

Abstract
Silk fibroin from the domesticated silkworm Bombyx mori is a naturally occurring biopolymer with charged hydrophilic terminal regions that end-cap a hydrophobic core consisting of repeating sequences of glycine, alanine, and serine residues. Taking inspiration from mussels that produce proteins rich in L-3,4-dihydroxyphenylalanine (DOPA) to adhere to a variety of organic and inorganic surfaces, the silk fibroin was functionalized with catechol groups. Silk fibroin was selected for its high molecular weight, tunable mechanical and degradation properties, aqueous processability, and wide availability. The synthesis of catechol-functionalized silk fibroin polymers containing varying amounts of hydrophilic polyethylene glycol (PEG, 5000 g/mol) side chains was carried out to balance silk hydrophobicity with PEG hydrophilicity. The efficiency of the catechol functionalization reaction did not vary with PEG conjugation over the range studied, although tuning the amount of PEG conjugated was essential for aqueous solubility. Adhesive bonding and cell compatibility of the resulting materials were investigated, where it was found that incorporating as little as 6 wt % PEG prior to catechol functionalization resulted in complete aqueous solubility of the catechol conjugates and increased adhesive strength compared with silk lacking catechol functionalization. Furthermore, PEG-silk fibroin conjugates maintained their ability to form β-sheet secondary structures, which can be exploited to reduce swelling. Human mesenchymal stem cells (hMSCs) proliferated on the silks, regardless of PEG and catechol conjugation. These materials represent a protein-based approach to catechol-based adhesives, which we envision may find applicability as biodegradable adhesives and sealants.
Graphical Abstract
INTRODUCTION
Polymers that adhere to wet surfaces that are biocompatible, degradable, and devoid of animal-sourced materials are desired for applications as tissue adhesives. Freshwater and marine mussels produce mussel adhesive proteins (MAPs) that allow them to adhere in wet environments to both organic and inorganic surfaces.1–4 Many different MAPs have been identified, but their unifying characteristics include a basic isoelectric point, an extended chain conformation, and high levels of the amino acid L-3,4-dihydroxyphenylalanine (DOPA), which is produced by post-translational modification of tyrosine.5,6 DOPA6,7 is thought to play a key role in adhesion by facilitating the adsorption of MAPs to wet surfaces and by forming covalent bonds to cross-link the protein.
The ability of marine organisms to adhere in wet environments to a variety of surfaces has inspired researchers to investigate oxidative cross-linking of MAPs8,9 and the forces and mechanisms of adhesion10–12 as well as to try to recapitulate the adhesive properties in proteins generated via genetic engineering.13,14 Many approaches to water-soluble natural and synthetic polymers containing DOPA or catechol groups have been reported,15 including dextran and starch,16 poly(ethylene glycol)17–20 with various chain architectures, poly(acrylic acid),21 poly(ethylene glycol) with polycaprolactone22 and/or polylactide,23 poly(ethylene oxide)-poly-(propylene oxide) block copolymers,24 as well as synthetic polypeptides prepared by ring-opening polymerization.25 Antibacterial,26 stimuli-responsive,17 and self-healing27 properties have also been incorporated into hydrogels based on catechol-functionalized polymers, and enzymatically degradable sequences have been inserted into poly(ethylene glycol)-based materials to accommodate in vivo degradation by elastase over a period of months.28 Furthermore, adhesives and composites based on copolymers synthesized by anionic polymerization of 3,4-dihydroxystyrene with styrene29,30 have been reported, resulting in impressively high strength that rivals commercial glues. These polymers are not soluble in water, however, and were therefore used in organic solvents.
Silks are protein-based biopolymers produced by many insects and arachnids. Domesticated silkworms, such as Bombyx mori, are used to produce silk for the textile industry. Cocoons fabricated by the B. mori silkworm are comprised of two main protein types: sericins and fibroin. Fibroin consists of a heavy chain (~390 kDa) and a light chain (~26 kDa) present in a 1:1 ratio and linked by a disulfide bond.31,32 Sericins are a family of gluelike proteins ranging from 20 to 310 kDa that coat the fibroin chains.33 Purified (removal of sericin) silkworm fibers as well as purified regenerated fibroin, as a biomaterial, has provided an emerging biomaterial platform34–39 for many needs in medical devices, tissue engineering, and tissue regeneration.
Silk fibroin32,40 has an amino acid sequence consisting of repeats of glycine-alanine-glycine-alanine-glycine-serine (GA-GAGS) that self-assemble to form beta sheets. Formation of beta sheets may be triggered by various processing techniques that involve energy input (e.g., sonication41–43 or vortexing44), lowering the solution pH, or through dehydration of the protein by removal of water (e.g., by methanol treatment45). Beta sheets are highly crystalline and serve to physically cross-link the protein through intra- and intermolecular hydrogen bonding and van der Waals interactions. The β sheet imparts impressive mechanical properties to the fibroin and renders the material insoluble in water. β-Sheet content has also been shown to affect the mechanical properties of materials generated from silk fibroin, including swelling and degradation. Degradation is complete and the rate can be tuned45,46 to be longer in vivo47 than other commonly studied protein polymers for biomaterials (e.g., collagens and elastins).
Regenerated silk fibroin is a robust protein biopolymer that is easily isolated from readily available natural starting materials using aqueous processing techniques.48 In the present work the goal was to incorporate functional side chain groups that would cause the fibroin to adhere to surfaces as well as to form cross-linked networks within itself. Utility for these biocompatible and biodegradable materials could include as tissue sealants, encapsulation matrices for cell delivery or culture, and as aqueous adhesives in general. To this end, we designed a strategy to synthesize catechol-functionalized silk fibroin. Potential advantages of the silk conjugates compared with the existing, water-soluble catechol conjugates based primarily of poly(ethylene glycol) (PEG) backbones are that the silk conjugates are aqueously processable yet hydrophobic, which should lead to lower degrees of swelling in water than PEG-based conjugates as well as mechanical reinforcement of the adhesive material through physical cross-linking by β sheet formation between silk chains.
EXPERIMENTAL SECTION
Instruments
1H NMR was performed using an Advance III NMR (Bruker BioSpin, Billerica, MA) operating at 500 MHz and equipped with Topspin Software (version 2.1 Bruker). Fourier transform infrared spectroscopy was performed using a FT/IR-6200 Spectrometer (Jasco, Japan) that was equipped with a triglycine sulfate detector and run in attenuated total reflection (ATR) mode. Fourier self-deconvolution (FSD) of the infrared spectra covering the Amide I region (1595–1705 cm−1) was performed with Opus 5.0 software (Bruker Optics, Billerica, MA). Quantification of catechol conjugation and cell proliferation on the modified silks was achieved using chemical assays coupled to absorbance and fluorescence measurements obtained using a SpectraMax M2 plate reader (Molecular Devices, Sunnyvale, CA). Adhesion tests were performed on an Instron 3366 testing frame equipped with a 100 N load cell (Norwood, MA).
Reagents
Dopamine hydrochloride (puriss, > 98.5%), COMU (97%), anhydrous trimethylamine (Et3N, 99%), anhydrous dimethyl sulfoxide (DMSO), sodium carbonate (>99.5%), lithium bromide (>99%), sodium phosphate monobasic (>99%), chloroacetic acid (>99%), methoxypolyethylene glycol activated with cyanuric chloride (average Mw 5000 g/mol), sodium periodate (>99.8%), sodium molybdate (98%), sodium nitrite (>97%), deuterated dimethyl sulfoxide-d6 (d-DMSO, 99.96%), and deuterium oxide (D2O, 99.99%) were obtained from Aldrich Chemical (St. Louis, MO). Hydrochloric acid, sodium hydroxide, and regenerated cellulose dialysis membranes (MWCO 3500, 6000–8000, or 12 000–14 000 g/mol) were purchased from Fisher Scientific. Deionized water (DI) at 18 MOhm was obtained from an in-house purification system.
Purification of B. mori Silk Fibroin
Removal of sericins from B. mori silk fibroin was achieved by a previously published procedure, with slight modifications as noted.48 In brief, 5 g of cut cocoons was added to 2 L of 0.2 M NaHCO3 solution at 100 °C. The cocoons were continuously rotated and separated during a 10 min boiling time, which was shorter than the referenced procedure (30 min boiling time) to afford higher molecular weight fibroin. After 10 min, the degummed fibers were placed in 2 L of distilled water and rinsed for 20 min. This rinsing procedure was repeated two additional times to remove sericins (maximum content <10 wt %)49 from the fibroin, after which the fibers were dried overnight at room temperature. The yield of the dried degummed fibers was ~3.7 g or 74% on average.
To dissolve the silk fibroin, we added 1 g of degummed fibers to 5 mL of a 9.3 M LiBr solution. Compared with previous reports that use 4 mL of 9.3 M LiBr solution to dissolve silk boiled for 30 min, the concentration of silk fibers in LiBr solution was lower here because it was found that the silk fibroin boiled for 10 min was slightly more difficult to dissolve than the 30 min boiled fibroin. Thus, the volume of LiBr solution was increased in this work. Removal of salts proceeded as previously published, with the solution placed in 3500 MWCO dialysis tubing and dialyzed against 2 L of DI water per 12 mL of silk solution. The water was changed at regular intervals (1, 3, 6, 18, 30, 42 h) before removal from tubing and centrifugation (8700 rpm, 4 °C, 20 min). Settled particulate was removed by pouring off the silk solution into a new container, and the silk solution was centrifuged again. The clarified silk fibroin solution was then transferred to a new container, and the concentration of fibroin was determined by weighing a known volume of silk that was dried overnight at 55 °C. Typical concentration of the silk fibroin solution was 55 mg/mL.
Enrichment of Carboxyl Groups on Fibroin
To increase reactive groups for the dopamine reaction, the hydroxyl groups of serine were converted to carboxylic acid groups using an established procedure.50 In brief, the purified silk fibroin solution was added to a round-bottomed flask and diluted to 45 mg/mL using distilled water. Next, 10 M NaOH was added to the flask to yield a final concentration of 3 M NaOH. This solution was stirred for 2 to 3 min before chloroacetic acid was added to give a final concentration of 1 M. This solution was stirred at room temperature for 1 h before stopping with the addition of NaH2PO4 (6.9 mg/mL). The reaction was then placed on ice and neutralized to pH 7 by dropwise addition of 12 M HCl. The use of the ice bath prevented the silk solution from large temperature increases during titration. The solution was allowed to stir for 30 min before transferring to dialysis tubing and dialyzing against distilled water. Like the dialysis previously described, the solution was placed in 3500 MWCO dialysis tubing and dialyzed against DI water, which was changed at regular intervals (1, 3, 6, 18, 30, 42 h). The product of this reaction is named CarboxySF.
Because the concentration of CarboxySF post dialysis was very low, the dialysis tubing containing the silk solution was placed on a clean surface in a chemical fume hood and allowed to concentrate by removal of water for 48 h. Two alternatives for this concentration method were considered, including lyophilization followed by dissolution at a higher concentration or concentrating by dialyzing against a solution containing a high-molecular-weight poly(ethylene glycol) (PEG), which will draw water out of the tubing yet not permit PEG to enter the tubing. The use of the fume hood air flow was selected over the lyophilization/dissolution route because lyophilization can induce β-sheet formation in the silk, rendering it more difficult to resolubilize in water. Concentration by air flow was selected instead of dialyzing against a PEG solution to avoid analytical complications, as some of these derivatives will be conjugated with PEG in downstream reactions. The typical concentration of CarboxySF after this concentration step was 15 mg/mL.
Conjugation of Poly(ethylene glycol) to CarboxySF
Poly-(ethylene glycol) (PEG) was conjugated to tyrosine to increase the hydrophilicity of CarboxySF. Different amounts of PEG were conjugated to CarboxySF using the same procedure. Materials are named as CarboxySF(x), where x is the number of PEG chains conjugated per molecule of silk. As an example, the synthesis of CarboxySF(29) is described, where the following synthesis led to 29 PEG chains conjugated per molecule of silk fibroin. The degree of conjugation was quantified by 1H NMR, as described later. To synthesize CarboxySF(29), we added 395 mL of an aqueous solution of CarboxySF (11.4 mg/mL) to a round-bottomed flask. Commercially available sodium borate buffer mixture (BupH Borate Buffer, Fisher) was dissolved in the silk solution to give 0.1 M borate buffer, and the pH was adjusted to 9.5 using 10 M NaOH. The solution was cooled to 4 °C before adding 3.31 g of cyanuric-chloride-activated methoxypoly(ethylene glycol) (Aldrich, average Mw 5000 g/mol). The reaction was allowed to proceed for 2 h with stirring at 4 °C. To conjugate different amounts of PEG to the CarboxySF, the same conditions were employed but the PEG content was varied. Because the PEG reaction was found to reproducibly lead to 34% conjugation efficiency, the amount of PEG added to each reaction was 2.94 times larger than the theoretical amount needed to achieve the desired amount of PEG conjugation, and this was found to lead to highly reproducible reactions. A control reaction was prepared in the same manner, except that no cyanuric chloride activated methoxypoly-(ethylene glycol) was added to this reaction, to confirm that the reaction workup did not change the silk. All reactions were worked up by dialysis in 12 000–14 000 MWCO tubing. For the first two changes, the dialysis was conducted against phosphate buffered saline at 4 °C, and for the last three dialysis steps the dialysis was run against distilled water at 4 °C.
Lyophilization of Silk Solutions
Aqueous solutions of CarboxySF or CarboxySF(x) were placed in 50 mL polypropylene tubes and placed in a dry ice/isopropanol slurry for 2 h to freeze the solutions. The samples were then transferred to a lyophilizer, where they were dried for 4 days.
Conjugation of Dopamine to Fibroin
Lyophilized CarboxySF or CarboxySF(x) were dissolved in anhydrous dimethyl sulfoxide (DMSO, Aldrich) in a round-bottomed flask at room temperature and at a concentration of 67 mg/mL and purged with argon. No stirring was applied during the dissolution process, which took several hours to reach a homogeneous solution. Dopamine hydrochloride (Aldrich) was then added to the solution at a 10-fold molar excess relative to carboxylic acid groups on the modified silk. For example, for 10 g of CarboxySF, 25.1 g of dopamine hydrochloride was added to the solution, but for 10 g of CarboxySF(23), 21.1 g dopamine hydrochloride was added because of the PEG chain contribution to the modified silk’s molecular weight. Next, COMU (Aldrich) was dissolved in the solution at a 2-fold molar excess relative to carboxylic acid functional groups. The reaction vessel was once again purged with argon gas, and the solution was subjected to gentle heating by placement in a 40 °C water bath to facilitate complete dissolution of the reactants. Once a homogeneous solution was achieved, an 8-fold molar excess of triethylamine (Aldrich) was added to the solution. Once again, the vessel was purged with argon before being placed in a 40 °C incubator with gentle shaking (50 rpm). The reaction proceeded in this manner for 16 h before the reacting mixture was removed and transferred to dialysis tubing (6000–8000 MWCO). The reaction was dialyzed against degassed DMSO in a sealed container. Upon completion of the dialysis, which was judged by the absence of a change in the color of the solvent that typically occurred after 48 h of dialysis, the solution was transferred to 50 mL polypropylene tubes and frozen using a dry ice/isopropanol slurry for 2 h. The frozen samples were then transferred to a lyophilizer where they were dried for 5 days. A second dialysis was performed after lyophilization, where the catechol-functionalized silks were placed in dialysis tubing (20 000 MWCO) and dialyzed against 8 M urea (pH 6.1) for 2 h, phosphate-buffered saline (pH 6.1) 2 h, phosphate-buffered saline for 2 h (pH 6.1), and finally distilled water for 48 h (pH 6.0). The conditions for this second dialysis, which is referred to as a washing protocol or an extraction for clarity, were guided by a separate experiment, as described later.
To ensure that dopamine was chemically coupled to silk fibroin and not absorbed to the silk, we developed a washing protocol. CarboxySF and dopamine were dissolved in water at the same concentration that was used in the catechol coupling reaction (50 mg/mL CarboxySF, 125 mg/mL dopamine). The mixture was then placed in dialysis tubing (20 000 MWCO) and dialyzed against one of the following solutions, which were all adjusted to pH 5.9 to 6.1 and degassed with argon to reduce catechol oxidation. Each of the washing protocols consisted of three solutions that the solution was dialyzed against for 2 h. For Protocol A, these solutions were: DI water, DI water, DI water; for Protocol B: 8 M urea in water, DI water, DI water; for Protocol C: 8 M urea in water, phosphate-buffered saline, phosphate-buffered saline; for Protocol D: phosphate-buffered saline, DI water, DI water; for Protocol E: 0.5 vol % Tween 20 in water, DI water, DI water; and for Protocol F: 0.5 vol % Tween 80 in water, DI water, DI water. Following these dialysis steps, all of the solutions were dialyzed against DI water for 48 h before being frozen, lyophilized, and analyzed using NMR in deuterated DMSO. As a control, a portion of the solution before any dialysis was frozen, lyophilized, and analyzed by NMR to quantify the initial dopamine content in the silk solution.
Characterization Techniques
1H NMR was used to confirm carboxylic acid functionalization of serine residues, quantify PEG conjugation, and confirm dopamine conjugation to the silk. All 1H NMR spectroscopy was performed on a Bruker NMR operating at 500 MHz using TopSpin Software. Samples were run with a relaxation delay of 5 s, and 64 scans were averaged for each spectrum. Samples were dissolved at a concentration of 10 mg/mL in deuterated water or DMSO, as noted, and data analysis was performed using MestreC Software.
ATR-FTIR measurements were performed to study secondary structure in cast films of the materials. Scans were conducted from 4000 to 800 cm−1 at a rate of 4 cm−1/min, and 64 scans were averaged for each sample. To prepare samples, we dissolved 40 mg/mL of the silk in DI water, and 0.7 mL of this solution was deposited onto a 1 cm diameter dish fabricated from aluminum foil. “As-cast” samples were allowed to air-dry at room temperature for 7 days before measuring. Induction of β sheet was accomplished by methanol treatment. Methanol-treated samples were prepared by soaking films for 24 h in methanol (Fisher Scientific) and drying at room temperature for 48 h before measuring. All measurements were conducted in triplicate. The background spectra were collected under the same conditions and subtracted from the scan for each sample. Fourier self-deconvolution (FSD) of the infrared spectra covering the amide I region (1595–1705 cm−1) was performed with Opus 5.0 software (Bruker Optics, Billerica MA), as previously described.51
Quantification of catechol functionalization to the silk protein was accomplished by Arnow’s assay.52 In this assay, nitrate ions react with phenolic hydroxyl groups on catechols to produce a nitric oxide compound, which causes a solution containing catechols to change color. The procedure was adopted from the literature, but a smaller scale was used to permit measurement using a microplate reader. In brief, an aqueous solution of the silk product was prepared at a concentration of 10 mg/mL. Twenty uL of the silk solution was added to a well of a 96-well microplate. The following components were then added to the well in the prescribed order: 20 uL of 0.5 M HCl, 20 uL of a sodium nitrate/sodium molybdate solution, 20 uL of 1 M NaOH, and 20 uL of distilled water. To prepare the sodium nitrate/sodium molybdate solution, we dissolved 0.1 g sodium nitrate and 0.1 g of sodium molybdate in 1 mL distilled water. The addition of the sodium nitrate/sodium molybdate solution caused a yellow color to develop in samples containing catechol, while the addition of 1 M NaOH caused the solution to turn red. Absorbance at 500 nm was measured using a microplate reader. To account for changes in absorbance due to the silk protein, we also measured the appropriate silk product (CarboxySF or CarboxySF(x)) using the same procedure, and the resulting absorbance was subtracted from the absorbance of the silk dopamine conjugate. Finally, quantification of conjugation was achieved using a standard curve prepared from dopamine hydrochloride.
Adhesion Studies
CarboxySF, CarboxySF(5), and CarboxySF-(20) and their dopamine conjugates were dissolved in water at a concentration of 100 mg/mL silk. Concentrated phosphate-buffered saline (10× normal salt concentration) was added to the solution to give a solution with 2× phosphate-buffered saline content. An aqueous solution of sodium periodate was prepared (75 mM), which was diluted with water to 54 mM for CarboxySF, 35 mM for CarboxySF(6), and 26 mM for CarboxySF(29). This dilution was performed so that an equal volume of periodate solution could be added to the silk solutions, thus ensuring that the silk concentration was maintained at 50 mg/mL for all compositions, while maintaining the ratio of periodate to catechol at 2.1:1.18 Aluminum shims (10 cm L × 1.9 cm W) were placed on a flat surface, and 4 μL of silk solution was added to the shim followed by 4 μL of the appropriate periodate solution. The solutions were briefly mixed using the pipet tip before overlaying a second shim (overlay was 2.5 cm L × 1.9 cm W, total area of overlay was 4.8 × 10−4 m2). A 13 g weight was placed on top of the shims to ensure good contact, and these were allowed to cure 24 h at room temperature. Single lap shear testing (N = 7, at minimum, for each material) was performed by mounting the shims in the tension grips of the Instron equipped with a 100 N load cell and programming the instrument to move the grips apart at a rate of 1 mm/s. The test ended when the adhesive bond ruptured, and this peak load was divided by the bonded area to give adhesion for each of these materials.
Cell Culture Studies
Films of the modified silks were prepared by casting from aqueous solutions in a 48-well tissue culture plate. At least six films were cast from each material. The films were allowed to dry on the benchtop for 4 days before water vapor annealing for 18 h at room temperature to induce β-sheet formation and render the film insoluble. Films were sterilized by immersion of the plates in 70% ethanol for 45 min. The ethanol was removed from the plate, which was allowed to dry under sterile conditions. The wells were then washed five times with sterile PBS before the addition of sterile medium (Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 1× antibiotic-antimycotic). The wells were washed five times with sterile medium before soaking overnight in a 37 °C incubator with a humidified 5% CO2 atmosphere. The films were then washed three times additional times with sterile medium to ensure that there was no residual ethanol before hMSCs (Passage 4) were seeded on the films at 4000 cells/cm2. Cell viability and metabolic activity were measured using Alamar Blue reagent (Life Technologies), which is converted to a fluorescent product when metabolized by mammalian cells and thus is used to assay live cell function. To assay the cells, we added 150 uL of working solution (300 uL of Alamar Blue added to 3 mL of sterile medium) to each well, including acellular control wells. The plate was incubated for 2 h at 37°C in the 5% CO2 incubator before 80 uL of solution was sampled from each well and read using a microplate reader operating in fluorescence mode (excitation 560 nm, emission 590 nm). The residual Alamar Blue working solution was aspirated from the wells, which were then washed three times with sterile phosphate-buffered saline before the addition of fresh sterile medium. Each well was measured at 1, 8, and 15 days post-seeding to track cell proliferation over time.
Statistical Analysis
Univariate analysis of variance (ANOVA) was conducted using a software package (SigmaPlot v.12.5) to determine statistical differences at the 95% confidence level (p < 0.05). Multiple comparisons was made using the Holm–Sidak method.
RESULTS AND DISCUSSION
Silk fibroin was isolated from B. mori cocoons and was enriched with carboxylic acid (COOH) functional groups on serine residues as outlined in Scheme 1 Reaction i. Because the heavy chain of silk fibroin contains 5525 amino acids and serine residues account for ~11.9% of these, this reaction could theoretically increase COOH functionality from 77 groups per chain to 737 groups. Although we cannot quantify the degree of conversion of the hydroxyl groups, 1H NMR spectra show a shift of the protons on the beta carbon of serine due to change in chemical environment. Because the reaction outlined in Scheme 1 results in a very dilute product post dialysis, the CarboxySF was concentrated by air flow in a chemical fume hood for ~48 h. The volume of solution removed from the tubing after the concentration step was about a third of the volume at the start of the concentration step. The solution was then lyophilized directly or conjugated with poly(ethylene glycol) (PEG) to tailor hydrophilicity.
Scheme 1. Synthesis of Dopamine-Modified Silk by (i) Carboxylic Acid Enrichment; (ii) PEGylation of Carboxylated Silk; and (iii) Dopamine Functionalizationa.

aReaction conditions (i): 1M ClCH2COOH, 3M NaOH, 25°C, 30 min. NaH2PO4 (6 mg/mL), 25°C, 30 min. Neutralize, purify by dialysis. Reaction conditions (ii): Methoxy(poly(ethylene glycol)) activated with cyanuric chloride, 0.1 M Na2B4O7 pH 9.5, 4°C, 2 h. Purify by dialysis. Reaction conditions (iii): Dopamine hydrochloride, COMU, Et3N, DMSO, 40 °C, 16 h. Purify by dialysis.
Scheme 1Reaction ii outlines the conjugation of PEG to silk fibroin. The amount of cyanuric chloride activated PEG fed into the reaction was varied to conjugate different amounts of PEG to CarboxySF. Efficiency of the conjugation reactions, the number of chains reacted per the number of sites (tyrosine, histidine, and lysine residues) that could potentially react, was found to be uniformly 34% regardless of the amount of PEG in the feed of the reaction.
One of the goals with designing these adhesive materials was to preserve the ability of the fibroin to form β-sheet structures to afford the potential for strengthening of the material through induction of physical cross-links. To this end, films of silk without carboxyl functionalization (Silk (No COOH)), carboxy silk (CarboxySF), and the PEG conjugated carboxy silks (CarboxySF(x)) were prepared by dissolving at 40 mg/mL, casting onto an aluminum foil substrate, and air drying for 7 days. Spectra of as-cast samples are shown in Figure 1a, where both as-cast CarboxySF and CarboxySF(x) adopted a random-coil conformation, as evidenced by a broad peaks at 1650 and 1540 cm−1. The same films were then treated with methanol for 24 h and then dried on the bench for 48 h before repeating the measurement (Figure 1b). Methanol-treated CarboxySF and CarboxySF(x) both showed the same changes: (1) A decrease in the peak at 1650 cm−1 and the appearance of a sharp peak at 1625 cm−1 and (2) a decrease in the 1540 cm−1 peak and the intensification of a sharper peak at 1515 cm−1. Both of these changes indicated the sample formed β-sheet secondary structures.53 Importantly, conjugation of PEG did not affect this transition, indicating that the PEGylated samples had the ability to be triggered to form physical cross-links in the form of beta sheets. Fourier self-deconvolution (FSD) of the infrared spectra covering the amide I region (1595–1705 cm−1) was performed as previously described51 to quantify the β-sheet content in the films. The secondary structure of regenerated silk fibroin before any functionalization reactions carried out was 39% β sheet using this method. The silk derivatives were also found to form beta sheets, where the content was between 35 and 39% for CarboxySF and PEG derivatives of CarboxySF (Table 1). These data suggest that the carboxylation and PEGylation reactions may slightly decrease the amount of β-sheet secondary structure; however, these differences were not statistically significant in the range studied. CarboxySF, CarboxySF(6), and CarboxySF(29) were selected for study in the catechol-silk reactions to give a range of PEG substitutions, while maintaining silk as the major component in the material and the capability to form beta sheets in the material.
Figure 1.

ATR-FTIR spectra of products from PEGylation reactions on carboxylated silk fibroin: (a) “as cast” samples and (b) samples treated with methanol for 24 h to induce β sheet, where SF(no COOH) is silk fibroin without carboxyl modification and CarboxySF(x) is carboxylated silk fibroin with x PEG chains attached per molecule of fibroin. Vertical reference lines mark 1650, 1625, 1540, and 1515 cm−1.
Table 1.
Average Beta Sheet Formation of Carboxylated and PEGylated Silks, As Determined by Deconvolution of ATR-FTIR Spectra Acquired after Treatment with Methanol for 24 h
| material | average β sheet, % (range, %) |
|---|---|
| silk (no COOH) | 39.3 (1.98) |
| carboxy-SF(0) | 35.2 (4.70) |
| carboxy-SF(6) | 38.7 (5.32) |
| carboxy-SF(12) | 36.1 (3.99) |
| carboxy-SF(29) | 38.4 (0.61) |
| carboxy-SF(71) | 35.5 (3.67) |
To ensure that dopamine was chemically coupled to silk fibroin and not absorbed, an aqueous solution of CarboxySF and dopamine was prepared, dialyzed against different solvents before being lyophilized, and analyzed using 1H NMR. Importantly, the silk did not precipitate and the solution did not turn brown (which may indicate catechol oxidation) during the dialysis for Protocols A–D. Precipitate formed during dialysis when Protocols E and F were used, and thus these protocols were not considered further as a viable option for the workup of silk-dopamine conjugates. Table 2 shows the results of this extraction study, where it was found that the wash steps in Protocol C were most effective at removing dopamine, as dopamine was not detected in the silk extracted using this method using 1H NMR. Additionally, although no precipitation was observed in Protocols A–D, all solutions were slightly translucent except during the urea steps when the solutions were completely transparent. The change in the clarity of the solutions was attributed to protein unfolding in the presence of urea. Thus, while increasing PBS washing steps of Protocol D may result in removal of dopamine to a level achieved by Protocol C, Protocol C was selected for the final purification method of the silk-dopamine conjugates because the denaturing (unfolding) of silk proteins by urea may assist in removing unreacted dopamine.
Table 2.
Removal of Unconjugated Dopamine from CarboxySF(0) Using Different Sample Washing Methodsa
| washing method | dopamine removed (% of initial) |
|---|---|
| initial (no wash) | 0.00 |
| protocol A | 93.3 |
| protocol B | 93.2 |
| protocol C | 100 (no dopamine detected) |
| protocol D | 96.3 |
| protocol E | 93.9 |
| protocol F | 93.5 |
After each wash, samples were lyophilized before dissolving in deuterated DMSO, measuring with 1H NMR, and integrating the resonances from dopamine (at 2.64 and 2.91 ppm) in the resulting spectra to quantify the dopamine present in the sample.
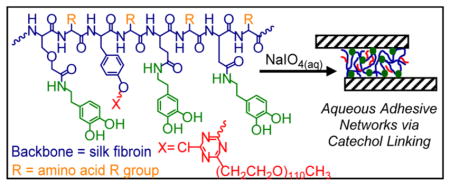
Conjugation of dopamine to CarboxySF and CarboxySF(x) is outlined in Scheme 1 Reaction 3. This reaction was performed in DMSO, an organic solvent, because it was found that the pH required to react dopamine with the silk fibroin under aqueous conditions led to two issues: precipitation of the fibroin or premature oxidation of the catechol. It was necessary to avoid premature oxidation of the catechol because the oxidation reaction is involved in the adhesion and cross-linking of catechol-based adhesives.18 DMSO was selected because it solubilizes CarboxySF and CarboxySF(x) and because regenerated cellulose dialysis tubing is stable in DMSO and thus purification using dialysis was possible. After dialysis against DMSO, the samples were lyophilized, dissolved back into water at 50 mg/mL, washed using Protocol C with the subsequent water dialysis steps, and lyophilized to give the final product. In each case, yield of the dopamine-silk conjugate was high, 80% at minimum. Figure 2 shows 1H NMR spectra of CarboxySF and the CarboxySF-dopamine conjugate. Examination of the CarboxySF-dopamine (Figure 2, trace (ii)) NMR spectrum shows a set of peaks between 6.45 and 6.64 ppm from the three aromatic protons of dopamine (Figure 2, trace (iii)) that are not present in CarboxySF (Figure 2, trace (i)). The upfield region of the spectra is more difficult to analyze due to the contributions of silk fibroin to the spectra, but it is possible to discern that the 2.32 ppm peak present in COMU (Figure 2, trace (iv)) is not present in CarboxySF-dopamine, indicating that the purification steps also resulted in the successful removal of COMU from the product.
Figure 2.
1H NMR spectra in deuterated DMSO of purified products of dopamine reactions run on 10 min boiled silk fibroin: (i) CarboxySF, (ii) CarboxySF-dopamine, (iii) dopamine hydrochloride, and (iv) COMU.
Aqueous solubility of dopamine conjugates was tested by attempting to prepare solutions of CarboxySF, CarboxySF-(20), CarboxySF(40), and their dopamine derivatives in water (pH 7.5) at a concentration of 80 mg/mL (Figure 3). This Figure shows that all of the silks were soluble in water prior to conjugation with dopamine; however, it is only the PEGylated silks that were fully soluble in water after conjugation with dopamine; CarboxySF-dopamine was only partially soluble in distilled water. Importantly, the addition of PEG to the silk chains resulted in a dopamine conjugate that was completely water-soluble, with as few as six PEG substitutions per molecule needed to render the dopamine conjugate soluble in water, which is highly desired for biological applications.
Figure 3.

Image of CarboxySF (a), CarboxySF(20) (b), CarboxySF-(40) (c), CarboxySF-dopamine (d), CarboxySF(20)-dopamine (e), and CarboxySF(40)-dopamine (f) dissolved at 80 mg/mL in DI water.
Quantification of dopamine by 1H NMR was challenging, as silk resonances overlap many of the aliphatic resonances of dopamine and quantitative integration of the well-separated protons in the aromatic region is not trivial, despite the long relaxation delays employed in these experiments. A colorimetric assay was thus used, in conjunction with a standard curve, to quantify the amount of dopamine conjugated to the fibroin. This procedure, first reported by Arnow,52 involves the reaction of nitrate ions in an acidic environment with phenolic alcohols to form a nitric oxide compound. Figure 4a shows the absorbance curves from CarboxySF(6)-dopamine and CarboxySF(6). By comparison with the dopamine standard curve, the amount of catechol in each sample was normalized to the silk content of each product (Figure 4b) to show that, while conjugation was successful for all products, conjugation efficiency was largest for CarboxySF(0) and there was no statistically significant difference between the two PEGylated CarboxySF that were studied. There are several possible reasons for this finding, including decreased accessibility to the reactive sites on silk due to the PEG chains, differences in the silk chain conformation in the solvent leading to decreased accessibility of reactive sites, or perhaps preparation-related reasons such as larger amounts of bound water that may poison the coupling reaction, despite the extensive drying that was applied to all samples.
Figure 4.

Catechol quantification using Arnow’s protocol. (a) UV–vis spectra of CarboxySF(6) and CarboxySF(6)-dopamine. (b) Dop-amine conjugated to the silk products, normalized to mole of product. + denotes significance (p < 0.001) relative to CarboxySF(6) and CarboxySF(29).
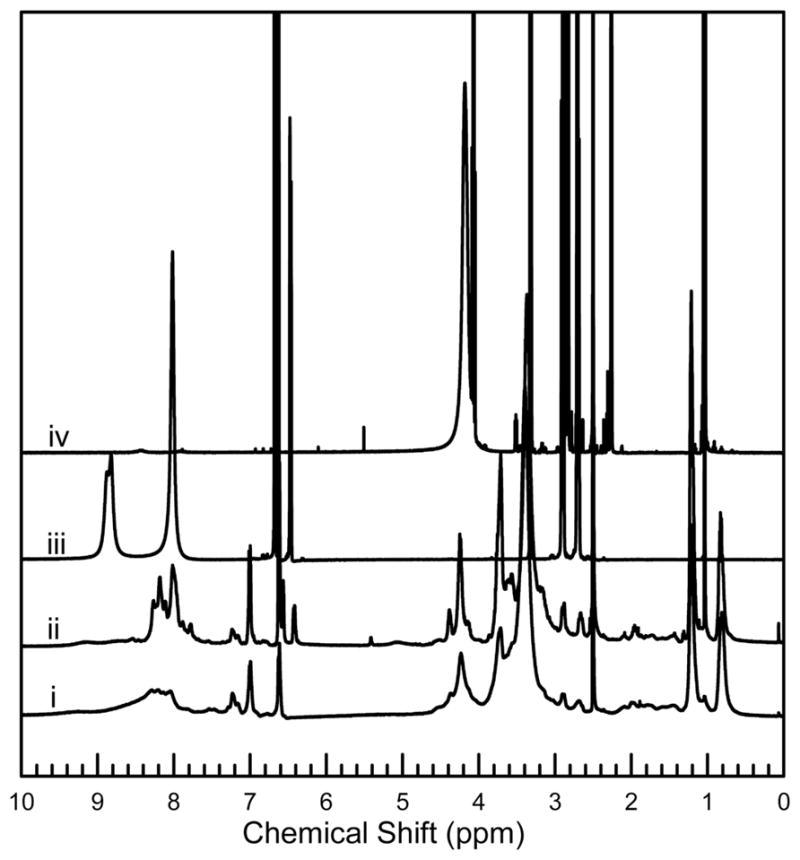
The adhesive bond of the modified silks to aluminum shims was quantified by single lap shear testing. The peak force that the adhesive was able to withstand was divided by bonded area to give the data reported in Figure 5, where it is shown that both the CarboxySF(0)-dopamine and CarboxySF(6)-dop-amine form a stronger adhesive bond to the shims than the same material that is not functionalized with dopamine. There is no statistically significant difference between the bond strength of CarboxySF(0)-dopamine and CarboxySF(6)-dopamine, but it is noted that CarboxySF(0) has a larger peak stress at break than CarboxySF(6). This indicates that silk itself, without catechol, can bond aluminum shims, a finding that is consistent with previous reports on silk hydrogels formed by electrogelation, although it is important to note that electrogelled silk hydrogel previously reported is reversible rather than permanent.54,55 While the bonding strengths between CarboxySF(0)-dopamine and CarboxySF(6)-dopamine are not statistically different, a potential advantage of the catechol coupling is the exploitation of the catechol’s ability to associate with a variety of materials. Furthermore, a small amount of PEG conjugation in CarboxySF(6)-dopamine renders the material completely soluble and therefore increases handleability compared with CarboxySF(0)-dopamine. Increasing the PEG content further increased the variability in the adhesive bond strength of the materials. The origin of this is not clear but may be for a variety of reasons, including steric or solvation effects. These studies thus emphasize the importance of balancing solubility with dopamine functionalization to maximize the strength of the adhesion bonding.
Figure 5.
Adhesion of dopamine-modified silks. Aluminum shims were adhered in a single lap configuration and pulled apart in tension. The average peak stress to break the bonded shims is plotted with error bars denoting standard deviation. + denotes significance at p < 0.001 and ++ denotes significance at p < 0.02.
Unique features of the silk-based dopamine adhesives are expected to arise from the structure of fibroin itself. The potential for β-sheet secondary structures to form in addition to the cross-linking between catechol residues is expected to serve as secondary cross-links in the material that will not only strengthen the adhesive but also hinder aqueous swelling of the polymer network. The hydrophobic nature of the silk core is also expected to reduce the degree of aqueous swelling of the silk cross-linked network and further assist in the mechanical strength of the polymer network. In this work, the tightly bonded shims did not permit adequate water penetration to conduct swelling studies, and thus quantification of the swelling behavior was deferred for future studies to quantify the properties of hydrogels formed from these materials. One recognized limitation of the adhesion studies is the lack of a representative poly(ethylene glycol)-based control material that could be used to compare the adhesive bond strengths. The challenge with obtaining such a material is that the average molecular weight between cross-links (Mc) of the silk-dopamine materials must be known so that a poly(ethylene glycol)-based material can be prepared with similar degrees of cross-linking, as the cross-linking will affect both the swelling and mechanical properties of the network and thus the utility of such a control. In the absence of the future mechanical studies of hydrogel networks that would permit an estimation of Mc from modulus and rubber elasticity theory, we thus compare the adhesive bond strengths of silk-dopamine conjugates with varying degrees of poly(ethylene glycol) substitution.
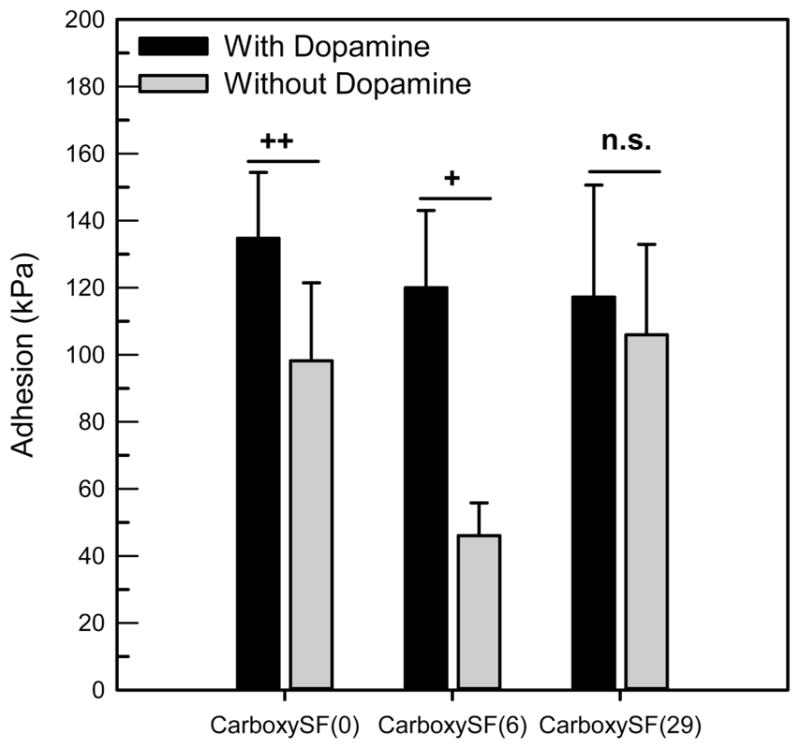
The cell compatibility of the silk and dopamine-functionalized silks was tested by seeding human mesenchymal stem cells (hMSCs). Assaying with Alamar Blue reagent permitted measurement of mitrochondrial activity in the cells, where an increase in fluorescence can be attributed to increased cell numbers if it is assumed that the cells’ mitochondrial activity is not changing between the different material substrates. Cells adhered and proliferated on all silk materials tested over the course of the experiment (15 days) (Figure 6). There were no clear trends in proliferation with the PEG functionalization and the dopamine functionalization, as the cells on all of the materials showed a statistically significant increase in fluorescence at each time point compared with their previous time point. These results thus show that all materials tested support the attachment and proliferation of hMSCs in vitro, although in vivo biocompatibility must still be explored in the future.
Figure 6.
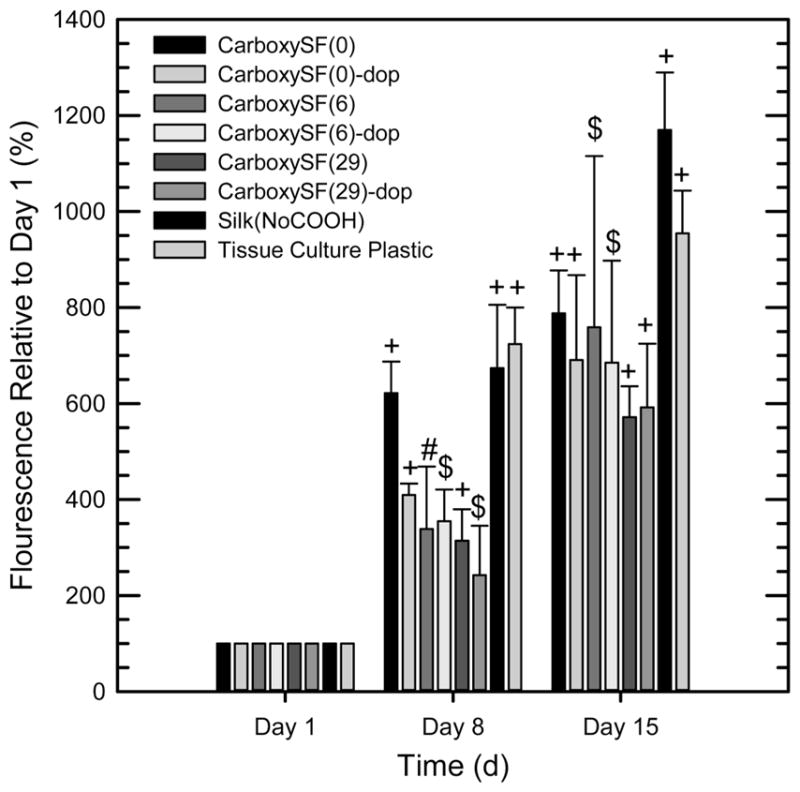
Cell proliferation on dopamine modified silks, as assayed using Alamar Blue. Significant difference between labeled time point and previous time point is denoted by symbol, where + denotes p < 0.001, $ denotes p < 0.05, and # denotes not significant (p > 0.05).
CONCLUSIONS
This work describes the synthesis of new silk fibroin conjugates that can be triggered to cross-link by the addition of an oxidant. The addition of poly(ethylene glycol) chains prior to dopamine conjugation yielded polymers with increased aqueous solubility, and this conjugation did not affect the ability of silk fibroin to form β-sheet structures. Dopamine was conjugated to the fibroin successfully. Tuning the amount of the hydrophilic PEG chains proved to be critical to maintaining solubility and the strength of the adhesive bond. All materials tested supported human mesenchymal cell attachment and proliferation throughout 15 days of in vitro culture. The additional option to induce β-sheet formation in these materials after bonding offers a path toward tunable control over the mechanics of the adhesion.
Acknowledgments
We thank the National Institutes of Health for funding support in the form of the Tissue Engineering Resource Center (NIH P41 P41 EB002520) and an NIH Ruth Kirschstein Postdoctoral Fellowship awarded to K.A.B. (F32-DK093194). We also thank the AFOSR for support of this work.
Footnotes
The authors declare no competing financial interest.
References
- 1.Stewart RJ, Ransom TC, Hlady V. J Polym Sci, Part B: Polym Phys. 2011;49:757–771. doi: 10.1002/polb.22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stewart RJ. Appl Microbiol Biotechnol. 2011;89:27–33. doi: 10.1007/s00253-010-2913-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilker JJ. Curr Opin Chem Biol. 2010;14:276–283. doi: 10.1016/j.cbpa.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Lee BP, Messersmith PB, Israelachvili JN, Waite JH. Annu Rev Mater Res. 2011;41:99–132. doi: 10.1146/annurev-matsci-062910-100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin Q, Gourdon D, Sun C, Holten-Andersen N, Anderson TH, Waite JH, Israelachvili JN. Proc Natl Acad Sci U S A. 2007;104:3782–3786. doi: 10.1073/pnas.0607852104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waite JH. Int J Biol Macromol. 1990;12:139–144. doi: 10.1016/0141-8130(90)90065-i. [DOI] [PubMed] [Google Scholar]
- 7.Waiter JH. Reverse engineering of bioadhesion in marine mussels. Ann N Y Acad Sci. 1999;875:301. doi: 10.1111/j.1749-6632.1999.tb08513.x. [DOI] [PubMed] [Google Scholar]
- 8.Monahan J, Wilker JJ. Langmuir. 2004;20:3724. doi: 10.1021/la0362728. [DOI] [PubMed] [Google Scholar]
- 9.Ninan L, Stroshine RL, Wilker JJ, Shi R. Acta Biomater. 2007;3:687. doi: 10.1016/j.actbio.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee H, Scherer NF, Messersmith PB. Proc Natl Acad Sci U S A. 2006;103:12999–13003. doi: 10.1073/pnas.0605552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cha HJ, Hwang DS, Lim S, White JD, Matos-Perez C, Wilker JJ. Biofouling. 2009;25:99–107. doi: 10.1080/08927010802563108. [DOI] [PubMed] [Google Scholar]
- 12.Wei W, Yu J, Broomell C, Israelachvili JN, Waite JH. J Am Chem Soc. 2013;135:377–383. doi: 10.1021/ja309590f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei W, Yu J, Gebbie MA, Tan Y, Martinez Rodriguez NR, Israelachvili JN, Waite JH. Langmuir. 2015;31:1105–1112. doi: 10.1021/la504316q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim BJ, Oh DX, Kim S, Seo JH, Hwang DS, Masic A, Han DK, Cha HJ. Biomacromolecules. 2014;15:1579–1585. doi: 10.1021/bm4017308. [DOI] [PubMed] [Google Scholar]
- 15.Faure E, Falentin-Daudré C, Jérôme C, Lyskawa J, Fournier D, Woisel P, Detrembleur C. Prog Polym Sci. 2013;38:236–270. [Google Scholar]
- 16.Hoffmann B, Volkmer E, Kokott A, Augat P, Ohnmacht M, Sedlmayr N, Schieker M, Claes L, Mutschler W, Ziegler G. J Mater Sci: Mater Med. 2009;20:2001–2009. doi: 10.1007/s10856-009-3782-5. [DOI] [PubMed] [Google Scholar]
- 17.Burke SA, Ritter-Jones M, Lee BP, Messersmith PB. Biomed Mater. 2007;2:203–210. doi: 10.1088/1748-6041/2/4/001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee BP, Dalsin JL, Messersmith PB. Biomacromolecules. 2002;3:1038–1047. doi: 10.1021/bm025546n. [DOI] [PubMed] [Google Scholar]
- 19.Cencer M, Murley M, Liu Y, Lee BP. Biomacromolecules. 2015;16:404–410. doi: 10.1021/bm5016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holten-Andersen N, Harrington MJ, Birkedal H, Lee BP, Messersmith PB, Lee KYC, Waite JH. Proc Natl Acad Sci U S A. 2011;108:2651–2655. doi: 10.1073/pnas.1015862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laulicht B, Mancini A, Geman N, Cho D, Estrellas K, Furtado S, Hopson R, Tripathi A, Mathiowitz E. Macromol Biosci. 2012;12:1555–1565. doi: 10.1002/mabi.201200179. [DOI] [PubMed] [Google Scholar]
- 22.Murphy JL, Vollenweider L, Xu F, Lee BP. Biomacromolecules. 2010;11:2976–2984. doi: 10.1021/bm1007794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee BP, Chao C, Nunalee FN, Motan E, Shull KR, Messersmith PB. Macromolecules. 2006;39:1740–1748. [Google Scholar]
- 24.Barrett DG, Bushnell GG, Messersmith PB. Adv Healthcare Mater. 2013;2:745–755. doi: 10.1002/adhm.201200316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu M, Deming TJ. Macromolecules. 1998;31:4739–4745. doi: 10.1021/ma980268z. [DOI] [PubMed] [Google Scholar]
- 26.Fullenkamp DE, Rivera JG, Gong Y-k, Lau KHA, He L, Varshney R, Messersmith PB. Biomaterials. 2012;33:3783–3791. doi: 10.1016/j.biomaterials.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krogsgaard M, Behrens MA, Pedersen JS, Birkedal H. Biomacromolecules. 2013;14:297–301. doi: 10.1021/bm301844u. [DOI] [PubMed] [Google Scholar]
- 28.Brubaker CE, Messersmith PB. Biomacromolecules. 2011;12:4326–4334. doi: 10.1021/bm201261d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meredith HJ, Jenkins CL, Wilker JJ. Adv Funct Mater. 2014;24:3259–3267. [Google Scholar]
- 30.Matos-Perez CR, White JD, Wilker JJ. J Am Chem Soc. 2012;134:9498–9505. doi: 10.1021/ja303369p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue S, Tanaka K, Arisaka F, Kimura S, Ohtomo K, Mizuno S. J Biol Chem. 2000;275:40517–40528. doi: 10.1074/jbc.M006897200. [DOI] [PubMed] [Google Scholar]
- 32.Yamaguchi K, Kikuchi Y, Takagi T, Kikuchi A, Oyama F, Shimura K, Mizuno S. J Mol Biol. 1989;210:127–139. doi: 10.1016/0022-2836(89)90295-7. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y-Q. Biotechnol Adv. 2002;20:91–100. doi: 10.1016/s0734-9750(02)00003-4. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, Reagan MR, Kaplan DL. Adv Drug Delivery Rev. 2009;61:988–1006. doi: 10.1016/j.addr.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Numata K, Kaplan DL. Adv Drug Delivery Rev. 2010;62:1497–1508. doi: 10.1016/j.addr.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yucel T, Lovett ML, Kaplan DL. J Controlled Release. 2014;190:381–397. doi: 10.1016/j.jconrel.2014.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kundu B, Rajkhowa R, Kundu SC, Wang X. Adv Drug Delivery Rev. 2013;65:457–470. doi: 10.1016/j.addr.2012.09.043. [DOI] [PubMed] [Google Scholar]
- 38.Tao H, Kaplan DL, Omenetto FG. Adv Mater. 2012;24:2824–2837. doi: 10.1002/adma.201104477. [DOI] [PubMed] [Google Scholar]
- 39.Vepari C, Kaplan DL. Prog Polym Sci. 2007;32:991–1007. doi: 10.1016/j.progpolymsci.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou CZ, Confalonieri F, Jacquet M, Perasso R, Li ZG, Janin J. Proteins: Struct, Funct, Genet. 2001;44:119–122. doi: 10.1002/prot.1078. [DOI] [PubMed] [Google Scholar]
- 41.Wang YY, Cheng YD, Liu Y, Zhao HJ, Li MZ. Adv Mater Res. 2011;175–176:143–148. [Google Scholar]
- 42.Samal SK, Kaplan DL, Chiellini E. Macromol Mater Eng. 2013;298:1201–1208. [Google Scholar]
- 43.Liu G, Xiong S, You R, Wang L, Li M. Adv Mater Res. 2013;634–638:1165–1169. [Google Scholar]
- 44.Yucel T, Cebe P, Kaplan DL. Biophys J. 2009;97:2044–2050. doi: 10.1016/j.bpj.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin HJ, Park J, Karageorgiou V, Kim UJ, Valluzzi R, Cebe P, Kaplan DL. Adv Funct Mater. 2005;15:1241–1247. [Google Scholar]
- 46.Lu Q, Hu X, Wang X, Kluge JA, Lu S, Cebe P, Kaplan DL. Acta Biomater. 2010;6:1380–1387. doi: 10.1016/j.actbio.2009.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bellas E, Panilaitis BJB, Glettig DL, Kirker-Head C, Yoo JJ, Marra KG, Rubin JP, Kaplan DL. Biomaterials. 2013;34:2960–2968. doi: 10.1016/j.biomaterials.2013.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rockwood DN, Preda RC, Yucel T, Wang X, Lovett ML, Kaplan DL. Nat Protoc. 2011;6:1612–1631. doi: 10.1038/nprot.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wray LS, Hu X, Gallego J, Georgakoudi I, Omenetto FG, Schmidt D, Kaplan DL. J Biomed Mater Res, Part B. 2011;99B:89–101. doi: 10.1002/jbm.b.31875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Serban MA, Kaplan DL. Biomacromolecules. 2010;11:3406–3412. doi: 10.1021/bm100925s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu X, Kaplan D, Cebe P. Macromolecules. 2006;39:6161–6170. [Google Scholar]
- 52.Arnow LE. Colorimetric Determination of the Components of 3,4-Dihydroxyphenylalaninetyrosine Mixtures. J Biol Chem. 1937;118:531–537. [Google Scholar]
- 53.Murphy AR, John PS, Kaplan DL. Biomaterials. 2008;29:2829–2838. doi: 10.1016/j.biomaterials.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elia R, Michelson CD, Perera AL, Harsono M, Leisk GG, Kugel G, Kaplan DL. J Biomater Appl. 2015;29:1247–1255. doi: 10.1177/0885328214561536. [DOI] [PubMed] [Google Scholar]
- 55.Yucel T, Kojic N, Leisk GG, Lo TJ, Kaplan DL. J Struct Biol. 2010;170:406–412. doi: 10.1016/j.jsb.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]