

Abstract
Proteins are often enantioselective towards their binding partners. When designing small molecules to interact with these targets, one should consider stereoselectivity. As considerations for exploring structure space evolve, chirality is increasingly important. Binding affinity for a chiral drug can differ for diastereomers and between enantiomers. For the virtual screening and computational design stage of drug development, this problem can be compounded by incomplete stereochemical information in structure libraries leading to a “coin toss” as to whether or not the “ideal” chiral structure is present. Creating every stereoisomer for each chiral compound in a structure library leads to an exponential increase in the number of structures resulting in potentially unmanageable file sizes and screening times. Therefore, only key chiral structures, enantiomeric pairs based on relative stereochemistry need be included, and lead to a compromise between exploration of chemical space and maintaining manageable libraries. In clinical environments, enantiomers of chiral drugs can have reduced, no, or even deleterious effects. This underscores the need to avoid mixtures of compounds and focus on chiral synthesis. Governmental regulations emphasizing the need to monitor chirality in drug development have increased. The United States Food and Drug Administration issued guidelines and policies in 1992 concerning the development of chiral compounds. These guidelines require that absolute stereochemistry be known for compounds with chiral centers and that this information should be established early in drug development in order that the analysis can be considered valid. From exploration of structure space to governmental regulations it is clear that the question of chirality in drug design is of vital importance.
Keywords: Chiral, virtual screening, drug design, drug discovery, FDA guidelines, enantiomer, in silico, computational chemistry
1. Introduction
The purpose of a drug discovery and development project is to find new therapeutic agents that target a key enzyme, protein-protein interaction, receptor-ligand, or protein-nucleic acid interaction of relevance in a disease of interest in order to mitigate the course of the disease. Finding compounds that can, for example, bind to the active site of an enzyme and inhibit its normal activity is merely the initial goal in most drug discovery projects. Typically, tens of thousands or even hundreds of thousands of different compounds must be screened in order to find a few promising compounds. The most promising compounds (lead compounds) can then be modified by medicinal chemists to develop even more potent variants while also addressing chemical and metabolic stability, safety, solubility, specificity, bioavailability, excretion and other characteristics as needed. Experimentally screening large numbers of compounds to find a few promising candidates is a costly and time consuming process and is just the first step in bringing a new drug to market. Chemists will attempt synthesis and modification of the promising compounds in order to perform structure-activity relationship (SAR) analysis around the compound's scaffold to optimize the compound for desired characteristics, while minimizing detrimental aspects. However, before committing time and resources to this lead optimization process, it is important to ensure that the candidate has been correctly identified, including elimination of any effects from impurities that may have resulted during the synthesis of experimental samples and eliminating any ambiguities in structural variations, such as degradation products and stereochemical variants. The less experimental effort spent on unsuccessful or mis-identified compounds, the more time and costs can be reduced. Therefore, initial drug discovery projects are steadily relying upon the use of computational (virtual) screening and computational molecular modeling to identify and eliminate less promising compounds.
In an increasing number of drug discovery projects, virtual 3D compound libraries are employed for computer-based screening against an X-ray or NMR structure of a target protein. This approach can greatly speed-up the discovery process while reducing costs by reducing the amount of “wet lab” experimental work required. Increased sophistication in modeling and screening software and increased availability of protein structures has greatly improved the efficacy and reliability of virtual screening. 3D coordinates of protein structures are available from the RCSB Protein Data Bank (www.pdb.org) [1] which has over 64,000 protein structure files (as of April 2010) so that the coordinates of most targets are available or can be obtained through homology modeling [2]. Additionally, collections of chemical structures can be assembled from commercial vendors so that a drug discovery project can rely upon virtual screening of millions of diverse compounds before experimental screening is attempted. Then, experimental screening can be used on a small subset of compounds enriched in potential binding characteristics for the target protein, as determined by the virtual screening. However, to make the screening relevant in addressing stereochemical issues that lay ahead in the process, each chiral compound should be properly represented in the 3D compound database used for virtual screening so that the drug discovery team knows which stereoisomer of the compound to pursue.
As of 1992, governmental regulations on drug safety and efficacy have become stricter with regard to compounds that have stereochemistry [3]. This has added complexity to the drug discovery process from initial virtual and experimental screening, through the rational design and synthesis, to clinical trial data analysis and manufacturing quality controls. When a compound can exist in several different stereochemical configurations, experimental work must be able to identify the stereoisomers present and ascribe biological effects to each of the stereochemical entities present since one stereoisomer may have quite different effects compared to another. Therefore, so that optimization studies are being performed on a valid lead compound and one that is relevant for the subsequent patenting and approval process, absolute stereochemistry should be characterized early in the process.
This discussion will provide a brief overview of stereochemistry in general and guidelines from a number of regulatory bodies including the FDA (U.S.A.), European Medicines Agency (E.U.), Health Canada (Canada) and International Conference on Harmonization of Technical Requirements (other nations such as Japan). Next, given the importance of deciding on stereochemistry issues early in the drug discovery process, often as early as initial project design and screening, an examination of some of the practical concerns of stereochemistry in drug discovery campaigns is provided.
2. Chirality
2.1. Chirality Defined
Chirality can be defined as the potential of a molecule to occur in two asymmetric forms that are non-superimposable mirror images of each other without changing the atomic composition, atom-atom connections, or bond orders Fig. (la). This phenomenon generally occurs due to a difference in the three-dimensional orientation of four different substituents attached to a single central atom, creating what can be considered left-hand and right-hand versions of the same molecule. These two versions of the molecule are referred to as enantiomers. When attempting to superimpose these versions, there will always be at least one substituent attached to the chiral atom that cannot be superimposed. In order to differentiate the two enantiomers, the Cahn-Ingold-Prelog system, or simply the R / S notation is employed, as recommended by the International Union of Pure and Applied Chemistry (IUPAC) [4] Fig. (lb). The D / L notation used for amino acids and sugars is restricted to those two molecular types with the D / L notation standing for dextrorotatory (clockwise) and levorotatory (counter-clockwise) optical rotation of polarized light. This convention is not in general use now, having been replaced by the R / S notation for chirality and + / - notation for optical rotation [5]. Enantiomers are usually described as having identical physical properties in achiral environments with the exception of the rotation of plane polarized light. In fact, plane polarized light is comprised of left- and right-handed components of circularly polarized light which is chiral and the phenomenon of optical rotation is due to slight differences in the way in which chiral molecules interact with these components. For a thorough presentation on chirality notations and examples, we refer the reader to the IUPAC home page (http://goldbook.iupac.org/) and the article by Caldwell & Wainer [5]. Molecules that are super-imposable on their mirror images are referred to as achiral.
Fig. (1).
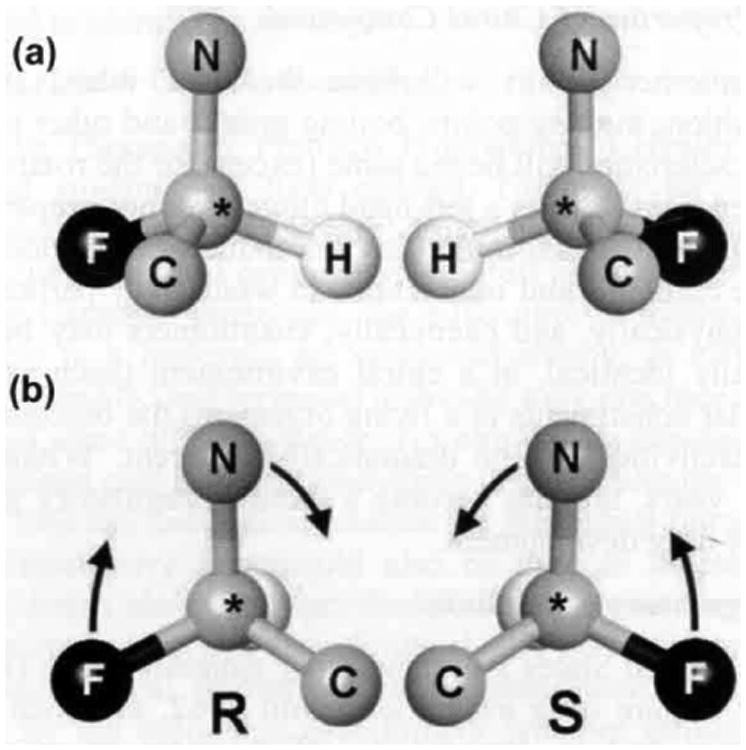
a) Basic chirality. These two molecules have the same atoms and the same atom-atom connections but they cannot be fully superimposed. They are therefore referred to as enantiomers and appear as mirror images in three-dimensional depictions. The central atom (*) is therefore considered to be a chiral center. (Note that bond lengths and atomic diameters have been simplified in order to focus on the basic concepts in these depictions.) (b) R / S conformations. In order to differentiate enantiomer pairs, R (rectus) and S (sinister) are used. To determine the R or S notation for the chiral molecules of Figure 1, the substituent atoms attached to the chiral atom are prioritized based on their atomic number with the higher number being the higher priority (therefore, F>N>C>H). The molecule is rotated until the lowest priority substituent, in this case H, is behind the chiral center. The chiral center is R if the three remaining substituents go clockwise from highest (F) to lowest priority (C). The chiral center is S if the three remaining stituents go counter-clockwise from highest to lowest priority.
When both enantiomers of a compound are present at equal concentrations in a sample, it is referred to as a racemate or a racemic mixture. Kinetic and thermodynamic resolution can be employed to separate enantiomers as can chiral chromatographic methods. Stereoselective chemical processes like asymmetric synthesis (sometimes referred to as chiral synthesis) or stereoselective enzymes can favor one enantiomer over the other.
As more complex molecules are considered, chirality can still occur, with more complex substituents attached to a chiral center or multiple chiral centers. These molecules, when not enantiomers of each other, are referred to as diastereoisomers. Likewise, such molecules can have different physical properties like boiling points, melting points, etc.
When a potential chiral center has two identical atoms attached to it, the next “level” of atoms needs to be considered in order to establish priority and therefore nomenclature. For example, if two carbons are attached to the chiral center and one of those carbons has only hydrogens attached while the other carbon has an oxygen atom attached, then the carbon-oxygen substituent has the higher priority over the carbon-hydrogens in determining the R or S orientation.
2.1.1. Properties of Chiral Compounds
Enantiomeric pairs will have the same mass, atomic composition, melting points, boiling points, and other physical characteristics will be the same (except for the rotation of polarized light) but, as a left-hand glove does not properly fit the right hand, chiral molecules can induce stereoselectivity into the reactions and interactions in which they participate. While physically, and chemically, enantiomers may behave essentially identical, in a chiral environment (such as biomolecular constituents in a living organism) the outcomes of their reactivities can be dramatically different. Within the past 22 years, this has become a focus in regulatory guidelines for drug development.
2.2. Regulatory Guidelines
The United States Food and Drug Administration (FDA) did not require drug evaluations until 1962, at which time new drugs had to demonstrate effectiveness to meet approval. It was not until 1988 that the content requirements for drug applications were defined to include dosage-response data and demographics regarding adverse reactions [6]. Around this time, chirality became a concern since many drugs that were succeeding in the clinic were chiral but were being marketed as racemates. The enantiomers may differ in their effects and so the exact composition of a racemate was of concern. Enantiomers that have the desired effect are termed eutomers. Enantiomers that do not have the desired effect or even a detrimental effect are termed distomers. Due to this, there are several potential scenarios that could exist:
The enantiomers that make up the racemic mixture have similar effects.
The enantiomers that make up the racemic mixture have differing effects and there can be enatiomeric drift in a chiral environment, such as in a biological setting, so that the original racemic 50:50 ratio shifts, which then can increase or decrease the dosage effect.
The compound samples at various steps in chiral-based manufacturing and/or selection processes can have a variable percentage of each stereoisomer leading to quality control issues.
The racemate contains an effective eutomer but the distomer has detrimental effects that must be eliminated.
An enantiomerically pure compound undergoes partial or complete racemization in vivo or ex vivo resulting in formation of at least some of the distomer.
One example that demonstrates the possibilities is the anti-inflammatory drug, ibuprofen. Oral administration of the R stereoisomer of ibuprofen will result in a mean of 63 ± 6% of the R form interconverting to the S form; whereas the S form shows no interconversion to the R form [7]. Therefore, the pharmacokinetics of each stereoisomer is required to understand the mixture's effects. Interestingly, most of the non-steroidal anti-inflammatory drugs (NSAIDs) have a characteristic pattern where the S configuration possesses most of the inhibitory effect on prostaglandin activity, with the R configuration being inert [8].
Since the manufacturing processes and the body's metabolism of the drug can alter the compound, including interconversion of enantiomers, it is essential to document these actions in animal studies, manufacturing steps, and pharmacokinetic studies from early clinical trials. Then data on the compound for safety and efficacy can be considered accurate and complete. If the marketed drug will be a racemate or mixture of enantiomers then the effects of each enantiomer must be documented [3]. The sooner in the drug discovery process the properties of each enantiomer are determined, the better so that costly experiments and studies do not have to be repeated.
2.2.1. FDA Guidelines
Guidelines regarding stereochemistry were published by the U.S. Food and Drug Administration in 1992 in a document entitled ‘Development of new stereoisomeric drugs’ [3].
These guidelines have altered the marketing and patenting opportunities and strategies for successful drugs. They compel drug discovery projects to consider stereochemistry as early as possible in the search for a new drug candidate in order to have complete and accurate data characterizing the drug candidate. In the FDA's 1992 guidelines it requires that absolute stereochemistry be known for compounds with chiral centers and that this information should be established early in drug discovery and development in order that analysis is considered thorough and valid for inclusion in the drug approval application. The FDA guidelines suggest that at each step of the manufacturing process, in animal testing, and in clinical trials there should be a means established and data collected for stereo-specific enantiomer identification and/or stereo-specific assaying of activity. The enantiomers, when determined to have differing effects individually will require that “specialized chiral techniques for their correct identification, characterization, separation and measurement” should be employed [3]. The means of identification and quantification can include measuring optical rotation, chiral chromatography, optical rotary dispersion, circular dichroism, and NMR with chiral shift reagents [6].
The guidelines do not give definitive percentages for considering a single enantiomer as pure but they do state that the enantiomers in what was originally a racemate cannot be assumed to remain equimolar (1:1) due to possible interconversion or differential metabolic degradation rates in a chiral environment. Therefore, the enantiomeric composition should be assessed in order to have valid data. Additionally, since in a chiral environment interconversions can occur and enantiomers can degrade or be metabolized at differing rates or bioavailability can differ, data on each enantiomer should be obtained at each step in the development process. Doing so allows, for example, Phase I clinical data to be compared to animal studies. Pharmacokinetic properties can vary between the two enantiomers and between each enantiomer relative to a racemate and, therefore, besides determining the differences in biological activity of the individual enantiomers and the racemate, other properties should be assayed, such as absorption, distribution, biological interconversions & modifications, and excretion rates. The enantiomers can differ in their pharmacological and toxicological effects and this must be documented [3]. As far as manufacturing processes and quality control of the products, the FDA suggests that, at each step, the “identity, strength, quality, and purity” be assured and the enantiomeric composition known [3]. The stability of the drug substances and products should be assessed to determine if racemization or degradation is occurring. Labeling should be unique and appropriately describe the stereochemistry of the drug [3].
The FDA leaves the decision to develop a drug as a racemate or as a single enantiomer to the developers. However, the rationale behind the decision to develop the drug as a racemate or as a single enantiomer must be included in the drug approval application. Therefore, as drug development proceeds for a promising compound, the company can strategize as to how best to implement the drug. If they want a slow effect from the drug conversion of an effect-less distomer to an effective eutomer could be a useful strategy. As a result, the developers may opt to develop the drug as a racemate. Later, as the drug patent is about to expire, they can use a patent on the eutomer, perhaps formulating it with another slower acting drug so that they can lower the risk of rare adverse effects that might have been attributed to the eutomer. This chiral switching from the racemate to a single enantiomer can extend the drug's patent protection, which is typically 20 years (from the date of filing) in the U.S.A., for an additional 5 years [9]. Having the patent on a racemate does not guarantee patent protection of the individual enantiomers and so developers need to file for patent protection of the single enantiomer(s) [5], This brings up several issues, however. If the racemate was safe and effective, the single enantiomer drug needs to provide an improvement so that it can compete against the influx of generic racemate drugs that will be marketed by competitors when the racemate patent expires. Also, during the patent protected life of the racemate, other drug companies may have come up with better drugs against the same target. The company with the original racemate patent may want to reformulate the drug with the single enantiomer and some additional component(s) that improves the single enantiomer's effect; such as improving bioavailability so that an even lower dose of the single enantiomer is needed. Agranat, et al., provide an excellent review of some of the racemate to single enantiomer switches and the consequences of the switch [10]. There has been an involved discussion as to how the FDA should address chiral switching and patent protection extension since the chiral switch may not provide enough novelty or additional benefit to exclude other companies with generic versions. A number of legal cases have occurred in this area and several reviews have been published in recent years [11-13].
2.2.2. European Medicines Agency Guidelines
The European Union has adopted guidelines from the European Medicines Agency in 1994 entitled ‘Investigation of chiral active substances’ [14]. These are additional guidelines to be followed along with previous directives (65/65/EEC and 75/318/EEC) that apply to quality, safety, and efficacy for drugs. For the most part the EMA guidelines of 1994 read similar to the FDA guidelines of 1992. The EMA does give more detail to suitable toxicity testing when a racemate is to be switched to a single enantiomer, calling for testing up to 3 months with repeated doses in an appropriate species using the racemate as a positive control for the single enantiomer dosing and to test for pre- and post-natal development effects. The EMA guidelines and directives also state that, for manufacturing processes, the starting materials, intermediates, and final products need to be fully characterized as to their identity and purity since interconversions of stereoisomers and conversions (chiral to achiral. achiral to chiral) can occur [14].
2.2.3. Health Canada Guidelines
The Therapeutic Products Programme of Health Canada issued guidance in 2000 entitled ‘Guidance for industry: stereochemical issues in chiral drug development’ after soliciting questions and comments to a draft posted in 1998 [15]. In the Canadian guidelines, the difficulties of multiple chiral centers in a molecule are addressed stating, in effect, that each scientific and technical difficulty may preclude application of some of the guidance. For chiral drug substances, the guidelines call for enantioselective tests for identity and purity. This can use optical rotation for the testing but a second enantioselective test should also be used to assess purity. Also, limits should be specified for the distomer in a single enantiomer drug and these limits should be met for preclinical and clinical studies. As far as manufacturing processes used for the individual enantiomers, whether through chiral synthesis or separation or both, the steps must be described in full and identity and purity testing performed for key intermediates and final products.
2.2.4. ICH Guidelines
Other countries have adopted the 1999 guidelines from the International Conference on Harmonization of Technical Requirements (ICH) [16]. For example, in Japan, although the Japanese government has not issued a policy statement on chiral drugs, it adopted in 2001 the guidelines for chiral drug quality as stated by the ICH for the Registration of Pharmaceutical Human Use, the guidelines being ‘Specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances’ [16]. In the case of the ICH guidelines, the concept of impurities is addressed with the distomers being considered an impurity and, therefore, the contribution of enantiomers to a racemate's efficacy and safety must be analyzed and the occurrence of interconversion must also be determined.
2.3. Trends in Chiral Drugs
2.3.1. Chiral Switching
Based on these new guidelines in the 1990's, most drug companies and research institutes have begun focusing on single enantiomers early on when they identify a chiral drug candidate. For those drugs that had been marketed as race-mates, the owners have been switching the drugs to the active enantiomer [17]. This strategy is referred to as chiral switching [17]. This gives extension to the patent protection of the drug as the racemate comes off patent, a patent on the eutomer can extend the drug's patent life.
The American Medical Association issued guidelines in 1995 for naming conventions for when chiral switching occurs [18]. These guidelines list a number of examples, such as the racemate omeprazole (Prilosec) first launched in 1988 but was switched to esomeprazole (Nexium) in 1999 in Europe and 2001 in the United States as the original patents expired [10]. Caner, et al. published a report on the trends in chiral drugs, with an emphasis on single enantiomer development, that shows a 20-year period (1983-2002), which provides 10-year bracketing of the 1992 FDA guidelines that influenced the drug industry [17]. According to their review, FDA approvals of new molecular entities in the 1983-1992 period were approximately 27% racemates, 33% single enantiomers, and 40% achiral, whereas in the 1993-2002 period FDA approvals were approximately 15% racemates, 47% single enantiomers, and 38% achiral. Agranat, et al., provide a review of the chiral switching strategy with many examples of well-known drugs, such as the aforementioned anti-gastric proton pump drug, omeprazole [10].
As marketing and patenting issues are addressed, the capabilities of the different enantiomers of a drug candidate must be considered in order to develop the best strategy for protecting intellectual property of the drug and extending the scope (patent life and content) of revenue generation from the drug. Racemic drugs will still be marketed but the trend is towards single enantiomer drugs due to regulatory pressures and advances in synthetic & manufacturing capabilities in dealing with chirality. The rationale in deciding whether to develop a racemic drug or a single enantiomer drug is evaluated as part of the drug approval process and so developers must prepare for this decision early in the drug discovery and development process.
2.3.2. The Chiral Drug Market
Many of the top selling drugs have been marketed as single enantiomer drugs, such as Fluticasone (GlaxoSmith-Kline) for respiratory therapeutics and Pravastatin (Bristol-Myers Squibb) for cardiovascular therapeutics [17]. As those authors demonstrated, there was a very definite change towards single enantiomer drugs in the 10 years following issuance of the FDA 1992 guidelines. Worldwide sales of single enantiomer drugs were reported as growing at 13% annually in 2000 and totally $133 billion. [19] The predictions were for $200 billion by 2008. Obtaining more recent sales data is difficult since it is proprietary and is the basis of sensitive strategic decisions made by individual pharmaceutical companies who are hesitant to share the data. However, this information serves to demonstrate the impact of chiral drugs on the pharmaceutical industry.
3. Practical Concerns of Stereochemistry in Early Drug Development
A common theme of the regulatory guidelines is that questions of stereochemistry should be addressed early in a drug discovery project. During the approval process, justifications will need to be provided with regards to the development of a racemic mixture or an enantiomerically pure compound. An analysis of the target protein's chiral selectivity can help guide this decision. Towards this end, crystallography and molecular modeling can play a key role in deducing this behavior.
Crystallography has been used to exam the effects of chirality in drug binding and from this three different scenarios have been proposed [20]. The commonly expected result would be for only one member of an enantiomeric pair to bind successfully into a site. Additionally, it is possible for the enantiomeric partners to share a binding site, or to adopt dramatically different binding poses that prevent the opposite partner from binding [20, 21]. Intriguingly, this same report demonstrates an extraordinary situation where both enantiomers can simultaneously occupy the same binding site [20]. While differences in binding can be slight, the effects on binding affinity can be dramatic. For example, when Fokkens, et at., examined the binding of their compound in thrombin using modeling while the (-) enantiomer showed only slight steric crowding in the binding site this seemed to account for a loss of greater than 800 fold binding affinity in the physical assay [21]. These types of events are discussed below.
Molecular modeling and virtual screening can serve well in the early stages of a drug discovery project with initial screening and de novo design. When pursuing molecular modeling, the investigator relies on the presence of 3D structures of the protein and of the compounds in question, either novel structures or existing libraries. However, as has been shown in crystallographic studies and physical screening studies the behavior of diasteriomers in their binding to protein targets can differ dramatically. These differences can be demonstrated in crystallography and modeling.
Of the two, physical screening and virtual screening, molecular modeling combined with virtual screening is easier to perform in the early stages of a drug discovery campaign and can nicely complement physical screening of compound libraries. Furthermore, virtual screening and modeling can be performed even if no lead compounds are known. This section will examine the effects of chirality on physical screening and then focus on the considerations of using virtual screening as a tool early in the drug discovery process.
3.1. Examination of Chiral Selectivity with Physical Methods
In some situations, an enzyme may be able to handle either enantiomer but use a quite different spatial means of accommodating each enantiomer. Mentel, et al. (2009) were able to obtain structures of PhzA/B enzyme from Burkholderia cepacia R18194 co-crystallized with either the R form alone, the S form alone, or with the racemate of a compound [20]. They found different residues were involved in the active site in docking the R form versus the S form. At saturating concentrations of the racemate, they found that both the R form and S form were accommodated in the active site, with the R form in the same pose as the R form alone and the S form adopting a different pose and location in the active site from the S form alone. KD values were in the low μM for the R form alone, S form alone, and racemate[20]. This suggests that either enantiomer or both could potentially serve as starting scaffolds for inhibitor development.
Aller, et al. (2009) also presented co-crystallization data in which an exporter protein, P-glycoprotein (Pgp), has sufficient conformational flexibility to accommodate a wide range of ligand sizes. It co-crystallized with one molecule of cyclic-tris-(R)-valineselenazole (QZ59-RRR) but co-crystallized with two molecules of the enantiomeric partner, cyclic-tris-(S)-valineselenazole (QZ59-SSS) [22]. And so either of the enantiomers could serve as the basis for drug development but with one (the S form) being more of a dimeric scaffold compared to the other. In those cases where both enantiomers bind the active site but with differing modes and/or affinities, we can expect to see experimentally a biphasic curve emerge as increasing concentrations of the racemate are used. Below the saturating concentration of the racemate, both enantiomers can bind freely. As the saturation concentration is approached, the stronger enantiomer will bind longer. The greater the difference between the enantiomers in their binding strength, the sooner the inflection point will be observed in the curve [21].
3.2. Examination of Chiral Selectivity with Computational Methods
In drug discovery programs virtual screening is maturing into a powerful tool [23]. Several software applications are available that will perform virtual screening including: GLIDE [24], Autodock [25], and Gold [26]. Essentially, virtual screening examines the geometric and charge “fit” of a small molecule for a designated binding site on a target protein. Once bound, an approximate free energy of binding can be calculated. When applied to a library of small molecules, the library can be reordered such that the “most likely” binding partners move to the top of the list. The result is an identified subset of the library that is enriched in potential binding partners for the protein. Virtual screening is also useful when physical screening is difficult, expensive, and/or inefficient for large libraries. One such example was the screening for S-adenosylmethionine decarboxylase (AdoMetDC) inhibitors [27]. Physical screening of AdoMetDC requires a radioactive assay, measuring release of radiolabeled CO2 as SAM (AdoMet) is converted to decarboxylated SAM. By using virtual screening, only a subset (133 selected from the top scoring 300 compounds) of the 1,990 that comprised the NCI Diversity set required physical screening and the results of this screen yielded an active compound [27]. This particular campaign demonstrated the efficacy and utility of virtual screening as a labor, cost, and time saving device in a drug discovery project.
Virtual screening projects, however, can provide misleading results if a library contains errors or does not fully represent the chemical space occupied by the individual molecules. For example, a single compound with potential ionizable groups (within a reasonable pH range) should be represented in a library as several different structures. Each of the structures would possess a different ionization state. A compound with a single carboxyl group, for example, that is substantially ionized near pH 7.0 should be represented in the library as the protonated and deprotonated forms of the compound. Likewise, tautomers should be considered as well as alternative ring conformations if appropriate. Of particular interest are stereoisomers. For the most part, compound library preparation programs (e.g. Schrödinger's Ligprep application [24], Corina [28], and Concord [29]) can enumerate alternative structures for ionization states, tautomers, and ring conformations. However, a question arises with regards to stereochemistry.
3.2.1. Characteristics of Chemical Structure Libraries
Due to the costs and difficulty of chiral selective synthesis, optically active compounds are often synthesized as racemates. When the relative stereochemistry of one compound is provided, the enantiomer is generally implied to be present in the mixture as well. However, compound structure databases frequently contain only one particular stereoisomer structure, if they contain any three-dimensional information at all. Commonly, library files are provided as MDL SDfiles [30]. These types of files may or may not contain 3D information. Even if the file does contain stereochemical information, that information is likely to be for one diastereomer and may not account for the diastereomers of the mixture found in the physical-compound library, assuming the sample is not enantiomerically pure. Certainly, any given entry in an SDfile cannot account for more than one structure. Furthermore in cases of molecules with multiple chiral centers, if stereochemical information is provided it should be considered relative stereochemical notation and not absolute stereochemical notation. The existence of the enantiomer is typically implied in such cases as the enantiomer is unlikely to be literally listed in the file and diastereomers may need to be added as well (discussed below).
For example, Fig. (2a) is a segment of the SDfile from the NCI Diversity Set II (http://dtpsearch.ncifcrf.gov/FTP/divii.sdf) database. This compound, NSC-479, possesses a single chiral carbon (denoted with * on the 2D structure). The file segment shows the X, Y, Z coordinates of the atoms and their type (first 4 columns) and the atom block (remaining 12 columns). The atom block contains codes for various characteristics as described in Fig. (3) of Dalby, et al.[30]. In particular, the atom stereo parity column (indicated in Fig. (2a, 2b, 2c) can have 1 of 4 values: i) 0 indicating not stereo, ii) 1 odd R configuration, iii) 2 even S configuration or iv) 3 indicating either odd or even or unmarked. In this case Fig. (2a), there is no Z coordinate and stereo information is missing (as shown in the figure).
Fig. (2).
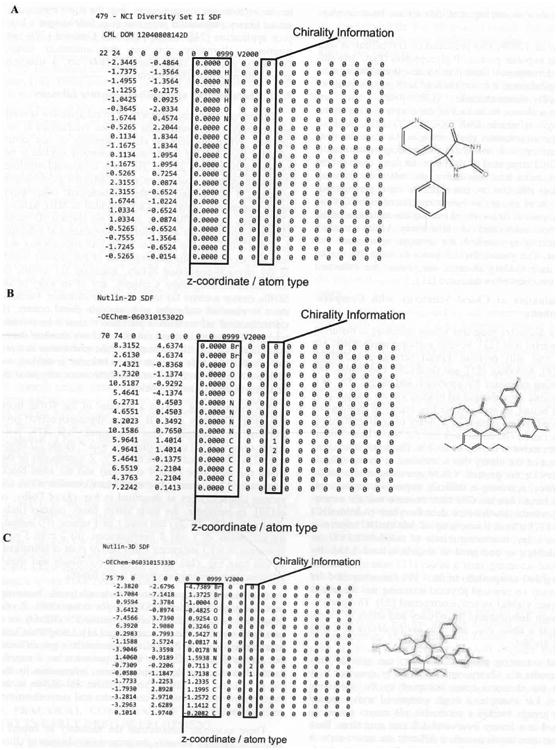
(a) Sample of the SDF for the NCI Diversity Set II. Compound 479 is a chiral compound. The SDFile for this compound contains no chiral information. The (*) denotes the chiral site. (b). 2D SDfile for Nutlin 2 from PubChem lacking Z coordinate information. Relative stereochemistry information is present in the atomic parameters block, (c). 3D sdf for Nutlin 2 from PubChem possessesing Z coordinate information. Relative stereochemistry information is present in the atomic parameters block.
Fig. (3).

a) Comparison of the docking pose of the R,S Nutlin (from crystal structure; grey carbons) to the enantiomer (S,R; blue). When seeded into the NCI Diversity Set II for docking, the original form of nutlin (R,S) ranks 51 and the enantiomer ranks 3,482 (out of 3,718 docked structures). GScores are -6.70 kcal/mol (R,S) vs -3.95 (S,R). RMSD = 7.07. (b) Line structure of R,S Nutlin. (c) Line structure of S,R Nutlin. (d) Comparison of the docking pose of the R,R Atorvastatin (from crystal structure; grey carbons) to the enantiomer (R,R; blue). When seeded into the NCI Diversity Set II for docking, the S,S Atorvastatin ranks 1 and the enantiomer ranks 2 (out of 3,718 docked structures). GScores are -8.80 kcal/mol (S,S) vs -8.25 (R,R). RMSD = 0.70. (e) Line structure of R,R Atorvastatin. (f) Line structure of S,S Atorvastatin. (g) Comparison of the docking pose of the R,S Sertraline (from crystal structure; grey carbons) to the enantiomer (S,R: blue). Wlien seeded into the NCI Diversity Set II for docking, the R,S Sertraline ranks 67 and the enantiomer ranks 407 (out of 3,879 docked structures). GScores are -7.05 kcal/mol (R,S) vs -6.29 (S,R). RMSD = 2.64. (h) Line structure of R,S Sertraline, (i) Line structure of S, R Sertraline.
PubChem (http://pubchem.ncbi.nlm.nih.gov/), however, provides slightly different SDfiles for compounds. Previously, we examined the effects of nutlin-2's chirality in a simulated virtual screening campaign [31]. Using PubChem as a source for nutlin-2's structure can result in two different but similar SDfiles. The 2D SDfile possesses no Z coordinate information but does contain stereo information for the chiral carbons Fig. (2b). The PubChem 3D SDfile is the most complete containing Z coordinates and stereochemistry information Fig. (2c).
These examples demonstrate the necessity to inspect a structural database to verify the presence or absence of chiral information. Given the large number of structural databases available from myriad sources and the lack of consistency of including or omitting stereochemistry information, investigators are well served to inspect databases prior to use in a virtual screening campaign. Furthermore, when chiral information is present, it is important to note that this information is not necessarily absolute chirality but should be considered relative chirality particularly in cases where a compound has multiple chiral centers. Even if the structure is known to contain absolute chirality the addition of enantiomeric partners to the library is prudent as the structures make ideal decoy molecules for enantioselective targets (discussed below).
3.2.2. Effects of Stereoisomers on Library Management
Lacking stereochemical information and alternative stereoisomers is not a short-coming of these databases. To include every possible member of a set of stereoisomers, required to thoroughly explore potential chiral space, increases the size of the library per molecule at a rate of 2n, where n is the number of stereocenters present in the molecule. In some cases, this can result in a tremendous increase in the size of the structure library. For example, the drug fluticasone furorate possesses nine chiral carbons [32]. Adding all possible stereoisomers for this molecule, could increase the number of structures to 512.
Although not every stereoisomer is necessarily chemically feasible this “brute force” approach to exploration of chiral space can result in dramatic increases in library size. The NCI Diversity Set II SDfile structure library contains 1,364 structures. If this library is prepped for docking (i.e. using Schrodinger's Ligprep application) and every chemically feasible structure is generated for every molecule the final structure count rises to 17,571. This breaks down as: 1,364 original structures (∼8% of the size of the new library), 652 structures representing alternate tautomers and ionization states (∼4% of the size of the new library), and 15,555 are alternative stereoisomers (∼88% of the size of the new library). In terms of data storage, a 7.0 megabyte file increases to 28.1 megabytes. In the same vein, preparing the fully enumerated file required 14 hours. Whereas the simple enumeration (no additional stereoisomers generated) required 23 minutes. Clearly, in larger libraries this problem will be more pronounced even if the specific increases in time required for preparation will vary between computers.
3.2.3. Effects of Including Enantiomers in Virtual Screening
Lacking a priori knowledge of which compounds would serve as actives from a library, a virtual screening campaign relies on a thorough exploration of the chemical space represented by the structure database. This includes alternate tautomers, ionization states, and stereoisomers. However thorough exploration of all the potential stereoisomers in a library can be prohibitive in terms of time, data storage, and library management. Thus, it is appropriate to find a “middle ground”. While the ideal situation is to include every feasible stereoisomer, analysis of the effects of stereoisomer structures on docking poses suggests that enantiomeric pairs are sufficient for exploration of chemical space [31]. The ability to include only enantiomeric pairs depends on the quality of the structure library [31]. If the library possesses no stereochemical information, then the “brute force” method will be needed to sufficiently explore chemical space. However, a library that possesses stereochemical information, albeit relative stereochemistry, is more suitable to enantiomeric extrapolation and confidence can be maintained that additional diastereomers are not relevant to the structure library. This provides a compromise between fully exploring chemical space and keeping structure libraries manageable in terms of numbers of structures, data storage, and time requirements for preparations and dockings.
Previously we reported that the docking of nutlin-2 to MDM2 was strongly dependent on which enantiomer was docked [31]. We examined nutlin-2 (PDB: 1RV1, [33]) again along with two other top selling chiral drugs available on the market: Atorvastatin (PDB: 1HWK, [34]) and Sertraline (PDB: 3GWU, [35]). These compounds and their enantiomers give a variety of outcomes based on the degree to which the chiral carbon(s) affects the overall geometry of the compound.
3.2.4. Nutlin and Enantiomer Docking to MDM2
R,S nutlin and the enantiomer S,R nutlin were seeded into the NCI Diversity Set II structure library file and docked to the MDM2 structure with which they were originally co-crystalized Fig. (3a-c). After docking, consistent with earlier observations, the R,S nutlin (the co-crystalized form) docked at rank 51, whereas the enantiomer docked at rank 3,482 out of 3,915 structures. Likewise, the estimated binding energy worsened from -6.70 kcal/mol to -3.95 kcal/mol. The RMSD between the two docking poses was 7.07. This coincides with what was previously reported and shows that when chirality has a strong influence on the geometry of the molecule the docking pose will vary dramatically between enantiomers [31].
3.2.5. Atorvastatin and Enantiomer Docking to HMG-CoA Reductase
R,R Atorvastatin and the enantiomer S,S Atorvastatin were seeded into the NCI Diversity Set II structure library file and docked to the HMG-CoA Reductase structure with which they were originally co-crystallized. Atorvastatin Fig. (3d-f) differs from nutlin in that the chiral carbons are in a highly flexible chain as opposed to a ring system Fig. (3a-c) vs. Fig. (3d-f). This suggests that geometric changes caused by a change in chirality could be absorbed due to the flexibility of the chain, which bears out in docking results. R,R Atorvastatin and S,S Atorvastatin bind very similarly to HMG-CoA Reductase with essentially no increase in rank or energy (Rank 1 vs. Rank 2 and -8.80 kcal/mol vs. -8.25 kcal/mol respectively). Additionally, the docking poses are almost identical with an RMSD of 0.70 Fig. (3d).
3.2.6. Sertraline and Enantiomer Docking to LeucineTransporter
R,S Sertraline and the enantiomer S,R Sertraline were seeded into the NCI Diversity Set II structure library file and docked to the leucine transporter structure with which they were originally co-crystalized Fig. (3g-i). In this case, the chiral carbon has some influence on the overall geometry of the molecule and its resulting docking pose. While the pose of the enantiomer is similar to that of the co-crystalized compound (RMSD = 2.64) and the estimated free energy of binding is not very different (-7.25 kcal/mol versus -6.29 kcal/mol) the ranking within the library is dramatically changed. R,S Sertraline comes in at rank 67 (out of 3,879 docked structures) while the S,R enantiomer ranks 407. This demonstrates that a moderate difference in binding pose due to chirality can result in dramatic ranking shifts within a library.
These observations provide insight into the behavior of enantiomeric pairs within a structure library during docking. Clearly, there is no hard and fast rule to how an enantiomeric pair will behave. It is equally clear that when a chiral atom influences a compound's geometry or binding pose (i.e. these changes are not simply “absorbed” by a flexible chain) that the results for each member of the pair can be dramatically different. Thus, important lead compounds can be missed as false negatives if the appropriate enantiomer was not included in the structure library. Additionally, even when absolute stereochemistry is known and the target is enantioselective then enantiomers serve as excellent decoy molecules during docking and can be added to a structure database such as the Directory of Useful Decoys [36].
3.2.7. Generation of Enantiomeric Pairs for Structure Library Files
Computational software is likely to either create all possible stereoisomers (or some user imposed limit thereof) or to generate structures based on the chiral notations in the input structure file. For example, Ligprep from Schrödinger [24] can “retain specified chiralities”, “determine chiralities from 3D structure”, or “generate all combinations” (with a user limit added to the total number of stereoisomers generated). However, none of these is going to give only the enantiomeric partner of a compound. Using PubChem's nutlin SDfiles as an example gives the results outlined in Table 1. As is apparent, Ligprep will either generate a single structure based on the chiral information from the SDfile or will generate all possible stereoisomers. Thus, a specific method of generating only enantiomeric partners should be employed to avoid the complications of library growth and time requirements of the docking simulation as discussed in section 2.2.2.
Table I. Structures Returned When Preparing Nutlin for Docking Based on Chirality Handling in Ligprep.
| File Type | Chirality Handing Style | Structure |
|---|---|---|
| 2D SDF | Retain Specified Chiralities | 1 |
| 3D SDF | Retain Specified Chiralities | 1 |
| 2D SDF | Determine Chiralities | 4 |
| 3D SDF | Determine Chiralities | 1 |
| 2D SDF | Generate All Combinations | 4 |
| 3D SDF | Generate All Combinations | 4 |
3.2.8. Specifically Generating Enantiomeric Partners to Compounds with Chiral Centers
If a file of small molecule 3D structures lacks the enantiomeric partners for the chiral molecules, the missing structures can be generated by creating a fairly simple script or program to parse through the file, identify each chiral structure, and then output a copy of that structure to a second file, multiplying the Z coordinate by -1, as shown in Fig. (4). The two files can then be concatenated if desired to create a single file with both stereo enantiomers represented. Each situation will be dependent on the type of file format involved and the programming ability of the user.
Fig. (4).
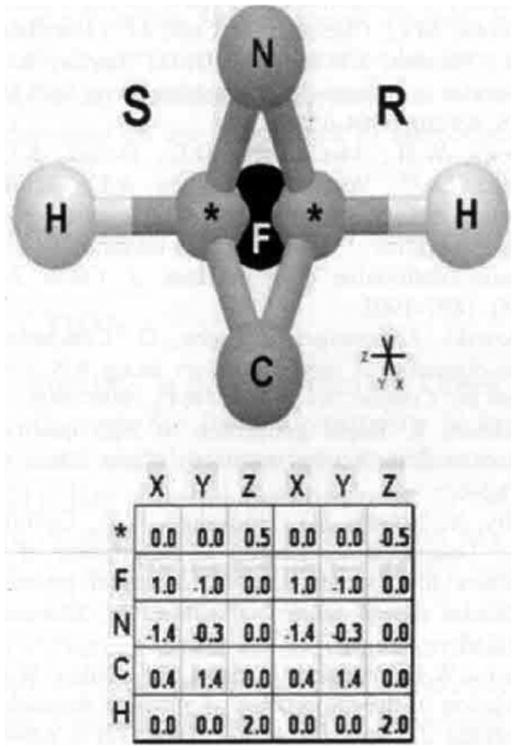
Generating a stereo enantiomer. If the coordinates for one chiral molecule are available, the other stereo enantiomer can be created by simply multiplying the Z coordinate by -1. When three of the four substituent first-level atoms of a chiral center are superimposed, it simplifies the relationship between the enantiomers such that you only need to multiple the Z coordinate of every atom by -1 to generate the missing stereo enantiomer. The relation is then ZS = |-ZR| for each atom.
As an example, we generated enantiomers for the NCI Diversity Set I, which contains 1,990 compounds of which 806 had chiral centers [31]. We first ran the file through Schrödinger's LigPrep to generate the different tautomers, ring conformations, and ionization states, which increased the number of structures to 2,392. This resulted in a file in Schrödinger's Maestro format (.mae extension). Stereo enantiomers were then created from the LigPrep processed file using a Perl script that we coded. This script parsed through each record of a structure to interrogate a field that flagged chirality. Of the 806 structures extracted from the NCI Diversity Set, 794 were successfully converted into the enantiomeric partner. The structures that failed this conversion we believe were large meso-compounds that presented difficulties for our simple PERL script to handle. As meso-compounds they have a symmetry which means that, inspite of having chiral centers, the two partners are superimposable and, therefore, would rank the same in virtual docking. The purpose of the exercise was to compare rankings when the enantiomeric partners differed. The successfully generated isomers were appended to the 2,392 NCI Diversity Set structures in the LigPrep file. This comprehensive file was then used in Schrödinger's Glide to dock and score the structures [31]. The purpose of our work was to study and report the differences in Glide Score rankings that can occur between enantiomers. We found that, in our example, approximately 25% of potential leads may be missed if the enantiomeric partners were not considered.
3. Conclusions
Chirality in drug discovery and development has increased in importance since guidelines for chiral compounds in drug development and approval were issued by regulatory agencies in the 1992-2000 timeframe. The guidelines were issued due to the potential differing activities of enantiomers in chiral compounds. Although enantiomers will have the same properties in achiral environments, the effects can be quite different in biological environments where enzymes and receptors can be chirally selective. In some cases, the presence of the distomer in a racemic mixture can affect the results due to detrimental effects of the distomer or its conversion to the eutomer configuration. The composition of the racemic mixture and its potential to change with time or depending on the system and tests (ex. animal studies versus clinical trials) meant that the more active enantiomer in a single enantiomer drug would be a better option in many cases if scientifically and technically possible.
As a result of the guidelines and regulations, the onus is on the investigators initiating drug discovery projects to determine early in the project whether to pursue racemates or enantiomerically pure compounds; with emphasis on enantiomerically pure compounds. To this end there are several important considerations in the early stages of a drug discovery project. While, crystallography is the gold standard for examining protein structure and compound binding there are limitations and costs associated with it. For example, there is a need for a relatively potent compound in order to achieve co-crystallization. A potent relevant compound may not be available in the early stages. As a result, very powerful tools available for lead discovery and refinement are virtual screening and molecular modeling.
Virtual screening and molecular modeling methods enable the rapid identification of lead compounds using calculated free energies of binding. However, due to the vagaries of diastereomers and their binding modes it is vital to start with accurate stereochemical depictions of compounds in virtual compound libraries used as an initial step in identifying potential drug candidates. Ideally, investigators should incorporate every possible stereoisomer in their databases. However, realistically this can lead to significant increases in time and management issues for those databases. Alternatively, if the original compound database does contain stereochemical information, then generation of enantiomeric partners is sufficient for exploration of chemical space based on the library and for including decoy molecules. In many cases, missing enantiomers can be generated with a minimal programming effort by a drug discovery team. The underlying key to virtual screening is to balance exploration of chemical space, data storage, and time needed to screen.
All of these issues from biological, intellectual property, regulatory, and laboratory (both virtual and physical) development strategies underscore the importance of considering stereochemistry, in gereral, and chirality, in particular, before embarking on and during a drug discovery campaign.
References
- 1.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein databank. Nucleic Acids Res. 2000;28, I:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grant MA. Protein structure prediction in structure-based ligand design and virtual screening. Comb Chem High Throughput Screen. 2009;72(10):940–960. doi: 10.2174/138620709789824718. [DOI] [PubMed] [Google Scholar]
- 3.Administration F.a.D. Development of new stereoisomeric drugs. 2005 Jul 6; [cited 2010 April 23]; www.fda.gov/Drugs/GuidanceComplianceRegulatorylnformation/Guidances/ucm122883.htm.
- 4.Nic M, Jirat J, Kosata B. IUPAC Gold Book Compendium of chemical terminology. 2005 [cited 2010 April 26]; http://goldbook.iupac.org.
- 5.Caldwell J, Wainer IW. Stereochemistry: definitions and a note on nomenclature. Hum Psvchopharmacol. 2001;16(S2):SI05–S107. doi: 10.1002/hup.334. [DOI] [PubMed] [Google Scholar]
- 6.Sahajwalla C. In: Regulatory Considerations in Drug Development of Stereoisomers, Chiralitv in Drug Design and Development. Reddy I, Mehvar R, editors. CRC Press; New York: 2004. [Google Scholar]
- 7.Lee EJ, Williams K, Day R, Graham G, Champion D. Stereoselective disposition of ibuprofen enantiomers in man. Br J Clin Pharmacol. 1985;79(5):669–674. doi: 10.1111/j.1365-2125.1985.tb02694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hardikar M. Chiral non-steroidal anti-inflammatory drugs – a review. J Indian Med Assoc. 2008;106:615–624. [PubMed] [Google Scholar]
- 9.Rouhi A. Chirality at work. Chem Engin News. 2003;81:56–61. [Google Scholar]
- 10.Agranat I, Caner H, Caldwell J. Putting chirality to work: the strategy of chiral switches. Nat Rev Drug Discov. 2002;7(10):753–768. doi: 10.1038/nrd915. [DOI] [PubMed] [Google Scholar]
- 11.Gupta H, Kumar S, Roy S, Gaud RS. Patent protection strategies. J Pharm BioAllied Sci. 2010;2(1):2–7. doi: 10.4103/0975-7406.62694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faget K. Why FDCA Section 505(u) Should not concern us greatly. Mich Telecomm Tech L Rev. 2009:453–466. [Google Scholar]
- 13.Yoshitani R, Cooper E. Pharmaceutical reformulation: the growth of life cycle management. Houston J Health Law Policv. 2007;7:379–410. [Google Scholar]
- 14.Agency EM. Investigation of chiral active substances. 1994 [cited 2010 April 25]; www.ema.europa.eu/pdfs/human/qwp/3cc29aeu.pdf.
- 15.Health Canada. Therapeutic Products Programme, Health Canada. 2007
- 16.Harmonization, I.C.o. Specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances, Q6A, Step 4. 1999 [cited 2010 April 25]; www.ich.org/cache/compo/363-272-1.html#Q6A. [PubMed]
- 17.Caner H, Groner E, Levy L. Trends in the development of chiral drugs. Drug Discov Today. 2004;9:105–110. doi: 10.1016/s1359-6446(03)02904-0. [DOI] [PubMed] [Google Scholar]
- 18.AMA. Geometric isomerism and chirality: the USAN perspective. 1995 [cited 2010 April 25]; www.ama-assn.org/ama/pub/about-ama/our-people/coalitions-consortiums/united-states-adopted-names-council/name-guidelines/geometric-isomerism-chirality-the-usan-perspective.shtmI.
- 19.Stinson S. Chiral drugs. Chem Eng News. 2000;78:55–78. [Google Scholar]
- 20.Mentel M, Blankenfeldt W, Breinbauer R. The active site of an enzyme can host both enantiomers of a racemic ligand simultaneously. Angew Chem Int Ed Engl. 2009;48:9084–9087. doi: 10.1002/anie.200902997. [DOI] [PubMed] [Google Scholar]
- 21.Fokkens J, Klebe G. A simple protocol to estimate differences in protein binding affinity for enantiomers without prior resolution of racemates. Angew Chem Int Ed Engl. 2006;45(6):985–989. doi: 10.1002/anie.200502302. [DOI] [PubMed] [Google Scholar]
- 22.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323(5922):1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh S, Nie A, An J, Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr Opin Chem Biol. 2006;70(3):194–202. doi: 10.1016/j.cbpa.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Schrodinger. Schrodinger Software Suite. Schrodinger LLC; New York: 2006. [Google Scholar]
- 25.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;50(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verdonk ML, Chessari G, Cole JC, Hartshorn MJ, Murray CW, Nissink JW, Taylor RD, Taylor R. Modeling water molecules in protein-ligand docking using GOLD. J Med Chem. 2005;48(20):6504–6515. doi: 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
- 27.Brooks WH, Mc Closkey DE, Daniel KG, Ealick SE, Secrist JA, 3rd, Waud WR, Pegg AE, Guida WC. In silico chemical library screening and experimental validation of a novel 9-aminoacridine based lead-inhibitor of human S-adenosylmethionine decarboxylase. J Chem Inf Model. 2007;47(5):1897–1905. doi: 10.1021/ci700005t. [DOI] [PubMed] [Google Scholar]
- 28.Sadowski J, Gasteiger J, Klebe G. Comparison of automatic three-dimensional model builders using 639 x-ray structures. H Chem Inf Comput Sci. 1994;54(4):1000–1008. [Google Scholar]
- 29.Pearlman R. Rapid generation of high quality approximate 3-dimensional molecular structures. Chem Des Auto News. 1987;2(1):5–6. [Google Scholar]
- 30.Dalby A, Nourse JG, Hounshell WD, Gushurst AKI, Grier DL, Leland BA, Laufer J. Description of several chemical structure file formats used by computer programs developed at molecular design limited. J Chem Inf Comput Sci. 1992;32:244–255. [Google Scholar]
- 31.Brooks WH, Daniel KG, Sung SS, Guida WC. Computational validation of the importance of absolute stereochemistry in virtual screening. J Chem Inf Model. 2008;48(3):639–645. doi: 10.1021/ci700358r. [DOI] [PubMed] [Google Scholar]
- 32.Biggadike K, Bledsoe RK, Hassell AM, Kirk BE, McLay IM, Shewchuk LM, Stewart EL. X-ray crystal structure of the novel enhanced-affinity glucocorticoid agonist fluticasone furoate in the glucocorticoid receptor-ligand binding domain. J Med Chem. 2008;57(12):3349–3352. doi: 10.1021/jm800279t. [DOI] [PubMed] [Google Scholar]
- 33.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammloft U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 34.Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292(5519):1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith ME, Wang DN. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat Struct Mol Biol. 2009;76(6):652–657. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shoichet BK. DUD - A Directory of Useful Decoys. 2010 Apr 20th; http://dud.docking.org/