

Abstract
The soluble form of guanylate cyclase (sGC) and cGMP signaling are major regulators of pulmonary vasodilation and vascular remodeling that protect the pulmonary circulation from hypertension development. Nitric oxide, reactive oxygen species, thiol and heme redox, and heme biosynthesis control mechanisms regulating the production of cGMP by sGC. In addition, a cGMP-independent mechanism regulates protein kinase G through thiol oxidation in manner controlled by peroxide metabolism and NADPH redox. Multiple aspects of these regulatory processes contribute to physiological and pathophysiological regulation of the pulmonary circulation, and create potentially novel therapeutic targets for the treatment of pulmonary vascular disease.
X.1 Introduction
The earliest studies detected evidence suggesting the cyclic GMP generating activity of the cytosolic or soluble form of guanylate cyclase (sGC) from lung and/or other partially purified tissue preparations was modulated by redox processes influenced by autooxidation, hydrogen peroxide, lipid peroxides, thiols, superoxide dismutase, ascorbate and drugs potentially releasing nitric oxide (NO) (1). Subsequently, the formation of nitrosothiols (RSNO) and the availability of ferrous (Fe2+) heme were proposed for explaining sGC sites mediating activation by NO (2, 3). Furchgott, Ignarro and Murad received the Nobel Prize in Physiology and Medicine in 1998 for identifying nitric oxide (NO) as the endothelium-derived relaxing factor (EDRF), which appeared to function as a physiological regulator of sGC. The initial work of Louis Ignarro evolved from studies conducted in bovine pulmonary arteries (PA) (4) and the similarities between superoxide inhibition of EDRF and NO was a key factor used by Ignarro LJ et al. (5) in identifying NO. A major interest of our lab has been elucidating aspects of multiple additional mechanisms through which redox can control sGC and cGMP signaling in PA (6–8). Some of these mechanisms seem to participate in pulmonary artery hypoxic pulmonary vasoconstriction (HPV) (6) and changes that occur in pulmonary hypertension (PH) (9, 10). There is now substantial evidence for a loss of endothelium-derived nitric oxide (EDNO) (11) and perhaps its ability to stimulate sGC (12, 13) in various forms of PH. NO and drugs including the phosphodiesterase-5 (PDE-5) inhibitor Sildenafil and the sGC stimulator Riociguat are now used to treat PH. The properties of cyclic guanosine monophosphate (cGMP) signaling suggest that it may normally function to attenuate vascular pathophysiological actions of stimuli promoting pulmonary hypertension development.
X.2 Organization of cGMP signaling in pulmonary arteries
Different redox systems can regulate sGC- and/or cGMP-associated signaling mechanisms, which in turn leads to relaxation of vascular smooth muscle (VSM) in pulmonary arteries. In smooth muscle tissue, cGMP is well established as an activator of type 1 and 2 forms of Protein Kinase G (PKG) present in vascular smooth muscle. More recently, a thiol oxidation resulting in a disulfide bond between the two subunits of PKG1α has been identified as a cGMP-independent activator of this system (14). Activation of PKG is known to promote the opening of calcium-activated potassium channels which leads to cell hyperpolarization and relaxation. PKG activates sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump on sarcoplasmic reticulum (SR) which pumps calcium back to sarcoplasmic reticulum (SR). As this store of calcium fills, extracellular calcium influx is also likely to be decreased. Thus, PKG signaling decreases intracellular calcium through multiple mechanisms, and this leads to smooth muscle relaxation. PKG inhibits Rho Kinase (a kinase which inhibits Myosin light chain (MLC) Phosphatase) and leads to relaxation of smooth muscle (15). While there may be differences in the systems activated by cGMP versus thiol oxidation activation of PKG due to different docking properties of these active forms of PKG (16), both of these activation mechanisms show many similarities in the way PKG regulates vascular smooth muscle relaxation and remodeling processes (17, 18).
Some of the cyclic nucleotide metabolizing phosphodiesterases are cGMP selective, and the type 5 isoform of this enzyme (PDE5) appears to be a major cGMP-selective phosphodiesterase in vascular smooth muscle. Thus, PDE5 may normally function in the pulmonary vasculature to remove cGMP generated in response to prevailing NO levels, by converting it to GMP. Under these conditions, inhibition of PDE5 causes smooth muscle relaxation by increasing cGMP, which decreases the levels and actions of calcium through PKG. NO may also activate K+ channels independent of cGMP, which would also lead to hyperpolarization and relaxation. Therefore, inhibitors cGMP-dependent phosphodiesterase, by increasing intracellular cGMP, enhance smooth muscle relaxation associated with lowering pulmonary arterial pressure under conditions promoting pulmonary hypertension.
NO/cGMP signaling pathway also has important pro-apoptotic and antimitogenic effects on vascular smooth muscle (VSM) and endothelial cells which play an essential role in pulmonary vascular remodeling. It has been documented that activation of the NO/cGMP pathway inhibits the proliferation of bronchial smooth muscle and vascular smooth muscle cells from the systemic (19–21) and pulmonary circulations (22–24). Actions such as PKG-mediated inhibition of Rho kinase activation could be a factor in processes such as inhibition of myosin light chain phosphatase activity associated with decreasing the sensitivity of the contractile apparatus to calcium and in attenuating smooth muscle growth-associated remodeling processes promoted by Rho kinase. Figure 1 shows some relationships associated with actions of cGMP signaling mechanisms that potentially contribute to pulmonary vascular function and disease processes.
Fig. 1.
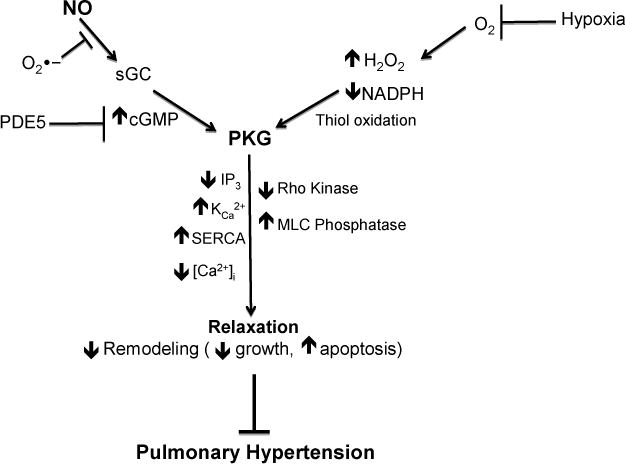
cGMP-related signaling mechanisms regulating pulmonary vascular relaxation and remodeling.
X.3 Redox modulation of cGMP signaling in pulmonary arteries
There appear to be many redox regulatory interactions influencing cGMP signaling that have been detected in pulmonary arteries. The discussion of these pathways are organized around considering how different individual cellular redox processes are designed to function, and potentially regulate aspects of cGMP signaling, in relation to other systems they could influence in physiological and pathophysiological regulation of the pulmonary vasculature. Redox regulation of the sGC heme and thiols can occur directly by systems such as NO, and reactive oxygen and NO-derived species. The function of redox systems such as NADPH/NADP and NADH/NAD, and related thiol redox control mechanisms in subcellular regions are key factors indirectly controlling the function and depletion of sGC through interactions illustrated in Figure 2 and processes described in Table 1. While the activity of PKG enzymes are controlled by redox systems influencing changes the levels of cGMP, the activity of PKG1α is also directly controlled by systems regulating the disulfide between its subunits, which activates this form of PKG in a cGMP-independent manner. While the expression of key enzymes participating in cGMP signaling such as PDE5 (25) may also be regulated by redox, it is currently difficult to discuss the mechanisms involved due to limitations in what is known.
Fig. 2.
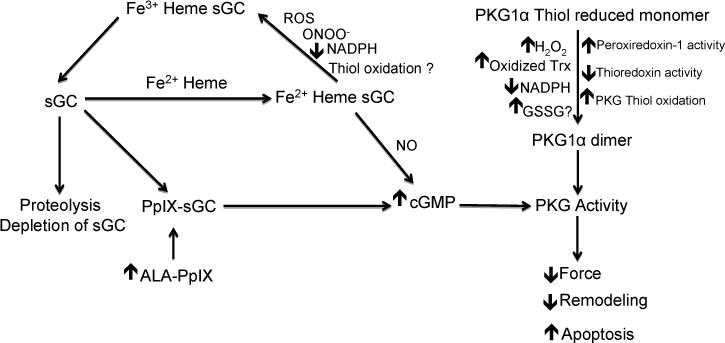
Nitric oxide, reactive oxygen species, thiol and heme redox, and heme biosynthesis control mechanisms that regulate the production of cGMP by sGC and/or a cGMP-independent mechanism regulating protein kinase G.
Table 1.
Redox processes modulating vascular cGMP signaling
| Redox Species & Systems | Mechanisms Regulating cGMP Signaling |
|---|---|
| Nitric oxide (NO) |
|
| Superoxide |
|
| Hydrogen Peroxide (H2O2) |
|
| Reactive Nitrogen Species (RNS) |
|
| Heme |
|
| Thiols |
|
| NADPH |
|
X.3.1 Redox regulation of sGC through its heme group
The ferrous or Fe2+ heme form of sGC is known to bind and be required for activation by low nanomolar concentrations of NO. This activation appears to occur through NO binding disrupting a histidine bond of sGC to Fe2+ of its heme (26, 27). When the heme of sGC is oxidized to its ferric Fe3+ form, it does not readily bind or become activated by the low levels of NO present in the vasculature. Oxidant stress conditions could potentially oxidize the heme of sGC, and, the product of NO reacting with superoxide, peroxynitrite is a well documented oxidant of the heme of sGC (28, 29). Oxidation of the sGC heme to its ferric Fe3+ form may result in the release of heme and formation of heme-free form of sGC. Heme-free sGC seems to preferentially bind some sGC activator drugs at the site normally occupied by heme. While sGC appears to undergo proteolytic depletion once its heme is oxidized, this process appears to be prevented when agents such as sGC activators bind the sGC heme site (29–31). These sGC activator drugs may actually be mimicking a potentially endogenous mechanism of activating heme-free sGC through binding protoporphyrin IX (PpIX) at the heme site (30). PpIX is normally used as the substrate for insertion of Fe2+ by ferrochelatase, during biosynthesis of heme in mitochondria. When there is a deficiency in Fe2+ needed for heme biosynthesis, ferrochelatase (FECH) can insert Zn into PpIX generating zinc-protoporphyrin IX (Zn-PpIX). Zn-PpIX could potentially bind the heme site of sGC in a manner that might prevent both its stimulation by NO and its degradation by proteolysis. While heat shock protein-90 appears to participate in the binding of heme to sGC (32), the role of this protein under conditions promoting sGC heme oxidation and depletion remain to be better defined. While the oxidation of heme of sGC under pathophysiological conditions and its association with enhanced sGC stimulation by sGC activators under these conditions are rather well documented processes, most of the hypothesized relationships between the function of ferrochelatase in heme biosynthesis and sGC regulation remain to be investigated.
X.3.2 Redox regulation of sGC through modulation of its thiol groups
There appear to be multiple ways through which thiol groups can influence the activity of sGC, and sGC has an unusually large number of thiols (33). Thiols potentially have a major influence of the generation of NO from potential NO-donors through processes such as forming S-nitrosothiols (RSNO) which release NO. Cysteine is one of the least abundant residues in proteins, but it is also one of the most conserved. Cysteine can undergo a variety of thiol oxidations, including disulfide (S-S), sulfenic acid (S–OH), nitrosylation (RSNO) and sulfhydration reactions. The ability of NO to stimulate sGC by its Fe2+heme-dependent activation mechanism appears to require certain specific thiols, and oxidation or modification of these thiols may prevent activation by NO.
Various forms of sGC thiol oxidation can play a role in the development of cardiovascular diseases by decreasing the NO-dependent production of cGMP and thus the vascular reactivity. This thiol-based resistance to NO (increased peripheral resistance) appears to be detected in hypertension (33). Studies have shown that the disulfide inducer diamide and the attenuation of nicotinamide adenine dinucleotide phosphate generation (NADPH) via the pentose phosphate pathway inhibitor 6-aminonicotinamide decreases the relaxation of pulmonary arteries and NO-stimulated sGC activity (34). Dithiotheritol (DTT), the thiol reductant, can reverse these inhibitory effects. Perturbations to the redox status of cells in the pulmonary vasculature due to activation of reactive oxygen species (ROS) - generating enzymes, such as xanthine oxidase, NADPH oxidase (NOX), or via disrupted electron transport chain (ETC) function in mitochondria promote pulmonary vasculopathy that is characterized by intimal thickening, impaired NO•-dependent vasodilation and perivascular fibrosis (35). In general, the types of sGC thiol modifications under these types pathophysiological conditions remain to be defined, and it is likely they go beyond NADPH redox controlled reversible disulfide regulation of its sensitivity to stimulation by NO, to more difficult to reverse modifications of sGC thiols that could impair the function of this system.
X.3.3 Redox regulation of PKG1α through modulation of its thiol groups
It was reported by Eaton and colleagues that exposure of arteries to hydrogen peroxide could promote relaxation by activating protein kinase G 1α (PKG1α) through formation of a disulfide bond between its two subunits, in a manner independent of cGMP (14). Inhibition of thioredoxin reductase was also observed to enhance this relaxation. Our lab studied this processes in pulmonary arteries and identified peroxiredoxin-1 as a peroxide metabolizing enzyme which potentially promotes PKG1α disulfide formation and activation. In addition, we provided evidence that the control of cytosolic NADPH oxidation resulting from decreased glucose-6-phosphate dehydrogenase (G6PD) activity and the pentose phosphate pathway (PPP) could function to promote thiol oxidation-mediated PKG1α activation (7). This potentially occurs through decreasing NADPH availability needed to support the activity of thioredoxin reductase for reversing the actions of oxidant processes promoting PKG1α activation by its disulfide form.
X.3.4 Coordination of the regulation of cGMP signaling by reactive oxygen and NO-derived species
Multiple interactions of reactive oxygen and NO-derived species are described in Table 1. When the levels of ROS are low, the sGC system is likely to be most sensitive to stimulation by NO as a result of thiols and heme being maintained in their reduced forms. As superoxide levels increase in any extracellular or subcellular region, it is likely to scavenge and attenuate the actions of NO by reacting with it to form peroxynitrite. This is because NO is a dissolved gas which readily diffuses across extracellular and intracellular membranes. Increased superoxide formation drives the generation of peroxide, which could stimulate both sGC generation of cGMP and/or promote cGMP-independent peroxide activation of PKG1α generally as a result of the actions of peroxide metabolizing enzymes such as catalase and peroxiredoxin-1, respectively, causing redox changes that influence sGC and/or PKG1α. Under these conditions, sGC stimulation by NO might also be attenuated by peroxide promoting thiol oxidation. While peroxynitrite is also known to oxidize the sGC heme and thiols, the importance of this interaction in pathophysiological dysfunction needs to be better defined. As the levels of reactive oxygen and/or NO-derived species increase, many other signaling mechanisms beyond those associated with promoting cGMP signaling become activated in various subcellular regions, and the actions of these other regulatory systems are likely to dominate the biological effects that are seen. There are many different processes controlling cellular systems generating ROS, and the stimuli activating these oxidases often defines the roles of various potential signaling mechanisms in the responses observed.
X.3.5 Coordination of the regulation of cGMP signaling by subcellular thiol redox systems
Cellular thiol redox systems such as glutathione and thioredoxin are oxidized through the functions of hydrogen peroxide (H2O2) metabolizing enzymes such as glutathione peroxidases and peroxiredoxins and enzymes including glutathione and thioredoxin reductases maintain these systems in their reduced forms based on the availability of co-factors such as NADPH. Thus, the redox status of thiol influencing sGC and PKG signaling in the subcellular regions these enzymes are located should dominate regulation of these systems by processes described previously in sections X.3.2 and X.3.3. Again, as described for ROS, there are many different processes controlling cellular thiol redox systems, and subcellular metabolic perturbations often define the roles of various potential signaling mechanisms in the responses observed.
X.3.6 Coordination of the regulation of cGMP signaling by subcellular NADH and NADPH redox systems
The redox status of NADH and NADPH in subcellular regions are usually influenced by multiple regulatory processes and metabolism-associated factors. They have a potential influence on cGMP signaling through their roles in supporting the generation of ROS by enzymes such as NOX oxidases and the mitochondrial electron transport chain, and as co-factors for influencing thiol and heme redox regulation. When biological factors such as hypoxia or stimulation of oxidases regulate NADH or NADPH redox systems they have the potential to regulate cGMP signaling through the pathways described above.
X.4 Redox regulation of pulmonary vascular function through cGMP signaling
The properties of redox regulation of cGMP signaling in pulmonary arteries isolated from normal healthy animals and limited information available from studies on properties of vascular reactivity the pulmonary circulation in vitro or in vivo indicate that sGC normally exists in its NO responsive form, suggesting the sGC heme is in its NO-binding ferrous form, with sGC thiols influencing sensitivity to NO being reduced as well. Studies primarily from our laboratory have also documented what appears to be a basal partial activation of both sGC and PKG by endogenous H2O2 in pulmonary arteries under aerobic conditions, which are both attenuated by hypoxia associated with a hypoxic pulmonary vasoconstriction response (36). The unusual property of pulmonary arteries compared to systemic arteries driving this aerobic generation of vasodilator levels of peroxide seems to be increased activity of glucose-phosphate dehydrogenase for supporting higher rates of basal peroxide generation by a Nox oxidase, which may be Nox4 (37). There is often evidence for baseline levels of endothelium-derived NO somewhat depressing responses to pulmonary vasoconstrictors including hypoxia and for endogenous NO having a major influence on blood flow shunting responses by the pulmonary circulation at birth (38).
Many conditions or agents participating in the control pulmonary vascular function, such as the shear forces of flow, the actions of increased pressure, vasoactive mediators and hypoxia have often been observed to regulate cGMP signaling aspects of pulmonary vascular function through modulating endothelium-derived NO. Many of these conditions or agents are also likely to modulate redox systems in pulmonary arterial smooth muscle which could influence cGMP-related signaling by additional mechanisms. For example, there is a large literature of the effects of hypoxia being mediated by redox mechanisms regulating a variety of systems, including our focus (6, 36) on redox regulation of sGC/cGMP and cGMP-independent PKG mechanisms. Under these types of conditions, many redox changes that are occurring could potentially have prominent influences on processes such as the activation of contractile and remodeling mechanisms, in addition to influencing cGMP-related signaling. Only a few studies have examined relationships between the influence of physiological conditions on oxidant-redox systems and their simultaneous influence on multiple other redox-regulated processes, including assessing what is happening with cGMP-related signaling. This is probably due to how difficult it currently is to document how each redox system functions in subcellular regions and how these redox changes influence aspects of cGMP signaling versus other processes influenced by redox. In addition, as discussed in section X.2, PKG regulates many processes controlling vascular function, and many of these processes have been suggested to be redox regulated without considering if PKG is involved. Thus, the properties of these systems suggest hypothesizing that there may be many conditions where the redox regulation of cGMP signaling (beyond the role of endothelium-derived NO) has regulatory roles in physiological processes influencing pulmonary vascular function.
X.5 Pathophysiological regulation of pulmonary vascular function through altered cGMP signaling
The endothelium is a known semi-permeable layer between the vascular and extravascular fluid compartments. It is involved in the regulation of vascular tone, differentiation and growth. In pulmonary hypertension (PH), vascular responses to injury caused by increased pressure, flow (shear stress), hypoxia, drugs (dexfenfluramine) etc. are mediated partly through the endothelial cell dysfunction. The endothelium releases several vasoactive substances, such as endothelin-1 (ET-1), angiotensin II (Ang II), thromboxane A2 as well as the growth factors such as transforming growth factor-β, fibroblast growth factor-2, vascular endothelial growth factor, platelet-derived growth factor. These factors influence the growth of the underlying smooth muscle layer and thus result in vascular remodeling and the progression of PH (39–42). There are several intracellular posttranslational modifications such as phosphorylation (e.g., Ser1177, Ser65, Thr495), S-nitrosylation (e.g., Cys94, Cys99), palmitoylation etc. which can regulate endothelial Nitric Oxide Synthase (eNOS) activation (43). The extracellular signaling pathways such as G-protein-coupled receptor signal transduction, Akt/PKB (protein kinase B) signaling via sphingosine 1-phosphate and vascular endothelial growth factor via phosphatase calcineurin are involved in eNOS activation (44–46). Studies have shown that there is decreased pulmonary vascular eNOS activity in various animal models of pulmonary hypertension as well as in human patients with PH (47, 48). Loss of NO bioavailability is associated with increased pulmonary vascular smooth muscle cell mitogenesis and impaired endothelium-dependent and -independent vasodilation. Hypoxia plays an important role in development of PH. Hypoxia can induce posttranslational modifications of eNOS and/or caveolin-1 that in turn lead to a decrease in calcium sensing by eNOS. This causes dissociation of eNOS from its regulatory proteins (heat shock protein 90 and calmodulin) and thus hypoxia decreases eNOS activity (46). Many of these processes lead to conditions that are associated with a loss of the favorable effects of EDNO and its regulation of sGC, and a progression of PH development.
In humans, increased levels of ROS generation in pulmonary vasculature may occur as a pathological response to various conditions such as chronic hypoxia, increased pulmonary vascular blood flow (secondary to intracardiac shunt) or due to impaired antioxidant enzyme function (glutathione peroxidase deficiency in sickle cell anemia-associated PH) (49). ROS by oxidizing the enzyme cofactors such as tetrahydrobiopterin (BH4) may inhibit eNOS activity. Additionally, superoxide by reacting with NO generates peroxynitrite (ONOO-•) and inactivates NO. Furthermore, superoxide also reacts with nitrite (either derived from diet or as a stable NO by-product) and generates peroxynitrate (O2NOO), which is a potential NOS-independent source of NO (50, 51). This decrease in NO synthesis leads to a decreased activation of sGC and thus decreased cGMP levels.
Endothelin-1 (ET-1) regulates pulmonary vascular tone by its interaction with the vasoconstrictor endothelin-type A (ETA) and endothelin-type B (ETB) receptors in pulmonary vascular smooth muscle cells and vasodilatory ETB receptors in pulmonary vascular endothelium. Stimuli associated with pulmonary vascular injury, such as cytokines that mediate vascular inflammation (52) as well as stimuli such as increased ROS generation in the pulmonary vasculature (53), hypoxia (54) decreased levels of bioavailable NO (55) can significantly upregulate ET-1 gene expression levels in right ventricular cardiac myocytes, pulmonary arterial endothelial and vascular smooth muscle cells. ET-1 immunohistochemical analysis demonstrates significantly increased immunoreactivity in pulmonary arterial endothelial and smooth muscle cells of plexiform lesions compared to blood vessels harvested from normal controls (56). Thus, ET-1 is an important factor generated under conditions leading to PH that contributes to the progression of this disease process. Our studies have found that ET-1 promotes in pulmonary arteries a loss of mitochondrial superoxide dismutase (SOD) expression associated with increased mitochondrial superoxide, and activation other signaling associated with pulmonary arterial smooth muscle remodeling that appear to be prevented by using δ-aminolevulinic acid (ALA) to generate protoporphyrin ix (PpIX), an activator of sGC (10). Treatment of mice with ALA prevented the development of hypoxia-induced PH in mice associated with actions similar to its effects on the actions of ET-1 observed in isolated pulmonary arteries. These observations suggest cGMP-activated PKG signaling by an sGC activator has actions that go beyond vasodilation, which can prevent fundamental processes contributing to the progression of PH. A sGC activator has been observed to increase vasodilator actions in a neonatal model of pulmonary hypertension, suggesting sGC heme oxidation is potentially an additional factor in this disease process (12, 13).
The actions of Angiotensin II (Ang II) on its endothelial type-2 receptors in the pulmonary circulation releasing NO has been suggested to be a dominant factor in protecting the pulmonary circulation from Ang II generated by systemic hypertension acting on its type 1 receptor in pulmonary arteries (57, 58). In the regulation of systemic vascular reactivity, Ang II activation of its type 1 receptor and NO/cGMP normally function as a vasoconstrictors or vasodilators, respectively. The countervailing influences of Ang II and NO/cGMP on vascular smooth muscle cell (VSMC) growth have been well documented. Ang II stimulates, whereas NO/cGMP inhibits, VSMC growth, through different mechanisms. Studies have shown that infusion of Ang II into rats significantly decreased the expression of both sGC subunits in blood vessels, associated with decreased PKG-mediated phosphorylation of vasodilator-stimulated phospho protein (VASP) (59). The inhibitory effects of Ang II on sGC are likely mediated by increased production of superoxide in response to Ang II. These results suggest that a decrease in PKG activity occurred in response to Ang II treatment. Ang II may exhibit inhibitory effects on cGMP accumulation through an additional mechanism in systemic arteries potentially mediated by increased Ca2+ promoting activation of Ca2+/calmodulin-stimulated phosphodiesterase-1A1 (60). Our studies in isolated coronary arteries (61) suggest that Ang II may function to deplete sGC by increasing mitochondrial and extra-mitochondrial superoxide. Increased mitochondrial superoxide is associated with a depletion of mitochondrial SOD and ferrochelatase (which has an iron-sulfur center that could be disrupted by superoxide) and a depletion of heme. Whereas, increased cytosolic superoxide may promote sGC heme oxidation and depletion. Promoting PKG activation by the accumulation of PpIX or by 8-bromo-cGMP attenuated all of these actions of Ang II. Our ongoing studies in pulmonary arteries (62) suggest that the direct effects of Ang II on endothelium-rubbed pulmonary arteries are consistent with it having actions similar to those described for coronary arteries, except ferrochelatase and sGC are not depleted. Since Ang II (and ET-1), increased the detection of fluorescence associated with PpIX accumulation in pulmonary arteries (10, 63) there may disruption of mitochondrial iron availability needed for heme biosynthesis, and sGC might be protected by PpIX and/or Zn-PpIX binding to its heme site.
X.6 Therapeutic Targeting of PA cGMP signaling
Pulmonary hypertension is a syndrome in which vasoconstriction, inflammation, and remodeling of small pulmonary arteries increases pulmonary vascular resistance, leading to right ventricular hypertrophy and failure. While vasodilator-related therapies have shown beneficial effects in the treatment of this rather poorly understood disease, the high rate of mortality remains a major concern. Sildenafil, a selective inhibitor for cGMP phosphodiesterases, is beneficial in the treatment of pulmonary hypertension (11). Tadalafil, a phosphodiesterase type 5 inhibitor, gained FDA approval in 2003 and is indicated for patients with pulmonary arterial hypertension to improve exercise capacity. Drugs stimulating sGC either through heme-dependent actions potentially enhancing the actions of endogenous NO (stimulators) or by directly activating sGC in a heme-independent manner (activators) have also showed potential for treatment the of pulmonary hypertension in humans (64). These studies lead to the sGC stimulator Riociguat recently being approved for treating some forms of PH. While the direct activators of sGC, which bind its heme site where PpIX binds, evolved into clinical trials for Cinaciguat, these were stopped during phase IIb studies due to adverse effects associated with it promoting systemic hypotension (65). Endothelin-1 Receptor Antagonists Bosentan, Ambrisentan and Macitentan are also used for treatment of pulmonary hypertension. Prostacyclin analogues such as Epoprostenol, Treprostinil and Iloprost are used as a therapy for pulmonary hypertension. The mechanism of action of prostacyclin analogues includes direct vasodilation of pulmonary and systemic arterial vascular beds, inhibition of platelet aggregation and anti-proliferative effects (66). Thus activation of cGMP signaling plays an important role for treatment of pulmonary hypertension. Clinical trials with dehydroepiandrosterone (DHEA), an activator of PKG through dimerization (67), treatment has reported significant improvement in 6-minute walk test, pulmonary hemodynamics and diffusing capacity of the lung of patients with chronic obstructive pulmonary disease-associated PH, without worsening gas exchange, as do other pharmacological treatments of PH (68). Another potential therapy for the treatment of pulmonary hypertension can be δ-aminolevulinic acid (ALA). ALA, the biosynthetic precursor to heme, by increasing PpIX, shows promising beneficial effects for the treatment of this disease in the mouse model of hypoxia-induced hypertension (9, 10). The heme-independent activators of sGC act in a manner similar to PpIX by binding the heme site of sGC after its endogenous heme has been oxidized by vascular disease-associated oxidant stress (29). The use of ALA as a therapeutic approach uses mechanisms (outlined in Figure 2), which go beyond the drug activators of sGC in that it may take advantage of additional disease-associated cellular mechanisms such as using a potentially disrupted heme biosynthetic pathway to accumulate the PpIX activator of sGC. Perhaps once increased cGMP reversed processes such as mitochondrial oxidant stress, which contribute to disease progression, improved availability of heme might also be a contributing beneficial factor of this new therapeutic approach. Figure 3 shows various mechanisms by which therapeutic targeting of cGMP signaling is useful for the treatment of pulmonary vascular disease.
Fig. 3.
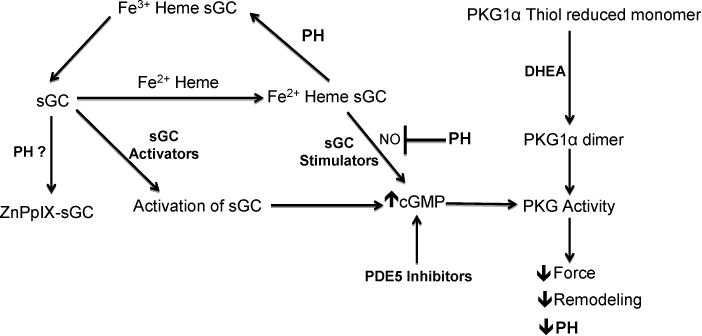
Therapeutic targeting of cGMP signaling for the treatment of pulmonary vascular disease.
X.7 Future Perspectives
The multiple different redox processes controlling sGC and PKG signaling suggest these systems are sensors for many aspects of different physiological and pathophysiological processes that remain to be defined. They are among the most sensitive sensors for signaling promoted by NO and peroxide, and inhibition of NO by superoxide. Routes to regulation by NADPH, NADH, thiol and heme redox, and heme biosynthesis provide routes through which aspects of metabolism can influence cGMP signaling. In addition to regulating smooth muscle contractile function, evidence is emerging for cGMP signaling having important roles in controlling aspects of signaling mechanisms modulating ROS generation, and inflammatory, fibrotic cell death signaling processes that participate in often beneficial actions against pathophysiological vascular remodeling. The significance and most aspects of these processes remain to be elucidated.
Acknowledgments
Recent studies from our lab have been funded by NIH grants R01HL115124 and R01HL129797.
Abbreviations
- ALA
δ-aminolevulinic acid
- Ang II
Angiotensin II
- Ca2+
Calcium
- cGMP
Cyclic Guanosine Monophosphate
- DHEA
Dehydroepiandrosterone
- EDNO
Endothelium-Derived Nitric Oxide
- EDRF
Endothelium-Derived Relaxing Factor
- ET-1
Endothelin-1
- eNOS
Endothelial Nitric Oxide Synthase
- FECH
Ferrochelatase
- Fe2+
Ferrous
- H2O2
Hydrogen Peroxide
- NOX
NADPH oxidase
- NAD
Nicotinamide adenine dinucleotide
- NADP
Nicotinamide adenine dinucleotide phosphate
- NO
Nitric Oxide
- PA
Pulmonary Arteries
- PKG
Protein Kinase G
- PpIX
Protoporphyrin IX
- PKG1α
Protein Kinase G 1α
- PH
Pulmonary Hypertension
- RNS
Reactive Nitrogen Species
- ROS
Reactive Oxygen Species
- sGC
Soluble Guanylate Cyclase
- SOD
Superoxide Dismutase
- VEGF
Vascular Endothelial Growth Factor
- VSM
Vascular Smooth Muscle
- VSMC
Vascular Smooth Muscle Cell
- Zn-PpIX
Zinc-Protoporphyrin IX
References
- 1.Waldman SA, Murad F. Cyclic GMP synthesis and function. Pharmacol Rev. 1987;39(3):163–196. [PubMed] [Google Scholar]
- 2.Craven PA, DeRubertis FR. Restoration of the responsiveness of purified guanylate cyclase to nitrosoguanidine, nitric oxide, and related activators by heme and hemeproteins. Evidence for involvement of the paramagnetic nitrosyl-heme complex in enzyme activation. J Biol Chem. 1978;253(23):8433–8443. [PubMed] [Google Scholar]
- 3.Ignarro LJ, Lippton H, Edwards JC, et al. Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of S-nitrosothiols as active intermediates. J Pharmacol Exp Ther. 1981;218(3):739–749. [PubMed] [Google Scholar]
- 4.Ignarro LJ, Burke TM, Wood KS, et al. Association between cyclic GMP accumulation and acetylcholine-elicited relaxation of bovine intrapulmonary artery. J Pharmacol Exp Ther. 1984;228(3):682–690. [PubMed] [Google Scholar]
- 5.Ignarro LJ, Buga GM, Wood KS, et al. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84(24):9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neo BH, Kandhi S, Ahmad M, et al. Redox regulation of guanylate cyclase and protein kinase G in vascular responses to hypoxia. Respir Physiol Neurobiol. 2010;174(3):259–264. doi: 10.1016/j.resp.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neo BH, Patel D, Kandhi S, et al. Roles for cytosolic NADPH redox in regulating pulmonary artery relaxation by thiol oxidation-elicited subunit dimerization of protein kinase G1alpha. Am J Physiol Heart Circ Physiol. 2013;305(3):H330–43. doi: 10.1152/ajpheart.01010.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolin MS. Reactive oxygen species and the control of vascular function. Am J Physiol Heart Circ Physiol. 2009;296(3):H539–49. doi: 10.1152/ajpheart.01167.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel D, Alhawaj R, Wolin MS. Exposure of mice to chronic hypoxia attenuates pulmonary arterial contractile responses to acute hypoxia by increases in extracellular hydrogen peroxide. Am J Physiol Regul Integr Comp Physiol. 2014;307(4):R426–33. doi: 10.1152/ajpregu.00257.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alhawaj R, Patel D, Kelly MR, et al. Heme biosynthesis modulation via delta-aminolevulinic acid administration attenuates chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2015;308(7):L719–28. doi: 10.1152/ajplung.00155.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coggins MP, Bloch KD. Nitric oxide in the pulmonary vasculature. Arterioscler Thromb Vasc Biol. 2007;27(9):1877–1885. doi: 10.1161/ATVBAHA.107.142943. ATVBAHA.107.142943 [pii] [DOI] [PubMed] [Google Scholar]
- 12.Deruelle P, Grover TR, Abman SH. Pulmonary vascular effects of nitric oxide-cGMP augmentation in a model of chronic pulmonary hypertension in fetal and neonatal sheep. Am J Physiol Lung Cell Mol Physiol. 2005;289(5):L798–806. doi: 10.1152/ajplung.00119.2005. 00119.2005 [pii] [DOI] [PubMed] [Google Scholar]
- 13.Deruelle P, Grover TR, Storme L, et al. Effects of BAY 41-2272 a soluble guanylate cyclase activator, on pulmonary vascular reactivity in the ovine fetus. Am J Physiol Lung Cell Mol Physiol. 2005;288(4):L727–33. doi: 10.1152/ajplung.00409.2004. 00409.2004 [pii] [DOI] [PubMed] [Google Scholar]
- 14.Burgoyne JR, Madhani M, Cuello F, et al. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317(5843):1393–1397. doi: 10.1126/science.1144318. 1144318 [pii] [DOI] [PubMed] [Google Scholar]
- 15.Lincoln TM, Dey N, Sellak H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol (1985) 2001;91(3):1421–1430. doi: 10.1152/jappl.2001.91.3.1421. [DOI] [PubMed] [Google Scholar]
- 16.Rainer PP, Kass DA. Old dog, new tricks: novel cardiac targets and stress regulation by protein kinase G. Cardiovasc Res. 2016;111(2):154–162. doi: 10.1093/cvr/cvw107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chettimada S, Rawat DK, Dey N, et al. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am J Physiol Lung Cell Mol Physiol. 2012;303(1):L64–74. doi: 10.1152/ajplung.00002.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupte RS, Rawat DK, Chettimada S, et al. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem. 2010;285(25):19561–19571. doi: 10.1074/jbc.M109.092916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83(5):1774–1777. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolpakov V, Gordon D, Kulik TJ. Nitric oxide-generating compounds inhibit total protein and collagen synthesis in cultured vascular smooth muscle cells. Circ Res. 1995;76(2):305–309. doi: 10.1161/01.res.76.2.305. [DOI] [PubMed] [Google Scholar]
- 21.Tanner FC, Meier P, Greutert H, et al. Nitric oxide modulates expression of cell cycle regulatory proteins: a cytostatic strategy for inhibition of human vascular smooth muscle cell proliferation. Circulation. 2000;101(16):1982–1989. doi: 10.1161/01.cir.101.16.1982. [DOI] [PubMed] [Google Scholar]
- 22.Ambalavanan N, Mariani G, Bulger A, et al. Role of nitric oxide in regulating neonatal porcine pulmonary artery smooth muscle cell proliferation. Biol Neonate. 1999;76(5):291–300. doi: 10.1159/000014171. 14171 [pii] [DOI] [PubMed] [Google Scholar]
- 23.Jourdan KB, Evans TW, Lamb NJ, et al. Autocrine function of inducible nitric oxide synthase and cyclooxygenase-2 in proliferation of human and rat pulmonary artery smooth-muscle cells: species variation. Am J Respir Cell Mol Biol. 1999;21(1):105–110. doi: 10.1165/ajrcmb.21.1.3502. [DOI] [PubMed] [Google Scholar]
- 24.Krick S, Platoshyn O, Sweeney M, et al. Nitric oxide induces apoptosis by activating K+ channels in pulmonary vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2002;282(1):H184–93. doi: 10.1152/ajpheart.2002.282.1.H184. [DOI] [PubMed] [Google Scholar]
- 25.Farrow KN, Wedgwood S, Lee KJ, et al. Mitochondrial oxidant stress increases PDE5 activity in persistent pulmonary hypertension of the newborn. Respir Physiol Neurobiol. 2010;174(3):272–281. doi: 10.1016/j.resp.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ballou DP, Zhao Y, Brandish PE, et al. Revisiting the kinetics of nitric oxide (NO) binding to soluble guanylate cyclase: the simple NO-binding model is incorrect. Proc Natl Acad Sci U S A. 2002;99(19):12097–12101. doi: 10.1073/pnas.192209799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herzik MA, Jr, Jonnalagadda R, Kuriyan J, et al. Structural insights into the role of iron-histidine bond cleavage in nitric oxide-induced activation of H-NOX gas sensor proteins. Proc Natl Acad Sci U S A. 2014;111(40):E4156–64. doi: 10.1073/pnas.1416936111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrammel A, Behrends S, Schmidt K, et al. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50(1):1–5. [PubMed] [Google Scholar]
- 29.Stasch JP, Schmidt PM, Nedvetsky PI, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116(9):2552–2561. doi: 10.1172/JCI28371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123(20):2263–2273. doi: 10.1161/CIRCULATIONAHA.110.981738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meurer S, Pioch S, Pabst T, et al. Nitric oxide-independent vasodilator rescues heme-oxidized soluble guanylate cyclase from proteasomal degradation. Circ Res. 2009;105(1):33–41. doi: 10.1161/CIRCRESAHA.109.198234. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh A, Stuehr DJ. Regulation of sGC via hsp90, Cellular Heme, sGC Agonists, and NO: New Pathways and Clinical Perspectives. Antioxid Redox Signal. 2017;26(4):182–190. doi: 10.1089/ars.2016.6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beuve A. Thiol-Based Redox Modulation of Soluble Guanylyl Cyclase, the Nitric Oxide Receptor. Antioxid Redox Signal. 2017;26(3):137–149. doi: 10.1089/ars.2015.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolin MS, Ahmad M, Gupte SA. Oxidant and redox signaling in vascular oxygen sensing mechanisms: basic concepts, current controversies, and potential importance of cytosolic NADPH. Am J Physiol Lung Cell Mol Physiol. 2005;289(2):L159–73. doi: 10.1152/ajplung.00060.2005. 289/2/L159 [pii] [DOI] [PubMed] [Google Scholar]
- 35.Mittal M, Gu XQ, Pak O, et al. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med. 2012;52(6):1033–1042. doi: 10.1016/j.freeradbiomed.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Neo BH, Kandhi S, Wolin MS. Roles for redox mechanisms controlling protein kinase G in pulmonary and coronary artery responses to hypoxia. Am J Physiol Heart Circ Physiol. 2011;301(6):H2295–304. doi: 10.1152/ajpheart.00629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupte SA, Kaminski PM, Floyd B, et al. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol. 2005;288(1):H13–21. doi: 10.1152/ajpheart.00629.2004. [DOI] [PubMed] [Google Scholar]
- 38.Fineman JR, Soifer SJ, Heymann MA. Regulation of pulmonary vascular tone in the perinatal period. Annu Rev Physiol. 1995;57:115–134. doi: 10.1146/annurev.ph.57.030195.000555. [DOI] [PubMed] [Google Scholar]
- 39.Dschietzig T, Richter C, Bartsch C, et al. Flow-induced pressure differentially regulates endothelin-1, urotensin II, adrenomedullin, and relaxin in pulmonary vascular endothelium. Biochem Biophys Res Commun. 2001;289(1):245–251. doi: 10.1006/bbrc.2001.5946. [DOI] [PubMed] [Google Scholar]
- 40.Mata-Greenwood E, Meyrick B, Soifer SJ, et al. Expression of VEGF and its receptors Flt-1 and Flk-1/KDR is altered in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2003;285(1):L222–31. doi: 10.1152/ajplung.00388.2002. [DOI] [PubMed] [Google Scholar]
- 41.Mata-Greenwood E, Meyrick B, Steinhorn RH, et al. Alterations in TGF-beta1 expression in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2003;285(1):L209–21. doi: 10.1152/ajplung.00171.2002. [DOI] [PubMed] [Google Scholar]
- 42.Wedgwood S, Devol JM, Grobe A, et al. Fibroblast growth factor-2 expression is altered in lambs with increased pulmonary blood flow and pulmonary hypertension. Pediatr Res. 2007;61(1):32–36. doi: 10.1203/01.pdr.0000250013.77008.28. [DOI] [PubMed] [Google Scholar]
- 43.Dudzinski DM, Igarashi J, Greif D, et al. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 44.Cerqueira FM, Brandizzi LI, Cunha FM, et al. Serum from calorie-restricted rats activates vascular cell through enhanced insulin signaling mediated by adiponectin. PLoS One. 2012;7(2):e31155. doi: 10.1371/journal.pone.0031155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Egom EE, Mohamed TM, Mamas MA, et al. Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am J Physiol Heart Circ Physiol. 2011;301(4):H1487–95. doi: 10.1152/ajpheart.01003.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murata T, Sato K, Hori M, et al. Decreased endothelial nitric-oxide synthase (eNOS) activity resulting from abnormal interaction between eNOS and its regulatory proteins in hypoxia-induced pulmonary hypertension. J Biol Chem. 2002;277(46):44085–44092. doi: 10.1074/jbc.M205934200. [DOI] [PubMed] [Google Scholar]
- 47.Gangopahyay A, Oran M, Bauer EM, et al. Bone morphogenetic protein receptor II is a novel mediator of endothelial nitric-oxide synthase activation. J Biol Chem. 2011;286(38):33134–33140. doi: 10.1074/jbc.M111.274100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steudel W, Scherrer-Crosbie M, Bloch KD, et al. Sustained pulmonary hypertension and right ventricular hypertrophy after chronic hypoxia in mice with congenital deficiency of nitric oxide synthase 3. J Clin Invest. 1998;101(11):2468–2477. doi: 10.1172/JCI2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gizi A, Papassotiriou I, Apostolakou F, et al. Assessment of oxidative stress in patients with sickle cell disease: The glutathione system and the oxidant-antioxidant status. Blood Cells Mol Dis. 2011;46(3):220–225. doi: 10.1016/j.bcmd.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7(2):156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 51.Spiegelhalder B, Eisenbrand G, Preussmann R. Influence of dietary nitrate on nitrite content of human saliva: possible relevance to in vivo formation of N-nitroso compounds. Food Cosmet Toxicol. 1976;14(6):545–548. doi: 10.1016/s0015-6264(76)80005-3. [DOI] [PubMed] [Google Scholar]
- 52.Olave N, Nicola T, Zhang W, et al. Transforming growth factor-beta regulates endothelin-1 signaling in the newborn mouse lung during hypoxia exposure. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L857–65. doi: 10.1152/ajplung.00258.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.An SJ, Boyd R, Zhu M, et al. NADPH oxidase mediates angiotensin II-induced endothelin-1 expression in vascular adventitial fibroblasts. Cardiovasc Res. 2007;75(4):702–709. doi: 10.1016/j.cardiores.2007.02.015. S0008-6363(07)00066-1 [pii] [DOI] [PubMed] [Google Scholar]
- 54.Yamashita K, Discher DJ, Hu J, et al. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol Chem. 2001;276(16):12645–12653. doi: 10.1074/jbc.M011344200. [DOI] [PubMed] [Google Scholar]
- 55.Kourembanas S, McQuillan LP, Leung GK, et al. Nitric oxide regulates the expression of vasoconstrictors and growth factors by vascular endothelium under both normoxia and hypoxia. J Clin Invest. 1993;92(1):99–104. doi: 10.1172/JCI116604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giaid A, Yanagisawa M, Langleben D, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–1739. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 57.Olson S, Oeckler R, Li X, et al. Angiotensin II stimulates nitric oxide production in pulmonary artery endothelium via the type 2 receptor. Am J Physiol Lung Cell Mol Physiol. 2004;287(3):L559–68. doi: 10.1152/ajplung.00312.2003. [DOI] [PubMed] [Google Scholar]
- 58.Olson SC, Dowds TA, Pino PA, et al. ANG II stimulates endothelial nitric oxide synthase expression in bovine pulmonary artery endothelium. Am J Physiol. 1997;273(2 Pt 1):L315–21. doi: 10.1152/ajplung.1997.273.2.L315. [DOI] [PubMed] [Google Scholar]
- 59.Mollnau H, Wendt M, Szocs K, et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90(4):E58–65. doi: 10.1161/01.res.0000012569.55432.02. [DOI] [PubMed] [Google Scholar]
- 60.Kim D, Rybalkin SD, Pi X, et al. Upregulation of phosphodiesterase 1A1 expression is associated with the development of nitrate tolerance. Circulation. 2001;104(19):2338–2343. doi: 10.1161/hc4401.098432. [DOI] [PubMed] [Google Scholar]
- 61.Patel D, Alhawaj R, Kelly MR, et al. Potential role of mitochondrial superoxide decreasing ferrochelatase and heme in coronary artery soluble guanylate cyclase depletion by angiotensin II. Am J Physiol Heart Circ Physiol. 2016;310(11):H1439–47. doi: 10.1152/ajpheart.00859.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patel D, Kelly MR, Accarino JJO, et al. Pulmonary Arteries Show Differences in the Effects of Angiotensin II Stimulation of Mitochondrial Superoxide on Regulation of Heme Biosynthesis and Soluble Guanylate Cyclase Expression. The FASEB Journal. 2016;30(1 Supplement):774.9–774.9. [Google Scholar]
- 63.Patel D, Alhawaj R, Kelly M, et al. Aminolevulinic Acid Treatment of Pulmonary Arteries Attenuates Endothelin-1 and Angiotensin II Elicited Increases in Mitochondrial, but not Extra-Mitochondrial Superoxide. The FASEB Journal. 2015;29(1 Supplement) [Google Scholar]
- 64.Ghofrani HA, Grimminger F. Soluble guanylate cyclase stimulation: an emerging option in pulmonary hypertension therapy. Eur Respir Rev. 2009;18(111):35–41. doi: 10.1183/09059180.00011112. [DOI] [PubMed] [Google Scholar]
- 65.Dasgupta A, Bowman L, D’Arsigny CL, et al. Soluble guanylate cyclase: a new therapeutic target for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Clin Pharmacol Ther. 2015;97(1):88–102. doi: 10.1002/cpt.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250–2294. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- 67.Patel D, Kandhi S, Kelly M, et al. Dehydroepiandrosterone promotes pulmonary artery relaxation by NADPH oxidation-elicited subunit dimerization of protein kinase G 1alpha. Am J Physiol Lung Cell Mol Physiol. 2014;306(4):L383–91. doi: 10.1152/ajplung.00301.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dumas de La Roque E, Savineau JP, Metivier AC, et al. Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): a pilot study. Ann Endocrinol (Paris) 2012;73(1):20–25. doi: 10.1016/j.ando.2011.12.005. [DOI] [PubMed] [Google Scholar]