

Summary
Clinical neurology has benefitted greatly from recent remarkable advances in molecular genetics. In 1991, we could approximate a patient's risk for Huntington disease (HD) based only on linkage analysis. Now, 20 years later, not only can we identify the HD mutation with certainty, we can do the same with several hundred diseases. Whole genome or exome sequencing will soon allow for one-step interrogation of multiple genes for an even larger range of diseases. The recognition of these genes and their associated proteins in combination with new technology has led to creative new approaches to treatment. The challenge for the practicing neurologist is to provide clinically relevant and accurate interpretation of the genetic test results, with successfully treating once “incurable” neurogenetic diseases our ultimate goal.
 |
The tremendous advances in the field of human genetics over the last 3 decades have been referred to as a genetic revolution. In actuality it has been a progressively developing evolution in which knowledge of molecular biology has advanced hand in hand with new technology. Two major landmarks were the first identification of human disease-associated genes in the mid-1980s and the sequencing of the human genome in 2001. These advances have directly affected the practice of medicine in general and the practice of neurology in particular. Here we briefly discuss 5 new things in the field of neurogenetics.
New genes, new diseases, new mechanisms
The first neurologic disease to have its underlying gene identified was Duchenne muscular dystrophy in 1986. Many more followed. In some cases, disease-causing mutations have been found in genes associated with well-established disease entities such as Huntington disease, myotonic muscular dystrophy, Charcot-Marie-Tooth (CMT) neuropathy, and Friedreich ataxia. These rare Mendelian diseases with high penetrance are shown on the left side of figure 1. Trinucleotide repeat expansions and gene duplication and deletion are other ways in which genes can be mutated. In other cases, entirely new diseases have been described on the basis of combining relatively unique phenotypes with their associated genetic basis. Examples of these new diseases are 1) PHARC (peripheral neuropathy, hearing loss, ataxia, retinitis pigmentosa, cataract) associated with mutations in the ABHD7 gene1; 2) sensory neuropathy, dementia, and hearing loss caused by mutations in the DNA methyltransferase 1 gene2; and 3) frontotemporal dementia/amyotrophic lateral sclerosis caused by an expanded nonexpressed hexanucleotide repeat in the C90RF72 gene.3,4 New genetic mechanisms underlying diseases have been discovered such as the derepression of the DUX4 gene in FSH muscular dystrophy5 and a group of diseases, such as myotonic dystrophy, caused by abnormalities in RNA processing.6 We now know that a single phenotype such as cerebellar ataxia or epilepsy can be caused by mutations in more than 30 different genes and, conversely, that different mutations in a single gene can cause numerous phenotypes. One example is the LMNA gene encoding lamin A/C, where both autosomal dominant and recessive mutations exist and can manifest in a diverse array of phenotypes including Emery-Dreyfus muscular dystrophy, congenital muscular dystrophy, CMT, Hutchinson-Gilford progeria, or dilated cardiomyopathy.
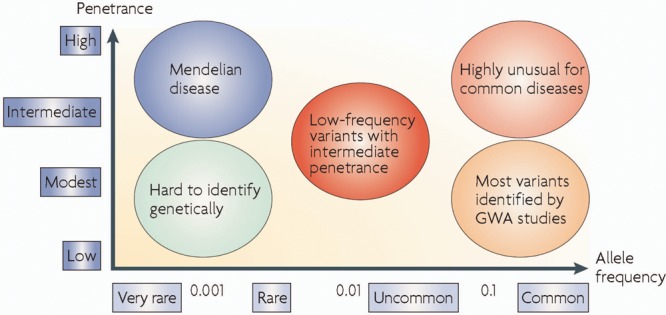
Spectrum of the effect of genetic variation
Figure 1. Genetic diseases and their associated genes can be divided into penetrance (from high to low on the y-axis) and frequency (very rare to common on the x-axis). Single gene Mendelian diseases (such as Huntington disease and muscular dystrophies) are rare, but have high penetrance. Diseases such as Alzheimer disease, Parkinson disease, and stroke are common, but their related risk genes have low penetrance. (From McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty, and challenges. Nat Rev Genet 2008;9:356–369. Reprinted with permission from Macmillan Publishers Ltd: Nature Reviews Genetics.)
DNA segments can now be sequenced in parallel in high throughput formats, allowing for the read of millions of base pairs in hours.
In many instances the newly identified genes code for proteins whose normal functions remain unclear, e.g., amyloid precursor protein associated with Alzheimer disease (AD), prion protein associated with familial Creutzfeldt-Jacob disease, and huntingtin protein associated with HD. Identifying the normal functions of these proteins and the ways in which mutations disturb these functions represent major efforts in biomedical science. However, the normal function of the wild-type protein may be obscure or perhaps irrelevant, as it may be the toxicity of the mutated version of the gene that causes disease. It is this further understanding that will pave the way toward treatments.
Clinical genetic testing
Discovery of new disease-associated genes has led to the clinical application of genetic testing. The explosion in genetic testing is shown in figure 2. In 1995, there were approximately 10 commercially available genetic tests relevant to neurology. Now there are several hundred tests related to all areas of clinical neurology including neuromuscular disorders, dementias, movement disorders, stroke, and white matter diseases. The availability of genetic testing has led to the expansion of disease phenotypes. For example, “senile chorea” has now been found generally to represent late-onset Huntington disease and a late-onset tremor ataxia phenotype (FXTAS) is now known to be part of the spectrum of the fragile X mental retardation syndrome. In addition, chromosomal microarray studies can now identify deletions or duplications in the genome (also known as copy number variants) often associated with childhood developmental disabilities or congenital brain anomalies. These studies have helped identify SHANK2 and SHANK3, synaptic scaffolding genes, as playing a role in autism.7 Microarray studies are now considered first-tier tests for individuals with developmental disabilities.8
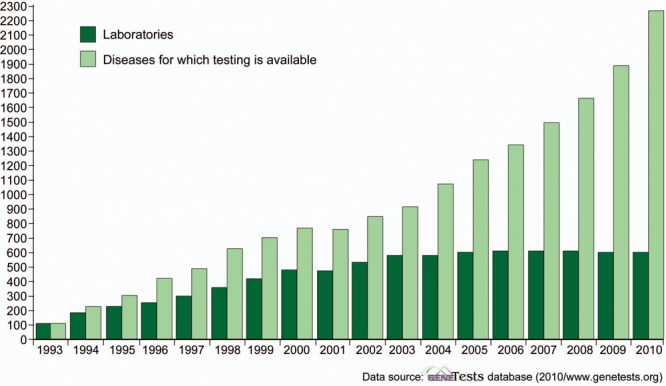
GeneTests: Growth of laboratory directory
Figure 2. From 1993 to 2010 there has been a considerable increase in the number of genetic testing laboratories and the volume of genetic diseases for which testing is available. (From www.genetests.org, 2011.)
In 1995, there were approximately 10 commercially available genetic tests relevant to neurology. Now there are several hundred tests related to all areas of clinical neurology.

The use of these genetic tests is complicated. The selection of appropriate tests depends on a detailed knowledge of differential diagnosis, relative frequency of disease subtypes in the population, test costs, test availability, and time required for the analysis. Attention must also be paid to the reasons for ordering the test. Sometimes a genetic test is used to make a specific diagnosis in an affected person. In fact genetic test results may represent the gold standard for a clinical diagnosis. Alternatively, the test may be used to identify a disease-associated mutation occurring in an asymptomatic family member who is at risk for the disease. Finally, interpretation of genetic test results can be problematic. Testing often reveals DNA changes whose clinical significance is unclear (variants of unknown significance). For these reasons genetic testing frequently requires the expertise of specialists in genetic medicine and genetic counseling. In some cases, consultation with genetic medicine specialists or genetic counselors may not be feasible or timely. When general neurologists pursue genetic testing, we recommend testing the most likely gene candidates first (which can often be identified through reviews in www.genetests.org). Some laboratories do have reflexive panels through which testing can be tiered; most common genes are tested first followed by more exhaustive gene testing if initial tests are negative. Genetic testing can raise complex issues regarding insurance, employment, and other legal, emotional, and socioeconomic factors. The US federal government has recently taken a significant step in protecting individuals from discrimination based upon genetic testing and information. The Genetic Information and Nondiscrimination Act of 2008 (GINA) took effect in 2009. GINA states that individuals may not be discriminated against by employers or health insurers based upon genetic information. There are important exclusions to this rule: 1) life, disability insurance, and long-term care are not included; 2) individuals in the US military, the Veterans Administration, and Indian Health Service systems are not protected; and 3) symptomatic individuals protected by the Americans with Disabilities Act are excluded. Clearly the merging of new genetic testing technology into our daily life logistics is challenging. Other countries are pursuing their own approaches to these issues, though discussion of alternatives is beyond the scope of this review.
Whole genome and whole exome sequencing
Sequencing of the first entire human genome required nearly a decade and 3 billion dollars. Recent technical advances have been so remarkable that the cost is now a few thousand dollars and the technology will soon be available in the practice of medicine. Next-generation sequencing technologies have overtaken traditional Sanger sequencing methods in the study of genome variation. DNA segments can now be sequenced in parallel in high throughput formats, allowing for the read of millions of base pairs in hours—about 100 times faster than earlier approaches used less than 10 years ago. Proof-of-principle studies have demonstrated that whole genome sequencing (WGS) can be successfully employed to identify causative mutations in previously known genes associated with neurologic diseases such as CMT.9 Alternatively, sequencing can be limited to the coding regions of the human genome (exome sequencing) which represents 2% of the entire genome. This approach has led to the identification of a new gene (DYNC1H1) associated with a dominant form of CMT2.10 Since the majority of monogenic diseases caused by single base variations are predicted to occur in exons, whole exome sequencing may be a more feasible approach. Exome sequencing is also being used in research to identify rare or new mutations associated with genetically complex diseases such as autism and AD as well as rare single gene Mendelian diseases. The clinical use of this technology will likely have important consequences for practicing neurologists. Genetic testing may become more straightforward and less expensive. Rather than having to select from a menu of hundreds of genes the clinician may be able to order one test that covers many disease subtypes and all the relevant genes. However, interpretation of the results may be extremely difficult. More variants of unknown significance will be identified; consequently it will be difficult to separate the disease-causing mutation from the genetic background noise (normal variants and polymorphisms). This has been referred to as “the thousand dollar genome with the million dollar analysis.” This interpretation problem will require a great deal of work and attention in molecular analysis.
Genome-wide association studies
Most of the newly discovered genes and diseases discussed here are single gene disorders showing classic Mendelian inheritance patterns of autosomal dominant, autosomal recessive, or X-linked. They are usually rare but have high penetrance. While these causative genes have been successfully identified, investigators are now seeking to recognize the gene variants that may confer risk to disease, perhaps in conjunction with other genetic variants or environmental influences. This has been conceptualized as a spectrum of the effect of genetic variation (figure 1). The monogenic diseases following Mendelian inheritance patterns tend to be caused by highly penetrant and rare mutations, whereas more common variants or polymorphisms (presenting with at least 1%–5% frequency in the general population) may contribute to complex genetic diseases such as sporadic AD and autism spectrum diseases.
Neurogenetics: Five new things
The discovery of new genes, diseases, and mechanisms has led to a greater understanding of disease pathogenesis.
Availability of genetic testing has led to expansion of disease phenotypes.
Whole genome and exome sequencing will likely make genetic testing less expensive but add layers of complexity to test interpretation.
Genome-wide association studies have led to the discovery of genetic variations clearly associated with specific diseases.
Gene replacement, stem cells, and RNA silencing are new approaches in neurogenetic therapeutics.
Genome-wide association studies analyze subsets of the 3 million or so DNA polymorphisms that make us each individuals. By testing 1 million or so single nucleotide polymorphisms (SNPs) in a single test of an individual's DNA, the pattern seen in patients with a common disease (e.g., AD, multiple sclerosis, macular degeneration) can be compared to normal controls, and patterns of polymorphisms associated with the disease state. The polymorphisms associated with the disease are often not pathogenic, but just mark a region of the chromosome where a certain variant of that region likely predisposes to disease. However, in some cases the SNPs can be directly pathogenic. In contrast to whole genome or exome sequencing, genotyping is not performed nucleotide by nucleotide, but only at the SNP locations.
The results of these GWAS studies have been successful. That is, genetic variants have been identified and confirmed that are clearly associated with specific diseases. For example, 10 such genes in addition to APOE have been found to be associated with AD.11 However, the apparent contribution of these genes to the pathophysiology of the associated diseases is relatively small. The incurred risk factors are generally not large enough to be useful with our current technology in the clinic setting. Nevertheless, the identification of these genes and their associated proteins should lead to a better understanding of the biological causes of the diseases.
New neurogenetic therapeutics
It is clear that the leap from disease gene discovery to therapy is daunting. For decades we have treated some neurogenetic diseases without a molecular approach such as the dietary treatments of PKU and Refsum disease and the replacement of pyridoxine in children with pyridoxine-dependent seizures. Now replacement of the abnormal protein with normal protein can be highly beneficial as demonstrated by protein replacement therapy in Pompe (acid maltase deficiency) muscular dystrophy.12 Development of new technologies and delivery methods has broadened our therapeutic approaches to genetic diseases. There are now a multitude of intriguing ideas on how to potentially treat neurogenetic diseases that may prove to be of great benefit in the near future. One such option is gene replacement therapy, which is being pursued in both animal models and humans with Duchenne muscular dystrophy.13 Another possibility is antisense oligonucleotide therapy that is currently in clinical trials for Duchenne muscular dystrophy. Figure 3 shows how this might be accomplished as demonstrated in a mouse model of myotonic dystrophy (DM1).14 Yet another approach is called RNA silencing or interference in which RNA produced by the mutated gene coding the abnormal protein is silenced or destroyed allowing the normal gene and protein (present in persons with autosomal dominant disorders) to pursue its normal function.15
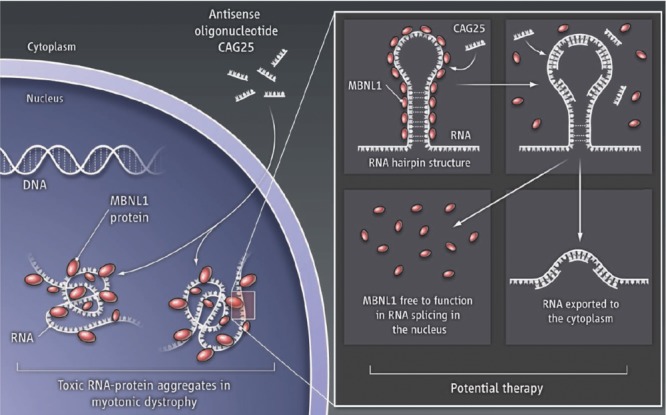
A potential therapeutic approach to myotonic muscular dystrophy (DM1)
Figure 3. Myotonic dystrophy type 1 can result from muscle blind protein (MBNL1) binding to the hairpin RNA produced by the abnormal CTG expansion present in DM1 thereby preventing the normal splicing of other genes (such as that coding for the chloride channel [CLCN1] resulting in myotonia). Normal function could be restored by using an antisense oligonucleotide (CAG25) to displace the muscle blind/RNA binding and free the RNA to be exported to the cytoplasm. (From Cooper TA. Molecular biology: neutralizing toxic RNA. Science 2009;325:272. Reprinted with permission from AAAS.)
Increased understanding of how mutant proteins produce malfunction of the relevant biochemical pathways has led to new ideas for treating 2 common neurogenetic diseases of childhood. The first is tuberous sclerosis (TSC) in which mutations in 2 tumor suppressor genes (hamartin and tuberin) fail to suppress mTOR leading to abnormal cellular growth. Successful treatment of subependymal giant cell astrocytomas in TSC patients with Everolimus, which inhibits mTOR, has been reported.16 The second is fragile X syndrome (FXMR), in which the abnormal fragile X–related protein (FMRP) fails to inhibit overexpression of metabotropic glutamate receptors. Clinical trials are now studying whether mGluR5 antagonists will benefit patients with FXMR.17
The field of stem cell therapeutics is clearly exciting but cannot be reviewed here in detail. An example of the potential of this approach is the successful treatment of the autosomal recessive leukodystrophy in Shiverer mice with pluripotent stem cells with implications for human pediatric myelin disorders.18
Practicing neurologists should follow these unfolding events with fascination and enthusiasm. Through continuing education we need to stay up-to-date on the latest developments in neurogenetics in order to provide our patients with the best diagnosis, care, and management.
Chromosomal microarray testing
A specialized test of patient DNA used for the detection of duplicated or deleted region of DNA that could cause a phenotype or disease.
Copy number variation
Any duplicated or deleted segment of DNA. Copy number variations may be benign or pathogenic depending on size and location in the genome.
Exome
Collective regions of DNA (exons) that are “expressed” into proteins.
Genome-wide association study
The unbiased, discovery approach to determining whether gene polymorphisms are associated with risk of disease.
Next generation sequencing
Technologies focused on sequencing DNA using high throughput programs that include “massively parallel sequencing” or multiple sequencing reactions. New technologies now cut DNA into many small pieces then sequence them many times.
Single nucleotide polymorphism
A polymorphism, or gene variant that involves only 1 nucleotide such as a change from C to T in the DNA sequence. Polymorphisms occur with high prevalence in the general population (at least 1%–5%). There are over 1 million SNPs thus far identified in the human genome.
Whole genome sequencing
A process that sequences base by base the entire genome of an individual. This method includes sequencing not only exomes, but also introns and nongene-associated DNA regions.
Correspondence to: tomnroz@uw.edu
Footnotes
Correspondence to: tomnroz@uw.edu
REFERENCES
- 1. Fiskerstrand T, Brahim DHB, Johansson S, et al.. Mutations in ABHD12 cause the neurodegenerative disease PHARC: an inborn error of endocannabinoid metabolism. Am J Hum Genet 2010;87:410–417. [DOI] [PMC free article] [PubMed]
- 2. Klein CJ, Botuyan MV, Wu Y, et al.. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 2011;43:595–600. [DOI] [PMC free article] [PubMed]
- 3. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al.. Expanded GGGGCC hexanucleotide repeat in noncoding region of C90RF72 causes chromosome 9p-linked FTD and ALS. Neuron Epub 2011. [DOI] [PMC free article] [PubMed]
- 4. Renton AE, Majounie E, Waite A, et al.. A hexanucleotide repeat expansion in C90RF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron Epub 2011. [DOI] [PMC free article] [PubMed]
- 5. Van der Maarel SM, Tawill R, Tapscott SJ. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol Med 2011;17:252–258. [DOI] [PMC free article] [PubMed]
- 6. Todd PK, Paulson HL. RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol 2010;67:291–300. [DOI] [PMC free article] [PubMed]
- 7. Berkel S, Marshall CR, Weiss B, et al.. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nature Genet 2010;42:489–491. [DOI] [PubMed]
- 8. Miller DT, Adam MP, Aradhya S, et al.. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749–764. [DOI] [PMC free article] [PubMed]
- 9. Lupski JR, Reid JG, Gonzaga-Juregui C, et al.. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med 2010;362:1181–1191. [DOI] [PMC free article] [PubMed]
- 10. Weedon MA, Hastings R, Caswell R, et al.. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 2011;89:308–312. [DOI] [PMC free article] [PubMed]
- 11. Naj AC, Jun G, Beecham GW, et al.. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 2011;43:436–441. [DOI] [PMC free article] [PubMed]
- 12. Kishnani PS, Corzo D, Nicolino M, et al.. Recombinant human acid α-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 2007;68:99–109. [DOI] [PubMed]
- 13. Goyenvalle A, Seto JT, Davies KE, Chamberlain J. Therapeutic approaches to muscular dystrophy. Hum Mol Genet 2011;20:R69–R78. [DOI] [PMC free article] [PubMed]
- 14. Wheeler TM, Sobczak K, Lueck JD, et al.. Reversal of RNA dominance by displacement of protein sequestered on triple repeat RNA. Science 2009;325:336–339. [DOI] [PMC free article] [PubMed]
- 15. Gonzalez-Alegre P, Paulson HL. Technology insight: therapeutic RNA interference: how far from the neurology clinic? Nat Clinic Practice Neurol 2007;3:394–404. [DOI] [PubMed]
- 16. Krueger DA, Care MM, Holland K, et al.. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 2010;363:1801–1811. [DOI] [PubMed]
- 17. Hampson DR, Adusei DC, Pacey LKK. The neurochemical basis for the treatment of autism spectrum disorders and fragile X syndrome. Biochem Pharmacol 2011;81:1078–1086. [DOI] [PubMed]
- 18. Goldman SA. Progenitor cell-based treatment of the pediatric myelin disorders. Arch Neurol. 2011;68:848–856. [DOI] [PMC free article] [PubMed]