

Abstract
This unit describes how to infect cells with vaccinia virus and then transfect them with a plasmid-transfer vector or PCR fragment to generate a recombinant virus. Selection and screening methods used to isolate recombinant viruses and a method for the amplification of recombinant viruses are described. Finally, a method for live immunostaining that has been used primarily for detection of recombinant modified vaccinia virus Ankara (MVA) is presented.
This unit first describes how to infect cells with vaccinia virus and then transfect them with a plasmid-transfer vector or PCR fragment to generate a recombinant virus (see Basic Protocol 1). Also presented are selection and screening methods used to isolate recombinant viruses (see Basic Protocol 2) and a method for the amplification of recombinant viruses (see Basic Protocol 3). Finally, a method for live immunostaining that has been used primarily for detection of recombinant modified vaccinia virus Ankara (MVA) is presented (see Basic Protocol 4).
HeLa S3 cells are recommended for large-scale growth of vaccinia virus. BS-C-1 cells may be used for xanthine-guanine phosphoribosyltransferase (XGPRT) and plaque size selection, fluorescent protein screening, transfection and determination of virus titer (UNIT 5.12; Cotter et al., 2017). For thymidine kinase (TK) selection, HuTK− 143B cells are used. With MVA, all steps are carried out in CEF or BHK-21 cells (UNIT 5.12; Cotter et al., 2017).
CAUTION: Proceed carefully and follow biosafety level 2 (BL-2) practices when working with standard vaccinia virus (see UNIT 5.12 (Cotter et al. (2017) and Moss and Earl (2002) for safety precautions).
NOTE: Carry out all procedures for preparation of virus in a biosafety cabinet.
BASIC PROTOCOL 1 GENERATION OF A VACCINIA VIRUS VECTOR BY HOMOLOGOUS RECOMBINATION
The foreign gene of interest is subcloned into a plasmid transfer vector (Fig. 15.3.1) or assembled as a PCR fragment so that it is flanked by DNA from the vaccinia virus genome for insertion into a non-essential site. This recombinant plasmid is then transfected into BS-C-1 or other cells that have been infected with vaccinia virus. Homologous recombination occurs between the vaccinia virus genome and homologous sequences (Fig. 15.3.1). The recombinant virus is obtained in a cell lysate, which is then subjected to several rounds of plaque purification using appropriate selection and/or screening protocols (see Basic Protocol 2). For MVA, the same transfection procedure is used and either CEF or BHK-21 (UNIT 5.12; Cotter et al., 2017) cells are substituted for BS-C-1 cells. For live immunostaining to detect recombinant MVA or standard virus, see Basic Protocol 4.
Figure 5.13.1.
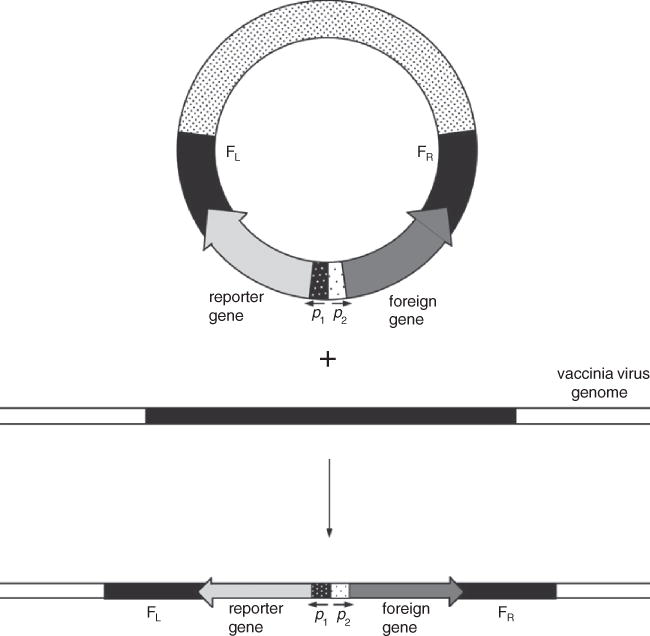
Homologous recombination between a transfected plasmid and the vaccinia virus genome. The resultant recombinant virus stably incorporates a reporter gene as well as the foreign gene of interest. FL=left flank, FR=right flank, p=promoter
Materials
pRB21, pSC11, pSC65, pLW-44, pLAS-1, pLW-73, or other suitable vector (Table 15.3.1)
BS-C-1, BHK-21, or CEF cells (UNIT 5.12; Cotter et al., 2017)
Complete MEM-8 and -2.5 media (UNIT 5.12; Cotter et al., 2017)
Vaccinia virus stock (UNIT 5.12; Cotter et al., 2017)
Liposomal transfection reagent (such as Lipofectamine-2000, Life Technologies)
Transfection buffer (see Reagents and Solutions)
2.5 M CaCl2
OptiMEM medium (Life Technologies)
Dry ice/ethanol bath
12-well tissue culture plates
12 × 75–mm polystyrene tubes
Disposable scraper, plunger from a 1 ml syringe, or rubber policeman, sterile
2-ml sterile microcentrifuge tubes (optional – use Sarstedt 2 ml screw-cap tubes)
Additional reagents and equipment for subcloning, isolation of plasmid, PCR amplification, and tissue culture
Table 5.13.1.
Vaccinia Virus Transfer Vectors
| Vectora | Promoterb | Cloning sitesc | Insertion sitesd | Selection/screening | Reference |
|---|---|---|---|---|---|
| pGS20 | p7.5 (E/L) | BamHI; SmaI | TK | TK− | Mackett et al., 1984 |
| pSC11 | p7.5 (E/L) | SmaI; MCS | TK | TK−,β-gal | Chakrabarti et al., 1985; Earl et al., 1990; Bacik et al., 1994 |
| pMJ601, pMJ602 | psyn (L) | MCS | TK | TK−, β-gal | Davison and Moss, 1990 |
| pRB21 | psyn (E/L) | MCS | F12L/F13L | Plaque | Blasco and Moss, 1995 |
| pMC02 | psyn (E/L) | MCS | TK | TK−, GUS | Carroll and Moss, 1995 |
| pSC59 | psyn (E/L) | MCS | TK | TK− | Chakrabarti et al., 1997 |
| pSC65 | psyn (E/L) | MCS | TK | TK−, β-gal | Chakrabarti et al., 1997 |
| pLW-44 | pmH5 (E/L) | MSC | Del II | GFP | Bisht et al., 2004 |
| pLAS-1 | pmH5 (E/L) | MSC | Del III | Transient GFPe | Wyatt et al., 2008 |
| pLAS-2 | pmH5 (E/L) | MSC | Del II | Transient GFPe | Wyatt et al., 2008 |
| pLW-73 | pmH5 (E/L) | MSC | I8R/G1L | Transient GFPe | Wyatt et al., 2009 |
| pLW-76 | pmH5 (E/L) | MSC | Del III rstr | Transient GFPe | L.Wyatt and B. Moss, unpubl. observ. |
| pLW-7 | psyn (E/L) | MCS | Del III | Transient gpte | Wyatt et al., 1996 |
| pMC03 | psyn (E/L) | MCS | Del III | GUS | Carroll and Moss, 1995 |
| pLW-9 | pmH5 (E/L) | MCS | Del III | Transient gpte | Wyatt et al., 1996 |
| pLW-17 | pmH5 (E/L) | MCS | Del II | None | L. Wyatt and B. Moss, unpub. observ. |
| pLW-21 | psyn (E/L) | MCS | Del II | None | L. Wyatt and B. Moss, unpub. observ. |
| pLW-22 | psyn (E/L) | MCS | Del II | β-gal | Ourmanov et al.,2000 |
| pLW-24 | p7.5 (E/L) | MCS | Del II | None | L. Wyatt and B. Moss, unpub. observ. |
pRB21 was specifically designed for use with vaccinia virus vRB12, which has a deletion in the F13L gene. The plasmids pLW-44, pLAS-1, pLAS-2, pLW-73, and pLW-76 were designed for MVA.
Abbreviations: E, early; L, late; E/L, early and late.
SmaI digestion gives a blunt end for cloning any fragment that has been blunt-ended. MCS signifies multiple cloning sites.
Abbreviations: TK, thymidine kinase locus; F12L/F13L, between F12L and F13L open reading frames; Del II and III, sites of natural deletion in MVA, MCS, multiple cloning site.
Transient selection in which gpt or GFP gene is deleted from recombinant vaccinia virus during recombination; see Background Information.
NOTE: All reagents and equipment coming into contact with live cells must be sterile, and aseptic technique should be used accordingly. Culture incubations should be performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified.
Prepare recombinant plasmid DNA
-
1a
Subclone the gene of interest into the multiple cloning site (MCS) in pRB21, pSC11, pSC65, pLW-44, pLAS-1, pLW-73, or other suitable vector and isolate plasmid using standard procedures.
Proceed to step 6.
Prepare PCR fragment
1b. Amplify the 2 flanking regions of the vaccinia virus genome between which the foreign gene will be inserted.
2b. Amplify the foreign gene of interest and relevant vaccinia virus promoter.
3b. Join the DNA containing the flanking regions and foreign gene by overlap PCR.
4b. Verify the size integrity of the PCR fragment by gel electrophoresis.
-
5b. Gel purify the DNA fragment.
Proceed to step 6.
Prepare vaccinia virus-infected cells
-
6. Seed wells of a 12-well tissue culture plate with 2.5 × 105 BS-C-1, BHK-21, or CEF cells in complete MEM-8 medium. Incubate to near confluency (usually overnight).
UNIT 5.12 (Cotter et al., 2017) details the culture of these cells.
-
7. Thaw an aliquot of vaccinia virus and sonicate in ice-water. Cool on ice and repeat sonication. (see UNIT 5.12; Cotter et al., 2017).
Virus stocks are usually at a titer of ~1 × 109 pfu/ml, but may be significantly lower depending on the source.
Vortexing usually breaks up any clumps of cells. However, if there are still visible clumps, chill to 0°C and sonicate 30 sec on ice. Sonication can be repeated several times but the sample should be allowed to cool on ice between sonications.
-
8. Dilute sonicated virus in complete MEM-2.5 to 0.5 × 105 pfu/ml. Aspirate medium from confluent monolayer of cells and infect with 0.5 ml diluted vaccinia virus (0.05 pfu/cell). Incubate 2 hr at 37°C.
Approximately 30 min before the end of the infection period, prepare DNA according to the liposomal reagent or CaCl2 method as described below.
Prepare DNA
For liposomal reagent method (Lipofectamine)
9a. Place 0.25 ml OptiMEM into each of 2 polystyrene tubes. To one tube add 5 μl of Lipofectamine-2000. To the other tube add 0.5 to 1 μg of the recombinant plasmid (from step 1a) or PCR fragment (from step 5b) (in <25 μl). Leave 5 min at room temperature.
10a. Add the DNA solution to the lipofectamine solution and leave another 30 min at room temperature.
For CaCl2 method
-
9b. Place 0.5 ml transfection buffer into a 12 × 75–mm polystyrene tube and add 0.5 to 1 μg of the recombinant plasmid (from step 1a) or PCR fragment (from step 5b) (in <25 μl).
A discussion of calcium phosphate transfection can be found in Critical Parameters.
-
10b. Slowly add 13 μl of 2.5 M CaCl2 and mix gently. Leave 20 to 30 min at room temperature.
Gentle mixing is essential; see Critical Parameters. A fine precipitate should appear.
Transfect cells
11. Aspirate virus inoculum from monolayer of cells (step 8) and wash twice with PBS.
12a. For lipofectamine method: Add the DNA/lipofectamine solution (step 10a) to the cells and incubate 4 hr at 37°C.
12b. For CaCl2 method: Add the precipitated DNA suspension (step 10b) to the cells and leave 30 min at room temperature, then add 1.5 ml complete MEM-8 and incubate 3 to 4 hr at 37°C.
13. Aspirate medium, replace with 1 ml MEM-2.5, and incubate 2 days at 37°C.
14. Dislodge cells with a disposable scraper, plunger from a 1ml syringe or sterile rubber policeman and transfer to a 2-ml sterile microcentrifuge tube.
15. Lyse the cell suspension by performing three freeze-thaw cycles, each time by freezing in a dry ice/ethanol bath, thawing in a 37°C water bath, and vortexing.
16. Store the cell lysate at −80°C until needed in the selection and screening procedure (see Basic Protocol 2).
BASIC PROTOCOL 2 SELECTION AND SCREENING OF RECOMBINANT VIRUS PLAQUES
For standard vaccinia strains, procedures are described involving xanthine-guanine phosphoribosyltransferase (XGPRT; Falkner and Moss, 1988) or thymidine kinase (TK; Mackett et al., 1984) for selecting virus plaques that contain recombinant DNA. A newer and simpler drug-free method (plaque selection) is based on repair of a mutation that caused the parental virus to form pin-point plaques (Blasco and Moss, 1995; see Background Information regarding choice of procedure). In addition, green fluorescent protein (GFP), β-galactosidase (Chakrabarti et al., 1985), or β-glucuronidase (GUS) screening (Carroll and Moss, 1995) can be used alone or in conjunction with TK selection to discriminate TK− recombinants from spontaneous TK− mutants. For each method, recombinant virus (obtained in the transfection; see Basic Protocol 1) is used to infect a monolayer culture of cells. For vaccinia virus, BS-C-1 cells are used because large plaques are obtained; for TK selection, it is necessary to use a cell line such as HuTK− 143B that is deficient in thymidine kinase. For MVA, CEF or BHK-21 cells are necessary. Medium containing 2.5% methylcellulose and the appropriate selective drugs, if applicable, is then pipetted onto the infected cell monolayer. Because of cell-to-cell spread of virus, each productively infected cell gives rise to an infected area on the monolayer. With vaccinia infection the cells are rounded and dead, and thus appear as colorless plaques. With MVA there is a build-up of cells resulting in appearance of a focus. If GFP (or other fluorescent protein) is used, fluorescent (recombinant) plaques are visualized with a fluorescence microscope. If β-galactosidase (or GUS) screening is used, the substrate Xgal (or Xgluc) is included in the overlay. Plaques containing infected cells that have expressed β-galactosidase or GUS turn blue; thus, blue (recombinant) plaques can be distinguished from clear (parental) plaques. Live immunostaining can also be employed (see Basic Protocol 4). A sterile toothpick or pipet tip is used to remove infected cells from the plaques/foci. The virus is placed in medium and is released by freeze-thaw cycling and sonication. Several rounds of plaque purification are used to ensure the absence of residual non-recombinant virus.
TK selection has not been used with MVA.
Materials
BS-C-1, HuTK− 143B, BHK-21, or CEF confluent monolayer cells (UNIT 5.12; Cotter et al., 2017). and appropriate complete medium
Complete MEM-2.5 medium (UNIT 5.12; Cotter et al., 2017).)
-
Selective agents (for XGPRT selection; filter sterilize, and store at −20°C):
10 mg/ml (400×) mycophenolic acid (MPA; Calbiochem) in 0.1 N NaOH
10 mg/ml (40×) xanthine in 0.1 N NaOH
10 mg/ml (670×) hypoxanthine in 0.1 N NaOH
Transfected cell lysate (see Basic Protocol 1)
5 mg/ml 5-bromodeoxyuridine (BrdU) in H2O (for TK selection; filter sterilize and store at −20°C)
4% Xgal in dimethylformamide (optional, for β-galactosidase screening
2% Xgluc in dimethylformamide (optional, for GUS screening)
Dry ice/ethanol bath
6-well, 35-mm tissue culture plates
Cup sonicator (e.g., Ultrasonic Processor VCX-750 from Sonics and Materials)
Fluorescence microscope (for GFP or other fluorescent protein screening)
Pipet tips or toothpicks, sterile
Sterile microcentrifuge tubes (optional – use Sarstedt 2 ml screw-cap tubes)
Additional reagents and equipment for tissue culture and counting cells and serial dilution of virus.
NOTE: All reagents and equipment coming into contact with live cells must be sterile, and aseptic technique should be used accordingly. Culture incubations should be performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified.
Prepare the cells
-
Trypsinize confluent monolayer culture and resuspend in appropriate complete medium as in UNIT 5.12; Cotter et al., 2017, steps 4 to 7 of Basic Protocol 1.
For XGPRT selection, plaque selection, color or GFP screening, use BS-C-1 cells.
For TK selection, use HuTK− 143B cells.
For MVA, use BHK-21 or CEF cells.
-
Count cells using a cell counter or hemacytometer.
The authors use a Nexcelom Cellometer cell counter
Plate 5 × 105 cells/well in a 6-well tissue culture plate (2 ml/well final). Incubate until confluent (this should take <24 hr).
-
Prepare cells as follows.
For XGPRT selection, preincubate monolayer for 12 to 24 hr in filter-sterilized complete MEM-2.5 containing 1/400 vol 10 mg/ml MPA, 1/40 vol 10 mg/ml xanthine, and 1/670 vol 10 mg/ml hypoxanthine.
For plaque or TK selection or color screening methods, do not preincubate.
Prepare the lysate and infect cells
5. Thaw the transfected cell lysate and sonicate 20 to 30 sec in ice-water to break up clumps. Repeat, keeping lysate on ice between steps.
6. Add 100, 10, 1, or 0.1 μl of lysate to duplicate wells containing 1 ml complete MEM-2.5. Gently swirl to mix. Incubate 2 hr
Perform methylcellulose overlay
-
7. Prepare selective medium containing 2.5% methylcellulose
For XGPRT selection, include MPA, xanthine, and hypoxanthine
For TK selection, include 1/100 vol of 5 mg/ml BrdU.
For β-galactosidase or GUS screening, add 1/240 vol 4% Xgal or 1/200 vol 2% Xgluc, respectively.
For plaque selection or fluorescent protein screening, make no additions.
8. Aspirate the virus inoculum from the infected cells (from step 6). Overlay each well with 2 ml appropriate selective medium containing 2.5% methylcellulose. Incubate 2 days.
Obtain the plaques
9. Add 0.5 ml complete MEM-2.5 to sterile microcentrifuge tubes. When incubation period is complete (step 8), pick well-separated plaques by scraping and suction with a pipet tip or scraping with a toothpick. Transfer to a tube containing 0.5 ml complete MEM-2.5. Repeat for six to twelve plaques with separate pipet tips or toothpicks, placing each in a separate tube.
10. Vortex each virus-containing tube, then perform three freeze-thaw cycles, each time by freezing in a dry ice/ethanol bath, thawing in a 37°C water bath, and vortexing.
-
11. Place tubes containing virus into a cup sonicator containing ice-water and sonicate 20 to 30 sec at full power.
If TK selection only has been used, plaque isolates should be tested by PCR, DNA dot-blot hybridization, or immunostaining because some plaques will contain spontaneous TK− mutations and not recombinant virus.
Carry out several rounds of plaque purification
-
12. Prepare monolayers of the appropriate cell line as described in steps 1 to 4.
One 6-well plate is needed for each plaque isolate.
-
13. Add 100, 10, 1, or 0.1 μl of lysate to duplicate wells containing 1 ml complete MEM-2.5. Gently swirl to mix. Incubate 2 hr. This should be performed with several plaque isolates.
If XGPRT selection is used, cells must be preincubated with selective drugs and serial dilutions of the viral isolates must also contain selective drugs (step 4, substep a). Note that the parental non-recombinant virus will form tiny plaques in the presence of drug.
14. Aspirate medium from cell monolayers and overlay with selective medium (if appropriate) containing 2.5% methylcellulose.
15. Repeat steps 5 to 11 for three or more rounds of plaque purification to ensure a clonally pure recombinant virus.
BASIC PROTOCOL 3 AMPLIFICATION OF VIRUS FROM A PLAQUE
Recombinant virus from a plaque (obtained after the selection and screening protocols; see Basic Protocol 2) is amplified by infection of successively larger numbers of cells. Medium containing drugs for XGPRT or TK selection is usually used, up to and including the infection of cells in a 25-cm2 flask. Freeze-thaw cycling is carried out to release the recombinant virus from the cells. The titer of the vaccinia virus or MVA stock is determined as described in UNIT 5.12; Cotter et al., 2017.
Materials
Resuspended recombinant plaque (see Basic Protocol 2)
Confluent monolayer cultures of appropriate cells in both a 6-well and a 25-cm2 tissue culture flask (UNIT 5.12; Cotter et al., 2017)
Complete MEM-2.5 and -8 media (UNIT 5.12; Cotter et al., 2017).
-
Selective agents (for XGPRT selection; filter sterilize, and store at −20°C):
10 mg/ml (400×) mycophenolic acid (MPA; Calbiochem) in 0.1 N NaOH
10 mg/ml (40×) xanthine in 0.1 N NaOH
10 mg/ml (670×) hypoxanthine in 0.1 N NaOH
5 mg/ml 5-bromodeoxyuridine (BrdU) in H2O (for TK selection; filter sterilize and store at −20°C)
Dry ice/ethanol bath
Spinner culture of HeLa S3 cells (UNIT 5.12; Cotter et al., 2017)
Cup sonicator (e.g., Ultrasonic Processor VCX-750 from Sonics and Materials)
15-ml conical centrifuge tubes
Sorvall centrifuge with H-6000A rotor (or equivalent)
162-cm2 tissue culture flask
Additional reagents and equipment for tissue culture and counting cells (APPENDIX 3C; Phelan, 2006)
NOTE: All reagents and equipment coming into contact with live cells must be sterile, and aseptic technique should be used accordingly. Incubations should be performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified.
Infect a monolayer culture of cells with virus from a plaque
Place tube containing recombinant virus from a resuspended plaque into a cup sonicator containing ice-water and sonicate 20 to 30 sec at full power. Cool on ice and repeat.
-
Dilute 0.25 ml of lysate from step 1 with 0.75 ml complete MEM-2.5 containing selective agents, if appropriate (see step 3). Infect appropriate confluent monolayer culture in 6-well plate and incubate 30 min.
If XGPRT selection is used, the monolayer culture is preincubated for 12 to 24 hr in complete MEM-2.5 containing MPA, xanthine, and hypoxanthine (see Basic Protocol 2, step 4, substep a). Infection should also be done in the presence of these drugs.
-
Overlay with 1 ml complete MEM-2.5 medium containing the appropriate selective agents.
For XGPRT selection, include 1/400 vol 10 mg/ml MPA, 1/40 vol 10 mg/ml xanthine, and 1/670 vol 10 mg/ml hypoxanthine.
For TK selection, include 1/200 vol of 5 mg/ml BrdU.
Incubate 2 days or until cytopathic effect (cell rounding) is obvious.
Remove and discard 1 ml of the medium covering the cell monolayer. Scrape cells, transfer to microcentrifuge tube.
Perform three freeze-thaw cycles, each time by freezing in a dry ice/ethanol bath, thawing in a 37°C water bath, and vortexing.
Place tube containing cell suspension into a cup sonicator and sonicate 20 to 30 sec at full power.
Scale up the culture
7. Dilute 0.5 ml of lysate from step 6 with 1.5 ml complete MEM-2.5 containing selective agents (see steps 2 and 3). Infect appropriate confluent monolayer culture in a 25-cm2 flask and incubate 30 min.
8. Overlay with 3 ml complete MEM-2.5 containing the appropriate selective agents (step 3). Incubate 2 days or until cytopathic effect is obvious.
9. Scrape cells, transfer to 15-ml conical centrifuge tube, and centrifuge 5 min at 1800 × g (2500 rpm in H-6000A rotor). Resuspend cells in 1 ml complete MEM-2.5 and repeat freeze-thaw cycling and sonication as described in steps 5 and 6.
10. Dilute 0.5 ml of lysate from step 9 with 4.5 ml complete MEM-2.5. Infect appropriate confluent monolayer culture in a 162-cm2 flask and incubate 30 min.
11. Overlay with 25 ml complete MEM-2.5 (selection is not required at this step) and incubate 2-3 days or until cytopathic effect is obvious.
12. Detach cells from the flask by shaking or scraping if necessary. Transfer to centrifuge tube by pipetting, then centrifuge 5 min at 1800 × g. Aspirate and discard supernatant.
13. Resuspend cells in 2 ml complete MEM-2.5 and carry out freeze-thaw cycling three times as described in step 5.
14. Determine titer of the viral stock as described in UNIT 5.12; Cotter et al., 2017. Freeze viral stock at −80°C.
Test for sterility
15. Place 10 μl of lysate from step 13 into 2 ml bacterial growth medium (such as LB) in a 15 ml tube. Shake at 37°C for 1-2 days. Examine the turbidity of the liquid to ensure the absence of bacterial growth.
BASIC PROTOCOL 4 LIVE IMMUNOSTAINING AND AMPLIFICATION OF MVA RECOMBINANTS
Live immunostaining was developed for modified vaccinia virus Ankara (MVA) because this strain does not form discrete, easily recognizable plaques and because this technique helps to avoid the need for incorporation of selectable or screening marker genes. Immunostaining can be used for recombinant proteins that are expressed on the cell surface or in the cytoplasm. These protocols are also applicable to standard strains of vaccinia virus.
The strong adherence of chicken embryo fibroblasts (CEF) to concavalin A–coated plastic dishes make them superior to BHK-21 or other cell lines that the authors have tried.
Materials
162-cm2 flask of confluent CEF (UNIT 5.12; Cotter et al., 2017)
Complete MEM-2.5, and -8 media (UNIT 5.12; Cotter et al., 2017)
Transfected cell lysate (see Basic Protocol 1)
Dry ice/ethanol bath
Primary antibody to protein product of foreign gene
Horseradish peroxidase–conjugated secondary antibody (to species of primary antibody)
Concanavalin A–coated 6-well tissue culture plates (see Support Protocol 3)
Cup sonicator (e.g., Ultrasonic Processor VCX-750 from Sonics and Materials)
Inverted microscope
Sterile toothpicks
Cell scraper or plunger of 1-ml syringe
75- and 162-cm2 tissue culture flasks
Additional reagents and equipment for culture, trypsinization, and immunostaining of CEF cells, titering of MVA, and preparation of MVA stocks (UNIT 5.12; Cotter et al., 2017)
NOTE: All reagents and equipment coming into contact with live cells must be sterile, and aseptic technique should be used accordingly. Incubations should be performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified.
Prepare, infect, and immunostain CEF cells
Trypsinize confluent CEF monolayer cultures, seed cells in concanavalin A–coated 6-well tissue culture plates, and incubate until nearly confluent (UNIT 5.12; Cotter et al., 2017).
Thaw transfected cell lysate (Basic Protocol 1) and sonicate in a cup sonicator in ice-water for 30 sec at full power. Add 100, 10, 1, or 0.1 μl of lysate to duplicate wells of CEF containing 1 ml of complete MEM-2.5 medium. Gently swirl to mix. Incubate 2 hr.
Aspirate virus inoculum and overlay with complete MEM-2.5 containing 2.5% methylcellulose. Incubate 3 days.
-
Immunostain the unfixed cells using antibody to the protein product of the foreign gene (see UNIT 5.12; Cotter et al., 2017, Support Protocol 3, steps 7 to 13).
If the protein of interest is expressed intracellularly, remove the fluid from the wells and place plate at −80°C for 1 hr. Allow to thaw and begin staining as described in UNIT 5.12; Cotter et al., 2017. This procedure ruptures the cells in situ and allows the antibody to penetrate.
Isolate recombinant virus
5. Examine the cell monolayer using an inverted microscope. Touch immunostaining foci with sterile toothpicks. Place toothpicks individually in small vials containing 0.5 ml of complete MEM-2.5, and break off each toothpick so that the sterile part is inside the tube.
6. Perform three freeze/thaw cycles on each tube, each time by freezing in a dry ice/ethanol bath, thawing in a 37°C water bath, and vortexing. Place tube into a cup sonicator containing ice-water and sonicate 30 sec at full power.
-
7. Replate onto new CEF monolayers and plaque purify the MVA recombinant a second time as in steps 2 to 6. Plaque purify an additional three times.
In this protocol, plaque-purification is done under a liquid overlay containing methlycellulose. To check stability and purity, an extra plate can be held for 3 days and then fixed and immunostained as in UNIT 5.12; Cotter et al., 2017, Support Protocol 3. The presence of nonstaining foci, which may be detected at this time because of cytopathic effects, are due to wild-type virus or an unstable recombinant.
Amplify plaque-purified virus
8. Infect 1 well of a 6-well plate of CEF or BHK-21 cells with 0.25 ml of plaque-purified MVA recombinant in a total volume of 2 ml MEM-2.5. Incubate 3 days.
9. Remove and discard 1 ml of medium covering the cell monolayer. Dislodge cells into the 1 ml of remaining medium with a cell scraper (or plunger of a 1 ml syringe) and transfer to a vial. Carry out three freeze/thaw cycles as in step 6 to produce a lysate.
10. Amplify the virus by inoculating 0.5 ml of lysate into 75-cm2 flask of CEF or BHK-21 cells containing 15 ml MEM-2.5. After 3 days harvest and lyse cells as in step 9.
11. Inoculate a 162-cm2 flask of CEF or BHK-21 cells containing 30 ml MEM-2.5 with 0.5 ml of lysate from step 10.
12. Determine titer and prepare large stock of recombinant MVA (UNIT 5.12; Cotter et al., 2017, Support Protocol 3 and Basic Protocol 5). Freeze viral stock in aliquots at −80°C.
SUPPORT PROTOCOL 1 PCR AMPLIFICATION and DNA SEQUENCING OF INSERTED GENE
This protocol describes PCR amplification of the DNA fragment inserted into the recombinant virus. The size of the DNA fragment is confirmed by gel electrophoresis and the integrity of the DNA sequence is then verified by sequencing.
Materials
Titered lysate of recombinant virus (see Basic Protocol 3)
6-well, 35-mm2 tissue culture plate with appropriate confluent monolayer cells (UNIT 5.12; Cotter et al., 2017)
2 ml screw-cap tubes
Qiagen QIAamp DNA blood mini Kit (catalog # 51104)
Appropriate oligonucleotides at the ends of the inserted DNA and as necessary within the inserted gene to allow sequencing of the entire insert
Agarose gel electrophoresis apparatus and gel and means of visualizing DNA
PCR thermal cycler
Infect a monolayer of cells
Infect a monolayer of cells in one well of a 6-well, 35-mm2 tissue culture plate with a multiplicity of infection of 5 plaque forming units of the recombinant virus.
Incubate overnight at 37°C.
Scrape cells with the plunger of a 1 ml syringe and transfer to a 2 ml screw-cap tube.
Follow manufacturer’s instructions (Qiagen) for preparation of DNA.
Amplify the DNA insert using appropriate oligonucleotides and run on agarose gel to verify the size of the insert
Verify nucleotide sequence of the DNA fragment using standard methods.
SUPPORT PROTOCOL 3 COATING PLATES WITH CONCANAVALIN A
This protocol describes the preparation of concanavalin A–coated plates to be used growing CEF monolayers for live immunostaining as in Basic Protocol 4.
Materials
Concanavalin A (Sigma)
Phosphate-buffered saline (PBS)
6-well, 35-mm tissue culture dishes
Plastic bags for storage
NOTE: All reagents and equipment coming into contact with live cells must be sterile, and aseptic technique should be used accordingly.
Add 12 mg concanavalin A to 120 ml sterile PBS for a final concentration of 100 μg/ml.
Add 1 ml of the PBS/concanavalin A solution to each well of twenty 6-well plates. Incubate 1 hr at room temperature.
Aspirate the liquid from each well and rinse with 2 ml PBS.
Remove the fluid from each well and let plates dry by storing open in a biological safety hood (to keep sterile).
Store plates in a plastic bag (to preserve sterility) at room temperature (will keep for months).
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps.
Transfection buffer
0.14 M NaCl
5 mM KCl
1 mM Na2HPO4·2H2O
0.1% (w/v) dextrose
20 mM HEPES
Adjust to pH 7.05 with 0.5 M NaOH and filter sterilize
Store indefinitely at −20°C
The pH of this buffer is critical and should be between 7.0 and 7.1.
COMMENTARY
Background Information
Vaccinia virus has numerous advantages as an expression vector. The virus is easily propagated and has a wide host range in cultured cells and experimental animals; the large genome has the capacity to incorporate at least 25,000 bp (Smith and Moss, 1983); transcription occurs in the cytoplasm bypassing steps in mRNA processing and nuclear-cytoplasmic transport; high levels of expression can be achieved; and efficient recombination facilitates insertion of genes. The basic strategy for construction of recombinant vaccinia viruses using plasmid transfer vectors was described more than 30 years ago (Mackett et al., 1984) and is still the most popular method. An alternative strategy is to prepare the DNA by polymerase chain reaction without cloning in bacteria. Still another option involves recombineering artificial chromosomes (Domi and Moss, 2005). Although the Western Reserve (WR) strain of vaccinia virus has been used most extensively for construction of recombinant viruses, more attenuated strains such as Modified Vaccinia Virus Ankara may be preferred primarily for safety reasons and vaccine purposes.
Plasmid-transfer vectors have three essential components: an expression cassette consisting of a natural or synthetic vaccinia virus promoter, restriction endonuclease sites for insertion of foreign genes, and flanking vaccinia virus DNA that determines the site of homologous recombination. An additional component may provide antibiotic selection or color screening. Various transfer vectors are listed in Table 5.13.1 and one is presented in detail in Figure 15.3.1.
Homologous recombination (Fig. 15.3.1) is the usual way of generating recombinant vaccinia viruses. Initially, recombination may result from a single cross-over event, resulting in integration of the circular plasmid and the creation of a tandem duplication; however, this intermediate is highly unstable. A second recombination event then occurs between the tandem repeats, resolving the structure into a small circular DNA molecule and either a wild-type or recombinant viral genome. An alternative method of direct ligation using an unique restriction endonuclese site in the vaccinia genome has also been described (Pfleiderer et al., 1995; Merchlinsky et al., 1997). This method avoids an E. coli cloning step and can potentially be used for direct cloning of cDNA libraries in vaccinia virus.
Considerable attention has been devoted to promoters because they affect both the time and level of expression. One commonly used promoter, p7.5, contains tandem late and early promoters (Cochran et al., 1985) and provides a moderate level of expression throughout infection. The pmH5 promoter provides higher levels of both early and late expression than p7.5 (Wyatt et al., 1996). Strong natural late promoters include p11 (Bertholet et al., 1985) and pCAE (Patel et al., 1988). About a two-fold increase in expression may be achieved with the synthetic late promoter in pMJ601 and pMJ602 (Davison and Moss, 1990) or the synthetic early/late compound promoter in pSC59 and pSC65 (Chakrabarti et al., 1997; Table 15.3.1).
The ability to synthesize many different kinds of proteins, including those with transmembrane domains, is one advantage of the vaccinia virus expression system. Nevertheless, very high expression of certain genes might adversely affect virus replication. If difficulty is experienced in obtaining recombinant vaccinia with strong promoters, the weaker p7.5 or pmH5 promoter or the inducible vaccinia virus/bacteriophage T7 hybrid system should be tried (ELROY-STEIN AND MOSS, 2001).
Many different methods for selecting recombinant viruses are available. The two simplest, and therefore preferable, methods are selection based on restoration of plaque size or co-expression of a fluorescent protein marker such as green fluorescent protein (GFP). Plaque size selection (Blasco and Moss, 1995) provides an extremely simple method of selecting recombinant vaccinia viruses that involves neither drugs nor reporter genes. The parental virus (vRB12) contains a deletion of most of the F13L gene, which prevents vaccinia virus from making normal-size plaques. The transfer vector pRB21 contains a segment of the F13L gene adjacent to the gene of interest, so that recombinant viruses will have a functional F13L gene and will make normal-size plaques that are easily distinguished from the pin-point plaques of vRB12. Therefore, one merely needs to pick large plaques to isolate recombinant viruses. However, it is important to check that the virus is a double cross-over recombinant. Co-insertion of GFP or other fluorescent protein marker under control of a vaccinia promoter adjacent to the gene of interest allows easy identification of recombinant plaques using a fluorescence microscope. An example is plasmid pLW-44 (Table 15.3.1) in which GFP is stably inserted into the recombinant virus. A modification of this method, transient GFP selection, is performed using a transfer vector in which a portion of one of the two vaccinia virus flanking regions that surround the cassette containing GFP and the foreign gene is also inserted between the GFP and foreign gene (Fig. 15.3.2). Thus, the GFP gene is flanked by direct repeats from one vaccinia virus flank. GFP-expressing plaques are isolated through several rounds of plaque purification to eliminate parental virus. However, the GFP gene is inherently unstable as recombination will readily occur between the two repeat sequences that surround the gene. Subsequent selection of non-GFP-expressing plaques allows isolation of recombinants that express only the foreign gene. Several rounds of plaque purification are necessary to ensure a pure clonal isolate. It is essential that these plaque isolates be characterized by immunostaining, PCR, or other methods to ensure that the desired gene has been incorporated.
Figure 5.13.2.
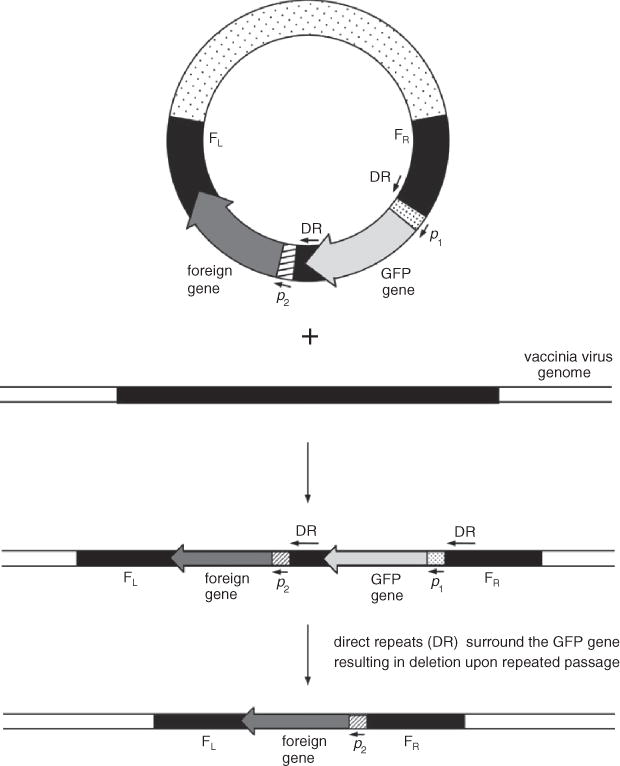
Homologous recombination between a transfected plasmid and the vaccinia virus genome demonstrating transient GFP selection. In the first recombination event, both GFP and the foreign gene are integrated into the vaccinia virus genome. Instability due to the presence of two direct repeats flanking the GFP gene result in a second recombination event upon passage that eliminates the GFP gene but maintains the foreign gene. FL=left flank, FR=right flank, p=promoter
The TK selection method is based on the insertional inactivation of the thymidine kinase gene (Mortensen and Kingston, 2009). In the presence of active TK, added BrdU is phosphorylated and incorporated into viral DNA, where it causes lethal mutations. If TK− cells are used, then TK− virus will replicate normally in the presence of BrdU, whereas TK+ virus will not. Because the product of a single cross-over event is still TK+, only double cross-over events are selected. However, not all TK− viral plaques will be recombinants because spontaneous TK− mutants arise at a frequency of 1:10,000. Depending on the transfection efficiency, recombinants may comprise 10% to >90% of the TK− plaques.
β-galactosidase or β-glucuronidase (GUS) screening is based on the coinsertion of the E. coli lacZ or GUS gene, under the control of a vaccinia virus promoter, into the vaccinia virus genome along with the gene of interest. If the TK gene is insertionally inactivated by such an event, recombinant viruses will be TK− and will make blue plaques on medium containing Xgal or Xgluc. This combination of color screening and TK selection will discriminate TK− recombinants from spontaneous TK− mutants. However, this method has been largely superceded by using fluorescent markers that do not require staining.
XGPRT selection uses mycophenolic acid (MPA), an inhibitor of purine metabolism (see Mortensen and Kingston, 2009). Because MPA blocks the pathway for GMP synthesis, it interferes with the replication of vaccinia and severely reduces the size of vaccinia plaques. This effect can be overcome, however, by expression of the E. coli gpt gene in the presence of xanthine and hypoxanthine (i.e., XGPRT can use either of these as substrate for synthesis of GMP). Thus, coexpression of XGPRT provides a useful selection system. Usually the XGPRT gene is placed adjacent to the gene of interest and within the vaccinia virus DNA flanking sequences. Unlike the TK− situation, the viral products arising from both single and double cross-over events will be selected. Therefore, it is important to do successive plaque isolations so that the single cross-over events will be resolved. A reverse-selection procedure can be used to delete the XGPRT gene from a recombinant vaccinia virus or replace it with another gene (Isaacs et al., 1990). Another procedure, transient XGPRT selection, is performed using a transfer vector with the XGPRT gene outside of the vaccinia virus DNA flanking sequences (Falkner and Moss, 1990). Under these conditions, only the single cross-over recombinant virus will express XGPRT, providing substantial enrichment over the parental virus. However, when MPA selection is removed, the desired double cross-over recombinant virus without the XGPRT gene will have to be differentiated from the parental virus by PCR, immunostaining, or other methods.
To avoid further impairment of MVA, foreign genes have been targeted to existing deletion sites. Deletion II encompasses parts of the HindIII N-K fragments; deletion III lies within the HindIII A fragment (Meyer et al., 1991). The TK gene has been successfully used as an insertion site for MVA (Carroll and Moss, 1997; T. Blanchard, pers. comm.), although there have been reports of some difficulties in isolating stable TK− MVA (Scheiflinger et al., 1996). Since there are no MVA-permissive TK− cell lines, the main attraction for using the TK site is the large number of available transfer vectors that target this site. When some foreign genes were placed under the control of the very strong synthetic early/late promoter, the resulting recombinant MVA were difficult to isolate or unstable (Wyatt et al., 1996). To date, the examples have been membrane proteins. In most cases, stable recombinants could be made by using a promoter of lower strength, e.g., pmH5 or p7.5. When a recombinant protein is somewhat toxic, non-expressing mutants that arise spontaneously can quickly overgrow the culture. To partially circumvent this problem, which has occurred with recombinant genes inserted in deletions II and III, a transfer vector, pLW-73, that directs recombination between two essential genes, I8R and G1L, was developed (Table 15.3.1). Thus, instability resulting from any deletion of the inserted gene that includes part of either of the two essential flanking genes will result in a non-viable virus that cannot overgrow the culture.
Critical Parameters
Calcium phosphate transfections are described in detail in Kingston, Chen, and Rose, 2003. As discussed there, obtaining a very fine precipitate of calcium phosphate and DNA is critical for efficient transfection. Several factors can influence the quality of the precipitate. First, the pH of the transfection buffer should be between 7.0 and 7.1. Second, the CaCl2 should be added slowly and mixed gently (not vortexed), only until full mixing is achieved. The tube should then be left undisturbed until the solution is layered on the cells. Cationic lipids are simpler to use and provide similar or better transfection efficiencies. Although the Lipofectamine procedure was described in this unit, other liposomal or cationic lipids are also effective.
During plaque purification and amplification of a new recombinant virus, it is important to maintain the appropriate selective pressure to prevent growth of any contaminating wild-type virus. After isolation, selection is not required. Check the purity of a recombinant virus by PCR amplification from the flanking DNA sequences. It is also advisable to perform DNA sequencing of the insert to ensure that no mutations have arisen, especially if a PCR product was used for transfection of the foreign gene. Immunostaining is another method that can be used to verify that all plaques/foci express the gene of interest. Non-expressing plaques/foci can be identified by microscopic visualization or double immunostaining. The latter technique requires an antibody to the expressed protein and one to vaccinia with different species specificities. This method has been used routinely with MVA recombinants.
Freezing vaccinia virus stocks causes clumping of particles; thus, stocks should be sonicated after thawing. This is particularly important when plaque-purifying a virus, as each plaque should have arisen from a single virus.
It is prudent to save half of each stock at each step in case of contamination or failure at a succeeding step. The authors strongly recommend preparation of a seed stock of the final recombinant virus that will be adequate for future needs. A common mistake is to continually passage the virus.
If immunostaining is used instead of selection or color screening, the antibody must give a good signal to initially pick out the few small foci of cells infected with recombinant virus among a great excess of cells infected with the parental nonstaining virus. Before using an antibody for isolation of recombinant MVA, it can be tested by staining the MVA-infected cells after the first transfection step.
Anticipated Results
Approximately 1–5 × 1010 pfu of purified virus should be obtained per liter of 5 × 108 HeLa cells.
Depending on the efficiency of the transfection, single, well-isolated plaques should be visible in cells infected with one of the recommended virus dilutions. With TK selection, from 10% to 90% of the plaques will contain recombinant virus. If GFP, β-galactosidase or GUS screening is also used, only fluorescent or blue recombinant virus plaques should be picked. With XGPRT selection, all plaques picked should contain recombinant virus. If the titer of recombinant virus is low, amplification can be achieved by a round of growth in the presence of MPA prior to plaquing (use the procedure described in amplification of a plaque).
Cytopathic effects should be clearly visible at each step of amplification of vaccinia vius except with final infection (infected HeLa cells do not exhibit clear cytopathic effects). The titer of the final crude stock should be 1-2 × 109 pfu/ml.
Time Considerations
The infection/transfection procedure for generation of recombinant virus takes ~6 hr and cells are harvested after 2 days.
Each round of selection and plaque purification takes 2-3 days, although little working time is required. Plaques can be picked and reinfections performed on the same day.
Amplification of a single plaque isolate to a small high-titer crude stock will take ~7 days. Large stocks should then be prepared as described in UNIT 5.12; Cotter et al., 2017.
Transfection of cells with the plasmid transfer vector requires ~2 hr to complete, and the cells are harvested after 2 days. Each round of isolation and plaque purification requires about 30 min for infection, 2 days for incubation, 3 hr for color or immunostaining, and 2 hr for isolating new recombinants and freeze-thaw cycling. Recombinant isolates can be picked and reinfections performed on the same day. From completion of plaque purification to the preparation and titering of a virus stock takes another 10 days.
Acknowledgments
The work was supported by the Division of Intramural Research, NIAID, NIH.
Literature Cited
- Bacik I, Cox JH, Anderson R, Yewdell JW, Bennink JR. TAP (transporter associated with antigen processing)–independent presentation of endogenously synthesized peptides is enhanced by endoplasmic reticulum insertion sequences located at the amino- but not carboxyl-terminus of the peptide. J Immunol. 1994;152:381–387. [PubMed] [Google Scholar]
- Bertholet C, Drillien R, Wittek R. One hundred base pairs of 5′ flanking sequence of a vaccinia virus late gene are sufficient to temporally regulate late transcription. Proc Natl Acad Sci USA. 1985;82:2096–2100. doi: 10.1073/pnas.82.7.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisht H, Roberts A, Vogel L, Bukreyev A, Collins PL, Murphy BR, Subbarao K, Moss B. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc Natl Acad Sci USA. 2004;101:6641–6646. doi: 10.1073/pnas.0401939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco R, Moss B. Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene. 1995;158:157–162. doi: 10.1016/0378-1119(95)00149-z. [DOI] [PubMed] [Google Scholar]
- Carroll MW, Moss B. E. coli β-glucuronidase (GUS) as a marker for recombinant vaccinia viruses. BioTechniques. 1995;19:352–355. [PubMed] [Google Scholar]
- Carroll MW, Moss B. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: Propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology. 1997;238:198–211. doi: 10.1006/viro.1997.8845. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Brechling K, Moss B. Vaccinia virus expression vector: Coexpression of beta-galatosidase provides visual screening of recombinant virus plaques. Mol Cell Biol. 1985;5:3403–3409. doi: 10.1128/mcb.5.12.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Sisler JR, Moss B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. BioTechniques. 1997;23:1094–1097. doi: 10.2144/97236st07. [DOI] [PubMed] [Google Scholar]
- Cochran MA, Puckett C, Moss B. In vitro mutagenesis of the promoter region for a vaccinia virus gene: Evidence for tandem early and late regulatory signals. J Virol. 1985;54:30–37. doi: 10.1128/jvi.54.1.30-37.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter CA, Earl PL, Wyatt LS, Moss B. Preparation of cell cultures and vaccinia virus stocks. Curr Protoc Protein Sci. 2017;89:5.12.1–5.12.18. doi: 10.1002/cpps.34. [DOI] [PubMed] [Google Scholar]
- Davison AJ, Moss B. New vaccinia virus recombination plasmids incorporating a synthetic late promoter for high level expression of foreign proteins. Nucl Acids Res. 1990;18:4285–4286. doi: 10.1093/nar/18.14.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domi A, Moss B. Engineering of a vaccinia virus bacterial artificial chromosome in Escherichia coli by bacteriophage lambda-based recombination. Nature Meth. 2005;2:95–97. doi: 10.1038/nmeth734. [DOI] [PubMed] [Google Scholar]
- Earl P, Koenig S, Moss B. Biological and immunological properties of human immunodeficiency virus type 1 envelope glycoprotein: Analysis of proteins with truncations and deletions expressed by recombinant vaccinia viruses. J Virol. 1990;65:31–41. doi: 10.1128/jvi.65.1.31-41.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elroy-Stein O, Moss B. Gene Expression Using the Vaccinia Virus/T7 RNA Polymerase Hybrid System. Curr Protoc Mol Biol. 2001;43(III):16.19, 16.19.1–16.19.11. doi: 10.1002/0471142727.mb1619s43. [DOI] [PubMed] [Google Scholar]
- Falkner FG, Moss B. Escherichia coli gpt gene provides dominant selection for vaccinia virus open reading frame expression vectors. J Virol. 1988;62:1849–1854. doi: 10.1128/jvi.62.6.1849-1854.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkner FG, Moss B. Transient dominant selection of recombinant vaccinia viruses. J Virol. 1990;64:3108–3111. doi: 10.1128/jvi.64.6.3108-3111.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs SN, Kotwal GJ, Moss B. Reverse guanine phosphoribosyltransferase selection of recombinant vaccinia viruses. Virology. 1990;178:626–630. doi: 10.1016/0042-6822(90)90367-z. [DOI] [PubMed] [Google Scholar]
- Kingston RE, Chen CA, Rose JK. Calcium Phosphate Transfection. Curr Protoc Mol Biol. 2003;63(I):9.1, 9.1.1–9.1.11. doi: 10.1002/0471142727.mb0901s63. [DOI] [PubMed] [Google Scholar]
- Mackett M, Smith GL, Moss B. General method for production and selection of infectious vaccinia virus recombinants expressing foreign genes. J Virol. 1984;49:857–864. doi: 10.1128/jvi.49.3.857-864.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Sutter G, Mayr A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol. 1991;72:1031–1038. doi: 10.1099/0022-1317-72-5-1031. [DOI] [PubMed] [Google Scholar]
- Merchlinsky M, Eckert D, Smith E, Zauderer M. Construction and characterization of vaccinia direct ligation vectors. Virology. 1997;238:444–451. doi: 10.1006/viro.1997.8828. [DOI] [PubMed] [Google Scholar]
- Mortensen RM, Kingston RE. Selection of Transfected Mammalian Cells. Curr Protoc Mol Biol. 2009;86(I):9.5, 9.5.1–9.5.13. doi: 10.1002/0471142727.mb0905s86. [DOI] [PubMed] [Google Scholar]
- Ourmanov I, Brown CR, Moss B, Carroll M, Wyatt L, Pleteva L, Goldstein S, Venson D, Hirsch VM. Comparative efficacy of recombinant Modified Vaccinia Virus Ankara expressing Simian Immunodeficiency Virus (SIV) gag-pol and/or env in macaque challenged with pathogenic SIV. J Virol. 2000;74:2740–2751. doi: 10.1128/jvi.74.6.2740-2751.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DD, Ray CA, Drucker RP, Pickup DJ. A poxvirus-derived vector that directs high levels of expression of cloned genes in mammalian cells. Proc Natl Acad Sci USA. 1988;85:9431–9435. doi: 10.1073/pnas.85.24.9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleiderer M, Falkner FG, Dorner F. A novel vaccinia virus expression system allowing construction of recombinants without the need for selection markers, plasmids and bacterial hosts. J Gen Virol. 1995;76:2957–2962. doi: 10.1099/0022-1317-76-12-2957. [DOI] [PubMed] [Google Scholar]
- Scheiflinger F, Falkner FG, Dorner F. Evaluation of the thymidine kinase (tk) locus as an insertion site in the highly attenuated vaccinia MVA strain. Arch Virol. 1996;141:663–669. doi: 10.1007/BF01718324. [DOI] [PubMed] [Google Scholar]
- Smith GL, Moss B. Infectious poxvirus vectors have capacity for at least 25,000 base pairs of foreign DNA. Gene. 1983;25:21–28. doi: 10.1016/0378-1119(83)90163-4. [DOI] [PubMed] [Google Scholar]
- Sutter G, Wyatt LS, Foley PL, Bennink JR, Moss B. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine. 1994;12:1032–1040. doi: 10.1016/0264-410x(94)90341-7. [DOI] [PubMed] [Google Scholar]
- Wyatt LS, Shors ST, Murphy BR, Moss B. Development of a replication-deficient recombinant vaccinia virus vaccine effective against parainfluenza virus 3 infection in an animal model. Vaccine. 1996;14:1451–1458. doi: 10.1016/s0264-410x(96)00072-2. [DOI] [PubMed] [Google Scholar]
- Wyatt LS, Earl PL, Vogt J, Eller LA, Chandran D, Liu J, Robinson HL, Moss B. Correlation of immunogenicities and in vitro expression levels of recombinant modified vaccinia virus Ankara HIV vaccines. Vaccine. 2008;26:486–493. doi: 10.1016/j.vaccine.2007.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt LS, Earl PL, Xiao W, Americo JA, Cotter CA, Vogt J, Moss B. Elucidating and minimizing the loss by recombinant vaccinia virus of Human Immunodeficiency Virus gene expression resulting from spontaneous mutations and positive selection. J Virol. 2009;83:7176–7184. doi: 10.1128/JVI.00687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]