

Abstract
Background
The role of low dose methotrexate (LDM) in potential serious toxicities remains unclear despite its common use. Prior observational studies investigating LDM toxicity compared LDM to other active drugs. Prior placebo-controlled clinical trials of LDM in inflammatory conditions were not large enough to investigate toxicity. The Cardiovascular Inflammation Reduction Trial (CIRT) is an ongoing NIH-funded, randomized, double-blind, placebo controlled trial of LDM in the secondary prevention of cardiovascular disease. We describe here the rationale and design of the CIRT-Adverse Events (CIRT-AE) ancillary study which aims to investigate adverse events within CIRT.
Design
CIRT will randomize up to 7,000 participants with cardiovascular disease and no systemic rheumatic disease to either LDM (target dose 15–20 mg/week) or placebo for an average follow-up period of 3–5 years; subjects in both treatment arms receive folic acid 1 mg daily for six days each week. The primary endpoints of CIRT include recurrent vascular events, incident diabetes, and all-cause mortality, and the ancillary CIRT-AE study has been designed to adjudicate other clinically important adverse events including hepatic, gastrointestinal, respiratory, hematologic, infectious, mucocutaneous, oncologic, renal, neurologic, and musculoskeletal outcomes. Methotrexate polyglutamate levels and genome-wide single nucleotide polymorphisms will be examined for association with adverse events.
Summary
CIRT-AE will comprehensively evaluate potential LDM toxicities among subjects with cardiovascular disease within the context of a large, ongoing, double-blind, placebo-controlled trial. This information may lead to a personalized approach to monitoring LDM in clinical practice.
Keywords: methotrexate, toxicity, pharmacovigilance, genetics, polyglutamate
INTRODUCTION
Low dose methotrexate (LDM) is the most commonly prescribed disease-modifying anti-rheumatic drug (DMARD) in rheumatoid arthritis (RA), used by 4–7 million people worldwide, with typical doses ranging from 10 to 25 mg weekly1,2. LDM is considered the standard of care for moderate to severe RA and has remained a cornerstone of treatment even in the era of targeted biologic DMARDs3. LDM use reduces symptoms, prevents joint damage, delays disease progression, works synergistically with biologic DMARDs, and may reduce mortality in RA1,4–8. LDM is also commonly used in other systemic rheumatic diseases, such as systemic lupus erythematosus, psoriatic arthritis, idiopathic inflammatory myopathies, polymyalgia rheumatica, and systemic vasculitides9–11.
While LDM has been in wide clinical use for more than 30 years, no randomized trial of adequate sample size has compared LDM to placebo and therefore available data on its toxicity are sparse and primarily derived from observational studies. Previous observational studies of LDM toxicities were limited by small sample size and short follow-up, lack of a well-defined control group, potential confounding from indication, and lack of adequate control for concurrent medications or systemic rheumatic disease activity. Many earlier studies investigating LDM toxicities were performed prior to routine folic acid supplementation, which is known to mitigate side effects, so findings may not be relevant to the current standard of care. Moreover, prior work often grouped all adverse events together; use of a heterogeneous outcome limits the identification of distinct genetic or clinical risk factors for toxicity. Due to these previous limitations, a large double-blinded randomized controlled trial with lengthy follow-up and adjudicated adverse event outcomes is necessary to investigate the causal effect of LDM use on toxicities.
In May of 2013, the Cardiovascular Inflammation Reduction Trial (CIRT) began randomizing patients with known atherosclerotic disease into a multi-national, double-blinded, placebo-controlled trial addressing whether anti-inflammatory therapy with LDM might reduce recurrent cardiovascular event rates9. This ongoing trial provides the unique opportunity to carefully assess the incidence rates of a wide variety of adverse events potentially associated with LDM use in a contemporary and non-confounded setting.
Previous Literature on Low Dose Methotrexate Toxicities
LDM has been associated with toxicities in previous studies (Table 1)12–15. Since LDM has clearly demonstrated efficacy as the initial treatment for many rheumatic diseases, a large randomized placebo-controlled trial within this patient population would currently be unethical. Beyond that barrier, the large number of subjects and length of follow-up needed to investigate toxicity using a clinical trial would be infeasible to pursue within a rheumatic disease population. Therefore, most previous investigations of LDM toxicity were performed using observational study designs, so may have the typical limitations of being underpowered to find a true association and possible residual confounding explaining reported associations. Despite these limitations, the prior observational studies have been instrumental in guiding the clinical care for patients with rheumatic diseases prescribed LDM for the past three decades. While much is already known about LDM toxicity, using a large placebo-controlled randomized controlled trial of LDM may identify novel toxicities, quantify the risk for known toxicities, and identify risk factors for these toxicities to further optimize the treatment of patients with rheumatic diseases.
Table 1.
Overview of common low dose methotrexate toxicities, their features, prevalence, and risk factors in prior literature.
| Toxicity | Feature | Prevalence | Selected risk factors | References |
|---|---|---|---|---|
| Hepatic | Above 1x ULN Above 2x ULN Persistent elevations |
20–49% 1–17% 0.1% |
Alcohol consumption, lack of folate supplementation, obesity, older age, history of liver disorder, diabetes and/or metabolic syndrome | 13,22,23 |
| Pulmonary | Interstitial pneumonitis | 0.43–4.5% | Older age, diabetes, prior pleuropulmonary disease, hypoalbuminemia | 13,27–34 |
| Hematologic | Cytopenia | 5.2% | Low RBC folate, elevated mean corpuscular volume due to folate depletion, impaired renal function, hypoalbuminemia, infection | 13,42,72,73 |
| Infection | Pneumonia Any severe infection |
2% 3% |
Older age, diabetes, advanced age, corticosteroids | 36,38,74 |
| Mucocutaneous | Stomatitis Nausea Rash |
2–14% 5–31% 8.9% |
Higher doses of methotrexate | 47,48,75 |
Most of these studies were conducted before routine folic acid supplementation was standard of care.
ULN, upper limit of normal; RA, rheumatoid arthritis; RBC, red blood cell.
Gastrointestinal toxicities are most frequent, with a prevalence of 20–65%, and are usually characterized by mild or moderate severity (e.g., nausea, vomiting, diarrhea, abdominal pain)13,16–19. Liver toxicity is a common concern and up to 20% of LDM users are estimated to have at least one episode of elevated serum transaminases, and 3.7% discontinue LDM due to liver toxicity13,20–26.
Interstitial pneumonitis represents the most serious respiratory toxicity for LDM, presenting as an acute hypersensitivity reaction with cough, fever, and subacute dyspnea, with mortality rates estimated up to 17%27,28. LDM-induced pneumonitis is relatively rare with an estimated prevalence of 0.43% and up to 4.5% for less severe manifestations such as pleuritis and cough13,27–34. Pneumonitis typically occurs within the first year of treatment and occurs more frequently in patients with pre-existing lung disease13,28,29,35.
Serious infections, including pneumonia and septic arthritis, may be more common in patients with RA treated with LDM compared to non-use, although these findings may be confounded by indication36–38. Since studies performed in large administrative datasets have used LDM as the active comparator, the risk due to LDM compared to placebo is unclear39.
Pancytopenia is reported in up to 1.4% of patients on LDM and can be fatal, but most studies investigating LDM and myelosuppression were performed prior to the routine use of folic acid supplementation15,40–46. Mucocutaneous toxicities of LDM may occur commonly and include stomatitis, oral ulceration, alopecia, and rashes47,48. The risk of lymphoproliferative disorders is increased among RA patients compared to the general population, primarily in the setting of high disease activity and some have ascribed this risk to LDM, although other RA-specific factors may also contribute13,49–52.
Context for Cardiovascular Inflammation Reduction Trial Adverse Events (CIRT-AE)
CIRT is a randomized, double-blinded, placebo-controlled, multi-center trial investigating whether direct inhibition of inflammation with LDM reduces rates of cardiovascular events and mortality among subjects with stable cardiovascular disease (ClinicalTrials.gov number NCT01594333)9. The primary aim of CIRT is to evaluate whether LDM (target dose 15–20 mg/week) reduces rates of recurrent non-fatal myocardial infarction, non-fatal stroke, and cardiovascular death among up to 7,000 men and women who have known coronary atherosclerosis (defined as a history of prior myocardial infarction or multi-vessel coronary disease) and who are at increased inflammatory risk due to the presence of either type 2 diabetes or metabolic syndrome. Potential participants with systemic inflammatory diseases or contraindications to LDM are not eligible. Subjects in both the LDM and placebo arms of CIRT receive folic acid 1 mg daily for six days per week. Prior to randomization, all subjects in CIRT undergo a 5–6 (maximum of 8) weeks open-label run-in period in which LDM dosing is increased from 5 to 15 mg/week. At randomization, subjects are assigned to receive 15 mg/week of either LDM or placebo. If this dose is tolerated for 4 months, the dose of study drug is increased to 20 mg/week. Estimated mean follow-up is planned for 3 years. Full details on the inclusion/exclusion criteria and study design of CIRT are described elsewhere (characteristics assessed in CIRT are listed in Table 2)53.
Table 2.
Covariates assessed in CIRT at baseline and/or during follow-up period.
| Category | Covariates |
|---|---|
| Sociodemographic |
|
| Clinical |
|
| Laboratory and Imaging Studies |
|
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; CBC, complete blood count, Cr, creatinine; EKG, electrocardiogram; HbA1c, hemoglobin A1c; hsCRP, high sensitivity C-reactive protein.
Herein, we describe the design and novel aspects utilized in CIRT-AE to prospectively identify, adjudicate, and classify adverse events (AEs) that occur both during the open-label run-in phase and in the post-randomization double-blind phase of CIRT. While AEs are standard to report in clinical trials, systematic and stringent adjudication and classification processes have not been typically pursued. CIRT-AE is positioned to efficiently determine whether LDM AEs occur above the background rate in this population at risk for cardiovascular disease by comparing to the placebo arm. We define AEs as the clinical outcomes occurring during CIRT follow-up and toxicities as the excess of AEs occurring in the LDM arm above the rate in the placebo arm in analyses. We detail analyses using CIRT-AE data to establish LDM toxicities as well as studies investigating the role of genetic polymorphisms and methotrexate polyglutamate levels in LDM toxicity.
STUDY DESIGN
Identification and Classification of Adverse Events
Subjects in CIRT undergo laboratory monitoring according to the American College of Rheumatology guidelines, including monitoring of the complete blood count, liver function tests, and serum creatinine every four weeks until a stable dosage is achieved, and then every eight weeks thereafter3. At the time of each laboratory test and follow-up, subjects complete a symptom checklist including dyspnea, cough, fever, nausea, abdominal pain, mouth pain, and hair loss. If no potential symptoms are reported and all laboratory results are within normal range, subjects continue study drug at doses guided by an automated titration algorithm developed by the CIRT investigators. The titration algorithm adjusts the dose of study drug based on pre-specified laboratory and clinical conditions (Figure 1)9. Subjects and sites do not have access to results of safety laboratory tests. To ensure consistent dose titration across all study sites and maintain blinding, subjects assigned to placebo also undergo temporary stops or dose reductions even with normal safety laboratory values and no clinical signs or symptoms of toxicity (i.e., “sham” dose changes). AEs are further categorized as serious adverse events (SAEs), defined as: death (also a primary endpoint in CIRT), all emergency department visits/inpatient admissions, life-threatening, resulting in disability/permanent injury, and those deemed serious by the site principal investigator.
Figure 1.

Overview of the titration algorithm, used to provide study drug dosing recommendations for subjects enrolled in the Cardiovascular Inflammation Reduction Trial (CIRT)53.
In addition to interim analyses of rates of the primary composite outcome, safety in CIRT is overseen by a fully independent Data and Safety Monitoring Board (DSMB). The DSMB has pre-specified interim analyses to evaluate early efficacy and futility9. To assist in the safe use of LDM, a team of rheumatologists at Brigham and Women’s Hospital in Boston, MA (currently JAS, MB, SYR, and DHS) who commonly prescribe LDM for use in systemic rheumatic diseases serve as CIRT medical monitors. Medical monitors in CIRT have access to individual participant laboratory results, clinical symptoms, and study drug dosage. If symptoms are reported and/or safety laboratory values are out of range on multiple occasions or critical values are reached, a medical monitor is alerted for manual review. Medical monitors review all SAEs and contact the site and data coordinating center to obtain further clinical information and communicate decisions about the use of study drug. For example, medical monitors recommend temporary stop of study drug for active infections, while on antibiotics, during hospitalizations, and during times of clinical instability. For some AEs, medical monitors recommend permanent stop of study drug. Examples of scenarios warranting permanent stop of study drug include new diagnosis of malignancy (except for resected non-melanoma skin cancers and other cancers considered cured after discussion with treating providers), cirrhosis, and systemic rheumatic disease or other inflammatory disorder where LDM may be clinically indicated. Medical monitors offer advice on the management of alopecia, nausea, or mucositis, all of which may necessitate higher doses of folic acid or dose reduction of study drug.
The process for identifying, adjudicating, and classifying AEs in CIRT-AE beyond mechanisms in place for CIRT is shown in Figure 2. Data on AEs are collected at every study visit in CIRT. All AEs are reported from sites to the data coordinating center and medical monitors. Sites collect all relevant medical records for AEs and submit them to site liaisons for further use in CIRT-AE or for use by the medical monitors in decisions about study drug.
Figure 2.
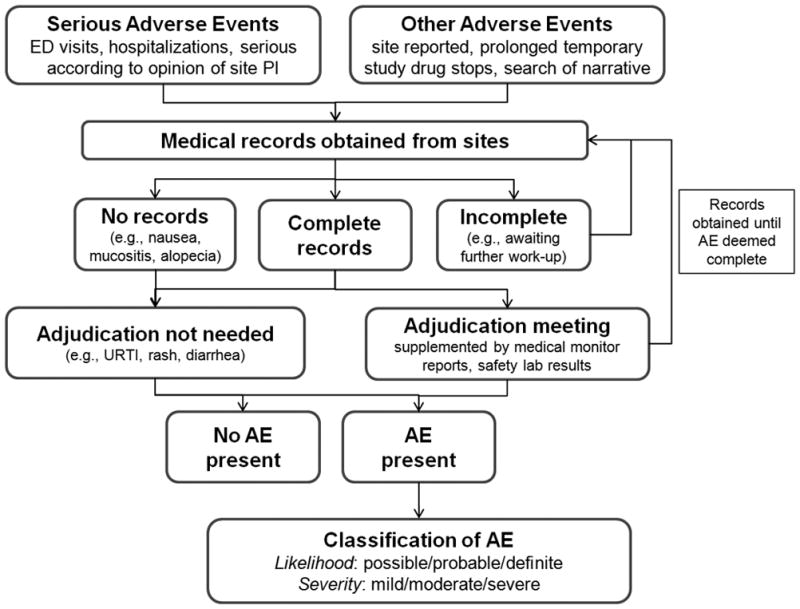
Identification, adjudication, and classification of adverse events in CIRT-AE. AE, adverse event; ED, emergency department; URTI, upper respiratory tract infection.
Sites provide medical records for all SAEs in CIRT, but CIRT-AE identifies other potentially important AEs that are not captured through the SAE mechanism. All subjects in CIRT are prompted to report potential adverse events including infections and antibiotic use. Site coordinators enter text describing the clinical status of subjects that may include AEs such as non-hospitalized infections (e.g., cellulitis or urinary tract, sinus, or upper respiratory tract infections treated in the outpatient setting), rash, stomatitis, gastrointestinal symptoms (including nausea, vomiting, or diarrhea), bleeding events, cancer, hepatic, alopecia, hematologic abnormalities, renal impairment, and neuromuscular symptoms. CIRT-AE staff regularly perform searches for terms relevant to these symptoms and screen each positive search for whether medical records should be pursued. For some AEs that are entirely subjective (e.g., nausea and alopecia), further records are not pursued and the description in the text is sufficient to classify these AEs. In addition, subjects on prolonged temporary study drug discontinuation are routinely queried to screen for the potential contribution of AEs. Even after permanent study drug discontinuation or primary endpoint occurrence, CIRT follows all participants for possible AEs until the conclusion of the trial. Once all relevant medical records have been collected, CIRT-AE staff systematically document events and prepare medical records for expert adjudication to further document and classify AEs.
Adjudication and Classification of Adverse Events in CIRT-AE
Medical records for AEs are adjudicated using standardized forms for hepatic, gastrointestinal, respiratory, hematologic, infectious, mucocutaneous, oncologic, renal, neurologic, and musculoskeletal events (included as Supplemental Material). Adjudication meetings include the principal investigator of CIRT-AE (DHS) and a subspecialty clinical expert except for mucocutaneous, neurologic, musculoskeletal, non-hepatic gastrointestinal, and renal AEs which are adjudicated and classified solely by DHS. In general, after reviewing all available medical records, AEs are classified by likelihood (i.e. possible, probable, or definite) as well as by severity (i.e. mild, moderate, or severe). More records are sought if the AE is not classifiable with the available records. Safety monitoring laboratory studies and study visit notes including medical monitor, site, and site liaison interactions are available for review to supplement records that were obtained from sites. Laboratory abnormalities that did not prompt further clinical work-up are not further adjudicated. When applicable, validated criteria are used to classify events54–57. In addition, details such as hospitalization, antimicrobial use, pertinent laboratory value results, pathologic diagnoses, imaging abnormalities, microbiologic results, symptoms, and dates of onset/diagnosis are systematically determined during this process. Some AEs may be categorized into multiple categories. For example, pneumonia is categorized in both pulmonary and infection categories to investigate the possible association of LDM assignment with each of these outcomes. For analyses investigating overall event rates, an AE such as this will only be counted once. Adjudicators are blinded to treatment assignment for post-randomization events.
PLANNED ANALYSES
Table 3 lists the planned analyses based on CIRT-AE. Broadly, CIRT-AE will pursue five general areas for investigation.
Table 3.
Analyses planned in CIRT-AE.
| Incidence rates and relative risk (LDM vs. placebo) of AEs to establish LDM toxicity | ||
|---|---|---|
| Exposures | Outcomes | Statistical analyses |
| Study drug assignment: Low dose methotrexate (LDM) Placebo (reference group) |
Any AE Severe AE Hepatic Infectious Pulmonary Hematologic Mucocutaneous Cancer Hemorrhagic Neurologic Musculoskeletal Renal Gastrointestinal Other systems and subgroups of categories pending sufficient numbers of outcomes |
-Cumulative incidence rates with 95% confidence intervals -Kaplan-Meier curves with log-rank tests -Cox proportional hazards model for first event analyses (reference: placebo) -Poisson models for number of event analyses (reference: placebo) |
| Risk factors for AEs | ||
| Exposures | Outcomes | Statistical analyses |
| Sociodemographics Comorbidities Cigarette smoking Alcohol intake Body mass index Hyperlipidemia Concurrent medications Baseline hepatic, renal, or pulmonary disease Genetics MTXglu level* Others pending sufficient numbers of outcomes in relevant exposure categories |
Any AE Severe AE Hepatic Infectious Pulmonary Hematologic Mucocutaneous Cancer Hemorrhagic Neurologic Musculoskeletal Renal Gastrointestinal MTXglu level* Other systems and subgroups of categories pending sufficient numbers of outcomes |
-Descriptive baseline analyses comparing those with toxicity to those without -Cox regression models for first event analyses -Poisson models for number of event analyses -Genome-wide association study controlling for population stratification by principal components -Investigation of candidate genes associated with LDM toxicity in prior studies -LDM-placebo interaction analyses if association found among both LDM and placebo arms -Mediation analyses for risk factors associated with both MTXglu levels and toxicity outcomes |
| Risk prediction rules for AEs | ||
| Development and validation | Outcomes | Statistical analyses |
| -Development group: two-third of LDM arm -Validation group: one-third of LDM arm -Predictors associated with AEs will be considered |
Any AE Severe AE Hepatic Infectious Pulmonary Hematologic Mucocutaneous Cancer Hemorrhagic Neurologic Musculoskeletal Renal Gastrointestinal Other systems and subgroups of categories pending sufficient numbers of outcomes |
-Exposure-treatment interactions to evaluate heterogeneity of treatment effect -Goodness-of-fit of models: Nagelkerke R2 and areas under the receiver operating characteristic curves (AUCs) -Re-classification in models: Integrated Discrimination Improvement and continuous Net Reclassification Improvement -Assignment of points for risk factors based on effect sizes in multivariable analyses; validating cutpoints for absolute risk categories of low/moderate/high risk of LDM toxicity |
MTXglu levels are measured among those who were taking LDM at the month 8 visit.
AE, adverse event; AUC, areas under the receiver operating characteristic curve; LDM, low dose methotrexate; MTXglu, methotrexate polyglutamate.
1. Causal role of LDM for specific toxicities
Since subjects in CIRT do not have diseases where LDM is currently indicated, characteristics of underlying systemic rheumatic disease are eliminated as confounders. CIRT-AE will determine the toxicities of LDM based on the imbalances in rates across the LDM and placebo arms as identified in CIRT. Unlike observational studies, CIRT-AE can rely on a direct placebo comparison within a double-blinded trial to make valid inferences about the causal effect of LDM on many clinically relevant AE outcomes. The excess in rates of AE outcomes for the LDM compared to the background placebo rate of AEs can therefore be directly attributed to LDM as toxicity.
For these analyses, allocation to LDM or placebo (reference) will be the primary comparison. The outcomes will include categories of AEs (any AE, severe AE, organ system events, and subgroups with sufficient number of outcomes, Table 3). Planned analyses include the effect of LDM on hepatic, gastrointestinal, respiratory, hematologic, infectious, mucocutaneous, oncologic, renal, neurologic, and musculoskeletal outcomes as well as composite measures of severe AE or any AE. The parent CIRT study is designed to investigate cardiovascular outcomes, incident diabetes, and death9. Given the large sample size of CIRT-AE, we will be able to determine the effect of LDM on uncommon AEs. We will describe the incidence rates and relative risk of LDM toxicity compared to toxicity occurring in the placebo arm. The run-in period and the post-randomization LDM arm will also be compared to test for differences in AE rates during initiation compared to maintenance of LDM as well as to assess the differences in event rates between those who were randomized and those who were not. Person-time will begin accruing at the date of randomization, and subjects will be censored at end of follow-up. Cumulative incidence and Kaplan-Meier curves with 95% confidence intervals will be compared using log-rank tests. Cox proportional hazards models will be used for time to first event analyses. Poisson models will be used for repeated event analyses.
2. Risk factors for AEs
Participants in CIRT will have detailed measurements at baseline and follow-up of many important covariates. These include sociodemographic factors, comorbidities, body mass index, lifestyle factors such as alcohol consumption and smoking, concurrent medications, and organ dysfunction (Table 2). Since excess weight may be an important factor for LDM toxicity (particularly hepatitis), we will investigate the effect of baseline body mass index categories for risk of AEs, investigate effect modification of body mass index for LDM toxicity, and perform subset analyses among those who are overweight or obese. Patients that reported alcohol abuse in the prior 3 years or were unwilling to limit alcohol consumption to <4 drinks per week were not eligible for CIRT. At the randomization visit, subjects were asked the frequency of alcoholic drinks they typically consumed (never or <1/month, 1–3 times/month, 1/week, 2–3 times/week, >3 times/week). Therefore, we will be able to categorize subjects as non-drinkers, light drinkers, and moderate drinkers at baseline for analyses to adjust for potential confounding or to study effect modification by alcohol. As detailed below, genetic factors and MTXglu levels will also be measured. Therefore, CIRT-AE will investigate whether these factors are associated with AEs. These analyses will be stratified by study drug assignment, LDM or placebo. If factors are also associated with AEs among those who received placebo as well as with LDM, then interaction terms will be considered. Multivariable regression models will be built to adjust for important confounders. In sensitivity analyses, we will also include the run-in period in which subjects received open-label LDM to study the association of risk factors for AEs with LDM exposure prior to randomization.
3. Methotrexate polyglutamate levels and AEs in LDM-treated subjects
Methotrexate enters cells through the reduced folate carrier and is activated to form methotrexate polyglutamate (MTXglu) by folylpolyglutamate synthase, an enzyme essential for folate homeostasis in the cytosol and mitochondria. The addition of glutamic acid residues enhances the intracellular retention of methotrexate and promotes the sustained inhibition of amino-imidazole carboxamide ribonucleotide transformylase and thymidylate synthase. Inhibition of these enzymes is important in de novo purine and pyrimidine biosynthesis, which results in a release of adenosine and the immunosuppressive effect of methotrexate.
MTXglu levels have not been strongly linked with toxicity in prior studies58,59. However, studies typically had <150 patients, so different toxicities were combined into a single composite outcome of toxicity. Some specific toxicities may be strongly related to MTXglu levels (i.e., hematologic and mucocutaneous) while others may be idiosyncratic or unrelated to drug levels. Therefore, combining heterogeneous toxicities into a single outcome may have reduced the potential for finding a true association. We will measure MTXglu levels on all subjects taking LDM eight months after randomization so that MTXglu levels stabilize at the expected maximum dosage60. Evaluation of MTXglu levels will also provide an objective method to assess overall adherence, aiding in interpretation of these studies. In CIRT-AE, MTXglu levels will be measured using the published liquid chromatography-electrospray ionization-tandem mass spectrometry-based assay to separately quantify MTXglu levels in red blood cells using stable-isotope-labeled internal standards61.
Since MTXglu levels may mediate toxicity, we will perform analyses investigating whether exposure categories are associated with MTXglu levels and whether MTXglu levels are associated with the outcome. In addition, we will perform a genome-wide association study (GWAS) to investigate whether genetic factors are associated with toxicity outcomes; a network analysis to evaluate potential biologic pathways involved in LDM analyses; and candidate gene analyses for prior genes associated with LDM toxicity in prior studies.
4. Genetic predictors of AEs
Prior pharmacogenomic studies have evaluated whether genetic factors are associated with both LDM efficacy and toxicity. Most of these studies have focused on candidate genes relevant to cellular pathways in LDM, such as folate, methionine, and adenosine metabolism as well as methotrexate polyglutamation62. In particular, single nucleotide polymorphisms (SNP) in the MTHFR (rs1801131 and rs1801133), SLC19A1 (RFC-1) (rs1051266), and ATIC (rs2372536) genes have been associated with LDM toxicity in prior studies62. The MTHFR 677C>T SNP results in a labile form of the MTHFR enzyme with decreased enzyme activity and a resultant elevation in plasma homocysteine levels63. Other candidate genes have plausible biologic mechanisms to be implicated in LDM toxicity.
However, an alternative, non-biased approach such as GWAS has the potential to find unanticipated genetics associations for LDM-dependent AEs. For example, SNPs in the major histocompatability complex often contribute to immune-mediated toxicity to medications64. In addition, measuring genome-wide information will allow for network analysis approaches. This might provide more biologic and clinical relevance than candidate gene studies since toxicity is likely to be a complex process involving many genetic factors beyond those in known candidate genes. Since CIRT-AE will likely have many AEs as outcomes, we will be able to investigate genetic associations with particular homogeneous sub-classifications, which may provide for improved power to detect a genetic basis for AEs.
To perform GWAS, we will genotype subjects who provide consent for genetic analysis and DNA samples (>6000 randomized samples anticipated) using the Illumina MEGA chip. This platform evaluates ~1.7 million multiethnic genome-wide markers capturing medically relevant variation including about ~21,000 pharmacogenomic variants reported in the literature that alter absorption, distribution, metabolism, and excretion ~25,000 variants across the entire Major Histocompatability Complex region, and is enriched for rare variants in genes with phenotype/disease associations and variants in coding regions including ~50,000 exome/loss of function variants curated from the Exome Aggregation Database. We will impute HLA alleles with our highly accurate and widely used SNP2HLA algorithm65. To ensure high quality genotypes, 5% of the genotyped samples will be reference DNAs with known genotypes randomly interspersed with study samples. We will exclude all SNPs with low call rates or those that are outside of Hardy-Weinberg equilibrium, or excessively rare. We will manually examine intensity scatter-pots for all SNP variants that emerge out of statistical analysis.
Genetic markers will be assessed using logistic regression and standard additive genetic models in PLINK66. Departures from the additive model will be assessed using the dominance deviation. For loci with multiple associated SNPs, we will use stepwise model selection to identify the primary SNPs. In addition, we will control for potential population stratification using the EIGENSTRAT method to derive ten principal components of race/ethnicity among the GWAS study participants67. We expect that these principal components will define clusters of individuals corresponding to self-reported European, African, and Asian ancestry. We will then apply EIGENSTRAT within each subpopulation to derive eigenvectors that will be included in the regression models for SNP association testing to adjust for any residual population substructure.
Power for genetic associations was estimated assuming a standard additive relationship between genome-wide SNP allele and the odds ratio (OR) of AEs for 1) gene*drug (i.e., LDM vs. placebo interaction) and 2) gene effect among those allocated to receive LDM. We assumed an AE frequency of 20% in the placebo arm and 26% in the LDM arm. In the gene*drug analysis, we have 80% power to detect an OR of 1.9 for minor allele frequency (MAF) of 25%; we can detect an OR of 3.85 for MAF of 5%. In the analysis restricted to those allocated to receive LDM, we have 80% power to detect an OR of 1.5 for MAF of 25%, and we can detect an OR of 2.1 for MAF of 5%.
We further performed estimates assuming significance thresholds among those targeted towards the HLA region (often implicated in immune-mediated AEs with relatively large effect sizes)64. The MEGA genotyping platform maps 13,468 SNPs to the HLA region. In the gene*drug targeted to HLA analysis, we have 80% power to detect an OR of 1.7 for MAF of 25%; we can detect an OR of 3.05 for MAF of 5%. In the analysis further restricted to those allocated to receive LDM, we have 80% power to detect an OR of 1.4 for MAF of 25%, and we can detect an OR of 1.9 for MAF of 5%.
Since detecting an association between a SNP and clinical outcome often requires a relatively large effect size, we will further evaluate for more modest associations between SNPs and an intermediate biomarker for toxicity, MTXglu. Since we will measure both genetics and MTXglu on subjects randomized to LDM, we are able to evaluate genetic associations for MTXglu levels. After initial genome-wide approaches, we will further restrict analyses to candidate genes with biologic plausibility on MTXglu to enhance the ability to detect a genetic association with this intermediate phenotype for toxicity.
5. Risk prediction rules for LDM toxicity
We will develop risk prediction rules for LDM toxicity incorporating significant findings from the above analyses, potentially including both clinical and genetic predictors. We will develop two different prediction rules. The first will use easily obtained demographic and clinical variables such as age, sex, race, comorbidities, medications, tobacco use, and body mass index. The second will use the MTXglu and genetic measures collected by CIRT-AE to maximize the predictive ability. Two-thirds of CIRT-AE will serve as the development group and one-third will serve as the validation group. We will perform multivariable regression to obtain effect size estimates to use in a weighted points system. We will validate cut-points to find absolute risk categories of low, medium, and high risk of any serious toxicity as well as for other clinically important definitions of LDM toxicity. We will perform exposure-treatment analyses to evaluate heterogeneity of treatment effect. The goodness-of-fit of the models will be tested using Nagelkerke R-squared and areas under the receiver operating characteristic curves. Re-classification in models will be tested using the Integrated Discrimination Improvement and continuous Net Reclassification Improvement68,69.
LIMITATIONS
Since patients with known systemic rheumatic diseases are excluded from CIRT, CIRT-AE may not directly apply to the rheumatic disease population. However, our study design using a randomized controlled trial will be able to establish the causal role of LDM in toxicities. Further, potential confounding effects from rheumatic disease states on AEs will not be present. Additionally, since those using other anti-inflammatory drugs are excluded in CIRT, CIRT-AE will not be able to assess for an interaction between LDM and other immunosuppressant medications, which are often used concomitantly in clinical practice for patients with systemic rheumatic diseases. We acknowledge that we are unable to investigate interactions between LDM and rheumatic disease-specific characteristics for risk of toxicity in CIRT-AE. For example, patients with interstitial lung disease at baseline are not eligible for CIRT, so we would be unable to investigate whether patients with rheumatoid arthritis and interstitial lung disease have a different LDM toxicity profile than patients with only rheumatoid arthritis. If LDM treatment becomes utilized as a secondary prevention therapy among patients with cardiovascular disease, metabolic syndrome or diabetes, it is critical to understand the true risk of toxicity from LDM among this population.
All subjects in CIRT receive study drug orally with a fixed protocol for folic acid supplementation and rate of study drug dose escalation, so these potential factors for LDM toxicity will not be able to be addressed in CIRT-AE. Subjects in CIRT are randomized to receive LDM or placebo at a dose of 15 mg/week. If this dose is tolerated for 4 months, the dose of study drug is then increased to 20 mg/week. Patients with rheumatic diseases may titrate to higher doses of LDM (but typically not more than 25 mg/week) over a shorter duration of time. However, doses of LDM of 15–20 mg/week are commonly used for patients in rheumatic diseases so our results are likely to be relevant. While LDM efficacy is shown to be dose-dependent in systemic rheumatic diseases, a switch to parenteral route may increase efficacy and have improved gastrointestinal tolerance but may also affect risk for toxicity70,71; however, this route of administration is not utilized in CIRT and thus will not be studied. Since all subjects receive folic acid supplementation, our study is not designed to evaluate the risk of non-use of folic acid or alternate folic acid or leucovorin supplementation strategies. All randomized subjects will have already tolerated LDM during the open-label run-in phase of CIRT. Therefore, results may not be generalizable to patients just prescribed LDM who may not tolerate the drug due to side effects such as fatigue and nausea. However, data on AEs occurring in these pre-randomized subjects are collected for potential analyses. Finally, the sample size and length of follow-up in CIRT were determined through power calculations for the primary composite outcome of cardiovascular events. Therefore, CIRT-AE, as a secondary study, may be underpowered in analyses for rare AE outcomes. However, many AE outcomes will occur frequently so we will have adequate power for the analyses we have detailed.
CONCLUSION
CIRT-AE will comprehensively investigate a wide variety of potential LDM toxicities using a large, placebo-controlled, double-blinded, randomized clinical trial with lengthy follow-up. AEs will be systematically identified, adjudicated, and classified to allow for studies investigating LDM toxicity. The very large sample size will allow us to develop and validate a LDM toxicity risk prediction rule for use in clinical practice. In addition, CIRT-AE will investigate the effects of genetic factors and MTXglu levels to further understand the mechanisms of LDM toxicities. Thus, CIRT-AE will advance the widely accepted goal of precision medicine in the use of LDM.
Supplementary Material
Acknowledgments
Funding/Support: This work was supported by the National Institutes of Health (grant number R01 HL119718). While the funder had input on the study design and conduct of CIRT, the authors had the final decision on study design, data collection, analysis, decision to publish, and preparation of this manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard University, its affiliated academic health care centers, or the National Institutes of Health.
The authors would like to acknowledge the participants, study sites, and staff in CIRT which enable these investigations.
References
- 1.Benucci M, Saviola G, Manfredi M, Sarzi-Puttini P, Atzeni F. Cost effectiveness analysis of disease-modifying antirheumatic drugs in rheumatoid arthritis. A systematic review literature. Int J Rheumatol. 2011;2011:845496. doi: 10.1155/2011/845496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ranganathan P, Culverhouse R, Marsh S, et al. Methotrexate (MTX) pathway gene polymorphisms and their effects on MTX toxicity in Caucasian and African American patients with rheumatoid arthritis. J Rheumatol. 2008;35(4):572–579. [PubMed] [Google Scholar]
- 3.Singh JA, Saag KG, Bridges SL, Jr, et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2016;68(1):1–26. doi: 10.1002/art.39480. [DOI] [PubMed] [Google Scholar]
- 4.Finckh A, Liang MH, van Herckenrode CM, de Pablo P. Long-term impact of early treatment on radiographic progression in rheumatoid arthritis: A meta-analysis. Arthritis Rheum. 2006;55(6):864–872. doi: 10.1002/art.22353. [DOI] [PubMed] [Google Scholar]
- 5.Weinblatt ME, Coblyn JS, Fox DA, et al. Efficacy of low-dose methotrexate in rheumatoid arthritis. N Engl J Med. 1985;312(13):818–822. doi: 10.1056/NEJM198503283121303. [DOI] [PubMed] [Google Scholar]
- 6.Weinblatt ME, Maier AL, Fraser PA, Coblyn JS. Longterm prospective study of methotrexate in rheumatoid arthritis: conclusion after 132 months of therapy. J Rheumatol. 1998;25(2):238–242. [PubMed] [Google Scholar]
- 7.Wasko MC, Dasgupta A, Hubert H, Fries JF, Ward MM. Propensity-adjusted association of methotrexate with overall survival in rheumatoid arthritis. Arthritis Rheum. 2013;65(2):334–342. doi: 10.1002/art.37723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi HK, Hernan MA, Seeger JD, Robins JM, Wolfe F. Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. Lancet. 2002;359(9313):1173–1177. doi: 10.1016/S0140-6736(02)08213-2. [DOI] [PubMed] [Google Scholar]
- 9.Everett BM, Pradhan AD, Solomon DH, et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166(2):199–207. e115. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sato EI. Methotrexate therapy in systemic lupus erythematosus. Lupus. 2001;10(3):162–164. doi: 10.1191/096120301666080831. [DOI] [PubMed] [Google Scholar]
- 11.Ash Z, Gaujoux-Viala C, Gossec L, et al. A systematic literature review of drug therapies for the treatment of psoriatic arthritis: current evidence and meta-analysis informing the EULAR recommendations for the management of psoriatic arthritis. Ann Rheum Dis. 2012;71(3):319–326. doi: 10.1136/ard.2011.150995. [DOI] [PubMed] [Google Scholar]
- 12.Ranganathan P, McLeod HL. Methotrexate pharmacogenetics: the first step toward individualized therapy in rheumatoid arthritis. Arthritis Rheum. 2006;54(5):1366–1377. doi: 10.1002/art.21762. [DOI] [PubMed] [Google Scholar]
- 13.Salliot C, van der Heijde D. Long-term safety of methotrexate monotherapy in patients with rheumatoid arthritis: a systematic literature research. Ann Rheum Dis. 2009;68(7):1100–1104. doi: 10.1136/ard.2008.093690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kremer JM, Lee JK. The safety and efficacy of the use of methotrexate in long-term therapy for rheumatoid arthritis. Arthritis Rheum. 1986;29(7):822–831. doi: 10.1002/art.1780290702. [DOI] [PubMed] [Google Scholar]
- 15.Hanrahan PS, Scrivens GA, Russell AS. Prospective long term follow-up of methotrexate therapy in rheumatoid arthritis: toxicity, efficacy and radiological progression. Br J Rheumatol. 1989;28(2):147–153. doi: 10.1093/rheumatology/28.2.147. [DOI] [PubMed] [Google Scholar]
- 16.Kremer JM, Phelps CT. Long-term prospective study of the use of methotrexate in the treatment of rheumatoid arthritis. Update after a mean of 90 months. Arthritis Rheum. 1992;35(2):138–145. doi: 10.1002/art.1780350203. [DOI] [PubMed] [Google Scholar]
- 17.Albrecht K, Muller-Ladner U. Side effects and management of side effects of methotrexate in rheumatoid arthritis. Clin Exp Rheumatol. 2010;28(5 Suppl 61):S95–101. [PubMed] [Google Scholar]
- 18.Verstappen SM, Jacobs JW, van der Veen MJ, et al. Intensive treatment with methotrexate in early rheumatoid arthritis: aiming for remission. Computer Assisted Management in Early Rheumatoid Arthritis (CAMERA, an open-label strategy trial) Ann Rheum Dis. 2007;66(11):1443–1449. doi: 10.1136/ard.2007.071092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yazici Y, Sokka T, Kautiainen H, Swearingen C, Kulman I, Pincus T. Long term safety of methotrexate in routine clinical care: discontinuation is unusual and rarely the result of laboratory abnormalities. Ann Rheum Dis. 2005;64(2):207–211. doi: 10.1136/ard.2004.023408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Visser K, van der Heijde DM. Risk and management of liver toxicity during methotrexate treatment in rheumatoid and psoriatic arthritis: a systematic review of the literature. Clin Exp Rheumatol. 2009;27(6):1017–1025. [PubMed] [Google Scholar]
- 21.Kremer JM, Alarcon GS, Lightfoot RW, Jr, et al. Methotrexate for rheumatoid arthritis. Suggested guidelines for monitoring liver toxicity. American College of Rheumatology.[see comment] Arthritis & Rheumatism. 1994;37(3):316–328. doi: 10.1002/art.1780370304. [DOI] [PubMed] [Google Scholar]
- 22.Curtis JR, Beukelman T, Onofrei A, et al. Elevated liver enzyme tests among patients with rheumatoid arthritis or psoriatic arthritis treated with methotrexate and/or leflunomide. Ann Rheum Dis. 2010;69(1):43–47. doi: 10.1136/ard.2008.101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kent PD, Luthra HS, Michet C., Jr Risk factors for methotrexate-induced abnormal laboratory monitoring results in patients with rheumatoid arthritis. J Rheumatol. 2004;31(9):1727–1731. [PubMed] [Google Scholar]
- 24.Bridges SL, Jr, Alarcon GS, Koopman WJ. Methotrexate-induced liver abnormalities in rheumatoid arthritis. J Rheumatol. 1989;16(9):1180–1183. [PubMed] [Google Scholar]
- 25.Brick JE, Moreland LW, Al-Kawas F, Chang WW, Layne RD, DiBartolomeo AG. Prospective analysis of liver biopsies before and after methotrexate therapy in rheumatoid patients. Semin Arthritis Rheum. 1989;19(1):31–44. doi: 10.1016/0049-0172(89)90085-1. [DOI] [PubMed] [Google Scholar]
- 26.Aponte J, Petrelli M. Histopathologic findings in the liver of rheumatoid arthritis patients treated with long-term bolus methotrexate. Arthritis Rheum. 1988;31(12):1457–1464. doi: 10.1002/art.1780311201. [DOI] [PubMed] [Google Scholar]
- 27.Hargreaves MR, Mowat AG, Benson MK. Acute pneumonitis associated with low dose methotrexate treatment for rheumatoid arthritis: report of five cases and review of published reports. Thorax. 1992;47(8):628–633. doi: 10.1136/thx.47.8.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Golden MR, Katz RS, Balk RA, Golden HE. The relationship of preexisting lung disease to the development of methotrexate pneumonitis in patients with rheumatoid arthritis. J Rheumatol. 1995;22(6):1043–1047. [PubMed] [Google Scholar]
- 29.Kremer JM, Alarcon GS, Weinblatt ME, et al. Clinical, laboratory, radiographic, and histopathologic features of methotrexate-associated lung injury in patients with rheumatoid arthritis: a multicenter study with literature review. Arthritis Rheum. 1997;40(10):1829–1837. doi: 10.1002/art.1780401016. [DOI] [PubMed] [Google Scholar]
- 30.Conway R, Low C, Coughlan RJ, O’Donnell MJ, Carey JJ. Methotrexate use and risk of lung disease in psoriasis, psoriatic arthritis, and inflammatory bowel disease: systematic literature review and meta-analysis of randomised controlled trials. BMJ. 2015 Mar 13;350(17):h1269–h1269. doi: 10.1136/bmj.h1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barrera P, Laan RF, van Riel PL, Dekhuijzen PN, Boerbooms AM, van de Putte LB. Methotrexate-related pulmonary complications in rheumatoid arthritis. Ann Rheum Dis. 1994;53(7):434–439. doi: 10.1136/ard.53.7.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hilliquin P, Renoux M, Perrot S, Puechal X, Menkes CJ. Occurrence of pulmonary complications during methotrexate therapy in rheumatoid arthritis. Br J Rheumatol. 1996;35(5):441–445. doi: 10.1093/rheumatology/35.5.441. [DOI] [PubMed] [Google Scholar]
- 33.Alarcon GS, Kremer JM, Macaluso M, et al. Risk factors for methotrexate-induced lung injury in patients with rheumatoid arthritis. A multicenter, case-control study. Methotrexate-Lung Study Group. Ann Intern Med. 1997;127(5):356–364. doi: 10.7326/0003-4819-127-5-199709010-00003. [DOI] [PubMed] [Google Scholar]
- 34.Dawson JK, Graham DR, Desmond J, Fewins HE, Lynch MP. Investigation of the chronic pulmonary effects of low-dose oral methotrexate in patients with rheumatoid arthritis: a prospective study incorporating HRCT scanning and pulmonary function tests. Rheumatology (Oxford) 2002;41(3):262–267. doi: 10.1093/rheumatology/41.3.262. [DOI] [PubMed] [Google Scholar]
- 35.Conway R, Low C, Coughlan RJ, O’Donnell MJ, Carey JJ. Methotrexate use and risk of lung disease in psoriasis, psoriatic arthritis, and inflammatory bowel disease: systematic literature review and meta-analysis of randomised controlled trials. BMJ. 2015;350:h1269. doi: 10.1136/bmj.h1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernatsky S, Hudson M, Suissa S. Anti-rheumatic drug use and risk of serious infections in rheumatoid arthritis. Rheumatology (Oxford) 2007;46(7):1157–1160. doi: 10.1093/rheumatology/kem076. [DOI] [PubMed] [Google Scholar]
- 37.van der Veen MJ, van der Heide A, Kruize AA, Bijlsma JW. Infection rate and use of antibiotics in patients with rheumatoid arthritis treated with methotrexate. Ann Rheum Dis. 1994;53(4):224–228. doi: 10.1136/ard.53.4.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edwards CJ, Cooper C, Fisher D, Field M, van Staa TP, Arden NK. The importance of the disease process and disease-modifying antirheumatic drug treatment in the development of septic arthritis in patients with rheumatoid arthritis. Arthritis Rheum. 2007;57(7):1151–1157. doi: 10.1002/art.23003. [DOI] [PubMed] [Google Scholar]
- 39.Schneeweiss S, Setoguchi S, Weinblatt ME, et al. Anti-tumor necrosis factor alpha therapy and the risk of serious bacterial infections in elderly patients with rheumatoid arthritis. Arthritis Rheum. 2007;56(6):1754–1764. doi: 10.1002/art.22600. [DOI] [PubMed] [Google Scholar]
- 40.Buchbinder R, Hall S, Sambrook PN, et al. Methotrexate therapy in rheumatoid arthritis: a life table review of 587 patients treated in community practice. J Rheumatol. 1993;20(4):639–644. [PubMed] [Google Scholar]
- 41.Weinblatt ME. Toxicity of low dose methotrexate in rheumatoid arthritis. J Rheumatol Suppl. 1985;12(Suppl 12):35–39. [PubMed] [Google Scholar]
- 42.Gutierrez-Urena S, Molina JF, Garcia CO, Cuellar ML, Espinoza LR. Pancytopenia secondary to methotrexate therapy in rheumatoid arthritis. Arthritis Rheum. 1996;39(2):272–276. doi: 10.1002/art.1780390214. [DOI] [PubMed] [Google Scholar]
- 43.Doolittle GC, Simpson KM, Lindsley HB. Methotrexate-associated, early-onset pancytopenia in rheumatoid arthritis. Arch Intern Med. 1989;149(6):1430–1431. [PubMed] [Google Scholar]
- 44.MacKinnon SK, Starkebaum G, Willkens RF. Pancytopenia associated with low dose pulse methotrexate in the treatment of rheumatoid arthritis. Semin Arthritis Rheum. 1985;15(2):119–126. doi: 10.1016/0049-0172(85)90029-0. [DOI] [PubMed] [Google Scholar]
- 45.Kivity S, Zafrir Y, Loebstein R, Pauzner R, Mouallem M, Mayan H. Clinical characteristics and risk factors for low dose methotrexate toxicity: a cohort of 28 patients. Autoimmun Rev. 2014;13(11):1109–1113. doi: 10.1016/j.autrev.2014.08.027. [DOI] [PubMed] [Google Scholar]
- 46.Mori S, Hidaka M, Kawakita T, et al. Factors Associated with Myelosuppression Related to Low-Dose Methotrexate Therapy for Inflammatory Rheumatic Diseases. PLoS One. 2016;11(4):e0154744. doi: 10.1371/journal.pone.0154744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalantzis A, Marshman Z, Falconer DT, Morgan PR, Odell EW. Oral effects of low-dose methotrexate treatment. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100(1):52–62. doi: 10.1016/j.tripleo.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 48.Varatharajan N, Lim IG, Anandacoomarasamy A, et al. Methotrexate: long-term safety and efficacy in an Australian consultant rheumatology practice. Intern Med J. 2009;39(4):228–236. doi: 10.1111/j.1445-5994.2009.01800.x. [DOI] [PubMed] [Google Scholar]
- 49.Baecklund E, Ekbom A, Sparen P, Feltelius N, Klareskog L. Disease activity and risk of lymphoma in patients with rheumatoid arthritis: nested case-control study. BMJ. 1998;317(7152):180–181. doi: 10.1136/bmj.317.7152.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamel OW, van de Rijn M, LeBrun DP, Weiss LM, Warnke RA, Dorfman RF. Lymphoid neoplasms in patients with rheumatoid arthritis and dermatomyositis: frequency of Epstein-Barr virus and other features associated with immunosuppression. Hum Pathol. 1994;25(7):638–643. doi: 10.1016/0046-8177(94)90295-x. [DOI] [PubMed] [Google Scholar]
- 51.Georgescu L, Quinn GC, Schwartzman S, Paget SA. Lymphoma in patients with rheumatoid arthritis: association with the disease state or methotrexate treatment. Semin Arthritis Rheum. 1997;26(6):794–804. doi: 10.1016/s0049-0172(97)80023-6. [DOI] [PubMed] [Google Scholar]
- 52.Solomon DH, Kremer JM, Fisher M, et al. Comparative cancer risk associated with methotrexate, other non-biologic and biologic disease-modifying anti-rheumatic drugs. Semin Arthritis Rheum. 2014;43(4):489–497. doi: 10.1016/j.semarthrit.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 53.Everett BM, Pradhan AD, Solomon DH, et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: A test of the inflammatory hypothesis of atherothrombosis. American Heart Journal. 2013;166(2):199–207. e115. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mehran R, Rao SV, Bhatt DL, et al. Standardized Bleeding Definitions for Cardiovascular Clinical Trials: A Consensus Report From the Bleeding Academic Research Consortium. Circulation. 2011;123(23):2736–2747. doi: 10.1161/CIRCULATIONAHA.110.009449. [DOI] [PubMed] [Google Scholar]
- 55.Searles G, McKendry RJ. Methotrexate pneumonitis in rheumatoid arthritis: potential risk factors. Four case reports and a review of the literature. J Rheumatol. 1987;14(6):1164–1171. [PubMed] [Google Scholar]
- 56. [Accessed September 12, 2016];National Cancer Institute Common Terminology Criteria for Adverse Events. doi: 10.1200/JOP.2015.006106. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf. [DOI] [PMC free article] [PubMed]
- 57.Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Inter Suppl. 2012;2:1–138. [Google Scholar]
- 58.Kremer JM. Toward a better understanding of methotrexate. Arthritis Rheum. 2004;50(5):1370–1382. doi: 10.1002/art.20278. [DOI] [PubMed] [Google Scholar]
- 59.Dervieux T, Orentas Lein D, Marcelletti J, et al. HPLC determination of erythrocyte methotrexate polyglutamates after low-dose methotrexate therapy in patients with rheumatoid arthritis. Clin Chem. 2003;49(10):1632–1641. doi: 10.1373/49.10.1632. [DOI] [PubMed] [Google Scholar]
- 60.Dervieux T, Furst D, Lein DO, et al. Polyglutamation of methotrexate with common polymorphisms in reduced folate carrier, aminoimidazole carboxamide ribonucleotide transformylase, and thymidylate synthase are associated with methotrexate effects in rheumatoid arthritis. Arthritis Rheum. 2004;50(9):2766–2774. doi: 10.1002/art.20460. [DOI] [PubMed] [Google Scholar]
- 61.den Boer E, Meesters RJ, van Zelst BD, et al. Measuring methotrexate polyglutamates in red blood cells: a new LC-MS/MS-based method. Anal Bioanal Chem. 2013;405(5):1673–1681. doi: 10.1007/s00216-012-6581-7. [DOI] [PubMed] [Google Scholar]
- 62.Malik F, Ranganathan P. Methotrexate pharmacogenetics in rheumatoid arthritis: a status report. Pharmacogenomics. 2013;14(3):305–314. doi: 10.2217/pgs.12.214. [DOI] [PubMed] [Google Scholar]
- 63.Kang SS, Zhou J, Wong PW, Kowalisyn J, Strokosch G. Intermediate homocysteinemia: a thermolabile variant of methylenetetrahydrofolate reductase. Am J Hum Genet. 1988;43(4):414–421. [PMC free article] [PubMed] [Google Scholar]
- 64.Nguyen DV, Vidal C, Li J, Fulton RB, Fernando SL. Validation of a rapid test for HLA-B*58:01/57:01 allele screening to detect individuals at risk for drug-induced hypersensitivity. Pharmacogenomics. 2016;17(5):473–480. doi: 10.2217/pgs.15.185. [DOI] [PubMed] [Google Scholar]
- 65.Jia X, Han B, Onengut-Gumuscu S, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One. 2013;8(6):e64683. doi: 10.1371/journal.pone.0064683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 68.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27(2):157–172. doi: 10.1002/sim.2929. discussion 207–112. [DOI] [PubMed] [Google Scholar]
- 69.Pencina MJ, D’Agostino RB, Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med. 2011;30(1):11–21. doi: 10.1002/sim.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Visser K, van der Heijde D. Optimal dosage and route of administration of methotrexate in rheumatoid arthritis: a systematic review of the literature. Ann Rheum Dis. 2009;68(7):1094–1099. doi: 10.1136/ard.2008.092668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oguey D, Kolliker F, Gerber NJ, Reichen J. Effect of food on the bioavailability of low-dose methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 1992;35(6):611–614. doi: 10.1002/art.1780350603. [DOI] [PubMed] [Google Scholar]
- 72.Franck H, Rau R, Herborn G. Thrombocytopenia in patients with rheumatoid arthritis on long-term treatment with low dose methotrexate. Clin Rheumatol. 1996;15(3):266–270. doi: 10.1007/BF02229705. [DOI] [PubMed] [Google Scholar]
- 73.White DH, Chapman PT, O’Donnell JL, James J, Frampton C, Stamp LK. Lack of association between elevated mean red cell volume and haematological toxicity in patients receiving long-term methotrexate for rheumatoid arthritis. Intern Med J. 2010;40(8):561–565. doi: 10.1111/j.1445-5994.2009.02059.x. [DOI] [PubMed] [Google Scholar]
- 74.Coyne P, Hamilton J, Heycock C, Saravanan V, Coulson E, Kelly CA. Acute lower respiratory tract infections in patients with rheumatoid arthritis. J Rheumatol. 2007;34(9):1832–1836. [PubMed] [Google Scholar]
- 75.Ortiz Z, Shea B, Suarez Almazor M, Moher D, Wells G, Tugwell P. Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. Cochrane Database Syst Rev. 2000;(2):CD000951. doi: 10.1002/14651858.CD000951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.