


Abstract
An essential coordinator of all DNA metabolic processes is Replication Protein A (RPA). RPA orchestrates these processes by binding to single-stranded DNA (ssDNA) and interacting with several other DNA binding proteins. Determining the real-time kinetics of single players such as RPA in the presence of multiple DNA processors to better understand the associated mechanistic events is technically challenging. To overcome this hurdle, we utilized non-canonical amino acids and bio-orthogonal chemistry to site-specifically incorporate a chemical fluorophore onto a single subunit of heterotrimeric RPA. Upon binding to ssDNA, this fluorescent RPA (RPAf) generates a quantifiable change in fluorescence, thus serving as a reporter of its dynamics on DNA in the presence of multiple other DNA binding proteins. Using RPAf, we describe the kinetics of facilitated self-exchange and exchange by Rad51 and mediator proteins during various stages in homologous recombination. RPAf is widely applicable to investigate its mechanism of action in processes such as DNA replication, repair and telomere maintenance.
INTRODUCTION
Enzymes that bind and function on DNA are necessary for all DNA metabolic processes such as replication, recombination, repair, and transcription. Multiple DNA binding proteins function together to catalyze these processes (1). Measuring the DNA binding properties of a single enzyme is relatively straightforward. However, when more than one enzyme is present in the reaction, determining the sequence of binding, and how the presence of one enzyme influences another is required to determine the overall mechanism of action. In multi-protein, steady-state experiments, the contribution of an individual enzyme is often probed by varying the concentration of the target enzyme relative to all other components (including the DNA substrate). Such experiments do not yield the microscopic rate constants of the individual steps in the reaction which are required to decipher the complete mechanism of action. Moreover, transient-kinetic tools are required to capture rapid conformational changes in proteins, and this information sheds light on how the various proteins interact with each other and the DNA template. The use of fluorescently-labeled DNA substrates serve as excellent tools to monitor overall reaction kinetics or to characterize the DNA binding/dissociation dynamics of a single protein (2–5). For example, short oligonucleotides have been labeled with fluorophores to monitor the outcome of DNA recombination (6), assembly/disassembly of Rad51 nucleoprotein filaments (7,8), track protein movement on DNA and RNA (9–11) and to capture DNA unwinding (12,13). Such an approach is limited by the read out from the DNA substrate and the contribution of individual enzymes cannot be interpreted when multiple proteins are present in a reaction.
To capture the sequence of binding and dynamics of each enzyme in a multi-protein reaction, one approach is to obtain a direct, quantifiable signal from a fluorescent label positioned on the individual protein that can undergo a change in fluorescence upon binding to DNA (9,14). The changes in fluorescence help ascertain enzyme dynamics on DNA and how such dynamics are influenced by the presence of other DNA binding proteins in the reaction. Ensemble and single-molecule based fluorescence spectroscopy serve as powerful tools to investigate enzyme function on nucleic acid substrates, but are reliant on the generation of proteins tagged with genetically encoded fluorophores such as eGFP or mCherry (15–17). These tags are large in size, have positional constraints, are likely to interfere with activity, (18) and potentially inhibit protein–protein interactions (19). The attachment of large genetically-encoded fluorophores is particularly problematic for smaller DNA binding proteins such as Rad51, RecA or Dmc1, that function by forming cooperative nucleoprotein filaments on DNA. Finally, attachment of genetically encoded fluorophores is often limited to the N- and C-terminal ends of the candidate proteins. Due to the positional constraints, and their size, they cannot be site-specifically introduced in internal regions of a protein, which is often necessary to capture conformational movements and for Förster Resonance Energy Transfer (FRET) studies (20). Small, chemical fluorophores circumvent many of these issues, and can be site-specifically placed anywhere in the primary sequence of a protein.
The most commonly used approach to site-specifically attach fluorescent probes on proteins relies on maleimide-thiol chemistry to cysteine residues (21). While robust, this approach suffers from the need to generate a single-cysteine bearing functional version of the target protein. This approach is not feasible in proteins where multiple cysteines are important for function or folding. In addition, screening and isolating modifiable cysteines in multi-cysteine proteins is time-intensive. This problem is circumvented using non-canonical amino acids (ncaa), an attractive strategy to engineer site-specific fluorescently labeled versions of proteins without the need to generate a single-cysteine version of a protein (22,23). The ncaa methodology uses an amber suppressor stop codon (UAG) to mark the site of incorporation in the protein of interest. Co-expression of the TAG-construct along with a cognate pair of tRNAAUG and tRNA synthetase, specific to ncaa recognition and incorporation, generates the site-specific ncaa carrying protein. This methodology has been successfully used to directly incorporate fluorescent-ncaa (24–26), or ncaa carrying functional groups that can subsequently be covalently attached to fluorophores using click-chemistry (27–30).
Using the ncaa approach, we sought to address the function of Replication Protein A (RPA), a key enzyme in various DNA metabolic processes (31,32). RPA is essential for survival and functions as a ssDNA binding protein during DNA replication, replication restart, repair, recombination, transcription, and telomere maintenance (33). In addition, RPA has also been shown to resolve R-loop and G-quadruplex secondary structures (34,35). Binding of RPA to ssDNA in the cell also serves as a signalling cue to initiate DNA repair processes acting as a binding platform for the recruitment of various DNA processing enzymes (36). Recent evidence also points to RPA facilitating the re-establishment of chromatin structures after DNA replication and repair (37). In each of these biologically important reactions, RPA functions in the presence of several other proteins capable of binding to DNA, e.g. polymerases, helicases and histones.
RPA plays an indispensable role in homologous recombination (HR), a critical DNA repair pathway that corrects double stranded DNA (dsDNA) breaks in the genome (33,36,38). Defective HR leads to genomic rearrangements and chromosomal defects often resulting in hereditary cancers and cancer-prone diseases such as Fanconi Anemia and Bloom's syndrome (39,40). In addition to DNA repair, HR is fundamental to the maintenance of gametic diversity during meiotic crossover events. During HR, dsDNA at the site of a break is resected to yield long stretches of ssDNA, which serve as a template for the nucleation of the Rad51 recombinase and formation of nucleoprotein filaments. Rad51 functions as the central engine in HR by performing ATP-dependent strand exchange (41,42). In the cell, the resected ssDNA is rapidly coated by RPA to protect it from nucleolytic degradation, and this step has been shown to activate the DNA damage response through several checkpoint kinases (36). To promote HR, RPA must first be displaced from the ssDNA, thus allowing formation of the Rad51 nucleoprotein filament. Pro-recombinogenic mediator proteins such as Rad52 and BRCA2 have been shown to promote Rad51 binding to ssDNA by displacing RPA (43–45). The Rad51 nucleoprotein filament then catalyzes strand exchange to drive HR. Similarly, in other instances where HR is inhibited, Rad51 filaments are disassembled by anti-recombinogenic mediators such as the Srs2 helicase (7,46,47). In Rad51 filament clearing reactions, RPA is proposed to sequester naked ssDNA once the Rad51 molecules are removed by Srs2 (48). While RPA has been shown to regulate several steps in HR, its precise mode of action in these events is not clearly understood.
The complexity of RPA function in multi-protein contexts necessitates the need for a site-specifically labeled fluorescent RPA probe to tease out its mechanism of action in the presence of other DNA binding proteins. The technical complexity in site-specifically labeling RPA arises from its heterotrimeric arrangement. RPA is composed of three subunits—RPA70, RPA32 and RPA14, and contains six oligosaccharide-oligonucleotide folds (OB-folds) and four DNA-binding domains (DBD) (49,50). Three of the DBD’s reside on the large RPA70 subunit and the fourth one lies in the RPA32 subunit (Figure 1A). The DBD’s are connected by flexible linkers that enable RPA to exist in multiple conformational states on DNA (51). The individual DBD’s play disparate roles in ssDNA binding and have been shown to modulate the strength of RPA–ssDNA interactions and/or its sliding/diffusion on DNA (52). DBD’s A and B in RPA70 are the dominant contributors that cooperatively bind to ssDNA with high affinity (Ka > 2 × 107 M−1) (53). The order of DBD binding and their orientation on ssDNA controls the polarity of RPA and associated activities (54–56). Our understanding of how RPA binds to DNA stems from elegant biophysical characterization of its kinetic and thermodynamic properties (52,57–60). How these properties are affected in the presence of other proteins such as the Rad51 recombinase or mediator proteins such as Rad52 or BRCA2 remains poorly resolved. Recent advancements in single-molecule fluorescence microscopy has enabled the study of RPA dynamics on long DNA substrates in the presence of multiple DNA binding proteins, (61–63) but are limited by resolution of the imaging technology. Thus far, these experiments have been reliant on genetically encoded fluorophores with the limitations described above. Our ncaa approach overcomes several of these limitations and enables the study of RPA dynamics in multi-protein reactions.
Figure 1.
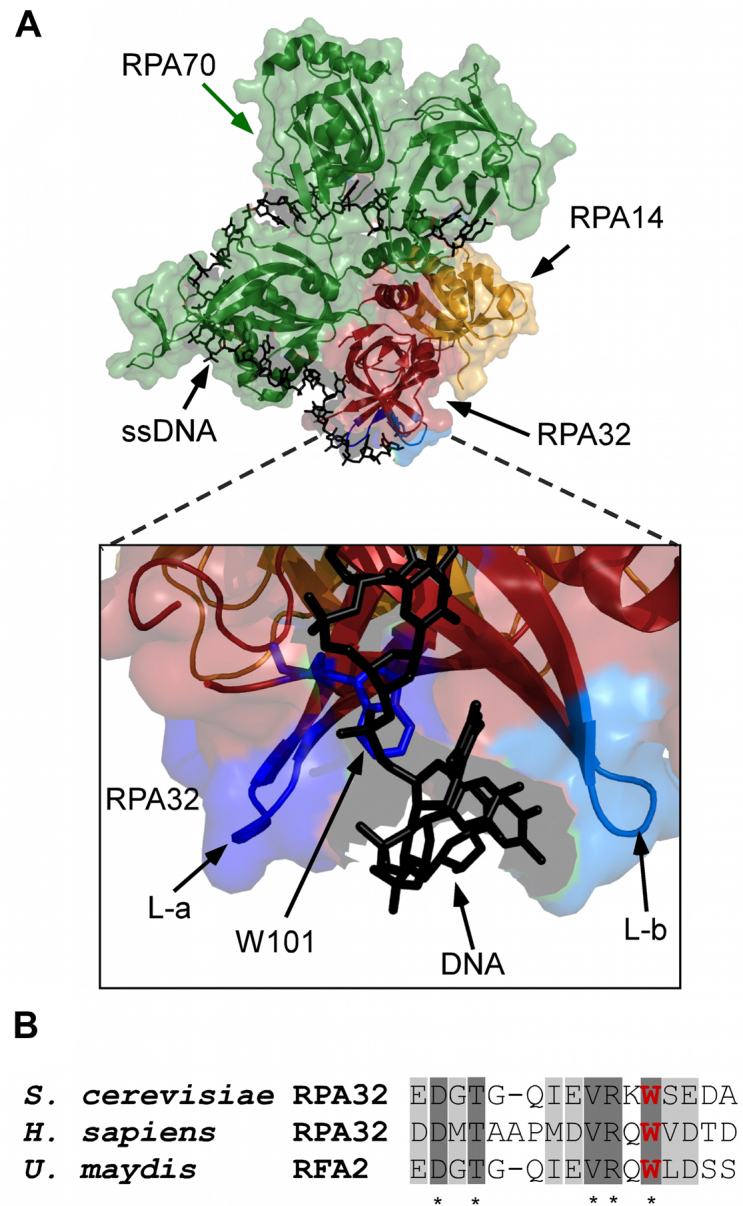
Position of non-canonical amino acid insertion in RPA. (A) Crystal structure of Ustilago maydis RPA bound to ssDNA is shown (PDB ID: 4GOP) with RPA70, RPA32 and RPA14 colored green, red and yellow, respectively. The zoomed-in image shows two loops, L-a and L-b, flanking the ssDNA (black sticks) and Trp-101 is shown as stick representation in blue. (B) Conservation of amino acid sequence in the region where Trp-101 resides in RPA32. W101 is highlighted in bold (red).
Here, we describe (a) the development of a Saccharomyces cerevisiae RPA probe (RPAf), labeled at a single site in the heterotrimeric complex using a combination of ncaa and bio-orthogonal chemistry. This methodology circumvents the need for maleimide chemistry to covalently attach fluorophores or create a single-cysteine version of the protein, thereby leaving all Cys residues in RPA intact and rendering a fully functional protein. (b) This approach also enables us to perform single molecule total internal reflection fluorescence (TIRF) microscopy-based investigation of protein dynamics without the need for genetically encoded fluorophores. (c) In addition, to facilitate cost-effective incorporation of this methodology, we describe a synthesis strategy for economical, large-scale production of the 4-azidophenylalanine ncaa. (d) We have identified a unique position in the RPA32 subunit which enables strategic attachment of a fluorescent reporter that does not affect RPA function, but yields a quantifiable change in fluorescence upon binding to ssDNA. (e) Finally, using RPAf, we describe the kinetics of RPA binding, dissociation, facilitated self-exchange, and facilitated exchange in the presence of Rad51 and mediator proteins.
MATERIALS AND METHODS
Materials
Standard laboratory chemicals and protease inhibitor cocktail (PIC) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Research Products International (Mt. Prospect, IL, USA). Q-sepharose, Heparin and S200 size exclusion chromatography resins were from GE Healthcare (Pittsburgh, PA, USA). Ni2+-NTA agarose was from Gold Biotechnology (St. Louis, MO). Biogel-P4 resin was from Bio-Rad Laboratories (Hercules, CA, USA). Enzymes for molecular Biology were from New England Biolabs (Ipswich, MA, USA). Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). Fmoc-4-amino-phenylalanine was from Angene International Ltd. (Nanjing, China). Commercial 4-azidophenylalanine was purchased from Chem-Impex International (Wood Dale, IL, USA). MB543 DBCO was purchased from Click Chemistry Tools (Scottsdale, AZ, USA). Alexa Fluor 594 DIBO alkyne and BL21Ai cells were purchased from ThermoFisher Scientific (Waltham, MA, USA).
Plasmids for protein overexpression and ncaa incorporation
The plasmid expressing all three subunits of RPA (p11d-tscRPA) was a kind gift from Dr Marc Wold (University of Iowa). The amber suppression stop codon (TAG) in RPA14 was substituted with an ochre stop codon (TAA), and a C-terminal 6X polyhistidine tag was incorporated in RPA32 using Q5 site directed mutagenesis (New England Biolabs, Ipswich, MA). Finally, a single TAG stop codon was introduced at position W101 in the RPA32 subunit marking the site for incorporation of p-azido-l-phenylalanine (4AZP). Plasmids for 4AZP incorporation are as described (29,30,64).
Synthesis of 4-azidophenylalanine (4AZP)
A detailed procedure for the economical synthesis of pure 4AZP from Fmoc-4-aminophenylalanine is described in the Supplemental Methods.
Expression and purification of proteins
Wild type RPA (RPAWT) was overexpressed in BL21Ai cells containing plasmid p11d-tscRPA and purified as described (65) with the following modifications. 4L Luria-broth cultures were grown for each protein preparation. Cells were induced with 0.4 mM IPTG and 0.05% (w/v) l-arabinose when they reached OD600 = 0.6, and grown for an additional 3 h at 37 °C. Harvested cells were resuspended in 120 ml cell resuspension buffer (30 mM HEPES, pH 7.8, 300 mM KCl, 0.1 mM EDTA, protease inhibitor cocktail, 1 mM PMSF, 10% (v/v) glycerol and 10 mM imidazole). Cells were lysed using 400 μg/ml lysozyme followed by sonication. Clarified lysates were fractionated on a Ni2+-NTA agarose column. Protein was eluted using cell resuspension buffer containing 400 mM imidazole. Fractions containing RPAWT were pooled and diluted three-fold with buffer H0 (30 mM HEPES, pH 7.8, 0.1 mM EDTA, 1 mM DTT and 10% (v/v) glycerol). The diluted protein sample was then fractionated over a Q-sepharose column equilibrated with buffer H100 (buffer H0 with 100 mM KCl). Protein was eluted with a linear gradient H100 – H400 (superscript denotes final KCl concentration in the buffer). Fractions containing RPAWT were pooled and diluted with H0 buffer to match the conductivity of buffer H100, and further fractionated over a Heparin column. Protein was eluted using a linear gradient H100–H1000, and fractions containing RPAWT were pooled and concentrated using an Amicon spin concentrator (30 kDa molecular weight cut-off). RPAWT was dialyzed into storage buffer (30 mM HEPES, pH 7.8, 30 mM KCl, 2 mM DTT and 10% (v/v) glycerol), flash frozen using liquid nitrogen, and stored at –80°C. RPAWT concentration was measured spectroscopically using ε280 = 98 500 M−1cm−1.
To obtain RPA carrying 4AZP (RPA4AZP), the p11d-tscRPA-TAG32-101 plasmid, which contains the TAG at position 101 in the RPA32 subunit, was cotransformed into BL21Ai cells with the plasmid pDule2-pCNF containing the orthogonal tRNAUAG and tRNA synthetase for 4AZP (Figure 2A) (29,30). Cotransformants were selected using both ampicillin (100 μg/ml) and spectinomycin (50 μg/ml). An overnight culture (50 ml) from a single colony was grown in LB media containing ampicillin and spectinomycin. 10 ml of the overnight culture was added to 1 L of minimal media. The minimal media for ncaa incorporation was prepared as previously described, (64) but lactose was excluded. Cells were grown at 37 °C until the OD600 reached 2.0 and then induced with 0.4 mM IPTG and 0.05% l-arabinose along with 1 mM 4AZP. The 4AZP solution was prepared by first dissolving 206 mg in 250 μl of 5 M NaOH (or 2 ml MeOH), vortexed extensively, and then adjusted to 8 ml with H2O, and the entire mixture was added to 1 L of media to achieve a 1 mM final concentration. Induction was carried out at 37 °C for 3 h. Cells were resuspended in cell resuspension buffer and purified as described above for RPAWT. Care should be taken not to add DTT during purification of RPA4AZP as it interferes with downstream click chemistry reactions. RPA4AZP was flash frozen and stored at –80°C. Srs2 and Rad51 were purified as described (7). Escherichia coli SSB was purified as described (66).
Figure 2.

Insertion of ncaa and bio-orthogonal labeling of RPA. (A) Plasmids used for the overexpression of RPA and ncaa components. The three subunits of RPA are cloned into a pET vector and the RPA32 subunit is engineered to carry a C-terminal polyhistidine tag and a TAG inserted for the incorporation of 4AZP. The genes for the tRNA that recognizes the amber suppressor codon and inserts 4AZP (tRNACUA) and its corresponding tRNA synthetase are engineered into the pDULE2-pCNF plasmid. (B) SDS-PAGE analysis of RPAWT, RPA4ZAP and the MB543-labeled RPA (RPAf)proteins are shown after coommassie staining (left) or fluorescence imaging (right). Site-specific fluorescence labeling of the RPA32 subunit is observed.
Bio-orthogonal labeling of RPA
∼2.5 ml of 20 μM RPA4AZP was incubated with 1.5 molar excess (30 μM) of DBCO-MB543 (an alkyne derivatized fluorophore) in labeling buffer (30 mM HEPES, pH 7.8, 300 mM KCl and 10% (v/v) glycerol) for 1 h at 4 °C. Labeled RPA (RPAf) was separated from excess dye using Biogel-P4 gel filtration (50 cm × 2 cm bed volume), in storage buffer (30 mM HEPES, pH 7.8, 30 mM KCl and 10% (v/v) glycerol). Labeling efficiency was calculated using absorption values at 280 and 550 nm and ε280 = 98500 and ε550 = 105000 for RPA and MB543 fluorophore, respectively. When measuring the concentration of RPAf, a correction factor of 0.127 was applied to the protein absorbance value at 280 nm to correct for the contribution of MB543 dye absorbance at 280 nm.
Fluorescence measurements
Fluorescence spectra were obtained using a PTI QM40 instrument (Horiba Scientific, Edison, NJ, USA). 100 nM of ssDNA [(dT)97] or plasmid dsDNA (100 nM nucleotides) were added to quartz cuvettes containing 2 ml solutions of 100 nM RPAf in reaction buffer (30 mM HEPES, pH 7.8, 100 mM KCl, 5 mM MgCl2, 1 mM β-mercaptoethanol and 6% (v/v) glycerol). Samples were excited at 535 nm and emission spectra (555–580 nm) were recorded. All experiments were performed at 25 °C.
DNA binding
The DNA binding activity of RPAf was measured using electromobility band shift analysis. 50 nM of 32P-labeled (dT)35 oligonucleotide was incubated with increasing concentrations of RPAWT, RPA4AZP or RPAf (0–1 μM) in reaction buffer for 10 min at 4 °C. 1 μl of DNA loading dye (50% (v/v) glycerol and 0.2% (w/v) bromophenol blue in 1 × TBE) was added to the samples and resolved using an 8% TBE–acrylamide gel (110 V, 25 °C). Gels were exposed overnight onto a phosphorimaging screen and scanned using a STORM scanner (GE Healthcare, Pittsburgh, PA, USA). Bound and unbound DNA signals were quantitated using ImageQuant software and the fraction ssDNA bound to RPA was calculated using the equation:
 |
(1) |
Stopped-flow assays
All stopped-flow experiments described below to monitor RPAf dynamics were performed on an Applied Photophysics SX20 instrument (Surrey, UK) in reaction buffer (30 mM HEPES, pH 7.8, 100 mM KCl, 5 mM MgCl2, 1 mM β-mercaptoethanol and 6% (v/v) glycerol) at 25 °C. Samples were excited at 535 nm and emission was monitored using a 555 nm cut-off filter (Newport corp., Irvine, CA, USA).
RPAf-ssDNA binding kinetics
To quantify RPAf binding to ssDNA, 100 nM RPAf was rapidly mixed with 30 nm (dT)97 oligonucleotide (post-mixing concentrations). Assuming a binding site size of 18–20 nt for RPA (59), we expect 4–5 RPA molecules to occupy each (dT)97 oligonucleotide in our experiments. Data were fit to a single-exponential plus linear equation to obtain observed rate constants:
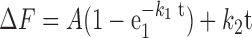 |
(2) |
where ΔF is change in RPAf fluorescence, A and k1 are the amplitude and observed rate of the exponential phase, k2 is the steady state rate and t is time. To measure non-specific changes in RPAf fluorescence in the presence of other proteins, 100 nM RPAf was mixed with Rad51 (0.97 μM) or Srs2 (200 nM). No change in fluorescence was observed. Post-mixing concentrations of proteins are noted.
RPAf facilitated exchange kinetics
RPAf-(dT)97 complexes were preformed using 200 nM RPAf and 60 nM (dT)97 in one syringe and rapidly mixed with increasing concentrations of RPAWT from the second syringe (100–1000 nM) and the change in RPAf fluorescence was monitored. Facilitated exchange experiments with Escherichia coli SSB were assayed similarly, where preformed RPAf-(dT)97 complexes were challenged with SSB (40–200 nM). Both data were analysed using Equation (2) to obtain observed rate constants. kobs,1 was plotted against [RPA] and a linear fit was used to generate a rate for the facilitated exchange processes.
RPAf kinetics during homologous recombination
To quantitate the dynamics of RPAf in the presence of Rad51, RPAf-(dT)97 complexes were performed as described above and challenged with Rad51 (0.97 μM; post-mixing concentration) in the absence or presence of ATP (5 mM). Assuming a binding site size of 3.3 nt/Rad51, there are ∼30 Rad51 binding sites per (dT)97 substrate. To saturate ∼900 nM Rad51 binding sites we used 970 nM Rad51 in our experiments. Rad51 displaces RPAf in the presence of ATP (Figure 6B) and the change in fluorescence was fit using a double exponential equation:
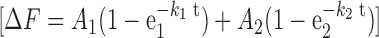 |
(3) |
Figure 6.

Dynamics of RPAf during stages of homologous recombination. (A) Schematic of Rad51 binding to RPAf–ssDNA complexes. (B) Preformed RPAf–ssDNA complexes are not disrupted by Rad51 in the absence of ATP (gray trace), but effectively displaced in the presence of ATP (orange trace). The data is well described by a double exponential fit yielding kobs,1 = 0.26 ± 0.08 s−1 and kobs,2 = 0.02 ± 0.004 s−1. (C) Schematic of stopped-flow experiments to observe the effect of Srs2 on RPAf–ssDNA complex stability. The green arrow depicts potential rebinding of RPAf in the reaction. (D) Increasing concentrations of Srs2 show a small change in the fluorescence signal, but no significant change in overall fluorescence is observed. Insert shows an exponential phase with an observed rate constant of 8.5 ± 0.8 s−1. (E) Schematic of events during filament disassembly. The green arrows denote removal of RPAf upon Rad51 rebinding. (F) In filament clearing experiments, a preformed Rad51 filament prevents RPAf from binding to ssDNA (blue trace), however, when Srs2 is present in the reaction, a gradual increase in fluorescence is observed (pink trace) highlighting clearing of Rad51 molecules from ssDNA by Srs2 followed by RPAf binding to ssDNA. The data displays a single exponential profile (dotted line) with kobs = 0.005 ± 0.001 s−1.
The analysis yields two observed rate constants. Similarly, displacement of RPAf by Srs2 was measured by challenging RPAf-(dT)97 complexes with increasing concentrations of Srs2 (100 or 200 nM; post-mixing). Data were well described by Equation (2) and yielded observed rate constants for the process. Finally, the ability of RPAf to disrupt Rad51-ssDNA nucleoprotein filaments in the presence or absence of Srs2 was investigated by premixing Rad51 (1.94 μM) with (dT)97 (60 nM) in one syringe and challenging the complex with RPAf (200 nM) in the absence or presence of Srs2 (200 nM) [all concentrations pre-mixing]. 5 mM ATP was present in both reactions. The filament clearing data in the presence of Srs2 is described by Equation (2).
Tryptophan quenching experiments to obtain RPA–ssDNA binding kinetics
Intrinsic tryptophan fluorescence was used to capture RPA binding to ssDNA in reaction buffer. 100 nM RPAWT or RPAf was rapidly mixed with 30 nM (dT)97 oligonucleotide and the change in Trp fluorescence was monitored by exciting the sample at 290 nm and measuring emission with a 350 nm cut-off filter. To obtain the association rates for RPAWT and RPAf, similar intrinsic tryptophan quenching stopped-flow experiments were performed with 100 nM RPAWT or RPAf (post-mixing concentrations) and increasing concentrations of (dT)35. All tryptophan quenching stopped-flow data were fit using Equation (2) to obtain the observed rate constants for RPA–DNA binding.
Single molecule DNA curtain assays to monitor RPA facilitated exchange
ssDNA curtains were prepared and visualized by total internal reflection fluorescence microscopy (TIRFM), as previously described, with the exception that GFP-RPA was replaced with RPAf (63,67). Briefly, a lipid bilayer is built upon a glass support with nanofabricated chromium barriers and pedestals inside a microfluidic flowcell that allows for buffer exchange. Long ssDNA were generated using rolling circle replication with Phi29 DNA polymerase from a circular ssDNA template (M13mp18) primed with a 5′-biotinylated oligonucleotide. ssDNA is then tethered to the lipid bilayer through a biotin-streptavidin-biotin linkage and pushed against the nanofabricated barrier with hydrodynamic force. Flowing HR buffer (30mM Tris-acetate, pH 7.5, 100 mM KCl, 5 mM Mg(OAc)2, 1 mM DTT, 2 mM ATP and 0.2 mg/ml BSA) containing 100 pM RPAf allows for visualization of ssDNA and a second attachment point is made through non-specific interaction of the 3′ end of ssDNA to a chromium pedestal to keep the ssDNA in the evanescent TIRF field. Presynaptic complex assembly was initiated by injecting 2 μM Rad51 in HR buffer lacking RPAf. Disassembly of the presynaptic complex was initiated by flushing the sample chamber with HR buffer with 100 pM RPAf lacking ATP as previously described (63,67).
RESULTS
Generation of fluorescent RPAf using non-canonical amino acids and bio-orthogonal chemistry
A fluorescent version of RPA that produces a change in signal upon binding to ssDNA would facilitate investigation of its binding dynamics in the presence of multiple DNA binding proteins. To obtain a fluorescent version of RPA (RPAf) that does not contain a large protein-based fluorophore, we used a combination of ncaa incorporation and bio-orthogonal chemistry. RPA is composed of three subunits – RPA70, RPA32 and RPA14, where the number corresponds to their respective molecular weights. There are 14 total cysteine residues in Saccharomyces cerevisiae RPA and the ncaa methodology circumvents the need to generate a cysteine-free version of the protein for site-specific labeling. We used the crystal structure of Ustilago maydis RPA as a guide for the positioning of the ncaa (Figure 1A), (49) and selected a position that resides close to the DNA binding interface in the RPA32 subunit. Both the RPA70 and RPA32 subunits interact with DNA through conserved DNA binding domains, and a greater degree of contacts reside in the larger RPA70 subunit (Figure 1A). We chose to insert the ncaa at position W101 in RPA32 due to its proximity to DNA in the crystal structure. Strong sequence conservation in this region is also observed (Figure 1B), suggesting that this region might be responsive to conformational changes upon binding to ssDNA. In addition, the region is situated away from terminal portions of RPA70 and RPA32, which are known to mediate protein–protein interactions (31).
We used p-azido-l-phenylalanine (4AZP) as the ncaa as it can be readily coupled to commercially available alkyne-fluorophores using click chemistry. A C-terminal polyhistidine tag was engineered into RPA32 to separate out 4AZP carrying RPA from prematurely truncated protein that is formed when the UAG is read as a stop codon during protein expression. The RPA plasmid was coexpressed with a cognate pair of amber suppressor tRNA and amino-acyl tRNA synthetase specific for the incorporation of 4AZP. RPA4AZP purified as a single complex (Figure 2A, B) and similar to the wild type RPA protein sedimented as a single species in sedimentation velocity experiments with apparent molecular weights consistent with a heterotrimer (Supplemental Figure 1).
Site specific incorporation of 4AZP at position W101 in RPA32 was confirmed by subjecting the protein to LC-MS analysis after tryptic digestion. Further MS-MS fragment analysis obtained using a linear ion trap generate spectra confirming the presence of peptides corresponding to RPA32 carrying 4AZP at position 101 (Supplemental Figure 2). The peptide contains amino acids RK right before the site of 4AZP incorporation. Since trypsin cuts after both R and K, we observe both the shortest peptide, with the K cut off, and a missed cleavage fragment, starting with K. The data also confirm the presence of peptides with 4AZP at position 101 and no tryptophan containing peptide (at 101) was observed. Peptides containing 4-aminophenylalanine at position 101 were also detected. Laser-induced degradation of azide (–N3) to amine (–NH2) occurs during MS analysis (68) and such chemical conversions do not occur during cell growth, as established for 4AZP incorporation in other systems (69–71).
Fluorescent RPA (RPAf) was generated by incubating RPA4AZP with dibenzocyclooctyne (DBCO) functionalized MB543 fluorescent dye, which covalently tethered the fluorophore onto the protein through strain-promoted azide-alkyne cycloaddition. Site-specific fluorescent labeling of RPAf was confirmed by analyzing the protein on SDS-PAGE, where only the RPA32 subunit is detected upon fluorescence imaging (Figure 2B). RPAf sediments as a stable trimer in sedimentation velocity experiments suggesting that fluorescence labeling does not alter the overall structure of the protein complex (Supplemental Figure 1). The copper-free coupling reaction yielded ∼50-65% labeling efficiencies and had no deleterious effects on the integrity of the protein, as evaluated by SDS-PAGE analysis (Figure 2B).
In reactions where copper-based click chemistry was attempted with 5-FAM-alkyne in the presence of 0.1 mM CuSO4, 0.5 mM Tris(3-hydroxypropyltriazolylmethyl)amine, 5 mM sodium ascorbate and 5 mM amino guanidine, no labeling of RPA4AZP was achieved and RPA4AZP showed severe degree of degradation over time in our reaction conditions (data not shown). For Saccharomyces cerevisiae RPA4AZP, DBCO functionalized fluorophores appear to be ideally suited under our reaction conditions because we do not observe non-specific labeling (Figure 2B). We also report a new procedure for the chemical synthesis of 4AZP from Fmoc-4-aminophenylalanine which enables more cost-efficient utilization of site-specific fluorescent labeling applications. This chromatography-free protocol utilizes reliable Sandmeyer chemistry that is easily scalable. Analytically pure material is obtained after recrystallization of the final product (Supplementary Methods).
ssDNA binding properties of RPAf
We next examined the spectral properties of RPAf to determine whether a change in fluorescence is observed upon binding to ssDNA. The excitation–emission spectra of RPAf show maximal fluorescence excitation and emission at 555 nm (λex) and 566 nm (λem), respectively (Figure 3A). RPA binds ssDNA selectively, rapidly, and with high affinity; and RPAf binding to ssDNA produces a ∼5% enhancement in total fluorescence (Figure 3B). No change in fluorescence is observed in the presence of dsDNA due to the lack of RPA binding. Next, to assess whether the positioning of the fluorophore interfered with the ssDNA binding properties of RPA, we compared the DNA binding affinities of RPAWT and RPAf using electrophoretic mobility shift analysis. Titration of increasing amounts of RPAWT or RPAf with a 32P-labeled (dT)35 oligonucleotide generated a RPA–ssDNA complex visible as a slower migrating band in the gel (Figure 3C). Quantitation of the bandshifts show that both proteins bind stoichiometrically to the (dT)35 ssDNA substrate (Figure 3D). RPA carrying the incorporated 4AZP (RPA4AZP) also binds to (dT)35 with similar affinity (Supplemental Figure 3). Therefore, positioning of the fluorophore at position 101 in the RPA32 subunit does not interfere with ssDNA binding affinity.
Figure 3.

ssDNA binding properties of RPAf. (A) Excitation and emission spectra of RPAf show maximum values at 555 nm (λex) and 566 nm (λem). (B) RPAf was excited at 535 nm and emission spectra were recorded in the absence or presence of ssDNA [(dT)97] orplasmid dsDNA. A ∼5% increase in fluorescence signal is observed when ssDNA is present in the reaction. (C) Electromobility band shift analysis (EMSA) of RPAWT (top) and RPAf (bottom) binding to 50 nM 32P-labeled (dT)35 oligonucleotides show bound and unbound complexes and (D) quantitation of the EMSA data show stoichiometric binding to ssDNA for both RPA and RPAf.
RPA binds rapidly to ssDNA, and on longer ssDNA substrates multiple RPA molecules bind and form a protein coated filament. Occluded ssDNA binding site sizes of 18–20 nt and 25–26 nt at low (0.02 M) versus high (1.5 M) NaCl concentrations have been reported for yeast RPA (59). Our reactions are performed at 100 mM KCl, where a 18–20 nt site size is expected. Thus, on a (dT)97 oligonucleotide, ∼4–5 RPA molecules could bind. To observe the kinetics of RPAf binding to ssDNA, we performed a stopped-flow fluorescence experiment by rapidly mixing RPAf with a (dT)97 oligonucleotide (Figure 4A). The reaction was excited at 535 nm and emission monitored using a 555 nm long pass filter. The data displays rapid (kobs = 23 ± 1.2 s−1) and slow phases (kobs = 0.003 ± 0.0006 s−1) of change in fluorescence signal upon binding to ssDNA (Figure 4B and C). To compare the ssDNA binding kinetics of RPAf with the RPAWT protein, we monitored changes in intrinsic tryptophan fluorescence of RPAWT and RPAf (Figure 4D and E). Both RPA proteins bind rapidly to (dT)97, and two binding phases are observed. The rapid phase shows kobs,1 = 28 ± 1.8 s−1 and 32 ± 3.8 for RPAWT and RPAf, respectively (Figure 4D and E). The slower phase shows kobs,2 = 0.014 ± 0.004 s−1 and 0.008 ± 0.003 for RPAWT and RPAf, respectively (Figure 4D and E).
Figure 4.

Kinetics of RPA binding to ssDNA. (A) Schematic of stopped-flow experiment to capture RPA–ssDNA binding kinetics. (B) A rapid change in RPAf fluorescence is observed upon binding to a (dT)97 ssDNA oligonucleotide (red trace), whereas no change in fluorescence is observed in the absence of DNA (black trace). (C) Fit of the stopped-flow data (dashed blue line) show the presence of a rapid (kobs,1 = 23 ± 1.2 s−1) and slow phase (kobs,2 = 0.003 ± 0.0006 s−1) for ssDNA dependent changes in RPAf fluorescence. Intrinsic tryptophan fluorescence changes in (D) RPAWT and (E) RPAf upon binding to ssDNA reveal rapid changes in fluorescence and fit of the data yield kobs,1 = 28 ± 1.8 s−1, kobs,2 = 0.014 ± 0.004 s−1 for RPAWT, and kobs,1 = 32 ± 3.8 s−1, kobs,2 = 0.008 ± 0.003 s−1 for RPAf, respectively. (F) Free Rad51 or Srs2 in the reaction do not affect the basal fluorescence of RPAf.
To test if the second phase reflected reorganization of multiple RPA molecules on the (dT)97 substrate, we performed these experiments on a shorter DNA substrate (dT)35. Given the occluded site-size of RPA (18–20 nt/RPA), less than two RPA molecules will bind to a (dT)35 oligonucleotide. If kobs,2 reports on Trp fluorescence changes associated with rearrangement of multiple RPA molecules on the (dT)97 substrate, then on a (dT)35 substrate kobs,2 should be significantly slower. We determined the association rate of RPAWT and RPAf by monitoring the change in Trp fluorescence as a function of increasing (dT)35 oligonucleotide concentration (Supplemental Figure 4). Under these conditions, kobs,2 is slower (0.002–0.006 s−1) compared to our observations on (dT)97 (0.014 and 0.008 s−1; Figure 4). More importantly, these rates do not change with increasing DNA concentration. The data also yield association rates for RPAWT and RPAf, and the values are very similar for both proteins: 5.2 ± 0.3 × 10−10 M−1s−1 versus 5.6 ± 0.5 × 10−10 M−1s−1 for RPAWT and RPAf, respectively (Supplemental Figure 4). These data suggest that the ssDNA binding kinetics are similar between RPAWT and RPAf protein. It should be noted that in the experiments with RPAf where fluorescence changes are monitored at 555 nm (Figure 4B and C), only conformational changes in RPA32, close to the single fluorophore, are observed upon binding to ssDNA; whereas, in the Trp-quenching experiments, global conformational changes in both RPA70 and RPA32 are captured. The smallest RPA14 subunit is not thought to interact with DNA (49). These experimental differences might account for the small variations in the two observed rate constants.
To test whether RPAf fluorescence is influenced by other proteins, we mixed it with Rad51 or Srs2 and monitored the change in fluorescence. We observe no change in the fluorescence signal when Rad51 or Srs2 is present in the reaction (Figure 4F). Rad51 and RPA have been shown to interact in the absence of DNA, (72) whereas no physical interactions have been reported between RPA and Srs2, but are known to work together in Rad51 clearing reactions (46). Both RPAWT and RPAf physically interact with Rad51 (Supplemental Figure 5). These experiments suggest that RPAf is fully functional, retains the ability to interact with Rad51, and can be used to investigate RPA–DNA binding dynamics in multi-protein reactions.
Facilitated exchange of RPAf by RPA and SSB
RPA bound on ssDNA has been shown to undergo facilitated exchange with free RPA in the reaction (61). To test whether RPAf can be utilized to capture such dynamics on ssDNA we performed stopped-flow experiments where preformed RPAf–ssDNA filaments were challenged with increasing concentrations of RPAWT or Rad51. In the first series of experiments, RPAf -(dT)97 complexes were preformed by incubating RPAf and (dT)97 oligonucleotides and rapidly mixed with increasing concentrations of RPAWT (unlabeled). The resulting change in fluorescence shows a rapid drop in fluorescence and both the observed rate constant and amplitude of the signal change increases with free RPA concentration (Figure 5A and B). A plot of the kobs versus free RPA concentration yields an apparent rate for the facilitated exchange process (0.7 ± 0.1 × 10−12 M−1 s−1; Figure 5C). A hypothetical model has been proposed for facilitated exchange where the four OB-folds of RPA can be remodelled individually, while allowing the complex to remain on the ssDNA (61). The precise mechanism of how this self-propagated facilitated exchange occurs is poorly understood. Escherichia coli SSB, the functional homolog of RPA in prokaryotes, can also catalyze facilitated exchange of RPA (61). In fact, we observe faster and effective displacement of RPAf by E. coli SSB with an apparent facilitated exchange rate of 47.3 ± 1.7 × 10−12 M−1 s−1 (Figure 5D–F). While both RPA and SSB are homologs, they are structurally unrelated and bind to DNA using different mechanisms (73). The differences between SSB-induced versus facilitated self-exchange of RPA remain to be explored.
Figure 5.

Facilitated exchange of RPAf on ssDNA by RPA and SSB. (A) Schematic of stopped-flow experiments to capture facilitated exchange of RPAf by unlabeled RPAWT. (B) Pre-formed RPAf-ssDNA complexes formed on (dT)97 ssDNA substrates are effectively displaced by unlabeled RPAWT. The rate of exchange increases with concentration of unlabeled RPAWT, and (C) yields an apparent observed rate of 0.7 ± 0.1 × 10−12 M−1 s−1 for facilitated exchange. (D) Schematic of facilitated exchange experiments with E. coli SSB, which (E) displaces or exchanges with RPAf more effectively than RPAWT with (F) an apparent observed rate of 47.3 ± 1.7 × 10−12 M−1 s−1.
Dynamics of RPAf during homologous recombination events
RPA-coated ssDNA serve as the foundation for the nucleation and growth of Rad51 presynaptic filaments in homologous recombination. Rad52 is a mediator protein that functions to promote the exchange of RPA for Rad51 on the DNA (43). However, in single molecule DNA curtain experiments, Rad51 displaces RPA in the absence of Rad52 (62). Rad51 binds to ssDNA in the presence of ATP and forms stable complexes (7). Rad51 binding to a Cy3-labeled DNA substrate results in an increase in fluorescence. Using this assay, we first measured the rate of Rad51 binding to free 5′-Cy3-(dT)79 oligonucleotide (kobs,1 = 1.3 ± 0.3 s−1; and kobs,2 = 0.008 ± 0.001 s−1; Supplemental Figure 6). To investigate RPAf dynamics during Rad51 nucleoprotein filament formation, we preformed RPAf-(dT)97 complexes and challenged them with Rad51 in the absence or presence of ATP (Figure 6A). In the absence of ATP, there is no change in the fluorescence signal, as yeast Rad51 does not form a complex with ssDNA in the absence of ATP and hence no nucleoprotein filament formation is expected, thus is not able to displace RPAf (Figure 6B). In the presence of ATP, Rad51 displaces RPAf as it forms a filament on the ssDNA substrate. The change in fluorescence signal shows RPAf being dissociated in two distinct steps with kobs,1 = 0.26 ± 0.08 s−1 and kobs,2 = 0.02 ± 0.004 s−1 (Figure 6B). Whether the two rate constants reflect Rad51 nucleation and growth, respectively, remains to be established. In the absence of other mediator proteins such as Rad52, our data show that Rad51 can form a stable nucleoprotein filament with rapid kinetics on RPA-coated ssDNA.
The Srs2 helicase/translocase is an anti-HR mediator and functions by disassembling Rad51 nucleoprotein filaments (47). Srs2 is a motor protein in yeast and uses ATP to translocate along ssDNA substrate and capable of unwinding dsDNA (7,14,74,75). Single stranded DNA binding proteins such as SSB and RPA can diffuse along ssDNA substrates and other DNA binding proteins have been shown to modulate this sliding behaviour to displace SSB/RPA from DNA (10,52). We tested if a translocase such as Srs2 would be able to displace RPAf from ssDNA. When preformed RPAf-(dT)97 complexes are rapidly mixed with Srs2 and ATP, an initial drop in fluorescence is observed followed by signal stabilization (Figure 6C and D). When the concentration of Srs2 is doubled in the reaction to 200 nM (post-mixing), the fluorescence signal does not appreciably change as in experiments with 100 nM Srs2. The early exponential drop in signal yields kobs = 8.3 ± 1.2 s−1 and 8.5 ± 0.8 s−1 for 100 and 200 nM Srs2 in the reaction, respectively (Figure 6D). These data suggest that there is displacement of RPAf by Srs2. However, Srs2 is either rapidly outcompeted on ssDNA by the dissociated RPAf, or RPAf rapidly rebinds the DNA after it's removed by Srs2.
RPA has been shown to promote Rad51 filament disassembly by Srs2 and is proposed to sequester the ssDNA substrate following Rad51 dissociation (47) To test this model, we preformed Rad51 nucleoprotein filaments on a (dT)97 substrate and rapidly mixed it with RPAf in the absence or presence of Srs2 (Figure 6E). In the absence of Srs2, no RPAf binding is observed, however, when Srs2 is present in the reaction, an increase in fluorescence is observed (kobs = 0.005 ± 0.002 s−1; Figure 6F). These data suggest that Srs2 displaces Rad51 and enables binding of RPAf to the free ssDNA. However, it should be noted that Rad51 displaces RPA (Figure 6B), hence the apparent rate for filament clearing would also be severely influenced by RPAf removal by Rad51. The presence of mediator proteins during homologous recombination might enable stabilization of RPA on the ssDNA.
Single molecule DNA curtain analysis of RPAf dynamics in HR
Using ncaa to generate fluorescent DNA binding proteins will be useful in single molecule analysis of enzyme reactions because it overcomes the need to attach genetically encoded fluorophores. This approach is vital to studying proteins such as Rad51 and RecA, which are rendered inactive when GFP is tethered to the termini (18). We have previously shown that GFP- or mCherry-tagged RPA can be used for preparing and visualizing ssDNA curtains for single molecule studies of HR intermediates (62). As a proof of principle, we next sought to determine whether RPAf could also be used in double-tethered ssDNA curtain measurements to visualize the RPAf-ssDNA complexes and monitor assembly of the Rad51 presynaptic complex (Figure 7A and B). For these experiments, long ssDNA substrates (∼50 knt) were loaded into a sample chamber and tethered to a lipid bilayer through a biotin–streptavidin linkage that also serves to prevent non-specific protein binding to the surface of the flowcell. Flushing the chamber with buffer containing 100 pM RPAf revealed ssDNA-RPAf complexes when visualized by total internal reflection fluorescence microscopy (TIRFM; Figure 7B). To further establish if these are bonafide ssDNA–RPAf complexes, we next tested whether Rad51 is able to displace RPAf. Injection of 2 μM wild type Rad51 (dark/unlabeled) in the presence of 2 mM ATP resulted in the rapid loss of fluorescence signal as RPAf is displaced from the ssDNA (Figure 7D). Consistent with previous findings, (62) these presynaptic Rad51 filaments remained stable on the ssDNA, even after flushing unbound Rad51 and introducing 100 pM free RPAf into solution for 30 min. Switching to a buffer that is identical except that it lacks ATP shows rapid return of RPAf signal as the Rad51-ADP dissociates and is replaced by RPAf (Figure 7D). These experiments highlight the utility of RPAf for use in single molecule experiments of recombination intermediates where large genetically-encoded fluorescent tags could potentially confound measurements or affect protein function.
Figure 7.

ssDNA curtains to visualize RPAf. (A) Schematic of a RPA-coated double-tethered ssDNA curtain. (B) Representative examples of individual ssDNA molecules bound by RPAf (shown in magenta). The 5′-biotin tether is oriented at the top of each window that are all 2.7 × 13.5 μm. (C) Schematic showing the ssDNA curtain experiment time course beginning in the top panel with an RPA-coated ssDNA molecule. Injecting 2 μM Rad51 displaces RPAf from the ssDNA to form the pre-synaptic filament that is resistant to rebinding of RPAf in the middle panel. Flushing ATP from the system results in spontaneous Rad51 dissociation and the re-binding of RPAf. (D) A representative kymograph of a ssDNA molecule through time. At the start, ssDNA is already coated by RPAf and buffer containing 100 pM RPAf is flowing through the chamber at 0.2 ml/min. Switching to a buffer lacking RPAf shows that bound RPAf remains stable until the introduction of 2 μM wild type Rad51 when flow is stopped. The loss of RPAf signal is evidence that Rad51 outcompetes the bound RPAf to form a pre-synaptic filament. Resuming flow with buffer containing 100 pM RPAf and 2 mM ATP shows the ssDNA remains dark as RPAf cannot displace Rad51. However, switching to buffer with 100 pM RPAf and no ATP shows assembly and disassembly of a wild type Rad51 filament on an RPAf-coated ssDNA molecule.
DISCUSSION
Multiple DNA binding proteins function together during all DNA metabolic process such as replication and repair. To investigate the mechanism of action of a single enzyme that functions in a multi-protein context, a quantifiable experimental signal from the enzyme of interest is required. In this study, we generated a fluorescent version of RPA (RPAf) utilizing the ncaa methodology. 4AZP was used as the ncaa and inserted at position 101 in the RPA32 subunit. W101 in one of two aromatic residues in RPA32 proposed to interact with DNA. However, mutation of both aromatic residues does not affect DNA replication as the mutant cells show normal growth rates (76). A slight sensitivity to UV damage is observed in the double mutant (77,78). The MB543 fluorescent dye was covalently attached to RPA4AZP using strain-promoted azide–alkyne cycloaddition. Upon binding to ssDNA, RPAf generates a robust and quantifiable change in florescence, and the DNA binding properties of RPAf are similar to RPAWT including stoichiometric and rapid binding to ssDNA (Figures 3 and 4; and Supplemental Figure 4). This approach leaves all the cysteine residues intact and generates functional RPA complexes. This methodology is an attractive alternative to using genetically-encoded fluorophores which are large, interfere with function, and have positional limitations for attachment.
RPA is a unique protein that functions as a control hub to recruit various proteins, thereby coordinating almost all DNA metabolic processes in the cell (79). By monitoring the change in fluorescence signal in RPAf we can selectively investigate its dynamics in multi-protein reactions such as HR. Here, we have captured the kinetics of RPA binding, dissociation, and facilitated exchange during HR in in the presence of the Rad51 recombinase and Srs2, an anti-recombination mediator. Multiple DNA binding proteins are required to orchestrate HR and are present together during various steps in the reaction. The 5′ ends flanking a dsDNA break are nucleolytically cleaved during resection to yield ssDNA overhangs that are sequestered by RPA (80,81). We show that RPAf binds rapidly to free ssDNA (kobs = 23 ± 1.2 s−1; Figure 4C) and self-exchanges at a 6–7-fold slower rate (kobs = 3–4 s−1; Figure 5C). These data show that RPA bound to ssDNA are stable, as described previously (82). During pre-synapsis, RPA bound to ssDNA needs to be displaced for Rad51 to bind and form the nucleoprotein filament. Pro-recombination mediator proteins such as Rad52 and BRCA2 are known to promote the binding of Rad51 on RPA-coated ssDNA (43–45). RPA interacts with both Rad52 and BRCA2 and assembly of Rad51 on RPA-coated ssDNA is regulated through post-translational modifications (83–85). The observed rate of Rad51 binding and filament formation on naked ssDNA is ∼1.3 s−1 (Supplemental Figure 6), whereas on RPAf coated DNA, Rad51 binds and displaces RPAf at ∼0.26 s−1 (Figure 6B). These data suggest that Rad51 displaces RPA at ∼15-fold slower rate compared to the RPA facilitated self-exchange process. The difference in rate constants suggest that the mechanism of Rad51 binding to an RPA-coated ssDNA is different compared to facilitated self-exchange. The four OB-folds of RPA bind to ssDNA with different affinities and can be individually remodeled by other proteins to gain access to ssDNA (51,61). Such differences in DNA binding affinities within the OB-folds allow RPA to be tightly bound on the ssDNA, while allowing access to other DNA binding proteins that they recruit during various DNA metabolic processes. However, for complete RPA displacement, all four OB-folds will have to dissociate from ssDNA.
During facilitated self-exchange, RPAf could dissociate from the ends or internal regions and the vacant ssDNA binding site can now be occupied by unlabeled RPA. On the other hand, RPA has also been shown to diffuse on ssDNA (52) and could be pushed off by free RPA or other proteins (63). Our data suggests that an active sliding-pushing off mechanism might be more applicable as we observe different observed rate constants for facilitated-self exchange (∼3.5 s−1; Figure 5C), facilitated exchange with SSB (∼17 s−1: Figure 5F), and displacement by Rad51 (0.26 s−1; Figure 6B). However, a passive mechanism, where the individual DNA binding domains of RPA dissociates followed by replacement with another DNA binding protein, cannot be ruled out. In our experiments, a (dT)97 ssDNA substrate is used where both ends of the DNA are free. Whether RPA can be removed effectively by other proteins in cases where the ends are blocked, as in a replication fork or dsDNA bound by histones, remains to be investigated. Sliding and diffusion models of RPA movement on ssDNA have been suggested and how these models fit into mechanisms of facilitated self-exchange versus Rad51-mediated displacement remain to be explored (52,61). While in vitro assays show that mediators such as Rad52 are not required to displace RPA from ssDNA, (62) mediators can enhance the rate of Rad51 nucleation and growth in HR reactions where other DNA structures and proteins are present.
Rad51 nucleoprotein filaments promote the search for homology and catalyze strand-exchange (synapsis) in HR followed by replication and strand resolution. The dynamics between Rad51 and RPA during pre-synapsis is of immense interest as mutations in Rad51, RPA, BRCA2 and Rad52 have all been linked to various cancerous states (86,87). To capture the kinetics of Rad51 nucleation and filament growth in the presence of RPA and mediator proteins, we are currently developing ncaa-based fluorescent Rad51 to directly investigate its dynamics in HR. In other scenarios, Rad51 filaments are displaced by anti-HR mediator proteins such as the Srs2 helicase. In the presence of ATP, Rad51 forms a stable nucleoprotein filament on ssDNA substrates that is refractory to RPA rebinding (Figure 7C). Previous studies have shown that Srs2 displaces Rad51 by stimulating ATP hydrolysis within the nucleoprotein filament and the reaction is enhanced in the presence of RPA (7,74,88). Based on these studies, current models posit RPA sequestering the ssDNA behind the Srs2 helicase as it clears Rad51 molecules (48). We have measured the binding of RPAf behind the Srs2 helicase during Rad51 filament clearing (Figure 6E). RPAf binding in this context occurs at kobs = 0.005 s−1, much slower than the rates of Srs2 filament clearing previously reported (7,14). Since Rad51 can displace RPAf in our experiments (Figure 6B), RPAf molecules that bind behind the Srs2 helicase will be exchanged by free Rad51 in the reaction. Thus, how RPA is stabilized during filament clearing remains to be established. One possibility is the contribution of a mediator protein that might stabilize RPA binding to DNA or post-translational modifications of Rad51 could prevent its rebinding or exchanging RPA. Interestingly, our results suggest that a motor protein such as Srs2 is capable of displacing RPAf from ssDNA. RPAf–ssDNA complexes challenged with Srs2 show a small, but rapid initial drop in RPAf fluorescence (kobs = 8.5 ± 0.8 s−1; Figure 6D) followed by a plateauing of the signal. This suggests that RPAf being displaced is either rapidly able to compete off Srs2 from ssDNA or binds to the free ssDNA behind the Srs2 helicase, causing the stabilization in fluorescence signal (Figure 6C and D). Single molecule fluorescence experiments will be required to better understand these mechanisms and the ncaa methodology can be applied to generate fluorescently labeled proteins.
We show the application of RPAf in single molecule DNA curtain assays where dynamics of RPA on long ∼50 knt substrates are captured (Figure 7). Assembly of RPAf molecules can be visualized and its displacement during facilitated self-exchange can be quantitated. Flowing in Rad51 with ATP displaces RPAf and formation of the nucleoprotein filament is observed. When ATP is washed out of the reaction, Rad51 dissociates and binding of RPAf is clearly visualized. The development of fluorescent Rad51 and mediator proteins will be applicable to monitoring their individual dynamics during HR and other DNA metabolic processes.
In summary, we have used RPAf to describe its dynamics during HR. Since RPA coordinates several DNA metabolic processes, this approach now presents a powerful experimental tool to investigate its dynamics in DNA replication, replication restart, nucleotide excision repair, dynamics on telomeric ends, and other such processes on DNA. The use of ncaa to generate fluorescently-labeled proteins should be broadly applicable to investigating other processes in multi-protein systems. In addition, this methodology enables click chemistry based attachment of dyes that will enable us to investigate conformational changes in this region using EPR and NMR specific probes.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Marc Wold (University of Iowa) for the RPA protein expression plasmid, and Drs. Michael Thomas and Michael Pereckas at the Medical College of Wisconsin for mass spectrometry analysis. We thank Dr. Tom Chang, Jaya Shrestha and Dr. Nicholas Dickenson at Utah State University for preliminary synthesis of 4AZP and assistance with setting up analytical ultracentrifugation experiments.
Footnotes
Present address: Eric Parker Davenport, Applied Genetic Technologies Corporation (AGTC), Alachua, FL 32615, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [7R15GM110671 to E.A., R15HL127636 to C.D., R35GM118026 to E.C.G]; National Science Foundation [MCB-1518265 to R.A.M]; Instrumentation used to obtain AUC data were supported by funds from the National Science Foundation [MRI-1625667]. Funding for open access charge: National Institutes of Health [7R15GM110671 to E.A.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Siebert G., Humphrey G.B.. Enzymology of the nucleus. Adv. Enzymol. Related Areas Mol. Biol. 1965; 27:239–288. [DOI] [PubMed] [Google Scholar]
- 2. Anderson B.J., Larkin C., Guja K., Schildbach J.F.. Using fluorophore-labeled oligonucleotides to measure affinities of protein-DNA interactions. Methods Enzymol. 2008; 450:253–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Valuchova S., Fulnecek J., Petrov A.P., Tripsianes K., Riha K.. A rapid method for detecting protein-nucleic acid interactions by protein induced fluorescence enhancement. Scientific Rep. 2016; 6:39653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Song D., Graham T.G., Loparo J.J.. A general approach to visualize protein binding and DNA conformation without protein labelling. Nat. Commun. 2016; 7:10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hwang H., Myong S.. Protein induced fluorescence enhancement (PIFE) for probing protein-nucleic acid interactions. Chem. Soc. Rev. 2014; 43:1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ragunathan K., Joo C., Ha T.. Real-time observation of strand exchange reaction with high spatiotemporal resolution. Structure. 2011; 19:1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Antony E., Tomko E.J., Xiao Q., Krejci L., Lohman T.M., Ellenberger T.. Srs2 disassembles Rad51 filaments by a protein–protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol. Cell. 2009; 35:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taylor M.R., Spirek M., Chaurasiya K.R., Ward J.D., Carzaniga R., Yu X., Egelman E.H., Collinson L.M., Rueda D., Krejci L. et al. . Rad51 paralogs remodel pre-synaptic Rad51 filaments to stimulate homologous recombination. Cell. 2015; 162:271–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fischer C.J., Lohman T.M.. ATP-dependent translocation of proteins along single-stranded DNA: models and methods of analysis of pre-steady state kinetics. J. Mol. Biol. 2004; 344:1265–1286. [DOI] [PubMed] [Google Scholar]
- 10. Rasnik I., Myong S., Cheng W., Lohman T.M., Ha T.. DNA-binding orientation and domain conformation of the E. coli rep helicase monomer bound to a partial duplex junction: single-molecule studies of fluorescently labeled enzymes. J. Mol. Biol. 2004; 336:395–408. [DOI] [PubMed] [Google Scholar]
- 11. Lucius A.L., Vindigni A., Gregorian R., Ali J.A., Taylor A.F., Smith G.R., Lohman T.M.. DNA unwinding step-size of E. coli RecBCD helicase determined from single turnover chemical quenched-flow kinetic studies. J. Mol. Biol. 2002; 324:409–428. [DOI] [PubMed] [Google Scholar]
- 12. Fischer C.J., Tomko E.J., Wu C.G., Lohman T.M.. Fluorescence methods to study DNA translocation and unwinding kinetics by nucleic acid motors. Methods Mol. Biol. 2012; 875:85–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bjornson K.P., Amaratunga M., Moore K.J., Lohman T.M.. Single-turnover kinetics of helicase-catalyzed DNA unwinding monitored continuously by fluorescence energy transfer. Biochemistry. 1994; 33:14306–14316. [DOI] [PubMed] [Google Scholar]
- 14. Davenport E.P., Harris D.F., Origanti S., Antony E.. Rad51 nucleoprotein filament disassembly captured using fluorescent Plasmodium falciparum SSB as a reporter for single-stranded DNA. PLoS One. 2016; 11:e0159242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collins B.E., Ye L.F., Duzdevich D., Greene E.C.. DNA curtains: novel tools for imaging protein-nucleic acid interactions at the single-molecule level. Methods Cell Biol. 2014; 123:217–234. [DOI] [PubMed] [Google Scholar]
- 16. Redding S., Greene E.C.. How do proteins locate specific targets in DNA. Chem. Phys. Lett. 2013; 570:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gorman J., Wang F., Redding S., Plys A.J., Fazio T., Wind S., Alani E.E., Greene E.C.. Single-molecule imaging reveals target-search mechanisms during DNA mismatch repair. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E3074–E3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fischer C.J., Wooten L., Tomko E.J., Lohman T.M.. Kinetics of motor protein translocation on single-stranded DNA. Methods Mol. Biol. 2010; 587:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lisby M., Barlow J.H., Burgess R.C., Rothstein R.. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004; 118:699–713. [DOI] [PubMed] [Google Scholar]
- 20. Hillisch A., Lorenz M., Diekmann S.. Recent advances in FRET: distance determination in protein-DNA complexes. Curr. Opin. Struct. Biol. 2001; 11:201–207. [DOI] [PubMed] [Google Scholar]
- 21. Fox C.F., Kennedy E.P.. Specific labeling and partial purification of the M protein, a component of the beta-galactoside transport system of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1965; 54:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu C.C., Schultz P.G.. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010; 79:413–444. [DOI] [PubMed] [Google Scholar]
- 23. Young T.S., Schultz P.G.. Beyond the canonical 20 amino acids: expanding the genetic lexicon. J. Biol. Chem. 2010; 285:11039–11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chatterjee A., Guo J., Lee H.S., Schultz P.G.. A genetically encoded fluorescent probe in mammalian cells. J. Am. Chem. Soc. 2013; 135:12540–12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee H.S., Guo J., Lemke E.A., Dimla R.D., Schultz P.G.. Genetic incorporation of a small, environmentally sensitive, fluorescent probe into proteins in Saccharomyces cerevisiae. J. Am. Chem. Soc. 2009; 131:12921–12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Summerer D., Chen S., Wu N., Deiters A., Chin J.W., Schultz P.G.. A genetically encoded fluorescent amino acid. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:9785–9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leisle L., Valiyaveetil F., Mehl R.A., Ahern C.A.. Incorporation of non-canonical amino acids. Adv. Exp. Med. Biol. 2015; 869:119–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blizzard R.J., Backus D.R., Brown W., Bazewicz C.G., Li Y., Mehl R.A.. Ideal bioorthogonal reactions using A site-specifically encoded tetrazine amino acid. J. Am. Chem. Soc. 2015; 137:10044–10047. [DOI] [PubMed] [Google Scholar]
- 29. Peeler J.C., Mehl R.A.. Site-specific incorporation of unnatural amino acids as probes for protein conformational changes. Methods Mol. Biol. 2012; 794:125–134. [DOI] [PubMed] [Google Scholar]
- 30. Miyake-Stoner S.J., Miller A.M., Hammill J.T., Peeler J.C., Hess K.R., Mehl R.A., Brewer S.H.. Probing protein folding using site-specifically encoded unnatural amino acids as FRET donors with tryptophan. Biochemistry. 2009; 48:5953–5962. [DOI] [PubMed] [Google Scholar]
- 31. Chen R., Wold M.S.. Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. BioEssays. 2014; 36:1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hass C.S., Lam K., Wold M.S.. Repair-specific functions of replication protein A. J. Biol. Chem. 2012; 287:3908–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Binz S.K., Sheehan A.M., Wold M.S.. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair. 2004; 3:1015–1024. [DOI] [PubMed] [Google Scholar]
- 34. Nguyen H.D., Yadav T., Giri S., Saez B., Graubert T.A., Zou L.. Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol. Cell. 2017; 65:832–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salas T.R., Petruseva I., Lavrik O., Bourdoncle A., Mergny J.L., Favre A., Saintome C.. Human replication protein A unfolds telomeric G-quadruplexes. Nucleic Acids Res. 2006; 34:4857–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zou Y., Liu Y., Wu X., Shell S.M.. Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses. J. Cell. Physiol. 2006; 208:267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu S., Xu Z., Leng H., Zheng P., Yang J., Chen K., Feng J., Li Q.. RPA binds histone H3-H4 and functions in DNA replication-coupled nucleosome assembly. Science. 2017; 355:415–420. [DOI] [PubMed] [Google Scholar]
- 38. Roy R., Kozlov A.G., Lohman T.M., Ha T.. SSB protein diffusion on single-stranded DNA stimulates RecA filament formation. Nature. 2009; 461:1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Daley J.M., Gaines W.A., Kwon Y., Sung P.. Regulation of DNA pairing in homologous recombination. Cold Spring Harbor Perspect. Biol. 2014; 6:a017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Daley J.M., Kwon Y., Niu H., Sung P.. Investigations of homologous recombination pathways and their regulation. Yale J. Biol. Med. 2013; 86:453–461. [PMC free article] [PubMed] [Google Scholar]
- 41. Kowalczykowski S.C., Dixon D.A., Eggleston A.K., Lauder S.D., Rehrauer W.M.. Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 1994; 58:401–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kowalczykowski S.C. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 2000; 25:156–165. [DOI] [PubMed] [Google Scholar]
- 43. Xie F., Wu C.G., Weiland E., Lohman T.M.. Asymmetric regulation of bipolar single-stranded DNA translocation by the two motors within Escherichia coli RecBCD helicase. J. Biol. Chem. 2013; 288:1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tomko E.J., Fischer C.J., Lohman T.M.. Single-stranded DNA translocation of E. coli UvrD monomer is tightly coupled to ATP hydrolysis. J. Mol. Biol. 2012; 418:32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu C.G., Bradford C., Lohman T.M.. Escherichia coli RecBC helicase has two translocase activities controlled by a single ATPase motor. Nat. Struct. Mol. Biol. 2010; 17:1210–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Seong C., Colavito S., Kwon Y., Sung P., Krejci L.. Regulation of Rad51 recombinase presynaptic filament assembly via interactions with the Rad52 mediator and the Srs2 anti-recombinase. J. Biol. Chem. 2009; 284:24363–24371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krejci L., Van Komen S., Li Y., Villemain J., Reddy M.S., Klein H., Ellenberger T., Sung P.. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003; 423:305–309. [DOI] [PubMed] [Google Scholar]
- 48. Macris M.A., Sung P.. Multifaceted role of the Saccharomyces cerevisiae Srs2 helicase in homologous recombination regulation. Biochem. Soc. Trans. 2005; 33:1447–1450. [DOI] [PubMed] [Google Scholar]
- 49. Fan J., Pavletich N.P.. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012; 26:2337–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bochkareva E., Belegu V., Korolev S., Bochkarev A.. Structure of the major single-stranded DNA-binding domain of replication protein A suggests a dynamic mechanism for DNA binding. EMBO J. 2001; 20:612–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen R., Subramanyam S., Elcock A.H., Spies M., Wold M.S.. Dynamic binding of replication protein a is required for DNA repair. Nucleic Acids Res. 2016; 44:5758–5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nguyen B., Sokoloski J., Galletto R., Elson E.L., Wold M.S., Lohman T.M.. Diffusion of human replication protein A along single-stranded DNA. J. Mol. Biol. 2014; 426:3246–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Arunkumar A.I., Stauffer M.E., Bochkareva E., Bochkarev A., Chazin W.J.. Independent and coordinated functions of replication protein A tandem high affinity single-stranded DNA binding domains. J. Biol. Chem. 2003; 278:41077–41082. [DOI] [PubMed] [Google Scholar]
- 54. de Laat W.L., Appeldoorn E., Sugasawa K., Weterings E., Jaspers N.G., Hoeijmakers J.H.. DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev. 1998; 12:2598–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Iftode C., Borowiec J.A.. 5′ → 3′ molecular polarity of human replication protein A (hRPA) binding to pseudo-origin DNA substrates. Biochemistry. 2000; 39:11970–11981. [DOI] [PubMed] [Google Scholar]
- 56. Kim C., Paulus B.F., Wold M.S.. Interactions of human replication protein A with oligonucleotides. Biochemistry. 1994; 33:14197–14206. [DOI] [PubMed] [Google Scholar]
- 57. Yuzhakov A., Kelman Z., Hurwitz J., O’Donnell M.. Multiple competition reactions for RPA order the assembly of the DNA polymerase delta holoenzyme. EMBO J. 1999; 18:6189–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Patrick S.M., Turchi J.J.. Stopped-flow kinetic analysis of replication protein A-binding DNA: damage recognition and affinity for single-stranded DNA reveal differential contributions of k(on) and k(off) rate constants. J. Biol. Chem. 2001; 276:22630–22637. [DOI] [PubMed] [Google Scholar]
- 59. Kumaran S., Kozlov A.G., Lohman T.M.. Saccharomyces cerevisiae replication protein A binds to single-stranded DNA in multiple salt-dependent modes. Biochemistry. 2006; 45:11958–11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim C., Snyder R.O., Wold M.S.. Binding properties of replication protein A from human and yeast cells. Mol. Cell. Biol. 1992; 12:3050–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gibb B., Ye L.F., Gergoudis S.C., Kwon Y., Niu H., Sung P., Greene E.C.. Concentration-dependent exchange of replication protein A on single-stranded DNA revealed by single-molecule imaging. PLoS One. 2014; 9:e87922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gibb B., Ye L.F., Kwon Y., Niu H., Sung P., Greene E.C.. Protein dynamics during presynaptic-complex assembly on individual single-stranded DNA molecules. Nat. Struct. Mol. Biol. 2014; 21:893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ma C.J., Gibb B., Kwon Y., Sung P., Greene E.C.. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017; 45:749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hammill J.T., Miyake-Stoner S., Hazen J.L., Jackson J.C., Mehl R.A.. Preparation of site-specifically labeled fluorinated proteins for 19F-NMR structural characterization. Nat. Protoc. 2007; 2:2601–2607. [DOI] [PubMed] [Google Scholar]
- 65. Sibenaller Z.A., Sorensen B.R., Wold M.S.. The 32- and 14-kilodalton subunits of replication protein A are responsible for species-specific interactions with single-stranded DNA. Biochemistry. 1998; 37:12496–12506. [DOI] [PubMed] [Google Scholar]
- 66. Lohman T.M., Green J.M., Beyer R.S.. Large-scale overproduction and rapid purification of the Escherichia coli ssb gene product. Expression of the ssb gene under lambda PL control. Biochemistry. 1986; 25:21–25. [DOI] [PubMed] [Google Scholar]
- 67. Ma C.J., Steinfeld J.B., Greene E.C.. Single-Stranded DNA Curtains for Studying Homologous Recombination. Methods Enzymol. 2017; 582:193–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li Y., Hoskins J.N., Sreerama S.G., Grayson S.M.. MALDI-TOF mass spectral characterization of polymers containing an azide group: evidence of metastable ions. Macromolecules. 2010; 43:6225–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shao N., Singh N.S., Slade S.E., Jones A.M., Balasubramanian M.K.. Site specific genetic incorporation of azidophenylalanine in Schizosaccharomyces pombe. Scientific Rep. 2015; 5:17196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu W., Brock A., Chen S., Schultz P.G.. Genetic incorporation of unnatural amino acids into proteins in mammalian cells. Nat. Methods. 2007; 4:239–244. [DOI] [PubMed] [Google Scholar]
- 71. Nehring S., Budisa N., Wiltschi B.. Performance analysis of orthogonal pairs designed for an expanded eukaryotic genetic code. PLoS One. 2012; 7:e31992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Stauffer M.E., Chazin W.J.. Physical interaction between replication protein A and Rad51 promotes exchange on single-stranded DNA. J. Biol. Chem. 2004; 279:25638–25645. [DOI] [PubMed] [Google Scholar]
- 73. Ferrari M.E., Bujalowski W., Lohman T.M.. Co-operative binding of Escherichia coli SSB tetramers to single-stranded DNA in the (SSB)35 binding mode. J. Mol. Biol. 1994; 236:106–123. [DOI] [PubMed] [Google Scholar]
- 74. Qiu Y., Antony E., Doganay S., Koh H.R., Lohman T.M., Myong S.. Srs2 prevents Rad51 filament formation by repetitive motion on DNA. Nat. Commun. 2013; 4:2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lytle A.K., Origanti S.S., Qiu Y., VonGermeten J., Myong S., Antony E.. Context-dependent remodeling of Rad51-DNA complexes by Srs2 is mediated by a specific protein–protein interaction. J. Mol. Biol. 2014; 426:1883–1897. [DOI] [PubMed] [Google Scholar]
- 76. Dickson A.M., Krasikova Y., Pestryakov P., Lavrik O., Wold M.S.. Essential functions of the 32 kDa subunit of yeast replication protein A. Nucleic Acids Res. 2009; 37:2313–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bastin-Shanower S.A., Brill S.J.. Functional analysis of the four DNA binding domains of replication protein A. The role of RPA2 in ssDNA binding. J. Biol. Chem. 2001; 276:36446–36453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Brill S.J., Bastin-Shanower S.. Identification and characterization of the fourth single-stranded-DNA binding domain of replication protein A. Mol. Cell. Biol. 1998; 18:7225–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sugitani N., Chazin W.J.. Characteristics and concepts of dynamic hub proteins in DNA processing machinery from studies of RPA. Prog. Biophys. Mol. Biol. 2015; 117:206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mazon G., Mimitou E.P., Symington L.S.. SnapShot: Homologous recombination in DNA double-strand break repair. Cell. 2010; 142:646. [DOI] [PubMed] [Google Scholar]
- 81. Mimitou E.P., Symington L.S.. Nucleases and helicases take center stage in homologous recombination. Trends Biochem. Sci. 2009; 34:264–272. [DOI] [PubMed] [Google Scholar]
- 82. Niedziela-Majka A., Chesnik M.A., Tomko E.J., Lohman T.M.. Bacillus stearothermophilus PcrA monomer is a single-stranded DNA translocase but not a processive helicase in vitro. J. Biol. Chem. 2007; 282:27076–27085. [DOI] [PubMed] [Google Scholar]
- 83. Kozlov A.G., Eggington J.M., Cox M.M., Lohman T.M.. Binding of the dimeric Deinococcus radiodurans single-stranded DNA binding protein to single-stranded DNA. Biochemistry. 2010; 49:8266–8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tomko E.J., Fischer C.J., Lohman T.M.. Ensemble methods for monitoring enzyme translocation along single stranded nucleic acids. Methods. 2010; 51:269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kozlov A.G., Cox M.M., Lohman T.M.. Regulation of single-stranded DNA binding by the C termini of Escherichia coli single-stranded DNA-binding (SSB) protein. J. Biol. Chem. 2010; 285:17246–17252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jeggo P.A., Pearl L.H., Carr A.M.. DNA repair, genome stability and cancer: a historical perspective. Nat. Rev. Cancer. 2016; 16:35–42. [DOI] [PubMed] [Google Scholar]
- 87. Jeggo P.A., Lobrich M.. How cancer cells hijack DNA double-strand break repair pathways to gain genomic instability. Biochem. J. 2015; 471:1–11. [DOI] [PubMed] [Google Scholar]
- 88. Van Komen S., Reddy M.S., Krejci L., Klein H., Sung P.. ATPase and DNA helicase activities of the Saccharomyces cerevisiae anti-recombinase Srs2. J. Biol. Chem. 2003; 278:44331–44337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.