (ab10983, Abcam), anti-PPARγ (H-100, Santa Cruz), anti-FLAG M2 (A8592, Sigma), anti-HA (Roche), or mouse anti-β-actin (A3854, Sigma) overnight. Anti-rabbit IgG (711-035-152, Jackson ImmunoResearch) was used as a secondary antibody for GTF2IRD1 and UCP1.
Metabolic Studies
Whole-body energy expenditure and associated metabolic parameters were performed using a temperature controlled Comprehensive Lab Animal Monitoring System (CLAMS) (Columbus Instruments). After mice were acclimatized to chambers under thermoneutrality (30°C), metabolic data were collected across different temperatures ranging from 30°C to 6°C. Serum level of insulin (Millipore), triglyceride (Thermo) and NEFA (Wako) were measured using commercially available kits. For glucose tolerance test experiments, male mice were fed a high-fat diet for 10 weeks. After 6 hours of fasting, the mice were injected intraperitoneally with glucose (1.2–1.5 g kg−1). For insulin tolerance test experiments, male mice under a high-fat diet for 10 weeks were used. After 3 hours of fasting, the mice were injected intraperitoneally with insulin (0.6 U kg−1).
Micropet/CT Scan Analysis And 18F-FDG Uptake Analysis
Mouse micro-PET/CT Scan was performed in wild-type and Gtf2ird Tg mice under ambient temperature following the established standard operating procedures that were approved by the UCSF Institutional Animal Care and Use Committee (IACUC) and Laboratory Animal Resource Center (LARC). Mice at 13 weeks of HFD were fasted for 5 h before each imaging session. A total of 100 μCi of 18F-FDG was administered via the tail vein, and PET scanning under 2% isoflurane anesthesia was started exactly at 55 minutes after FDG administration for 10 minutes, immediately followed by CT scanning. The data at a 10-minute time point were considered static and integrated for the data analysis. All mice were euthanized and dissected at 1 hour and 30 mins after injection. BAT, gastrocnemius muscle, and liver were collected and weighed. The radioactivity in the tissues was measured against known activity standards using a gamma-counter (Wizard 3; Perkin Elmer).
In Vivo Insulin Stimulation Assay
Mice kept under ambient temperature were anesthetized with Tribromoethanol (Avertin). 5U of human Insulin (Novo Noldisc) was injected into the inferior venae cavae. Livers (2min), BAT (4min) and soleus muscle (5min) were harvested after the injection and lysed in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% (w/v) glycerol, 100 mM NaF, 10 mM EGTA, 1 mM Na3VO4, 1% (w/v) Triton X-100, 5μM ZnCl2, 2 mM), with protease inhibitor cocktail (cOmplete, Roche) and phosphatase inhibitor cocktail 2 and 3 (Sigma). The lysates were isolated and separated by SDS–polyacrylamide gel electrophoresis (SDS–PAGE). Akt (Pan) antibody (Cell Signaling) and Phospho-Akt (Ser473) antibody (Cell Signaling) were used for western blotting.
Histological Analysis and Measurement of Fibrosis
For histology, all tissues were placed in 10% paraformaldehyde for 24 hr, followed by 70% ethanol until processing. Tissues were processed, embedded in paraffin, sectioned at 5 μm, and stained with Hematoxylin & Eosin (H&E). To assess adipose fibrosis, Masson’s trichrome, Picrosisius red staining and immunohistochemical staining with an anti-mouse endotrophin antibody (Park and Scherer, 2012; Sun et al., 2014) were performed. The endotrophin antibody was kindly provided by Dr. Philipp Scherer at UT South Western Medical Center. Images were acquired with a DM2000 digital camera (Leica). For the measurement of fibrosis, hydroxyproline content in adipose tissue was measured with a Hydroxyproline Assay Kit (Quickzyme Biosciences).
RNA-Sequencing Analysis
RNA-sequencing libraries were constructed from total RNA using Ovation RNA-sequencing system version 2 (NuGEN). The isolated RNA was reverse transcribed to cDNA using a combination of random hexameric and poly-T primers. The cDNA libraries were amplified using the Ultralow DR library kit (NuGEN) according to the manufacturer’s instructions. Quality of the libraries was determined by Bioanalyzer (Agilent Technologies). Subsequently, high-throughput sequencing was performed using a HiSeq 2500 instrument (Illumina) at the UCSF Genomics Core Facility. Raw reads for each library were mapped using TopHat version 2.0.8 against the mouse (mm10) genome. The mapped reads were converted to FPKM (fragments per kilobase of exon per million fragments mapped) by running Cuffdiff 2.1.1 to determine gene expression. Bioinformatic analyses using the Metascape pathway analysis (Tripathi et al., 2015) and Ingenuity Pathways Analysis (Kramer et al., 2014) were carried out to find molecular functions and upstream signaling pathways that were significantly associated with differentially expressed genes by GTF2IRD1.
Gene Expression Analysis
Total RNA in mouse tissues was isolated using RiboZol reagents (AMRESCO) and reversed transcribed using an iScript cDNA synthesis kit (Bio-Rad). RNA isolation from intact and aspirated human subcutaneous WAT samples was done using the RNeasy Lipid Tissue Mini kit (Qiagen, Valencia, CA). Total RNA (1 μg) was reverse transcribed using the SensiFAST cDNA Synthesis Kit (Bioline Reagents Ltd, UK). Quantitative real-time PCR (qRT-PCR) was performed using Sybr Green (Applied Biosystems by Life Science, Warrington, UK). Each sample was run in duplicate, and the quantity of a particular gene in each sample was normalized to both beta-actin and Cyclophiln B for human samples and to TATA-binding protein, 36B4 or 18srRNA for mouse samples. Relative mRNA levels were determined by the ΔΔCt method and expression normalized to an internal calibrator specific to each gene using the formula 2−ΔΔCT. For bone marrow-derived macrophages, relative mRNA abundance was normalized to the average of hypoxanthine quinine phosphoribosyl transferase and cyclophilin A. Primer sequences are provided in Table S2.
DNA Constructs and Viruses Production
The constructs were subcloned into a pMSCV-puro retroviral vector (Stratagene) or pcDNA3.1 (Invitrogen) using full-length Flag-tagged GTF2IRD1. For retrovirus production, Phoenix packaging cells were transfected at 70% confluence by calcium phosphate method with 10 μg of retroviral vectors. After 48 h, the viral supernatant was harvested and filtered. Cells were incubated overnight with the viral supernatant, supplemented with 8 μg/mL polybrene. Subsequently, puromycin (GFP and Flag-tagged GTF2IRD1), or G418 (shRNAs) were used for selection. The sequences used for retroviral shRNA expression vectors targeting GTF2IRD1 were described in Supplementary Table 2. The retroviral shRNA expression vectors targeting EHMT1 were previously described (Ohno et al., 2013). The corresponding double-stranded DNA sequences were ligated into pSUPER-Retro (GFP-Neo) (Oligoengine) for retroviral expression. Adenovirus for mouse GTF2IRD1 were obtained from Vector Biolabs (SKU#: ADV-260890).
In Vitro Glucose Uptake Assay
Brown adipocytes were plated and differentiated in 12-well plates. Five days after inducing adipocyte differentiation, differentiated brown adipocytes were washed three times with PBS, and incubated in RPMI-1640 medium containing 1% BSA for 2 hr, and subsequently incubated in 1 ml/well PBS containing 100 nM insulin for 30 minutes at 37°C. After washing in PBS, the cells were incubated in 1 ml PBS containing 0.1 mM 2-deoxyglucose and 0.5 mCi/ml of 2 deoxy D [3H] glucose for 5 min. After washing in ice-cold PBS twice, the cells were solubilized in 0.4 ml of 2% SDS. 3H-glucose uptake was measured in scintillation cocktails using Beckman LS 3801 scintillation counter. Nonspecific deoxyglucose uptake was measured in the presence of cytochalasin B and subtracted from the total uptake.
ChIP Assay
Immortalized brown adipocytes were transduced with Flag-tagged GTF2IRD1 or GFP control. ChIP assays were performed following the manufacturer’s instructions (Agarose ChIP kit; Pierce). Anti-GTF2IRD1 (NBP1-91973, Novus) and EHMT1 (R&D) were used to precipitate protein-bound DNA. Mouse IgG was used as a control. qRT-PCR analysis of recovered DNA fragments was performed using primers that span the putative GTF2IRD1-binding site A to C, respectively. qRT-PCR results were normalized to input values. Primer sequences used in the ChIP assays were provided in Supplementary Table 2.
Macrophage Assay
TNF-α, MCP-1 and IL6 concentrations secreted in the culture medium of isolated macrophages were measured by ELISA kits (eBioscience) according to the manufacturer’s instructions and normalized to protein mass. mRNA expression of pro-inflammatory genes in the isolated macrophages was measured by qRT-PCR.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mouse data are presented as mean ± SEM. Statistical significance was defined as P < 0.05 and determined by an unpaired Student’s t-test or one-way ANOVA throughout the study. Two-way ANOVA followed by post-hoc comparison was applied to determine the statistical difference in GTT and ITT. Human data are presented as means +/− SD, and all analyses were performed using features present in GraphPad Prism (version 7; GraphPad Software, Inc., San Diego, CA). The Shapiro-Wilks test was used to verify quantitative variables for normality distribution. Outliers were determined using the ROUT method with an average False Discovery Rate less than 1%. This process led to the exclusion of 4 outliers for final gene expression analysis. Mann-Whitney U test was used to compare differences between groups. Correlations between gene expression measurements and body composition parameters were examined with the Spearman’s rank test. The statistical parameters and the number of mice used per experiment are found in the figure legends.
DATA AND SOFTWARE AVAILABILITY
RNA-sequencing reads are available in ArrayExpress (www.ebi.ac.uk) under accession number E-MTAB-4163. Bioinformatic software used in the study are publically available.
Supplementary Material
List of DNA-binding transcriptional factors in the PRDM16 complex.
Primer sequences used in the study.
Highlights.
GTF2IRD1 is a transcription factor that forms a complex with PRDM16 and EHMT1.
A PRDM16-GTF2IRD1 complex cell-autonomously represses adipose tissue fibrosis.
Repression of adipose tissue fibrosis improves systemic glucose homeostasis.
GTF2IRD1 expression inversely correlates with subcutaneous WAT fibrosis in humans.
Acknowledgments
We are grateful to Dr. C. Paillart for his assistance in the CLAMS studies, Dr. Y. Seo for his support in 18F-FDG-PET/CT studies, and Dr. P. Scherer at UT Southwestern Medical Center, Dr. P. Seale at University of Pennsylvania, and Dr. L. Osborne and Dr. E. Tam at University of Toronto for sharing materials. This work was supported by the NIH (DK97441 and DK108822 to Kajimura; DK103175 and DK098722 to Koliwad; the UCSF Diabetes Research Center and Nutrition and Obesity Research Center P30 awards; and by a training fellowship 5T32 DK007418 to D.A.), and by a cohort development grant by the UCSF Department of Medicine (to Koliwad). Y.H. and K.I. were supported by the Manpei Suzuki Diabetes Foundation and the Uehara Memorial Foundation.
Footnotes
AUTHORS CONTRIBUTIONS
Conceptualization, Y.H., and S.K.; Investigation, Y.H., K.I., Y.C., D.A., D.S., K.S., T.H., J.C., and S.K.; Validation, P.M., Y.Y.; Data Curation, K.S., P.C.; Resources, D.A., K.S., and S.K.; Writing and Editing, S.K., and S.K.; Funding Acquisition, SK., and SK.; Supervision, Y.I., P.C., S.K., and S.K
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Chimge NO, Makeyev AV, Ruddle FH, Bayarsaihan D. Identification of the TFII-I family target genes in the vertebrate genome. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9006–9010. doi: 10.1073/pnas.0803051105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chondronikola M, Volpi E, Borsheim E, Porter C, Annamalai P, Enerback S, Lidell ME, Saraf MK, Labbe SM, Hurren NM, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes. 2014;63:4089–4099. doi: 10.2337/db14-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy L, Derynck R. Transforming growth factor-beta inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. The Journal of biological chemistry. 2003;278:9609–9619. doi: 10.1074/jbc.M212259200. [DOI] [PubMed] [Google Scholar]
- Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, et al. Ablation of PRDM16 and Beige Adipose Causes Metabolic Dysfunction and a Subcutaneous to Visceral Fat Switch. Cell. 2014;156:304–316. doi: 10.1016/j.cell.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crewe C, An YA, Scherer PE. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. The Journal of clinical investigation. 2017;127:74–82. doi: 10.1172/JCI88883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankel SN, Svard J, Mattha S, Claussnitzer M, Kloting N, Glunk V, Fandalyuk Z, Grytten E, Solsvik MH, Nielsen HJ, et al. COL6A3 expression in adipocytes associates with insulin resistance and depends on PPARgamma and adipocyte size. Obesity (Silver Spring, Md. 2014;22:1807–1813. doi: 10.1002/oby.20758. [DOI] [PubMed] [Google Scholar]
- Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, Basdevant A, Guerre-Millo M, Poitou C, Zucker JD, et al. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes. 2010;59:2817–2825. doi: 10.2337/db10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Enkhmandakh B, Makeyev AV, Erdenechimeg L, Ruddle FH, Chimge NO, Tussie-Luna MI, Roy AL, Bayarsaihan D. Essential functions of the Williams-Beuren syndrome-associated TFII-I genes in embryonic development. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:181–186. doi: 10.1073/pnas.0811531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier B, Murray B, Gutzwiller S, Marcaletti S, Marcellin D, Bergling S, Brachat S, Persohn E, Pierrel E, Bombard F, et al. Blockade of the activin receptor IIb activates functional brown adipogenesis and thermogenesis by inducing mitochondrial oxidative metabolism. Molecular and cellular biology. 2012;32:2871–2879. doi: 10.1128/MCB.06575-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Molecular and cellular biology. 2009;29:4467–4483. doi: 10.1128/MCB.00192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen MJ, Hoeks J, Brans B, van der Lans AA, Schaart G, van den Driessche JJ, Jorgensen JA, Boekschoten MV, Hesselink MK, Havekes B, et al. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nature medicine. 2015;21:863–865. doi: 10.1038/nm.3891. [DOI] [PubMed] [Google Scholar]
- Harms MJ, Ishibashi J, Wang W, Lim HW, Goyama S, Sato T, Kurokawa M, Won KJ, Seale P. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell metabolism. 2014;19:593–604. doi: 10.1016/j.cmet.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms MJ, Lim HW, Ho Y, Shapira SN, Ishibashi J, Rajakumari S, Steger DJ, Lazar MA, Won KJ, Seale P. PRDM16 binds MED1 and controls chromatin architecture to determine a brown fat transcriptional program. Genes & development. 2015;29:298–307. doi: 10.1101/gad.252734.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henegar C, Tordjman J, Achard V, Lacasa D, Cremer I, Guerre-Millo M, Poitou C, Basdevant A, Stich V, Viguerie N, et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome biology. 2008;9:R14. doi: 10.1186/gb-2008-9-1-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kang Q, Yoneshiro T, Camporez JP, Maki H, Homma M, Shinoda K, Lu X, Chen Y, Maretich P, et al. UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nature medicine. 2017 doi: 10.1038/nm.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T, Sakai J, Kajimura S. Transcriptional and epigenetic control of brown and beige adipose cell fate and function. Nature reviews. Molecular cell biology. 2016;17:480–495. doi: 10.1038/nrm.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwayama T, Steele C, Yao L, Dozmorov MG, Karamichos D, Wren JD, Olson LE. PDGFRalpha signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes & development. 2015;29:1106–1119. doi: 10.1101/gad.260554.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP, Spiegelman BM. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009;460:1154–1158. doi: 10.1038/nature08262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Spiegelman BM. Transcriptional control of brown fat development. Cell metabolism. 2010;11:257–262. doi: 10.1016/j.cmet.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, Spiegelman BM. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes & development. 2008;22:1397–1409. doi: 10.1101/gad.1666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Spiegelman BM, Seale P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell metabolism. 2015;22:546–559. doi: 10.1016/j.cmet.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keophiphath M, Achard V, Henegar C, Rouault C, Clement K, Lacasa D. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Molecular endocrinology (Baltimore, Md. 2009;23:11–24. doi: 10.1210/me.2008-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. The International Protein Index: an integrated database for proteomics experiments. Proteomics. 2004;4:1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Molecular and cellular biology. 2009;29:1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliwad SK, Streeper RS, Monetti M, Cornelissen I, Chan L, Terayama K, Naylor S, Rao M, Hubbard B, Farese RV., Jr DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. The Journal of clinical investigation. 2010;120:756–767. doi: 10.1172/JCI36066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koncarevic A, Kajimura S, Cornwall-Brady M, Andreucci A, Pullen A, Davies M, Sako D, Liu J, Kumar R, Burton R, et al. A novel therapeutic approach to treating obesity through modulation of TGFβ signaling. Endocrinology. 2012;153:3133–3146. doi: 10.1210/en.2012-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics (Oxford, England) 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey DE, Burk DH, Ali MR, Mostaedi R, Smith WH, Park J, Scherer PE, Seay SA, McCoin CS, Bonaldo P, et al. Contributions of adipose tissue architectural and tensile properties toward defining healthy and unhealthy obesity. American journal of physiology. 2014;306:E233–246. doi: 10.1152/ajpendo.00476.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62:864–874. doi: 10.2337/db12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liisberg Aune U, Ruiz L, Kajimura S. Isolation and differentiation of stromal vascular cells to beige/brite cells. Journal of visualized experiments : JoVE. 2013 doi: 10.3791/50191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeyev AV, Erdenechimeg L, Mungunsukh O, Roth JJ, Enkhmandakh B, Ruddle FH, Bayarsaihan D. GTF2IRD2 is located in the Williams-Beuren syndrome critical region 7q11.23 and encodes a protein with two TFII-I-like helix-loop-helix repeats. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11052–11057. doi: 10.1073/pnas.0404150101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni RG, Korman BD, Wei J, Wood TA, Graham LV, Whitfield ML, Scherer PE, Tourtellotte WG, Varga J. Myofibroblasts in Murine Cutaneous Fibrosis Originate From Adiponectin-Positive Intradermal Progenitors. Arthritis Rheumatol. 2015;67:1062–1073. doi: 10.1002/art.38990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelin G, Ferreira A, Liu Y, Atlan M, Aron-Wisnewsky J, Pelloux V, Botbol Y, Ambrosini M, Fradet M, Rouault C, et al. A PDGFRalpha-Mediated Switch toward CD9high Adipocyte Progenitors Controls Obesity-Induced Adipose Tissue Fibrosis. Cell metabolism. 2017 doi: 10.1016/j.cmet.2017.01.010. [DOI] [PubMed] [Google Scholar]
- Martinez-Martinez E, Calvier L, Rossignol P, Rousseau E, Fernandez-Celis A, Jurado-Lopez R, Laville M, Cachofeiro V, Lopez-Andres N. Galectin-3 inhibition prevents adipose tissue remodelling in obesity. International journal of obesity (2005) 2016;40:1034–1038. doi: 10.1038/ijo.2016.19. [DOI] [PubMed] [Google Scholar]
- McDonald ME, Li C, Bian H, Smith BD, Layne MD, Farmer SR. Myocardin-related transcription factor a regulates conversion of progenitors to beige adipocytes. Cell. 2015;160:105–118. doi: 10.1016/j.cell.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nature reviews. Nephrology. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- Muir LA, Neeley CK, Meyer KA, Baker NA, Brosius AM, Washabaugh AR, Varban OA, Finks JF, Zamarron BF, Flesher CG, et al. Adipose tissue fibrosis, hypertrophy, and hyperplasia: Correlations with diabetes in human obesity. Obesity (Silver Spring, Md. 2016;24:597–605. doi: 10.1002/oby.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Shinoda K, Ohyama K, Sharp LZ, Kajimura S. EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature. 2013;504:163–167. doi: 10.1038/nature12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Scherer PE. Adipocyte-derived endotrophin promotes malignant tumor progression. The Journal of clinical investigation. 2012;122:4243–4256. doi: 10.1172/JCI63930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasarica M, Gowronska-Kozak B, Burk D, Remedios I, Hymel D, Gimble J, Ravussin E, Bray GA, Smith SR. Adipose tissue collagen VI in obesity. The Journal of clinical endocrinology and metabolism. 2009;94:5155–5162. doi: 10.1210/jc.2009-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pober BR. Williams-Beuren syndrome. The New England journal of medicine. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- Rausch ME, Weisberg S, Vardhana P, Tortoriello DV. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. International journal of obesity (2005) 2008;32:451–463. doi: 10.1038/sj.ijo.0803744. [DOI] [PubMed] [Google Scholar]
- Reggio S, Rouault C, Poitou C, Bichet JC, Prifti E, Bouillot JL, Rizkalla S, Lacasa D, Tordjman J, Clement K. Increased Basement Membrane Components in Adipose Tissue During Obesity: Links With TGFbeta and Metabolic Phenotypes. The Journal of clinical endocrinology and metabolism. 2016;101:2578–2587. doi: 10.1210/jc.2015-4304. [DOI] [PubMed] [Google Scholar]
- Samad F, Yamamoto K, Pandey M, Loskutoff DJ. Elevated expression of transforming growth factor-beta in adipose tissue from obese mice. Molecular medicine. 1997;3:37–48. [PMC free article] [PubMed] [Google Scholar]
- Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, Spiegelman BM. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. The Journal of clinical investigation. 2011;121:96–105. doi: 10.1172/JCI44271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda K, Luijten IH, Hasegawa Y, Hong H, Sonne SB, Kim M, Xue R, Chondronikola M, Cypess AM, Tseng YH, et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nature medicine. 2015;21:389–394. doi: 10.1038/nm.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidossis L, Kajimura S. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. The Journal of clinical investigation. 2015;125:478–486. doi: 10.1172/JCI78362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer M, Yao-Borengasser A, Unal R, Rasouli N, Gurley CM, Zhu B, Peterson CA, Kern PA. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. American journal of physiology. 2010;299:E1016–1027. doi: 10.1152/ajpendo.00329.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Halberg N, Khan M, Magalang UJ, Scherer PE. Selective inhibition of hypoxia-inducible factor 1alpha ameliorates adipose tissue dysfunction. Molecular and cellular biology. 2013a;33:904–917. doi: 10.1128/MCB.00951-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Park J, Gupta OT, Holland WL, Auerbach P, Zhang N, Goncalves Marangoni R, Nicoloro SM, Czech MP, Varga J, et al. Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nature communications. 2014;5:3485. doi: 10.1038/ncomms4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Tordjman J, Clement K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell metabolism. 2013b;18:470–477. doi: 10.1016/j.cmet.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassabehji M, Hammond P, Karmiloff-Smith A, Thompson P, Thorgeirsson SS, Durkin ME, Popescu NC, Hutton T, Metcalfe K, Rucka A, et al. GTF2IRD1 in craniofacial development of humans and mice. Science (New York, N Y) 2005;310:1184–1187. doi: 10.1126/science.1116142. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Webb M, Beckett W, Hinsley T, Jowitt T, Sharrocks AD, Tassabehji M. GTF2IRD1 regulates transcription by binding an evolutionarily conserved DNA motif ‘GUCE’. FEBS letters. 2007;581:1233–1242. doi: 10.1016/j.febslet.2007.02.040. [DOI] [PubMed] [Google Scholar]
- Tipney HJ, Hinsley TA, Brass A, Metcalfe K, Donnai D, Tassabehji M. Isolation and characterisation of GTF2IRD2, a novel fusion gene and member of the TFII-I family of transcription factors, deleted in Williams-Beuren syndrome. European journal of human genetics : EJHG. 2004;12:551–560. doi: 10.1038/sj.ejhg.5201174. [DOI] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell host & microbe. 2015;18:723–735. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nature medicine. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Petrovic N, Cao R, Larsson O, Lim S, Chen S, Feldmann HM, Liang Z, Zhu Z, Nedergaard J, et al. Hypoxia-independent angiogenesis in adipose tissues during cold acclimation. Cell metabolism. 2009;9:99–109. doi: 10.1016/j.cmet.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, Zerfas P, Zhigang D, Wright EC, Stuelten C, et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell metabolism. 2011;14:67–79. doi: 10.1016/j.cmet.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. American journal of physiology. 2007;293:E1118–1128. doi: 10.1152/ajpendo.00435.2007. [DOI] [PubMed] [Google Scholar]
- Young EJ, Lipina T, Tam E, Mandel A, Clapcote SJ, Bechard AR, Chambers J, Mount HT, Fletcher PJ, Roder JC, et al. Reduced fear and aggression and altered serotonin metabolism in Gtf2ird1-targeted mice. Genes, brain, and behavior. 2008;7:224–234. doi: 10.1111/j.1601-183X.2007.00343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of DNA-binding transcriptional factors in the PRDM16 complex.
Primer sequences used in the study.