

Abstract
Targeting proteins within the N-type voltage-gated calcium channel (CaV2.2) complex has proven to be an effective strategy for developing novel pain therapeutics. We describe a novel peptide aptamer derived from the collapsin response mediator protein 2 (CRMP2), a CaV2.2-regulatory protein. Addition of a 14-carbon myristate group to the peptide (myr-tat-CBD3) tethered it to the membrane of primary sensory neurons near surface CaV2.2. Pull-down studies demonstrated that myr-tat-CBD3 peptide interfered with the CRMP2–CaV2.2 interaction. Quantitative confocal immunofluorescence revealed a pronounced reduction of CaV2.2 trafficking after myr-tat-CBD3 treatment and increased efficiency in disrupting CRMP2-CaV2.2 colocalization compared with peptide tat-CBD3. Consequently, myr-tat-CBD3 inhibited depolarization-induced calcium influx in sensory neurons. Voltage clamp electrophysiology experiments revealed a reduction of Ca2+, but not Na+, currents in sensory neurons after myr-tat-CBD3 exposure. Current clamp electrophysiology experiments demonstrated a reduction in excitability of small-diameter dorsal root ganglion neurons after exposure to myr-tat-CBD3. Myr-tat-CBD3 was effective in significantly attenuating carrageenan-induced thermal hypersensitivity and reversing thermal hypersensitivity induced by a surgical incision of the plantar surface of the rat hind paw, a model of postoperative pain. These effects are compared with those of tat-CBD3—the nonmyristoylated tat-conjugated CRMP2 peptide as well as scrambled versions of CBD3 and CBD3-lacking control peptides. Our results demonstrate that the myristoyl tag enhances intracellular delivery and local concentration of the CRMP2 peptide aptamer near membrane-delimited calcium channels resulting in pronounced interference with the calcium channel complex, superior suppression of calcium influx, and better antinociceptive potential.
Keywords: CaV2.2, CRMP2, N-myristoylated peptide, GPMVs, Trafficking, Calcium imaging, Inflammatory pain, Postoperative pain, Conditioned place preference
1. Introduction
N-type voltage-gated calcium channels (CaV2.2) are critical determinants of increased neuronal excitability and neurotransmission accompanying persistent neuropathic pain.14,20,58 Expressed in the presynaptic termini of primary afferent nociceptors in the spinal cord,83 CaV2.2 represent a possible control for synaptic activity. The potential of targeting CaV2.2 has been demonstrated with CaV2.2-blocking conotoxins on allodynia70 and inferred from altered pain behavior of CaV2.2 knockout mice.44,45 Furthermore, Ziconotide (Prialt, Jazz Pharmaceuticals, Palo Alto, CA),54 and Gabapentin (Neurontin, Pfizer, Inc., New York, NY) directly target different elements of CaV2.2. These drugs, however, present problematic side effects and difficult dosing regimens. To circumvent these problems, we have advanced a strategy targeting protein interactions modulating CaV2.2.29
The study of CaV2.2 trafficking modulators43 led us to identify axon/dendrite specification collapsin response mediator protein 2 (CRMP2) as a CaV2.2-binding partner,43 which enhances CaV2.2 membrane trafficking and facilitates synaptic transmission.10,19 CaV2.2 membrane trafficking was effectively disrupted by a 15-amino-acid region of CRMP2, named Ca2+ channel-binding domain 3 (CBD3).10 Systemic injection of CBD3 peptide conjugated to the cell-penetrating motif of the HIV transduction domain protein tat (tat-CBD3) reduced hypersensitive behavior in chemically induced inflammatory and neuropathic pain models.9,39,59,63 tat-CBD3 reduced CaV2.2-mediated currents by inhibiting CaV2.2–CRMP2 interaction,59 which resulted in reduced nociceptor excitability, diminished neuropeptide release from primary sensory neurons, and lowered excitatory synaptic transmission.9
A recent study suggested that CBD3 might be developed for gene therapy in chronic neuropathic pain.31 Adenoviral injection of CBD3 into rat lumbar dorsal root ganglia (DRG) protected them from nerve injury–induced neuropathic pain.31 Unfortunately, the 30% viral transduction efficiency coupled with an incomplete block of CaV2.2 resulted in limited efficacy.31 One improvement of CBD3 would be a membrane targeting strategy. The tethering of the CBD3 peptide to the membrane would allow a local enrichment near surface CaV2.2 leading to a greater inhibitory effect. Another benefit of a membrane-delimited CBD3 may be to allow separation of membrane-CRMP2 functions from those mediated by its interactions with cytoplasmic proteins (reviewed in Ref. 42). CRMP2 has been demonstrated in detergent-resistant membrane fractions65 where it may colocalize with surface CaV2.2 clusters64 thereby helping to organize functional CaV activity into macromolecular complexes.
We tested the efficacy of a CBD3 peptide harboring a myristoylated N-terminal glycine, a lipid modification of eukaryotic and viral proteins targeting them to membranes.3,6,88 Such a strategy has been successfully used before; N-myristoylation of a substrate analogue of protein kinase C enhanced its inhibitory potency compared with the free peptide.81 The myristoyl group is known to directly insert into membranes through hydrophobic interactions55 and allows for a greater range of cell types to be loaded.57
We report that myristoylated tat-CBD3 is membrane tethered, restricts the CaV2.2–CRMP2 interaction, blocks CaV2.2 trafficking, and strongly inhibits Ca2+ influx from primary sensory neurons. Intrathecal injection of myr-tat-CBD3 reduced carrageenan-induced inflammation and reversed paw incision–induced thermal and mechanical hypersensitivity without producing rewarding behavior.9
2. Methods
2.1. Animals
Pathogen-free adult male and female Sprague-Dawley rats (150–200 g; Harlan Laboratories, Madison, WI) were housed in temperature (23°C ± 3°C) and light (12-hour light–12-hour dark cycle; lights on at 07:00 hours) controlled rooms with standard rodent chow and water available ad libitum. The Institutional Animal Care and Use Committee of the College of Medicine at the University of Arizona approved these experiments. All procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health and the ethical guidelines of the International Association for the Study of Pain. Animals were randomly assigned to treatment or control groups for the behavioral experiments.
2.2. Materials
The following peptides were synthesized and HPLC purified by GenScript USA Inc (Piscataway, NJ): (1) N-terminal myristoylated peptide (myr-tat-CBD3; N-myristoyl-YGRKKRRQRRRARSRLAELRGVPRGL; tat sequence denoted in the underlined text), (2) a nonmyristoylated peptide (tat-CBD3; YGRKKRRQRRRARSRLAELRGVPRGL), (3) a random control peptide (YGRKKRRQRRRWEAKEMLYFEALVIE); 2 scramble CBD3 peptides, (4) myr-tat-CBD3scr1 (sequence: YGRKKRRQRRRAALRLLPRERGRGSV, the underlined sequence is a scramble version the CBD3 peptide), and (5) myr-tat-CBD3scr2 (sequence: YGRKKRRQRRRLPLVRRLEARGRGAS, the underlined sequence is another scrambled version of the original CBD3 peptide); (6) a myr-tat peptide (lacking the CBD3 cargo). Importantly, the scramble control peptides are exactly the same sequence and charge as the CBD3 peptide while the third control peptide addresses any confounds produced by the myristate or tat motifs in the absence of the CBD3 cargo. Additional fluorescently labeled peptides with fluorescein isothiocyanate (FITC) were generated. Myristoylated peptides were produced by incorporating a myristoylated glycine during solid phase synthesis. Peptides were dissolved in deionized water or DMSO (Sigma-Aldrich, St Louis, MO), aliquoted, and stored at −70°C. All chemicals, unless noted, were purchased from Sigma-Aldrich. Fura-2AM was obtained from Invitrogen (Grand Island, NY). Antibodies were obtained as follows: anti-β-tubulin monoclonal (Promega, Madison, WI) and anti-CaV2.2 polyclonal (OriGene Technologies Inc, Rockville, MD).8,9,39 The CRMP2-His-pET28B cDNA was provided by Dr Rihe Liu (University of North Carolina). The membrane potential and membrane order probe, di-4-ANNEPDHQ,38 was purchased from Life technologies (Grand Island, NY) and resuspended in ethanol. Two-percent carrageenan was prepared in Milli-Q water. Carrageenan was purchase from Sigma (St Louis, MO, Catalog Number C1867-25G).
2.3. Purification of recombinant CRMP2-6XHis and CRMP2-GST
Both fusion proteins were purified as described,87 with the following modifications. Briefly, after a 19-hour induction at 16°C with 0.5 mM IPTG, bacterial cells expressing recombinant CRMP2-6XHis or CRMP2-GST were resuspended in 50 mM NaH2PO4, pH 7.5, 500 mM NaCl, 10% glycerol, 0.5 mM TCEP, supplemented with complete EDTA-free protease inhibitors (Roche, Basel, Switzerland). Disruption of the bacteria was performed by 2 rounds of high-pressure homogenization at 10,000 PSI with an LM10 microfluidizer (Microfluidics, Westwood), and the lysate was centrifuged 45 minutes at 4500g at 4°C. The supernatant was loaded on a HisTrap HP column or a GST-Trap HP column (GE Healthcare, Uppsala, Sweden) equilibrated with 50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, and 0.5 mM TCEP. After a washing step with 50 mM HEPES pH 7.5, 20 mM imidazole, 300 mM NaCl, 10% glycerol, and 0.5 mM TCEP, CRMP2-6XHis was eluted with an imidazole gradient and CRMP2-GST was eluted with a glutathione gradient. The fractions of interest for CRMP2-6XHis were desalted using a PD10 desalting column (GE Healthcare), in 50 mM NaH2PO4, pH7, 10% glycerol, 0.5 mM TCEP, and loaded on a HiPrep Q XL column (GE Healthcare). Elution was performed with an NaCl gradient between 50 mM NaH2PO4, pH7.5 10% glycerol, 0.5 mM TCEP, and 50 mM NaH2PO4, 1 M NaCl, 10% glycerol, 0.5 mM TCEP. Finally, the fractions were loaded on a HiLoad Superdex size exclusion column (GE Healthcare) and eluted with 50 mM NaH2PO4, pH 7.5 10% glycerol, 0.5 mM TCEP. The fractions of interest for CRMP2-GST were loaded on a HiLoad Superdex size exclusion column (GE Healthcare) and eluted with 50 mM NaH2PO4, pH 7.5 10% glycerol, 0.5 mM TCEP. The eluted proteins were concentrated with Amicon Ultra 15 centrifugal filters (Regenerated cellulose 10,000 NMWL; Merck Millipore, Darmstadt, Germany) and flash frozen on dry ice and stored at −80°C until use. Protein concentration was determined by a Pierce assay using bovine serum albumin (BSA) as a standard. The purity of the protein was verified with SDS-PAGE and structure/activity by checking its ability to bind tubulin (data not shown). Purity of CRMP-6His was further confirmed through mass spectrometry: 2 prominent peaks, both corresponding to CRMP2, of molecular weights 65.5 kDa and 59.3 kDa, were deconvoluted.
2.4. Primary dorsal root ganglion neuronal cultures
Sensory DRG neurons from Sprague-Dawley rats were isolated as described previously.9,39,87 Dorsal root ganglia were excised aseptically and placed in Hank buffered salt solution (HBSS, Life Technologies) on ice. The ganglia were dissociated enzymatically with collagenase A (1 mg/mL, 25 minutes, Roche) and collagenase D (1 mg/mL, Roche) with papain (30 U/mL, Roche) for 20 minutes at 37°C. To eliminate debris, 70 μm (BD Biosciences) cell strainers were used. The dissociated cells were resuspended in DMEM/F12 (Life Technologies) containing 1× pen-strep (Life Technologies), 1× GlutaMax, 3 μg/mL 5-FDU (Sigma), 7 μg/mL uridine (Sigma), and 10% fetal bovine serum (Hyclone). The cells were seeded on poly-d-lysine–coated coverslips (BD Falcon) and incubated at 37°C in a humidified 95% air/5% CO2 incubator.
2.5. Formation of giant plasma membrane vesicles
Giant plasma membrane vesicles (GPMVs) were obtained as previously described.71 Giant plasma membrane vesicles represent a natural membrane model system with a cytoplasmic lumen devoid of cellular organelles and the actin cytoskeleton, low intracellular membranes content, and redolent of the protein and lipid composition of the plasma membrane, having a phospholipid/cholesterol ratio of ~2:1.5,16,32 Briefly, GPMVs were generated from DRG (Supplementary Fig. 1B, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A59) incubated at 37°C for 90 minutes in a vesiculation buffer containing HEPES, 150 mM NaCl, 2 mM CaCl2, pH 7.4, containing 2 mM N-ethyl maleimide. After GPMV formation, the buffer containing vesicles was recovered and conserved at 4°C for at least 2 hours to allow the vesicles to concentrate at the bottom of the tube. Because the GPMVs were stable for ~ 48 hours (data not shown), they were used the day after formation at the latest.
2.6. Peptide–giant plasma membrane vesicles incubation and labeling
All experiments for microscopic analyses of GPMVs were performed on 35 mm glass bottom dishes precoated with BSA (1 mg/mL for 1 hour at room temperature). Approximately 100 μL of GMPVs labeled with 5 μM di-4-ANEPPDHQ were incubated for 3 hours at room temperature with 20 μM of either tat-CBD3, myr-tat-CBD3, myr-tat, myr-tat-CBD3scr1, or myr-tat-CBD3scr2 peptides.
2.7. Fluorescence microscopy and image analysis
Fluorescence imaging was performed with an inverted microscope, Nikon Eclipse TE2000-U, using objective Nikon Super Fluor 20× 0.75 NA and a Photometrics-cooled CCD camera CoolSNAPHQ ES2 (Roper Scientific, Tucson, AZ) controlled by MetaFluor 6.3 software (Molecular Devices, Downingtown, PA). The excitation light was delivered by a Lambda-LS system (Sutter Instruments, Novato, CA). The excitation filters were controlled by a Lambda 10-2 optical filter change (Sutter Instruments). Twenty images of each condition were systematically recorded randomly, using an FITC filter (excitation and emission wavelength 488 nm and 500–550 nm, respectively), which accounted for the liquid-ordered contribution of di-4-ANEPPDHQ or the localization of FITC-peptides. The freeware image analysis program ImageJ (http://rsb.info.nih.gov/ij/) was used for quantifying lipid domains. All images were corrected for background by subtracting the average background fluorescence (areas within the field of view not containing vesicles) from the region of interest.
2.8. Uptake and efflux of tat-CBD3 and myr-tat-CBD3 from dorsal root ganglion neurons
Dorsal root ganglion neurons were seeded into 96-well plates at 1.5 × 104 cells per well in Dulbecco minimum essential media with 10% fetal bovine serum and cultured overnight at 37°C and 5% CO2. Then, 20 μM of either FITC conjugated tat-CBD3, myr-tat-CBD3, myr-tat, myr-tat-CBD3scr1, or myr-tat-CBD3scr2 peptide aptamers were added on DRG and allowed to penetrate the cells for 10 minutes. After 4 washes with phenol red–free MEM medium, the FITC fluorescence in the cells was measured using a Synergy 2 fluorescent plate reader (BioTek, Winooski, VT) at an excitation wavelength of 485 nm and emission wavelength of 530 nm. Efflux of the peptides from the cells was assessed by measuring FITC fluorescence in the media after 0, 10, 30, and 60 minutes. To correct for any differences in cell plating, at the end of the experiment, DRG were lysed with 20 mM Tris, pH 7.4, 50 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS protease inhibitor cocktail set III (Calbiochem), phosphatase inhibitor cocktail set III (Calbiochem), 50 U/mL benzonase, (Merck) and the protein quantity determined with a Pierce assay.
2.9. GST pull-down and Western blotting
Spinal cord lysates prepared from adult Sprague-Dawley rats were generated by homogenization and sonication in RIPA buffer (50 mM Tris-HCl, pH 7.4, 50 mM NaCl, 2 mM MgCl2, 1% [vol/vol] NP40, 0.5% [mass/vol] sodium deoxycholate, 0.1% [mass/vol] SDS). Protease inhibitors (Cat# B14002; Biotools, Houston, TX), phosphatase inhibitors (Cat# B15002, Biotools), and benzonase (Cat# 71206, Millipore, Billerica, MA). Protein concentrations were determined using the BCA protein assay (Cat# PI23225, Thermo Fisher Scientific, Waltham, MA). Glutathione magnetic beads (Cat# B23702, Biotools), preincubated with purified CRMP2-GST (~ 0.4 μM), were incubated overnight with 300 μg of total protein from spinal cord lysates at 4°C in the absence or presence of the indicated peptides with gentle rotation. Beads were washed 3 times with RIPA buffer before resuspension in Laemmli buffer and denaturation (5 minutes at 95°C) and immunoblotting.8,39,87
For examining the effect of peptides on CRMP2 phosphorylation state, DRG were treated for 2 hours with the indicated peptides and then lysed using RIPA buffer. Approximately 3 μg of total proteins were loaded on an SDS-PAGE and then transferred to polyvinylidene difluoride membranes and blocked at room temperature for 1 hour. Primary antibodies used for probing were CRMP2 (Cat# C2993, Sigma, St Louis, MO), CRMP2 pThr509/Thr514 (Cat# PB-043, Kinasource, Dundee, Scotland, United Kingdom), CRMP2 pSer522 (Cat# CP2191, ECM Biosciences, Versailles, KY), and CRMP2 pT555 (Cat# CP2251, ECM Biosciences). Immunoblots were revealed by enhanced luminescence (WBKLS0500, Millipore) before exposure to a photographic film. Films were scanned, digitized, and quantified using Un-Scan-It gel version 6.1 scanning software (Silk Scientific Inc, Orem, UT).
2.10. Enzyme-linked immunosorbent assay-based CRMP2-tubulin binding assay
These were performed exactly as described by Wilson et al.86 Briefly, 96-well plates (Nunc MaxiSorp, Thermo Scientific) were coated with tubulin (200 ng per well, Cytoskeleton Inc) and incubated at room temperature overnight. The next day, the plates were washed and blocked with 3% BSA to minimize nonspecific adsorptive binding to the plates. Escalating concentrations of CRMP2 were added to the plates. As a negative control, some wells received escalating concentrations of CRMP2 which was denatured by heating at 95°C for 5 minutes. The plates were incubated at room temperature with shaking for 2 hours. The plates were then washed with PBS containing 0.5% Tween-20 to eliminate unbound CRMP2. The bound CRMP2 was detected by CRMP2 primary antibody (150 ng/mL, Sigma) followed by HRP-conjugated secondary antibody (GE Healthcare). Tetramethylbenzidine (R&D Systems) was used as the colorimetric substrate. The optical density of each well was determined immediately, using a microplate reader (Multiskan Ascent, Thermo) set to 450 nm with a correction wavelength of 570 nm. Data were analyzed by nonlinear regression analysis using GraphPad Prism 5 (GraphPad, San Diego, CA).
To determine the effect of peptides on CRMP2-tubulin binding, 96-well plates were coated with tubulin and incubated overnight. The next day, the plates were washed and blocked with 3% BSA to minimize nonspecific adsorptive binding to the plate. After the wash, all the wells received 1 μM of CRMP2 and no peptide or escalating concentrations of peptides. The plates were then incubated at room temperature with shaking for 2 hours. This was followed with washes to eliminate the unbound CRMP2. CRMP2 bound to the plates was detected using the method described above.
2.11. Oligomerization of CRMP2
The oligomeric status of purified CRMP2 was analyzed by covalent cross-linking and silver-stained SDS-PAGE according to the method reported by Stenmark et al.72 with the following modifications: ~4.6 μM of CRMP2 was incubated in 100 mM HEPES pH 7.3, 50 mM NaCl in the absence or presence of 20 μM of either tat-CBD3 or myr-tat-CBD3. After 15 minutes at room temperature, escalating concentrations of glutaraldehyde (0.1, 1, and 10 mM) were added for 30 minutes to induce covalent cross-linking. The reaction was stopped with the addition of loading dye and heat denaturation, then the samples were resolved by SDS-PAGE and silver stained.
2.12. Immunocytochemistry, confocal microscopy, and colocalization analysis
Immunocytochemistry was performed on DRG incubated with vehicle (control) or peptides (20 μM) for the indicated times as described previously.10,19 Briefly, cells were fixed using 4% paraformaldehyde, 4% sucrose for 20 minutes at room temperature. Permeabilization was achieved by a 30-minute incubation in phosphate-buffered saline (PBS), 0.1% Triton X-100, following which nonspecific binding sites were saturated by PBS containing 3% BSA for 30 minutes at room temperature. Cell staining was performed with primary antibodies (CaV2.2 [OriGene, #TA308673, Rockville, USA] and CRMP2 [C4G, Immuno-Biological Laboratories, #11096, Minneapolis]) in PBS with 3% BSA overnight at 4°C. The cells were then washed thrice in PBS and incubated with PBS containing 3% BSA and secondary antibodies (Alexa 488 goat anti-mouse and Alexa 594 goat anti-rabbit secondary antibodies [Life Technologies]) for 1 hour at room temperature. Coverslips were mounted and stored at 4°C until analysis. Immunofluorescent micrographs were acquired on a Zeiss LSM 710 inverted upright microscope using a ×40, 1.3 numerical aperture oil immersion objective or on a Nikon C1si scanning confocal microscope using CFI Plan APO VC ×60 oil immersion objective with 1.4 numerical aperture. For all quantitative comparisons among cells under differing experimental conditions, camera gain and other relevant settings were kept constant. The freeware image analysis program ImageJ (http://rsb.info.nih.gov/ij/) was used for quantifying cellular fluorescence. Regions of interest (ie, cells) were defined by hand using ImageJ. All images were corrected for background by subtracting the average background fluorescence (areas within the field of view not containing cells) from the region of interest.
Colocalization analysis, including calculation of the Pearson coefficient and Manders overlap coefficient (MOC), was performed using a plug-in for ImageJ provided by Li et al.48 at the Wright Cell Imaging Facility, University Health Network Research, Canada (http://www.uhnresearch.ca/facilities/wcif/fdownload.html). Pearson correlation coefficient is used to describe the degree of overlap between 2 patterns. A major advantage of the Pearson overlap coefficient is that it is not sensitive to differences in signal intensities between the components of an image caused by variations in labelling by fluorophores, photobleaching, or different gain settings of the amplifiers. However, the Pearson coefficient is hard to interpret because of the strong influence of the ratio of the number of objects in the 2 components. Manders overlap coefficient remedies the disadvantages of the Pearson coefficient, where the numbers are proportional to the amount of fluorescence of the colocalizing objects in each channel and are dependent on the intensities of the signals. However, unlike the Pearson coefficient, the Manders coefficient is sensitive to background intensities.21,48,50 These analysis methods provide a significant advantage over qualitative methods, thus, we have opted to report both values. Images are presented without manipulation of contrast and brightness levels, and all colocalization values were generated using the raw, unadjusted data.
2.12. Live cell imaging
Dorsal root ganglion cultures were treated with tat-CBD3-FITC, myr-tat-CBD3-FITC, myr-tat-FITC, or myr-tat-CBD3scr1-FITC peptides for 5 minutes in serum-free media. The cells were then washed and supplemented with media. Fluorescent Z-stack images spanning the thickness of the neurons were acquired by confocal microscopy. Orthogonal views were generated from the Z-stacks using ImageJ. ImageJ was also used to generate the 360° 3-D projections that were exported to a digital video file.
2.13. Calcium imaging
Dorsal root ganglion neurons were loaded at 37°C with 3 μM Fura-2AM (Kd = 25 μM, λex 340, 380 nm/λemi 512 nm) to follow changes in intracellular calcium ([Ca2+]c) in a standard bath solution containing 139 mM NaCl, 3 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 10 mM NaHEPES, pH 7.4, 5 mM glucose exactly as previously described.9,39,40 Fluorescence imaging was performed with an inverted microscope, Nikon Eclipse TE2000-U, using an objective Nikon Super Fluor 20× 0.75 NA and a Photometrics-cooled CCD camera CoolSNAPHQ (Roper Scientific, Tucson, AZ) controlled by MetaFluor 6.3 software (Molecular Devices, Downingtown, PA). The excitation light was delivered by a Lambda-LS system (Sutter Instruments, Novato, CA). The excitation filters (340 ± 5 nm and 380 ± 7 nm) were controlled by a Lambda 10-2 optical filter change (Sutter Instruments). Fluorescence was recorded through a 505-nm dichroic mirror at 535 ± 25 nm. To minimize photobleaching and phototoxicity, images were taken every ~2.4 seconds during the time course of the experiment using the minimal exposure time that provided acceptable image quality. The changes in [Ca2+]c were monitored by following a ratio of F340/F380, calculated after subtracting the background from both channels.
2.14. Whole-cell voltage-clamp and current-clamp electrophysiology
Recordings were obtained from acutely dissociated DRG neurons as described previously.12 To isolate calcium currents, Na+ and K+ currents were blocked with 1 μM tetrodotoxin (TTX; Alomone Laboratories, Jerusalem, Israel) and 30 mM tetraethylammonium chloride (TEA-Cl; Sigma). Extracellular recording solution (at ~315 mOsm) consisted of the following (in mM): 110 N-methyl-d-glucamine (NMDG), 10 BaCl2, 30 TEA-Cl, 10 HEPES, 10 glucose, 0.001 TTX, 0.005 nifedipine, 0.0005 ω-conotoxin GVIA, and 0.0002 ω-agatoxin IVA. The intracellular recording solution (at ~305 mOsm) consisted of the following (in mM): contained 150 CsCl2, 10 HEPES, 5 Mg-ATP, 5 BAPTA, pH at 7.2 with KOH. The internal solution for recording Na+ currents contained the following (in mM): 110 CsCl, 5 MgSO4, 10 EGTA, 4 ATPNa2, and 25 HEPES (pH 7.2, 290–310 mOsm/L). For recording Na+ currents, the external solution contained the following (in mM): 100 NaCl, 10 tetraethylammonium chloride (TEA-Cl), 1 CaCl2, 1 CdCl2, 1 MgCl2, 10 d-glucose, 44-AP, 0.1 NiCl2, 10 HEPES (pH 7.3, 310–315 mOsm/L).
For current clamp recordings, the extracellular recording solution (at ~305 mOsm; pH 7.2) consisted of the following (in mM): 154 NaCl, 5.6 KCl, 2 CaCl2, 2.0 MgCl2, 1.0 Glucose, and 10 HEPES pH 8.0. The intracellular recording solution (at ~295 mOsm) consisted of the following (in mM): 140 KCl, 10 NaCl, 1 MgCl2, 1 EGTA, 10 HEPES (pH 7.2), and 1 ATP-Mg. To assess excitability, DRG neurons were held at their resting potentials (range, −40 to −62 mV), and a step of depolarizing current (1 second in duration) was applied to the neuron wherein the amplitude of the step was adjusted to produce 5 or more action potentials (APs) under control conditions. The stimulation amplitude (2 to 4 times the rheobase) then remained constant throughout the recording period for each individual neuron.25
Fire-polished recording pipettes, 2 to 5 MΩ resistances were used for all recordings. Whole-cell recordings were obtained with a HEKA EPC-10 USB (HEKA Instruments Inc., Lambrecht/Pfalz, Germany); data were acquired with a Patchmaster (HEKA) and analyzed with a Fitmaster (HEKA). Capacitive artifacts were fully compensated, and series resistance was compensated by ~70%. Recordings made from cells with greater than a 5 mV shift in series resistance compensation error were excluded from analysis. All experiments were performed at room temperature (~23°C). The currents were filtered at 5 kHz and sampled at 2 kHz using a Patchmaster (HEKA).
2.15. Carrageenan-induced acute inflammatory pain and paw edema
Acute inflammatory conditions were induced by injection of 50 μL 2% carrageenan in the dorsal surface of the hind paw. To test the antinociceptive activity of tat-CBD3 and myr-tat-CBD3 under acute inflammatory conditions, thermal paw withdrawal latency (PWL) was measured as described in the Thermal withdrawal latency section. In a separate experiment, hind paw edema was assessed by measuring paw thickness. Measurements of paw thickness were made before carrageenan administration and 3 hours after carrageenan administration. tat-CBD3, myr-tat-CBD, myr-tat, myr-tat-CBD3scr1, or myr-tat-CBD3scr2 peptides were then administered, and measurements were again made 3 hours after drug administration. Animals were randomly assigned to treatment conditions. All behavioral testing was performed with the experimenter blind to the treatment conditions.
2.16. Thermal withdrawal latency
Withdrawal latency to thermal stimulus was tested, as described by Quartilho et al.61 Before thermal testing, animals were allowed to acclimate in Plexiglas cages for 30 minutes. Baseline values were obtained before carrageenan treatment. Testing protocol consisted of measuring PWL in seconds by placing a mobile infrared heat source directly under the left paw hind paw of the animal. A positive response consisted of the animal lifting or licking the paw. A maximum cutoff was set at 30 seconds to avoid tissue damage. Drug or vehicle was administered 3 hours after carrageenan injection. Paw withdrawal latencies were measured every 15 minutes for the first hour and every 30 minutes for 4 more hours for a total of 5 hours of testing time. The contralateral paw was not tested.
2.17. Paw incision model of postoperative pain
An animal model of surgical pain was generated by plantar incision as previously described.7,74 Male Sprague-Dawley rats were anesthetized with isoflurane vaporized through a nose cone. The plantar aspect of the left hind paw was scrubbed with betadine and 70% alcohol 3 times. A 1-cm long incision, starting 0.5 cm from the heel and extending toward the toes, was made with a number-11 blade, through the skin and fascia of the plantar aspect of the left hind paw including the underlying muscle. The plantaris muscle was then elevated and longitudinally incised, leaving the muscle origin and insertion intact. After hemostasis with gentle pressure, the skin was closed with 2 mattress sutures of 5-0 nylon on a curved needle. Rats received an injection of gentamicin (1 mL/kg of 8 mg/mL solution, s.c.) and were allowed to recover from the anesthesia before returning to their home cage. Sham animals were anesthetized and the left hind paw scrubbed with betadine 3 times, then 70% ethanol, but no incision was made. Animals were allowed to recover for 24 hours, and then paw withdrawal thresholds were measured at 24 hours after surgery.61
2.18. Conditioned place preference/aversion
The conditioned place preference studies were performed as described previously.47 Three-chambered rat-conditioned place preference/aversion (CPP/CPA) apparatus (San Diego Instruments, San Diego, CA) with the following modifications were used: (1) end chamber of horizontal black and white striped walls, smooth floor; (2) end chamber of black walls, rough floor; and (3) middle chamber of neutral gray walls and metal rod flooring. Male Sprague-Dawley rats (200–225 g, Harlan) were implanted with intracerebroventricular cannula (i.c.v.47). Although we have previously observed penetration of tat-CBD3 into the brain after an intraperitoneal injection,9 here we chose the i.c.v. route so as to allow for the assessment of direct actions of the peptides on CNS regions associated with reward pathways. After a recovery period (7 days), rats were put in conditioned place preference boxes for 20-minute sessions and allowed to freely explore 2 end chambers and a middle transition zone with unique environmental cues in each end chamber. Baseline measurements were obtained for each rat on days 1 through 4. The average baseline per rat was determined across days and used to calculate the mean time spent in each chamber. Animals spending more than 80% of the total time in any chamber at baseline were excluded from the study. On days 5, 7, 9, 11, and 13, tat-CBD3 or myr-tat-CBD3 (10 μg in 5 μL, i.c.v.) were paired with one of the end chambers, whereas rats were confined to that chamber for 20 minutes. On days 6, 8, 10, and 12, animals received an injection of vehicle (saline 5 μL, i.c.v.) paired with the opposite chamber. On days 13 to 14, in a peptide-free state, the total time spent by the rat was recorded and compared with baseline values for each chamber (paired t test). Preference was instated if there was a significant increase in the total time spent in the putative conditioning chamber; likewise, aversion was noted by a significant decrease in total time.
2.19. Rotarod
After placement of the intrathecal catheters, the rats were trained to walk on a rotating rod (10 revolutions per minute; Rotamex 4/8 device) with a maximal cutoff time of 180 seconds.60 Training was initiated by placing the rats on a rotating rod and allowing them to walk on the rotating rod until they either fell off or 180 seconds was reached. This process was repeated 6 times, and the rats were allowed to recover for 24 hours before beginning the treatment session. Before treatment, the rats were run once on a moving rod to establish a baseline value. Saline or peptides were administered spinally through the intrathecal catheter. Assessment consisted of placing the rats on the moving rod and timing until either they fell off or reached a maximum of 180 seconds. This was repeated every 15 minutes for the first hour after drug administration and 30 minutes for the 4 hours after for a maximum time course of 5 hours.
2.20. Data analysis
GraphPad Software (La Jolla, CA) was used to determine the statistical significance. The statistical significance of differences between means was determined by Student t test or a 1- or 2-way analysis of variance (ANOVA) followed by post hoc comparisons (Dunnett or Tukey test). Groups were compared using ANOVA followed by pairwise comparisons using the Student t test with Bonferroni correction. A repeated-measures ANOVA was used when repeated measurements were made on the same animals. Significance was defined as P < 0.05.
3. Results
3.1. Rationale for generating a membrane-delimited CBD3 peptide
We previously showed that CRMP2 is a novel binding partner of CaV2.2.10,19 A 15-amino acid peptide derived from CRMP2 (designated CBD3 for calcium channel binding domain) uncoupled the CaV2.2–CRMP2 interaction leading to a decrement in Ca2+ current and neurotransmitter release and, consequently, suppressed persistent inflammatory and neuropathic hypersensitivity.9,63,85 Mutating single residues within the CBD3 peptide sequence or changing the cell-penetrating motif improved the efficacy of CBD3 in models of neuropathic pain.59,63 Here, we tested the hypothesis that tethering the CBD3 peptide to the membrane with myristate would confine the myristoylated peptide’s action(s) to uncoupling membrane CaV2.2–CRMP2 interactions, block Ca2+ influx in sensory neurons, and be effective in reducing pain-related behaviors in models of pain.
3.2. Addition of the 14-carbon myristate group targets the tat-CBD3 peptide into domains in the dorsal root ganglion plasma membrane that results in a lowered efflux from the cells
First, we synthesized a CBD3 peptide with the hydrophobic 14-carbon saturated fatty acid myristate added to the N-terminal; the protein transduction domain of the HIV-1 TAT protein was added as it was previously shown to confer intracellular delivery to the CBD3 peptide.9,85 The resulting peptide, designated myr-tat-CBD3, was further conjugated with FITC and applied onto dorsal root ganglion (DRG) neurons, and localization was assessed with live cell confocal fluorescent microscopy. Within 5 minutes of application, the fluorescence associated with the myr-tat-CBD3 peptide was localized exclusively to the membrane in DRG (Fig. 1A, top right panel). In contrast, fluorescence associated with tat-CBD3 was spatially distributed throughout the cytoplasm without any cytoplasmic rimming (Fig. 1A, top left panel). Orthogonal views (vertical and horizontal lines are planes of an optical section) of a confocal z-stack confirmed the diffuse staining of tat-CBD3 peptide (Fig. 1A, top left panel; Supplementary Information Movie S1, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A60) compared with the exclusively membrane-delimited cytoplasmic rimming of myr-tat-CBD3 peptide (Fig. 1A, top right panel; Supplementary Information Movie S2, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A61). Control peptides myr-tat and myr-tat-CBD3scr1 also exhibited membrane-delimited cytoplasmic rimming (Fig. 1A, bottom panels). Membrane staining, largely within plasma membrane domains, of myr-tat-CBD3 was recapitulated in GPMVs, which are a useful model to study peptide-lipid dynamics and the translocation of protein transduction domains across the plasma membrane (Supplementary Fig. 1, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A59). Thus, these results showed that retention of the myristoyl group on the peptide enhanced its localization to cell membranes.
Figure 1.
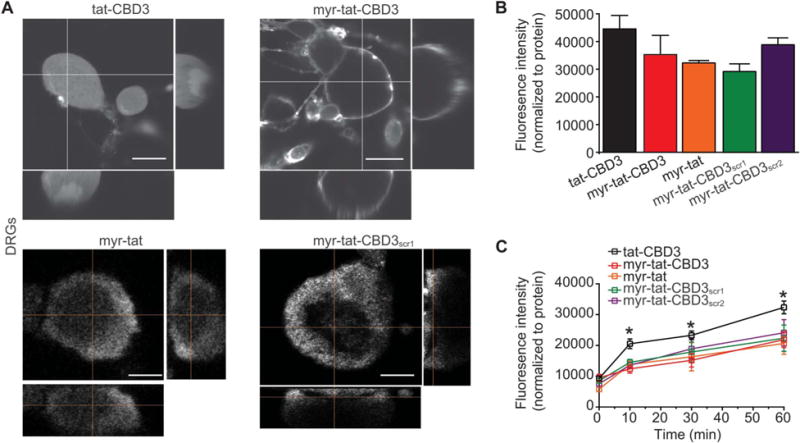
Preferential association of N-myristate-tat-CBD3 peptide in membrane-delimited domains results in a reduced efflux from DRG neurons compared with cytoplasmically localized tat-CBD3. (A), Representative confocal images of DRG neurons incubated for 10 minutes at 37°C with the fluorescein isothiocyanate (FITC)-labeled peptides. Orthogonal views (right, bottom; vertical and horizontal lines are planes of an optical section) of a confocal z-stack showing diffuse staining of tat-CBD3 (top left) compared with the exclusively membrane-delimited staining of myr-tat-CBD3 (top right). tat-CBD3 fluorescence is observed throughout the cytosol, whereas myr-tat-CBD3 labeling is delimited to the membrane of the DRG. Control peptides myr-tat (bottom left) and myr-tat-CBD3scr1 (bottom right) also exhibit membrane staining. Scale bar: 20 μm. n = 2 separate, individual experiments; the total number of neurons analyzed is 15 to 18. (B), Mean fluorescence uptake of peptides into DRG, normalized to the amount of protein per well, was similar between the indicated peptides. (C), Mean fluorescence efflux of peptides from DRG, normalized to the amount of protein per well, was significantly lower for myr-tat-CBD3 at 10, 30, and 60 minutes compared with tat-CBD3. The efflux of control myr-tat, myr-tat-CBD3scr1, or myr-tat-CBD3scr2 peptides was also significantly lower than tat-CBD3 (*P < 0.01). n = 2 separate, individual experiments; the total number of wells analyzed is 15 per condition.
3.3. Influx and efflux of tat-CBD3 and myr-tat-CBD3 peptides
The propensity of myr-tat-CBD3, but not tat-CBD3, to segregate into lipid domains may contribute to their accumulation into and efflux from DRG membranes. Therefore, we next quantitatively tested influx and rates of efflux of the 2 peptides in DRG. Dorsal root ganglia were incubated with 20 μM fluorescently labeled tat-CBD3, myr-tat-CBD3 peptides, or control peptides myr-tat, myr-tat-CBD3scr1, and myr-tat-CBD3scr2, and the FITC fluorescence was determined. To minimize any possible variability in uptake due to differences in cell plating, we normalized the fluorescence per well to the amount of protein. The fluorescence intensities were not different among either peptide-treated condition (Fig. 1B), demonstrating that influx is similar as all peptides are able to enter or stay in the membrane equally.
To interrogate the potential leakage of peptides from DRG, the media of DRG incubated with fluorescently labeled peptides was sampled immediately and 10, 30, and 60 minutes after peptide application. The fluorescence intensities were normalized to the amount of protein per well determined at the end of the experiment. The fluorescence intensity recorded with tat-CBD3-treated cells exhibiting a significantly greater efflux at 10, 30, and 60 minutes compared with myr-tat-CBD3–treated cells or control peptides (Fig. 1C). Collectively, these results demonstrate that myr-tat-CBD3 is retained in the cells longer than tat-CBD3.
3.4. N-myristate-tat-CBD3 peptide inhibits the CaV2.2–CRMP2 interaction
We previously showed that tat-CBD3 inhibited the CaV2.2–CRMP2 interaction.9 Therefore, we next tested whether the myristoylated CBD3 peptide could interfere with the CaV2.2–CRMP2 interaction. Rat spinal cord lysates were incubated with glutathione beads preadsorbed with purified CRMP2-GST in the presence of DMSO (0.3%, control), 10 μM of the control peptides (myr-tat, myr-tat-CBD3scr1, or myr-tat-CBD3scr2), or increasing concentrations (0.3–10 μM) of tat-CBD3 or myr-tat-CBD3 peptides, and then CRMP2-bound proteins were recovered by boiling in Laemmli buffer in reducing conditions, followed by immunoblotting. Probing of the CRMP2-enriched fraction with a CaV2.2 antibody demonstrated a robust interaction between CaV2.2 and CRMP2 (Fig. 2A; top blot, lane 2). Although the control peptides did not alter this interaction, the tat-CBD3 peptide was able to inhibit CaV2.2–CRMP2 interaction by ~43% at 10 μM. Myr-tat-CBD3, in a concentration-dependent manner, attenuated this interaction with greater than ~81% inhibition at 10 μM (Fig. 2A; top blot and Fig. 2B). These results demonstrate that the myristoyl group improves the ability of the peptide to break the CaV2.2–CRMP2 interaction.
Figure 2.
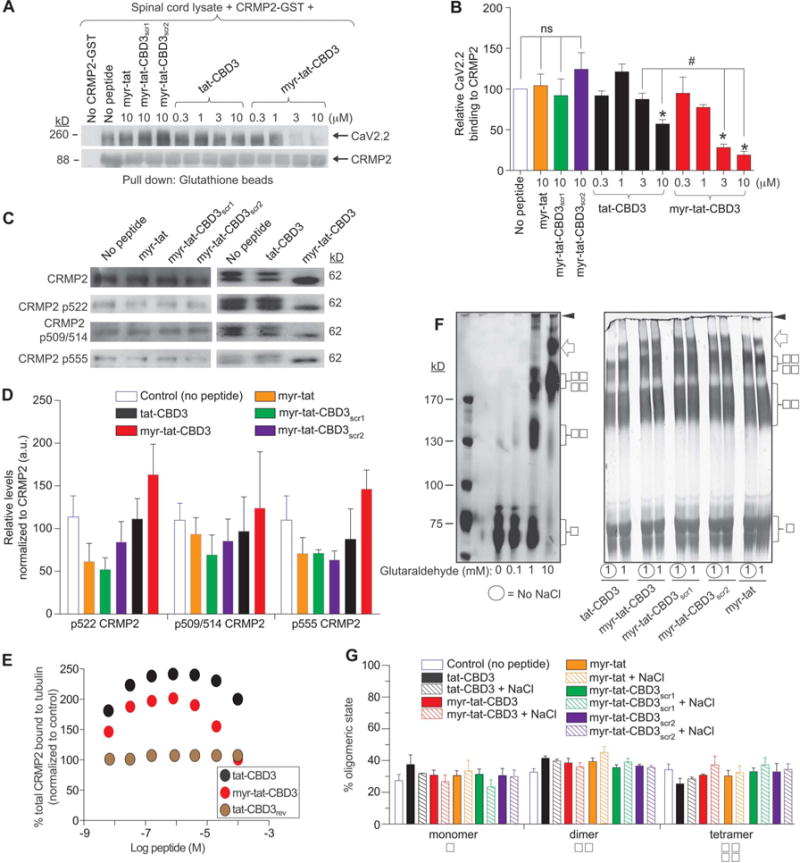
N-myristate-tat-CBD3 peptide inhibits the CaV2.2–CRMP2 interaction and augments CRMP2–tubulin interaction without effects on CRMP2 phosphorylation or oligomerization. (A), Spinal cord lysates were incubated with recombinant CRMP2-GST protein (0.4 μM) in the absence (no peptide, lane 2) or presence of increasing concentrations (as indicated) of control peptides (myr-tat, myr-tat-CBD3scr1, myr-tat-CBD3scr2) or tat-CBD3 or myr-tat-CBD3 peptides. The CRMP2-GST–bound proteins were recovered by incubation with glutathione sepharose beads and immunoblotted with CaV2.2 (top) and CRMP2 (bottom). Representative blots from 3 separate experiments are shown. (B), Summary of mean relative binding of CaV2.2 to CRMP2. CaV2.2–CRMP2 binding was reduced by myr-tat-CBD3 in a concentration-dependent manner (*P < 0.01). n = 3 separate, individual experiments. (C), Representative Western blots of lysates prepared from DRG incubated for 2 hours with vehicle (no peptide) or the indicated peptides (20 μM) probed with the indicated CRMP2 and phospho-specific CRMP2 antibodies. The positions of molecular weight markers (in kilodaltons, kD) are illustrated on the right. (D), Summary of the mean relative levels of phospho-CRMP2 (normalized to total CRMP2) in arbitrary units (au). None of the peptides affected the expression of phospho-CRMP2 forms (n = 3–6 wells per condition). (E), 96-well plates coated with 200 ng tubulin were incubated with increasing concentrations of peptides. The Y-axis displays the OD450 absorbance of the ELISA using CRMP2-specific antibodies. CRMP2 bound to tubulin with half-saturation concentration of ~607 nM.86 Binding of heat-denatured CRMP2 demonstrated nonsaturable background binding to tubulin. A control peptide, tat-CBD3rev, did not affect CRMP2-tubulin binding. Competitive binding assay revealed that the tat-CBD3 and myr-tat-CBD3 peptides increase the binding of CRMP2 to tubulin. All measurements were performed in sextuplicate and error bars indicate SEM. Most of the error bars are smaller than the symbols. (F), Covalent cross-linking of purified CRMP2 was induced with increasing concentrations of glutaraldehyde (0.1–10 mM) in the absence or presence of 50 mM NaCl. The analyzed molecular weights of the purified CRMP2 protein are 65.5 kDa and 59.3 kDa. Silver-stained sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of a typical cross-linking experiment is shown. The positions of monomer (
 ), dimer (
), dimer (
 ), and tetramer (
), and tetramer (
 ) are indicated on the right; the positions of molecular weight markers (kD) are on the left. Increasing the amount of cross-linker increases the amount of cross-linked oligomers with dimers and tetramers forming at 1 mM and predominantly tetramers at 10 mM or higher. The inclusion of tat-CBD3 or myr-tat-CBD3 or the control peptides (myr-tat, myr-tat-CBD3scr1, myr-tat-CBD3scr2) does not alter the extent of dimer or tetramer induced by 1 mM glutaraldehyde. A small amount of octameric CRMP2 is also observed (arrow). High-molecular weight aggregates that do not enter the gel are also observed (arrowhead). The peptides do not directly bind CRMP2 as the oligomeric profiles are similar irrespective of the presence of NaCl which tests if peptides disrupt the structure during the oligomerization of CRMP2. (G), The amount of protein found in oligomers (monomers, dimers, and tetramers) under 1 mM glutaraldehyde cross-linking condition was divided by the total CRMP2 in the same lane and plotted as a percentage of oligomers. n = 3 separate, individual experiments; the total number of samples analyzed is 4 to 6 per condition.
) are indicated on the right; the positions of molecular weight markers (kD) are on the left. Increasing the amount of cross-linker increases the amount of cross-linked oligomers with dimers and tetramers forming at 1 mM and predominantly tetramers at 10 mM or higher. The inclusion of tat-CBD3 or myr-tat-CBD3 or the control peptides (myr-tat, myr-tat-CBD3scr1, myr-tat-CBD3scr2) does not alter the extent of dimer or tetramer induced by 1 mM glutaraldehyde. A small amount of octameric CRMP2 is also observed (arrow). High-molecular weight aggregates that do not enter the gel are also observed (arrowhead). The peptides do not directly bind CRMP2 as the oligomeric profiles are similar irrespective of the presence of NaCl which tests if peptides disrupt the structure during the oligomerization of CRMP2. (G), The amount of protein found in oligomers (monomers, dimers, and tetramers) under 1 mM glutaraldehyde cross-linking condition was divided by the total CRMP2 in the same lane and plotted as a percentage of oligomers. n = 3 separate, individual experiments; the total number of samples analyzed is 4 to 6 per condition.
3.5. N-myristate-tat-CBD3 augments the CRMP2–tubulin interaction without effects on CRMP2 phosphorylation
We designed the membrane-delimited CBD3 to allow separation of membrane CRMP2 functions from those mediated by its interactions with cytoplasmic proteins (reviewed in Ref. 42). Because CRMP2-mediated cellular functions are regulated by its phosphorylation state, we next determined whether the peptides could alter the phosphorylation status of CRMP2. Immunoblot analyses of lysates prepared from DRG exposed to 20 μM of control (myr-tat, myr-tat-CBD3scr1, or myr-tat-CBD3scr2), tat-CBD3, or myr-tat-CBD3 peptides for 2 hours demonstrated no changes in expression of phosphorylated CRMP2 at sites 522 (by Cdk5), 509/514 (by GSK3β), or 555 (RhoK or CaM KII) (Fig. 2C, D). These results rule out a potential side effect of the myristate CBD3 peptide on CRMP2 phosphorylation. Aside from an interaction with CaV2.2, CRMP2 also interacts with tubulin,33 among other proteins.42 We had previously reported that 10 μM tat-CBD3 did not affect the CRMP2-tubulin interaction9; however, we did not address additional concentrations.76 Here, we used an enzyme-linked immunosorbent assay (ELISA)-based competition assay to determine the ability of CRMP2 to bind tubulin in the presence of increasing concentrations of a control peptide (tat-CBD3rev; CBD3 in reverse orientation), tat-CBD3 or myr-tat-CBD3 peptides. Although the tat-CBD3rev peptide had no effect, both tat-CBD3 and myr-tat-CBD3 peptides increased CRMP2 bound to tubulin with higher binding observed at lower concentrations of the peptides (Fig. 2E).
3.6. CRMP2 oligomerization in unaffected by tat-CBD3 or myr-tat-CBD3 peptides
CRMPs exists as tetramers79 and it has been demonstrated that semaphorin 3A-mediated CRMP2 signal transduction is contingent on CRMP2 tetramers.68 Moreover, mutagenesis of residues linked oligomerization to control of CRMP2-dependent neurite outgrowth.24 Therefore, we next asked whether the peptides could impact CRMP2 tetramerization. The cross-linking agent glutaraldehyde was used to examine the oligomerization status of purified CRMP2. Increasing concentrations of glutaraldehyde yielded progressively higher amounts of CRMP2 dimers and tetramers compared with monomers: at 0.01 mM glutaraldehyde, most of the CRMP2 remained monomeric; at 1 mM glutaraldehyde, CRMP2 was observed to be equally distributed among the oligomeric states; at 10 mM glutaraldehyde, CRMP2 was mostly tetrameric (Fig. 2F, left panel, Fig. 2G). Consequently, we examined the effects of the peptides on CRMP2 oligomerization in the presence of 1 mM glutaraldehyde. None of the peptides (tat-CBD3, myr-tat-CBD3, myr-tat, myr-tat-CBD3scr1, or myr-tat-CBD3scr2) changed the extent of monomers, dimers, or tetramers formed (Fig. 2F, right panel; Fig. 2G). Similar results were obtained with Western blotting with CRMP2-specific antibodies (data not shown). These results suggest that the peptides do not disrupt CRMP2 tetramerization. Modification of CRMP2 and/or tubulin conformation to a more open state with higher affinities for the other protein in the presence of the peptides might lead to increased CRMP2-tubulin binding. Such allosteric effects have been described with taxol, which promotes tubulin polymerization by enhancing its flexibility.56
3.7. Surface trafficking of CaV2.2 is inhibited by tat-CBD3 and myr-tat-CBD3 peptides
Although CBD3 has been previously demonstrated to interfere with the CaV2.2–CRMP2 interaction, the dynamics of the disruption over time by this peptide have never been studied. Having established that both peptides can inhibit the CaV2.2– CRMP2 interaction in vitro, we next asked whether this translated into a disruption in cells. An additional question was to determine whether the peptides affected the subcellular localization of both proteins. Thus, we used immunofluorescent microscopy to determine the time course of CBD3-mediated disruption of the CaV2.2–CRMP2 interaction. Sensory neuron soma were incubated with each peptide for 5 minutes, 30 minutes, 2 hours, and 48 hours and examined for their labeling of CaV2.2 and CRMP2 with specific antibodies. Visual inspection of the micrographs (Fig. 3A, B; control or myr-tat or myr-tat-CBD3scr1 panels) demonstrated that, in control cells treated with vehicle or with control peptides, the fluorescent signals for both proteins appeared to overlap at the plasma membrane of the DRG. As illustrated in Figure 3, prominent CaV2.2 staining (green) was observed within the membrane in the absence of peptide or with control peptides, and this staining colocalized strongly with CRMP2 (red). The Pearson correlation coefficient (PCC) and the MOC were used to quantify the degree of colocalization between the fluorophores for the 2 proteins; the 2 coefficients are mathematically similar but PCC relies on the deviation from the mean, whereas MOC uses absolute intensities. The high PCC and MOC values verified the membrane colocalization (Fig. 4). However, within only 5 minutes of tat-CBD3 or myr-tat-CBD3 peptide treatment, the CaV2.2 labeling from the membrane was reduced whereas CRMP2 labeling of the membrane was unaffected. This pattern of labeling was sustained for at least 2 hours and consistent with reduced PCC and MOC values (Fig. 4). The reduction in these parameters of colocalization was greater with the myr-tat-CBD3 peptide (Fig. 4). After 48 hours of either tat-CBD3 or myr-tat-CBD3 peptide treatment, CRMP2 labeling was largely absent from the membrane (Figs. 3A, B, and 4) appearing in puncta within the cytosol. CaV2.2 staining was similarly excluded from the membrane, particularly in cells treated with myr-tat-CBD3 (Fig. 3B, 48-hour panels). In contrast, cells treated with tat-CBD3 for 48 hours displayed a mixed pattern of CaV2.2 staining with cytosol punctate staining as well prominent membrane staining (Fig. 3A, 48-hour merged and inset panels). These observations were quantitatively verified by reduced PCC and MOC values after treatment with the peptides (Fig. 4A, B). These results demonstrate that by uncoupling the CaV2.2– CRMP2 interaction, both tat-CBD3 and myr-tat-CBD3 reduce surface trafficking of CaV2.2 (eg, 0.60 PCC and 0.65 MOC for the myr-tat-CBD3-treated DRG as noted by the lack of colocalization in the insets).
Figure 3.

tat-CBD3 and N-myristate-tat-CBD3 peptides induce changes in localization of CaV2.2 and CRMP2 in primary sensory neurons. Representative micrographs of DRG cells immunolabeled with CaV2.2 (green) and CRMP2 (red). The neurons are treated with (A) tat-CBD3 (20 μM) and (B) myr-tat-CBD3 (20 μM) for 5 minutes, 20 minutes, 2 hours and 48 hours. Control cells not treated with the peptides or treated with the control peptides myr-tat or myr-tat-CBD3scr1 (20 μM) display an overlap in membrane labeling of both CaV2.2 and CRMP2. Treatment with the peptides for 5 minutes, 30 minutes, and 2 hours reduces the membrane labeling of the CaV2.2 while retaining CRMP2 labeling at the membrane. At 48 hours, CaV2.2 and CRMP2 labeling is scattered in distinct puncta within the cytosol. Scale bar length is 20 μm. n = 2 separate, individual experiments; the total number of cells analyzed is 18 to 26 per time point.
Figure 4.
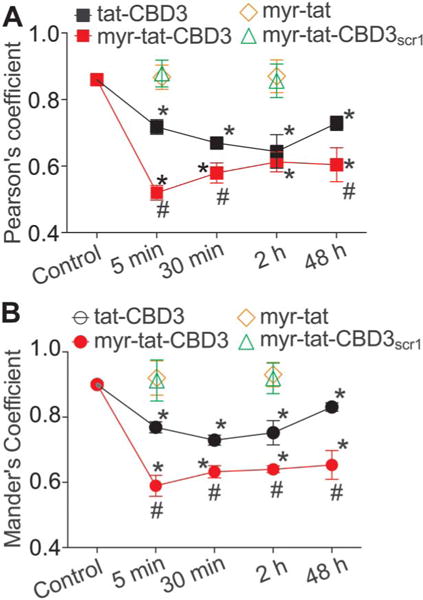
tat-CBD3 and N-myristate-tat-CBD3 peptides induce changes in colocalization of CRMP2 and CaV2.2. Quantification of colocalization of CaV2.2 with CRMP2 was carried using Pearson (A) and Manders (B) overlap method. The values in the graph are the overlap coefficients. Control cells not treated with a peptide or treated with myr-tat or myr-tat-CBD3scr1 peptides display high level of overlap as demonstrated by the high coefficient values. However, treatment with tat-CBD3 and myr-tat-CBD3 for 5 minutes, 30 minutes, 2 hours, and 48 hours reduces the overlap as indicated by the reduction of the coefficient values. *P < 0.05 compared with control. #P < 0.05 comparing tat-CBD3-treated vs myr-tat-CBD3-treated or with control peptides myr-tat-treated or myr-tat-CBD3scr1-treated DRG. n = 2 separate, individual experiments; the total number of cells analyzed is 18 to 26 per time point.
3.8. Myr-tat-CBD3 peptide reduces K+-evoked Ca2+ influx
We previously reported that uncoupling the CaV2.2–CRMP2 interaction with tat-CBD3 resulted in reduction of depolarization-induced Ca2+-influx in sensory neurons.9 Here, we asked whether the myr-tat-CBD3 peptide could work similarly. Calcium imaging experiments performed with Fura-2AM on adult mouse DRG neurons demonstrated that stimulation with high 90 mM KCl (a concentration where mostly CaV2 channels are recruited84) produced a transient rise in intracellular calcium ([Ca2+]c) (Fig. 5C, F, G). Peak calcium influx was recorded within 20 seconds of stimulation (Fig. 5G). Dorsal root ganglia incubated for 20 minutes with myr-tat-CBD3 peptide showed a concentration-dependent decrease in K+-stimulated [Ca2+]c influx (Fig. 5H). A concentration–response analysis indicated that the peptide inhibited Ca2+ influx in a concentration dependent manner with IC50 values of ~2.8 ± 0.7 μM (n = 4) (Fig. 5H). We previously reported an IC50 value of ~12.1 ± 0.6 μM for tat-CBD3.39 The myristoylated peptide was significantly better in its efficacy as maximum block by this peptide was ~50% greater than that conferred by tat-CBD3. However, 10 μM of myr-tat or myr-tat-CBD3scr1 or 30 μM of tat-CBD3rev control peptides had no effect on K+-stimulated [Ca2+]c influx (Fig. 5H).
Figure 5.
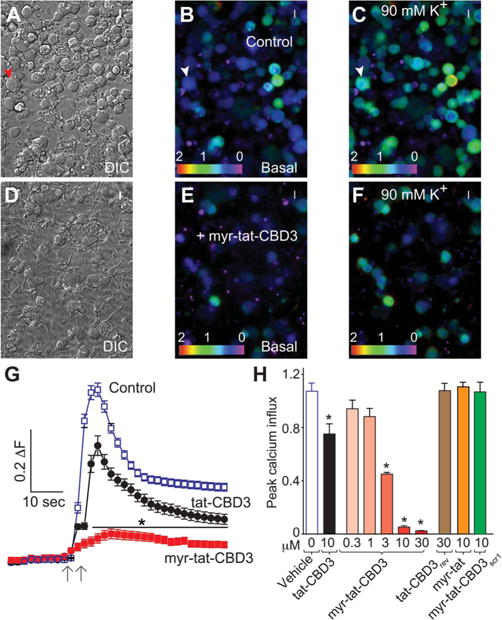
Myr-tat-CBD3 inhibits depolarization-evoked Ca2+ influx in dorsal root ganglion (DRG) neurons. (A, D), Differential interference contrast and pseudocolored fluorescent images of a field of DRG neurons visualized for Fura-2, before (basal; B, E) and after stimulation with KCl (90 mM K+; C, F). Arrowheads show a typical responding sensory neuron. Ca2+ imaging was performed on rat DRG neurons using the ratiometric Ca2+-sensitive dye Fura-2. After a 1-minute baseline measurement, neurons were stimulated with 90 mM KCl for 10 seconds. (G), The change in fluorescence (peak 2 basal; ΔF) and time as indicated by the scale bars for DRG neurons treated with vehicle control (blue), 20 μM tat-CBD3 (black), and 10 μM myr-tat-CBD3 (red) are shown. (H), Bar graphs show the peak fluorescence response (adjusted for background) of DRG incubated for 20 minutes with the indicated concentrations of myr-tat-CBD3 peptide, 10 μM control peptides (tat-CBD3rev, myr-tat, or myr-tat-CBD3scr1), or vehicle-treated DRG. Values represent the average 6 SEM from 2 to 4 separate imaging experiments from 29 to 37 cells per condition. Asterisks indicate statistical significance compared with untreated cells (*P < 0.05, 1-way analysis of variance with Dunnett post hoc test). Concentration-dependent effect of myr-tat-CBD3 peptide on depolarization-evoked Ca2+ influx in sensory neurons. Percent inhibition of peak Ca2+ influx normalized to the control is plotted for various concentrations of the indicated peptides. IC50 = 12.1 ± 0.6 μM (n = 4) for tat-CBD339 and 2.8 ± 0.7 μM for myr-tat-CBD3 (n = 4).
3.9. Myr-tat-CBD3 decreases Ca2+ currents without affecting Na+ currents in sensory neurons
We have previously shown that tat-CBD3 inhibits CaV2.2 currents in sensory neurons.9 Here, we investigated whether the myristoylated form could achieve a similar inhibition. Acute application (5–30 minutes) of myr-tat-CBD3 peptide was unable to inhibit currents (data not shown). In contrast, chronic application (~24 hours) of the myr-tat-CBD3 peptide inhibited Ca2+ currents by ~40% (Fig. 6AC); no changes were observed in activation (Fig. 6D) or inactivation of these currents (data not shown). Chronic application of control peptides myr-tat or myr-tat-CBD3scr1 did not inhibit the Ca2+ currents (Fig. 6A-C). To rule out off-target effects, we tested the effect of myr-tat-CBD3 on Na+ currents; chronic application of the peptides did not affect Na+ currents (Fig. 6E, F). Our findings demonstrate that a block of CaV2.2 currents after chronic treatment of the myr-tat-CBD3 peptide mirrors those of gabapentin, which is also ineffective in blocking currents after acute treatments.35
Figure 6.
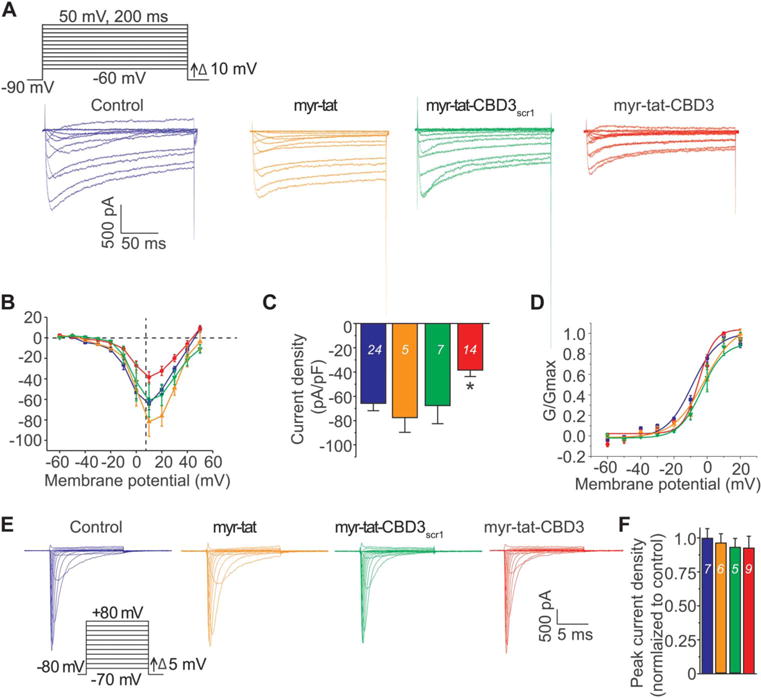
Chronic application of myr-tat-CBD3 specifically decreases Ca2+ currents with no effect on Na+ currents in sensory neurons. (A), Voltage protocol used to evoke Ca2+ currents. Currents were evoked by 200-millisecond prepulses between −90 mV and +50 mV. Representative family of current traces are illustrated from untreated (blue) neuron, 20 μM myr-tat–treated neurons (orange), 20 μM myr-tat-CBD3scr1–treated neurons (green), or 20 μM myr-tat-CBD3–treated neurons (red). (B), Summary of the current density vs membrane potential curves from sensory neurons in the absence or presence of 20 μM peptides applied overnight. (C), Peak current density, at +10 mV, for the indicated conditions. n = 14 to 16 cells, *P < 0.05; Student t test. (D), Boltzmann fits for activation for DRG treated as indicated. Values for V1/2, the voltage of half-activation, and slope factors (k) were not different between the 4 conditions. (E), Representative family of Na+ current traces recorded from a neuronal cell model (catecholamine A differentiated cell line) treated with vehicle or with the indicated peptides (20 μM) overnight before using the voltage protocol illustrated (bottom left). (F), Summary of the peak current in the absence or presence of tat-myr-CBD3. n = 5 to 8 cells (no statistical difference was observed between the means; Student t test).
3.10. Myr-tat-CBD3 suppresses excitability of small diameter dorsal root ganglion neurons
Although increases in Na+ channel density after nerve injury influence the hyperexcitability of sensory neurons, Ca2+ channel subtypes modulate nociceptive signal transduction by affecting synaptic transmission and may enhance cellular excitability by producing subthreshold membrane oscillations and spike firing within the sensory neurons.75 Both T- and R-type Ca2+ channels have been demonstrated to contribute to neuronal excitability.13,51 We previously reported that a variant of the CBD3 peptide inhibited excitability of dissociated small-diameter sensory neurons, which was attributed to inhibition of T- and R-type Ca2+ channels.59 Therefore, we investigated whether the myr-tat-CBD3 peptide could affect DRG excitability. The excitability was measured by injecting current pulses (nA) into the soma of small-diameter DRG neurons to elicit 6 to 10 APs under control conditions before the addition of peptides into the recording bath. Representative recordings (Fig. 7A-C) and grouped data show that the number of APs (APs; Fig. 7D) of small-diameter DRG neurons was significantly decreased by myr-tat-CBD3 but not by any of the other peptides tested. The rheobase (threshold current) was not affected by the control or tat-CBD3 peptides; because only 1 neuron exhibited a single AP in myr-tat-CBD3-treated cells, the rheobase value could not be reliably determined (Fig. 7E). The robust suppression of excitability by myr-tat-CBD3 peptide may suggest involvement of other channels as Ca2+ influx has been reported to be coupled to big conductance Ca2+-dependent K+ (BKCa) channels69 that contribute to the modulation of both excitability and AP duration in rat DRG sensory neurons.89 Whether myr-tat-CBD3 affects BKCa, or other channels that affect excitability, remains to be tested.
Figure 7.
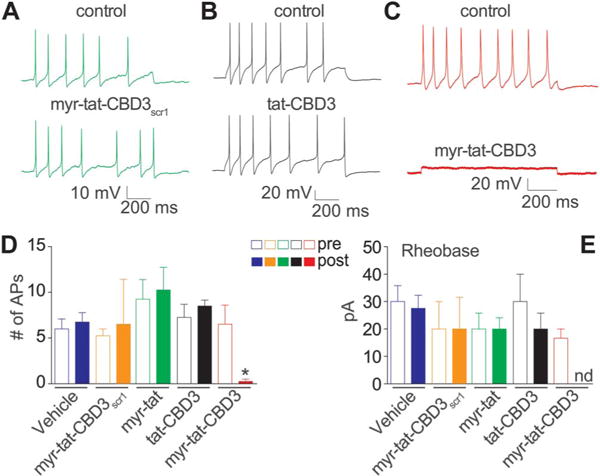
Myr-tat-CBD3 suppresses the excitability of small-diameter DRG neurons. (A-C), Representative recordings in response to a step of depolarizing current evoked action potentials (APs) under control conditions and after exposure to the indicated peptides. Summary of the actions of the peptides on the number of APs (D) and rheobase (E). Rheobase is the current required for eliciting the first AP. The AP number was significantly reduced after application of myr-tat-CBD3 but not any other peptides. Because only 1 AP was observed in 1 neuron (of 6) in myr-tat-CBD3-treated cells, the rheobase value could not be determined (nd). n = 4 to 6 cells per condition (*P < 0.001 when compared with vehicle or control peptides or tat-CBD3; 1-way analysis of variance with the Dunnett post hoc test).
3.11. tat-CBD3 and myr-tat-CBD3 peptides reverse carrageenan-induced inflammatory pain
Administration of carrageenan (50 μL of 2%) significantly decreased PWLs by ~76% 3 hours after carrageenan injection (Fig. 8A-C). The intrathecal administration of tat-CBD3 (20 μg/5 μL, i.t.) significantly attenuated carrageenan-induced thermal hypersensitivity at 90, 120, 210, 240, and 300 minutes after tat-CBD3 administration compared with the vehicle group (Fig. 8A). Administration of myr-tat-CBD3 (20 μg/5 μL i.t.) significantly attenuated carrageenan-induced thermal hypersensitivity at 15, 60, 90, 120, and 180 minutes after myr-tat-CBD3 administration compared with vehicle group (Fig. 8B). The administration of control peptides myr-tat or myr-tat-CBD3scr1 did not decrease carrageenan-induced thermal hypersensitivity. Administration of carrageenan (50 μL of 2%) produced a significant increase in paw thickness 3 hours after carrageenan injection (Fig. 8D). The carrageenan-induced edema was not blunted by either peptide (Fig. 8D).
Figure 8.
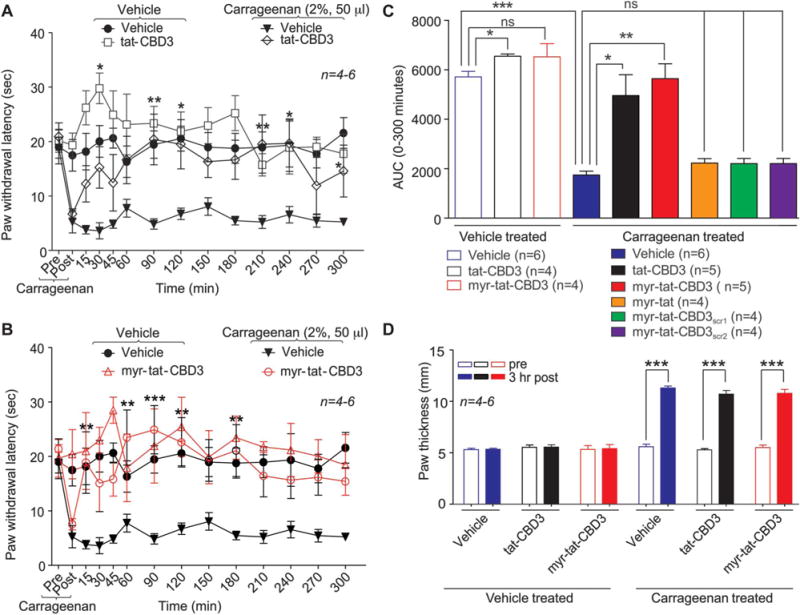
tat-CBD3 and N-myristate-tat-CBD3 peptides reduce carrageenan-induced inflammation. Spinal administration of (A) tat-CBD3 (20 μg/5 μL i.t.) and (B) myr-tat-CBD3 (20 μg/5 μL i.t.) significantly attenuate carrageenan (50 μL, 2%, intrapaw)-induced thermal hypersensitivities in rats. Paw withdrawal latencies were significantly decreased 3 hours after carrageenan injection. Administration of tat-CBD3 (20 μg/5 μL i.t.) significantly attenuated carrageenan induced thermal hypersensitivity 90, 120, 210, 240, and 300 minutes after tat-CBD3 administration compared with vehicle group (n = 5–6, *P < 0.05, **P < 0.01, ***P < 0.001). Administration of myr-tat-CBD3 (20 μg/5 μL i.t.) significantly attenuated carrageenan-induced thermal hypersensitivity 15, 60, 90, 120, and 180 minutes after myr-tat-CBD3 administration compared with the vehicle group (n = 5–6, *P < 0.05, **P < 0.01, ***P < 0.001). The administration of myr-tat, myr-tat-CBD3scr1, or myr-tat-CBD3scr2 control peptides did not significantly decrease carrageenan-induced thermal hypersensitivity. Data analyzed using 2-way analysis of variance followed by Bonferroni post tests where time was treated as “within subjects” factor, whereas treatment was treated as “between” subjects factor. (C), Summary of the data in panels A and B plotted as area under the curve (AUC) between 0 and 300 minutes. (D), Intrapaw administration of carrageenan (50 μL 2%) produced significant paw edema in rats 3 hours after carrageenan treatment. Data are expressed as mean ± SEM. ***P < 0.001 when compared with precarrageenan values.
3.12. tat-CBD3 and myr-tat-CBD3 peptides reverse post-surgical pain
Gabapentin has been shown to be antinociceptive in this model, implicating involvement of CaV2.2.30 Further involvement was proposed by a clinical study that reported analgesia after intrathecal administration of ziconotide to patients with acute postoperative pain: decreased patient controlled analgesia morphine equivalent consumption and visual analog pain intensity scores.4 Therefore, we tested the antinociceptive potential of tat-CBD3 and myr-tat-CBD3 on thermal hyperalgesia and tactile allodynia induced by an incision of the plantaris muscle of the rat hind paw, a model of postoperative surgical pain.7
An incision of the rat plantaris muscle led to an induction of thermal and tactile hypersensitivity (Fig. 9). Both nociceptive responses peaked within 24 hours after surgery and were maintained during the 5- to 6-hour experimental period in vehicle-treated animals. Spinal administration of tat-CBD3 and myr-tat-CBD3 peptides blocked the maintenance of thermal and tactile hypersensitivity for at least 4 hours (Fig. 9). The antihyperalgesic effect of tat-CBD3 disappeared after 4 hours (Fig. 9D), whereas that of myr-tat-CBD3 was sustained longer (Fig. 9E). The reversal of tactile hypersensitivity after injection of myr-tat-CBD3 was far better than tat-cbd3 with observed withdrawal thresholds: ~11 vs 5 g at 5 hours, respectively. Neither peptide demonstrated significant antinociceptive potential in sham-injured animals. The vehicle (saline) did not affect the nociceptive responses. Finally, none of the control peptides (myr-tat, myr-tat-CBD3scr1, or myr-tat-CBD3scr2) altered thermal and tactile hypersensitivities (Fig. 9C, F).
Figure 9.
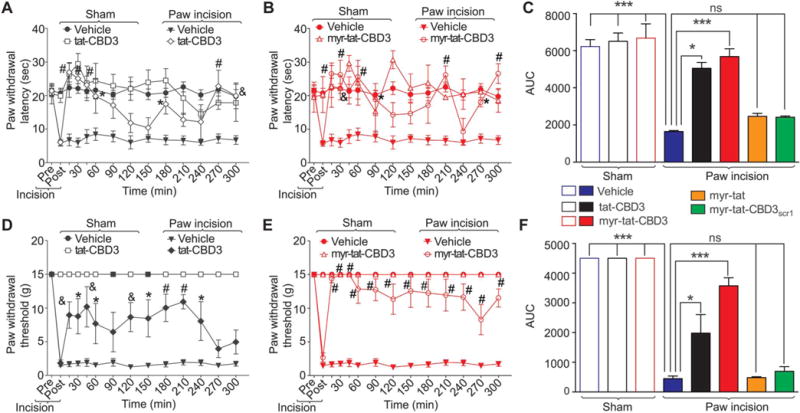
tat-CBD3 and N-myristate-tat-CBD3 peptides reduce plantar incision-induced thermal hyperalgesia and mechanical allodynia. Rats received a plantar incision on the left hind paw. Paw withdrawal latencies (PWLs) were significantly decreased 24 hours after incision. Peptides (20 μg/5 μL) or vehicle (saline) were injected into the intrathecal space and PWLs measured. Paw withdrawal latencies were significantly reversed at the indicated times after injection of tat-CBD3 (A) or myr-tat-CBD3 (B) (n = 5; *P < 0.05; &P < 0.01; and #P < 0.001; 2-way analysis of variance [ANOVA] with a Bonferroni post hoc test) where time was treated as “within subjects” factor, whereas treatment was treated as “between” subjects factor. Likewise, paw withdrawal thresholds (PWTs) were significantly decreased 24 hours after incision. Injection of tat-CBD3 (C) or myr-tat-CBD3 (D) significantly reversed PWTs at the indicated times (n = 5; *P < 0.05; &P < 0.01; and #P < 0.001; 2-way ANOVA with a Bonferroni post hoc test). Area under the curve (AUC), using the trapezoid method, for PWL (C; summary for data shown in A, B) and PWT (E; summary for data shown in D, E) are shown. There was a trending toward higher PWTs in animals injected with myr-tat-CBD3 compared with tat-CBD3 (P = 0.08) (F). Injection of myr-tat, myr-tat-CBD3scr1, or myr-tat-CBD3scr2 control peptides had no effect on thermal hyperalgesia and mechanical allodynia.
3.13. tat-CBD3 and myr-tat-CBD3 peptides do not result in conditioned place preference
Conditioned place preference (CPP) was used to determine the reward liability of tat-CBD3 and myr-tat-CBD3. Drug administration to rats was paired with a chamber for 5 exposures; total time spent in that paired chamber was measured 24 hours after the final exposure, in drug-free animals (Fig. 10A). At baseline, all rats spent 43% to 45% of the total time in the putative conditioning chamber over a 20-minute period (526.6 ± 52.8 seconds; Figure 10B). Unlike our previous studies demonstrating that morphine (i.c.v. dosing) results in a positive condition place preference indicative of the potential of rewarding behavior,47 tat-CBD3 and myr-tat-CBD3 (10 μg in 5 μL, i.c.v.) did not produce a significant increase in total time per chamber (45.7% ± 7.1% and 46.5% ± 5.9%, respectively; P = 0.76) nor a decrease suggesting lack of conditioned place aversion. The CPP experiments performed here used an ICV route of administration that allowed for direct delivery to the brain centers for rewarding behavior hence giving us the best ability to detect whether these peptides would be abused or avoided. These data suggest that both peptides act neutrally at supraspinal sites and are not likely to produce rewarding or aversive behaviors.
Figure 10.
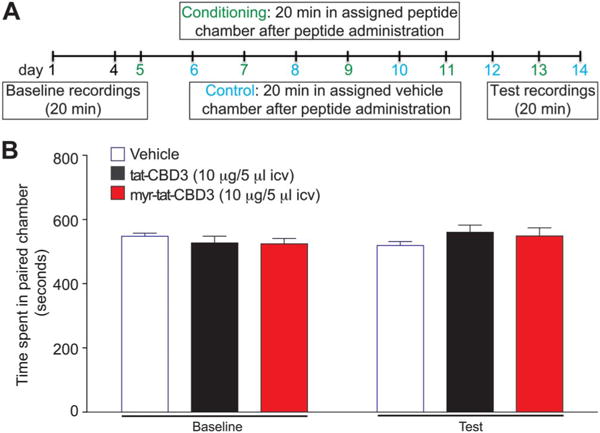
Intracerebroventricular administration of tat-CBD3 or myr-tat-CBD3 does not induce conditioned place preference in rats. (A) Schematic of the conditioned place preference (CPP) experimental timeline. (B) Animals were conditioned to associate with vehicle or drug (5 injections) with environmental cues. Baseline before peptide administration resulted in animals spending approximately equal time on both sides of the CPP chambers. Intracerebroventricular administration (10 μg/5 μL, i.c.v.) of either peptide did not result in an increase or decrease in total time spent in the putative conditioning chamber 24 hours after the final i.c.v. injection (P > 0.5, paired t test). Data represent mean time (in seconds) ± SEM. n = 11 rats per condition.
3.14. tat-CBD3 and myr-tat-CBD3 peptides do not produce motor impairment or paralysis in rats
tat-CBD3 (20 μg/5 μL i.t.) and myr-tat-CBD3 (20 μg/5 μL i.t.) peptides were evaluated for typical motor deficits reported with opioids including sedation. Rats were trained to walk on a rotating rod with a maximal cutoff time of 180 seconds before administration of drug or vehicle. The mean baseline latency for all animals was 180 ± 0 seconds. Vehicle-treated animals remained on the rotarod for an average of 180 ± 0 seconds at each time point over the course of 300 minutes. Animals treated with the control peptides myr-tat-CBD3scr1 or myr-tat-CBD3scr2 (20 μg/5 μL i.t.) remained on the rotarod for an average of 172.9 ± 11.0 seconds or 175.7 ± 12.2 seconds, respectively, over the course of 300 minutes (Fig. 11). Animals treated with tat-CBD3 (20 μg/5 μL i.t.) remained on the rotarod for an average of 180 ± 0 seconds over the course of 300 minutes (Fig. 11). Animals treated with myr-tat-CBD3 (20 μg/5 μL i.t.) remained on the rotarod with minimum value of 147.4 ± 20.0 seconds, not significantly different from vehicle or control peptide-treated animals and baseline values (Fig. 11).
Figure 11.

The spinal administration of tat-CBD3 and myr-tat-CBD3 did not produce significant deficits in motor coordination as tested using the rotarod performance test. tat-CBD3 (20 μg/5 μL i.t.) and myr-tat-CBD3 (20 μg/5 μL i.t.) was evaluated for motor deficits using the rotarod performance test. Vehicle-treated animals and tat-CBD3–treated animals remained on the rotarod for an average of 180 ± 0 seconds at each time point over the course of 300 minutes. Animals treated with myr-tat-CBD3 remained on the rotarod for at least an average of 147.4 ± 20.0 seconds, a value that was not significantly different from vehicle treated or control peptides-treated animals and baseline values. n = 5 to 6 rats per condition.
4. Discussion
The present findings make a compelling case for the use of a myristoylated CRMP2 peptide, which uncouples the CaV2.2–CRMP2 interaction, for the treatment of inflammatory and incision-induced pain. The myristoylated peptide potently and efficaciously inhibits CaV2.2 trafficking and activity in sensory neurons in vitro. CRMP2 and CaV2.2 upregulation have been reported in preclinical inflammatory and neuropathic models.20,41 The plasticity in expression associated with these targets in pain pathology supports the therapeutic targeting of the CaV2.2–CRMP2 axis for a variety of pain conditions. The pharmacological action of peptides observed in our in vitro experiments is linked to uncoupling of the CaV2.2–CRMP2 interaction culminating in alleviation of inflammatory and neuropathic pain.9,42,59,85,87 Sustained, but incomplete, relief of neuropathic pain has been successfully demonstrated for a nonmyristoylated CRMP2 peptide.31 We postulate that the myristoylated version described herein may afford greater efficacy and clinical utility as a gene therapy strategy for chronic pain.
Myristoylation is an irreversible protein lipidation that anchors modified proteins to membranes.3,6,88 We postulated that addition of myristate to the CRMP2 peptide would anchor it to the membrane, enhance its local concentration near CaV2.2, resulting in greater efficacy and limiting spurious effects. Indeed, myristoylation restricted CBD3 to the DRG membrane (Fig. 1A). Because myristoyl is insufficient for stable membrane association,62,67 inclusion of the protein transduction domain of the TAT protein, which harbors a basic motif, was necessary for myr-tat-CBD3 peptide incorporation into the membrane.
Myristoylation allows uptake of cargo into cells impermeant to tat57 possibly through flip-flop diffusion mechanisms26; tat is believed to deliver cargo through an endocytosis-dependent fashion. The uptake of myr-tat-CBD3 was similar to that of tat-CBD3 (Fig. 1B, C), but its leakage/efflux from cells was reduced. Coupled with myr-tat-CBD3 membrane rimming, this may account for the greater and long-lasting reduction in surface CaV2.2 trafficking (Figs. 3, 4). CaV2.2 channels are reportedly organized into cell surface clusters64 containing raft components necessary for CaV activity. Notably, CRMP2 binding to lipid rafts has been reported.65 Myristoylation has been shown to differentially target proteins to submembrane domains such as lipid rafts.52,53 The myr-tat-CBD3 peptide may modulate CaV2.2 clustering thereby limiting their activity and redistributing CRMP2 within the membrane. In support of this idea, using model membranes, we demonstrated that the myr-tat-CBD3 peptide distributes into membrane clusters and may induce a rearrangement of membrane components.
As previously described, CaV2.2–CRMP2 disruption by tat-CBD3 decreased surface CaV2.2 in DRG.9,39 Myr-tat-CBD3 peptide interfered with the CaV2.2–CRMP2 interaction more efficiently than tat-CBD3 as observed by a reduction in colocalization within minutes of incubation (Fig. 3). Myr-tat-CBD3 was also more efficient in disrupting CaV2.2–CRMP2 colocalization after chronic (48 hours) treatment compared with tat-CBD3 peptide. The greater impairment of CaV2.2 recycling was accompanied with a superior ability to block depolarization-triggered calcium influx (Fig. 5); a significantly higher extent of inhibition (~50%) of calcium influx in DRG neurons was concomitant with a >4-fold change in IC50 value compared with tat-CBD3. The ability to affect CaV2.2 trafficking after acute and chronic administration distinguishes the peptides from gabapentin, which inhibits CaV2.2 currents only after chronic incubation.36 The acute effect of the peptides on CaV2.2 trafficking observed in our studies is commensurate with the rapid (within minutes) removal of CaV channels from the plasma membrane.78 Although gabapentin is postulated to be only an inhibitor of CaV2.2 trafficking in neuropathic pain models, our data suggest that the peptides are able to inhibit both trafficking and calcium channel activity that may account for their superior antinociceptive actions.
The mechanism of action of myr-tat-CBD3 may involve effects on both channel trafficking and inhibition. The auxiliary subunit α2δ is upregulated after injury1,49 but no such increases have been reported for CaV2.2. It has been recently postulated that the α2δ subunit associates intimately with CaV2.2 in the endoplasmic reticulum and can then rapidly increase its surface trafficking, but not endocytosis.15 This interaction is targeted by gabapentin and results in a decrease of recycling of CaV2.2 explaining the delayed effect of this drug.35,36 The rapid effect (within 5 minutes) of the myr-tat-CBD3 peptide on CaV2.2 suggests a more direct action in triggering CaV2.2 endocytosis. In addition to this effect on surface CaV2.2 channels, the data from chronic application suggests that the myristoylated peptide is also able to target CaV2.2 on its peregrination to the membrane. The ability of the myr-tat-CBD3 to induce endocytosis within minutes of application also provides a valuable tool for studies examining trafficking of CaV channels in neurons. In addition, the ability of myr-tat-CBD3 to inhibit currents after chronic application (Figs. 3, 4, 6) may allow prolongation of the reduction in calcium influx to control long-term calcium dysregulation and nociceptive behaviors.
CaV2.2 channels are a nidus for neurotransmitter release and nociceptive neurotransmission. Plasticity in expression of presynaptic CaV2.2,20 and CRMP2,41 has been demonstrated in chronic states supporting an increased dependence of nociceptive transmission upon CaV2.2. However, the efficacy of peptides acutely in the incision model indicates that increased expression of CRMP2 or CaV2.2 may not be necessary. We and others9,31,42,59,85,87 demonstrate that targeting CaV2.2, with synthetic CBD3 peptides, is an efficient strategy for treating acute inflammatory and persistent neuropathic pain.
Targeting CaV2.2 has been a relatively successful strategy for treating preclinical and clinical pain. For instance, Phα1β, a neurotoxin from the spider Phoneutria nigriventer, which reversibly inhibits L-(CaV1.2), N, P/Q-(CaV2.1), and R-(CaV2.3) type calcium channels, decreased mechanical hypersensitivity for 3 hours after CFA treatment with a maximum effect at 2 hours.22 Intrathecal administration of 100 μg of ω-conotoxin MVIIA reduced both thermal hypersensitivity and mechanical allodynia for nearly 2 to 4 hours with a maximal effect at 2 hours.80 Although effective in inhibiting pain in humans and rodents, ziconotide (Prialt) has limited use because of a narrow therapeutic window and can only be given intrathecally.46 Intrathecal administration of gabapentin resulted in an increase in the withdrawal threshold evoked by application of von Frey filaments on the incised paw with a peak effect (~19% reversal of hypersensitivity) occurring 60 minutes after injection.18 Gabapentin is widely used clinically but has had mixed results concerning efficacy and often results in severe sedation among other side effects. These can be attributed to the controversy surrounding the purported mechanism of action of gabapentin as studies report different mechanisms.12,27,28,36,73 Recently, Huwentoxin-XVI, a toxin from the tarantula Ornithoctonus huwena, which inhibits N-type calcium channels, reduced postincision allodynia with a maximal effect at 2.5 hours23; however, this should be used cautiously in humans because of potential toxicities.
Our extended examination of CBD3 in a postoperative incisional pain model revealed that using a myr-tat-CBD3 approach was highly efficacious after a single administration. This model, in which a surgical incision in the animal’s hind paw causes thermal and tactile hypersensitivity, displays similarities to the human postoperative pain syndrome and relies on CaV2.2 for pain signal transmission.17 Mechanistically, p38, a Cdk5 substrate,77 is hyperphosphorylated in DRG during postsurgical pain,2 reflecting increased Cdk5 activity. We reported that Cdk5-mediated CRMP2 phosphorylation increases its binding to CaV2.2, which augments calcium influx11; thus, the increased interaction between CRMP2 and CaV2.2 may underlie the mechanism of antinociception in this particular model. Indeed, spinal administration of both tat-CBD3 and myr-tat-CBD3 reduced thermal and tactile hypersensitivity when administered after the surgery (Fig. 8). The absence of effects on hypersensitivity with a peptide lacking CBD3 (myr-tat) or 2 scrambled versions of the CBD3 peptide (myr-tat-CBD3scr1 or scr2) demonstrates specificity of the antinociceptive effect to the CBD3 sequence (Fig. 8). Reversal of thermal hypersensitivity was immediate and complete, lasting for ~2.5 hours with tat-CBD3 and ~4 hours with myr-tat-CBD3; similarly, reversal of tactile hypersensitivity was also immediate and sustained for the entire duration of the experiment.85 Thus, the extended duration of action may be linked to membrane tethering of the peptide. Our studies suggest that myr-tat-CBD3, by targeting the modulation of CaV2.2 rather than the calcium channels or their subunits directly, has potential application in the management of inflammatory and postoperative pain and may offer a longer duration of antinociception without noted side effects of motor impairment/sedation or reward liability when compared with clinical standards (ie, opioids or gabapentin).
The risks associated with narcotic dosing for pain have resulted in 36% of patients and 68% of primary care physicians reporting reservations about taking or prescribing opioids.37,66 Notably, opioid analgesics are among the most highly abused drugs in the United States causing overdose as measured by treatment center admission.34,82 In 2012, 7 million Americans aged 12 years and older abused prescription pain-relievers for nonmedical purposes. Data here demonstrate a novel efficacious compound to inhibit pain without demonstrating any addiction or motor deficits (Fig. 10, 11) providing an instructive example of how designing peptides tailored to limit membrane CaV2.2–CRMP2 interactions can have a great utility in the treatment of post-surgical and inflammatory pain.
Supplementary Material
Acknowledgments
This work was supported by a Career Development Award from the Arizona Health Science Center to M. Khanna, a National Scientist Development grant SDG5280023 from the American Heart Association, and a Neurofibromatosis New Investigator Award NF1000099 from the Department of Defense Congressionally Directed Military Medical Research and Development Program to R. Khanna.
The authors thank Dr Joel M. Brittain for helpful discussions and members of the Khanna Laboratory for technical assistance regarding this project; Dr Karl J. Dria (Department of Chemistry and Chemical Biology, Indiana University-Purdue University Indianapolis) for mass spectrometry analysis of purified CRMP2; Dr Pat W. Mantyh (Department of Pharmacology, University of Arizona) for generous use of a Zeiss confocal microscope, and Dr Brooke B. Massani (W. M. Keck Center for Surface and Interface Imaging, University of Arizona Chemistry & Biochemistry) for assistance with confocal imaging. L. François-Moutal, Y. Wang, and A. Moutal contributed equally to this study.
Appendix A. Supplemental Digital Content
Supplemental Digital Content associated with this article can be found online at http://links.lww.com/PAIN/A59, http://links.lww.com/PAIN/A60, http://links.lww.com/PAIN/A61.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.painjournalonline.com).
Conflict of interest statement
The authors have no conflicts of interest to declare.
References
- 1.Abe M, Kurihara T, Han W, Shinomiya K, Tanabe T. Changes in expression of voltage-dependent ion channel subunits in dorsal root ganglia of rats with radicular injury and pain. Spine (Phila Pa 1976) 2002;27:1517–24. doi: 10.1097/00007632-200207150-00007. [DOI] [PubMed] [Google Scholar]
- 2.Alkaitis MS, Solorzano C, Landry RP, Piomelli D, DeLeo JA, Romero-Sandoval EA. Evidence for a role of endocannabinoids, astrocytes and p38 phosphorylation in the resolution of postoperative pain. PLoS One. 2010;5:e10891. doi: 10.1371/journal.pone.0010891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asada A, Yamamoto N, Gohda M, Saito T, Hayashi N, Hisanaga S. Myristoylation of p39 and p35 is a determinant of cytoplasmic or nuclear localization of active cyclin-dependent kinase 5 complexes. J Neurochem. 2008;106:1325–36. doi: 10.1111/j.1471-4159.2008.05500.x. [DOI] [PubMed] [Google Scholar]
- 4.Atanassoff PG, Hartmannsgruber MW, Thrasher J, Wermeling D, Longton W, Gaeta R, Singh T, Mayo M, McGuire D, Luther RR. Ziconotide, a new N-type calcium channel blocker, administered intrathecally for acute postoperative pain. Reg Anesth Pain Med. 2000;25:274–8. doi: 10.1016/s1098-7339(00)90010-5. [DOI] [PubMed] [Google Scholar]
- 5.Bauer B, Davidson M, Orwar O. Proteomic analysis of plasma membrane vesicles. Angew Chem Int Ed Engl. 2009;48:1656–9. doi: 10.1002/anie.200803898. [DOI] [PubMed] [Google Scholar]
- 6.Bhatnagar RS, Gordon JI. Understanding covalent modifications of proteins by lipids: where cell biology and biophysics mingle. Trends Cell Biol. 1997;7:14–20. doi: 10.1016/S0962-8924(97)10044-7. [DOI] [PubMed] [Google Scholar]
- 7.Brennan TJ, Vandermeulen EP, Gebhart GF. Characterization of a rat model of incisional pain. PAIN. 1996;64:493–501. doi: 10.1016/0304-3959(95)01441-1. [DOI] [PubMed] [Google Scholar]
- 8.Brittain JM, Chen L, Wilson SM, Brustovetsky T, Gao X, Ashpole NM, Molosh AI, You H, Hudmon A, Shekhar A, White FA, Zamponi GW, Brustovetsky N, Chen J, Khanna R. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2) J Biol Chem. 2011;286:37778–92. doi: 10.1074/jbc.M111.255455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, Liu N, Xiong W, Ripsch MS, Wang Y, Fehrenbacher JC, Fitz SD, Khanna M, Park CK, Schmutzler BS, Cheon BM, Due MR, Brustovetsky T, Ashpole NM, Hudmon A, Meroueh SO, Hingtgen CM, Brustovetsky N, Ji RR, Hurley JH, Jin X, Shekhar A, Xu XM, Oxford GS, Vasko MR, White FA, Khanna R. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2)(+) channel complex. Nat Med. 2011;17:822–9. doi: 10.1038/nm.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem. 2009;284:31375–90. doi: 10.1074/jbc.M109.009951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brittain JM, Wang Y, Eruvwetere O, Khanna R. Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2. 2 FEBS Lett. 2012;586:3813–18. doi: 10.1016/j.febslet.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 12.Brown JT, Randall A. Gabapentin fails to alter P/Q-type Ca2+ channel-mediated synaptic transmission in the hippocampus in vitro. Synapse. 2005;55:262–9. doi: 10.1002/syn.20115. [DOI] [PubMed] [Google Scholar]
- 13.Cain SM, Snutch TP. Contributions of T-type calcium channel isoforms to neuronal firing. Channels (Austin) 2010;4:475–82. doi: 10.4161/chan.4.6.14106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao YQ. Voltage-gated calcium channels and pain. PAIN. 2006;126:5–9. doi: 10.1016/j.pain.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 15.Cassidy JS, Ferron L, Kadurin I, Pratt WS, Dolphin AC. Functional exofacially tagged N-type calcium channels elucidate the interaction with auxiliary alpha2delta-1 subunits. Proc Natl Acad Sci U S A. 2014;111:8979–84. doi: 10.1073/pnas.1403731111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charras GT, Yarrow JC, Horton MA, Mahadevan L, Mitchison TJ. Non-equilibration of hydrostatic pressure in blebbing cells. Nature. 2005;435:365–9. doi: 10.1038/nature03550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng JK, Chen CC, Yang JR, Chiou LC. The antiallodynic action target of intrathecal gabapentin: Ca2+ channels, KATP channels or N-methyl-d-aspartic acid receptors? Anesth Analg. 2006;102:182–7. doi: 10.1213/01.ane.0000189550.97536.83. [DOI] [PubMed] [Google Scholar]
- 18.Cheng JK, Pan HL, Eisenach JC. Antiallodynic effect of intrathecal gabapentin and its interaction with clonidine in a rat model of postoperative pain. Anesthesiology. 2000;92:1126–31. doi: 10.1097/00000542-200004000-00031. [DOI] [PubMed] [Google Scholar]
- 19.Chi XX, Schmutzler BS, Brittain JM, Hingtgen CM, Nicol GD, Khanna R. Regulation of N-type voltage-gated calcium (CaV2.2) channels and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J Cell Sci. 2009;23:4351–62. doi: 10.1242/jcs.053280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cizkova D, Marsala J, Lukacova N, Marsala M, Jergova S, Orendacova J, Yaksh TL. Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp Brain Res. 2002;147:456–63. doi: 10.1007/s00221-002-1217-3. [DOI] [PubMed] [Google Scholar]
- 21.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Souza AH, Lima MC, Drewes CC, da Silva JF, Torres KC, Pereira EM, de Castro Junior CJ, Vieira LB, Cordeiro MN, Richardson M, Gomez RS, Romano-Silva MA, Ferreira J, Gomez MV. Antiallodynic effect and side effects of Phalpha1beta, a neurotoxin from the spider Phoneutria nigriventer: comparison with omega-conotoxin MVIIA and morphine. Toxicon. 2011;58:626–33. doi: 10.1016/j.toxicon.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Deng M, Luo X, Xiao Y, Sun Z, Jiang L, Liu Z, Zeng X, Chen H, Tang J, Zeng W, Songping L. Huwentoxin-XVI, an analgesic, highly reversible mammalian N-type calcium channel antagonist from Chinese tarantula Ornithoctonus huwena. Neuropharmacology. 2014;79:657–67. doi: 10.1016/j.neuropharm.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Deo RC, Schmidt EF, Elhabazi A, Togashi H, Burley SK, Strittmatter SM. Structural bases for CRMP function in plexin-dependent semaphorin3A signaling. EMBO J. 2004;23:9–22. doi: 10.1038/sj.emboj.7600021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan JH, Wang Y, Duarte D, Vasko MR, Nicol GD, Hingtgen CM. Ras signaling pathways mediate NGF-induced enhancement of excitability of small-diameter capsaicin-sensitive sensory neurons from wildtype but not Nf1+/− mice. Neurosci Lett. 2011;496:70–4. doi: 10.1016/j.neulet.2011.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisele F, Kuhlmann J, Waldmann H. Synthesis and membrane binding properties of a lipopeptide fragment from influenza virus a hemagglutinin. Chemistry. 2002;8:3362–76. doi: 10.1002/1521-3765(20020802)8:15<3362::AID-CHEM3362>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 27.Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–92. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fehrenbacher JC, Taylor CP, Vasko MR. Pregabalin and gabapentin reduce release of substance P and CGRP from rat spinal tissues only after inflammation or activation of protein kinase C. PAIN. 2003;105:133–41. doi: 10.1016/s0304-3959(03)00173-8. [DOI] [PubMed] [Google Scholar]
- 29.Feldman P, Khanna R. Challenging the catechism of therapeutics for chronic neuropathic pain: Targeting CaV2. 2 interactions with CRMP2 peptides Neurosci Lett. 2013;557:27–36. doi: 10.1016/j.neulet.2013.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Field MJ, Holloman EF, McCleary S, Hughes J, Singh L. Evaluation of gabapentin and S-(+)-3-isobutylgaba in a rat model of postoperative pain. J Pharmacol Exp Ther. 1997;282:1242–6. [PubMed] [Google Scholar]
- 31.Fischer G, Pan B, Vilceanu D, Hogan QH, Yu H. Sustained relief of neuropathic pain by AAV-targeted expression of CBD3 peptide in rat dorsal root ganglion. Gene Ther. 2014;21:44–51. doi: 10.1038/gt.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fridriksson EK, Shipkova PA, Sheets ED, Holowka D, Baird B, McLafferty FW. Quantitative analysis of phospholipids in functionally important membrane domains from RBL-2H3 mast cells using tandem high-resolution mass spectrometry. Biochemistry. 1999;38:8056–63. doi: 10.1021/bi9828324. [DOI] [PubMed] [Google Scholar]
- 33.Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–91. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- 34.Hall AJ, Logan JE, Toblin RL, Kaplan JA, Kraner JC, Bixler D, Crosby AE, Paulozzi LJ. Patterns of abuse among unintentional pharmaceutical overdose fatalities. JAMA. 2008;300:2613–20. doi: 10.1001/jama.2008.802. [DOI] [PubMed] [Google Scholar]
- 35.Heblich F, Tran Van Minh A, Hendrich J, Watschinger K, Dolphin AC. Time course and specificity of the pharmacological disruption of the trafficking of voltage-gated calcium channels by gabapentin. Channels (Austin) 2008;2:4–9. doi: 10.4161/chan.2.1.6045. [DOI] [PubMed] [Google Scholar]
- 36.Hendrich J, Van Minh AT, Heblich F, Nieto-Rostro M, Watschinger K, Striessnig J, Wratten J, Davies A, Dolphin AC. Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc Natl Acad Sci U S A. 2008;105:3628–33. doi: 10.1073/pnas.0708930105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Institute of Medicine Report from the Committee on Advancing Pain Research C. Education Relieving pain in America, a blueprint for transforming prevention, care, education and research. The National Academies Press; 2011. [PubMed] [Google Scholar]
- 38.Jin L, Millard AC, Wuskell JP, Clark HA, Loew LM. Cholesterol-enriched lipid domains can be visualized by di-4-ANEPPDHQ with linear and nonlinear optics. Biophys J. 2005;89:L04–06. doi: 10.1529/biophysj.105.064816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ju W, Li Q, Allette YM, Ripsch MS, White FA, Khanna R. Suppression of pain-related behavior in two distinct rodent models of peripheral neuropathy by a homopolyarginine-conjugated CRMP2 peptide. J Neurochem. 2012;124:869–79. doi: 10.1111/jnc.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ju W, Li Q, Wilson SM, Brittain JM, Meroueh L, Khanna R. SUMOylation alters CRMP2 regulation of calcium influx in sensory neurons. Channels (Austin) 2013 May-Jun;7(3):153–9. doi: 10.4161/chan.24224. Epub 2013 Mar 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamiya YSK, Takiguchi M, Funakoshi K. CDK5, CRMP2 and NR2B in spinal dorsal horn and dorsal root ganglion have different role in pain signaling between neuropathic pain model and inflammatory pain model: 14AP4‐5. Eur J Anaesthesiol. 2013;30:214. [Google Scholar]
- 42.Khanna R, Wilson SM, Brittain JM, Weimer JM, Sultana R, Butterfield AD, Hensley K. Opening Pandoras’ jar: a primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neurons, and central disorders. Future Neurol. 2012;5:749–71. doi: 10.2217/FNL.12.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khanna R, Zougman A, Stanley EF. A proteomic screen for presynaptic terminal N-type calcium channel (CaV2.2) binding partners. J Biochem Mol Biol. 2007;40:302–14. doi: 10.5483/bmbrep.2007.40.3.302. [DOI] [PubMed] [Google Scholar]
- 44.Kim C, Jeon D, Kim YH, Lee CJ, Kim H, Shin HS. Deletion of N-type Ca2+ channel Cav2.2 results in hyperaggressive behaviors in mice. J Biol Chem. 2009 Jan 30;284(5):2738–45. doi: 10.1074/jbc.M807179200. Epub 2008 Nov 12. [DOI] [PubMed] [Google Scholar]
- 45.Kim C, Jun K, Lee T, Kim SS, McEnery MW, Chin H, Kim HL, Park JM, Kim DK, Jung SJ, Kim J, Shin HS. Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage-dependent calcium channel. Mol Cell Neurosci. 2001;18:235–45. doi: 10.1006/mcne.2001.1013. [DOI] [PubMed] [Google Scholar]
- 46.Klotz U. Ziconotide—a novel neuron-specific calcium channel blocker for the intrathecal treatment of severe chronic pain—a short review. Int J Clin Pharmacol Ther. 2006;44:478–83. doi: 10.5414/cpp44478. [DOI] [PubMed] [Google Scholar]
- 47.Largent-Milnes TM, Brookshire SW, Skinner DP, Jr, Hanlon KE, Giuvelis D, Yamamoto T, Davis P, Campos CR, Nair P, Deekonda S, Bilsky EJ, Porreca F, Hruby VJ, Vanderah TW. Building a better analgesic: multifunctional compounds that address injury-induced pathology to enhance analgesic efficacy while eliminating unwanted side effects. J Pharmacol Exp Ther. 2013;347:7–19. doi: 10.1124/jpet.113.205245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q, Lau A, Morris TJ, Guo L, Fordyce CB, Stanley EF. A syntaxin 1, Galpha (o), and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J Neurosci. 2004;24:4070–81. doi: 10.1523/JNEUROSCI.0346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, Yaksh TL. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21:1868–75. doi: 10.1523/JNEUROSCI.21-06-01868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manders EMM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-colour confocal images. J Microsc. 1993;169:375–82. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 51.Matthews EA, Bee LA, Stephens GJ, Dickenson AH. The Cav2.3 calcium channel antagonist SNX-482 reduces dorsal horn neuronal responses in a rat model of chronic neuropathic pain. Eur J Neurosci. 2007;25:3561–9. doi: 10.1111/j.1460-9568.2007.05605.x. [DOI] [PubMed] [Google Scholar]
- 52.McCabe JB, Berthiaume LG. Functional roles for fatty acylated amino-terminal domains in subcellular localization. Mol Biol Cell. 1999;10:3771–86. doi: 10.1091/mbc.10.11.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCabe JB, Berthiaume LG. N-terminal protein acylation confers localization to cholesterol, sphingolipid-enriched membranes but not to lipid rafts/caveolae. Mol Biol Cell. 2001;12:3601–17. doi: 10.1091/mbc.12.11.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McGivern JG. Ziconotide: a review of its pharmacology and use in the treatment of pain. Neuropsychiatr Dis Treat. 2007;3:69–85. doi: 10.2147/nedt.2007.3.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLaughlin S, Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci. 1995;20:272–6. doi: 10.1016/s0968-0004(00)89042-8. [DOI] [PubMed] [Google Scholar]
- 56.Mitra A, Sept D. Taxol allosterically alters the dynamics of the tubulin dimer and increases the flexibility of microtubules. Biophys J. 2008;95:3252–8. doi: 10.1529/biophysj.108.133884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nelson AR, Borland L, Allbritton NL, Sims CE. Myristoyl-based transport of peptides into living cells. Biochemistry. 2007;46:14771–81. doi: 10.1021/bi701295k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park J, Luo ZD. Calcium channel functions in pain processing. Channels (Austin) 2010;4:510–17. doi: 10.4161/chan.4.6.12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piekarz AD, Due MR, Khanna M, Wang B, Ripsch MS, Wang R, Meroueh SO, Vasko MR, White FA, Khanna R. CRMP-2 peptide mediated decrease of high and low voltage-activated calcium channels, attenuation of nociceptor excitability, and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Mol Pain. 2012;8:54. doi: 10.1186/1744-8069-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Podolsky AT, Sandweiss A, Hu J, Bilsky EJ, Cain JP, Kumirov VK, Lee YS, Hruby VJ, Vardanyan RS, Vanderah TW. Novel fentanyl-based dual mu/delta-opioid agonists for the treatment of acute and chronic pain. Life Sci. 2013;93:1010–16. doi: 10.1016/j.lfs.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quartilho A, Mata HP, Ibrahim MM, Vanderah TW, Porreca F, Makriyannis A, Malan TP., Jr Inhibition of inflammatory hyperalgesia by activation of peripheral CB2 cannabinoid receptors. Anesthesiology. 2003;99:955–60. doi: 10.1097/00000542-200310000-00031. [DOI] [PubMed] [Google Scholar]
- 62.Rebaud S, Simon A, Wang CK, Mason L, Blum L, Hofmann A, Girard-Egrot A. Comparison of VILIP-1 and VILIP-3 binding to phospholipid monolayers. PLoS One. 2014;9:e93948. doi: 10.1371/journal.pone.0093948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ripsch MS, Ballard CJ, Khanna M, Hurley JH, White FA, Khanna R. A peptide uncoupling CRMP-2 from the presynaptic Ca2+ channel complex demonstrate efficacy in animal models of migraine and AIDS therapy-induced neuropathy. Transl Neurosci. 2012;3:1–8. doi: 10.2478/s13380-012-0002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson P, Etheridge S, Song L, Armenise P, Jones OT, Fitzgerald EM. Formation of N-type (Cav2.2) voltage-gated calcium channel membrane microdomains: lipid raft association and clustering. Cell calcium. 2010;48:183–94. doi: 10.1016/j.ceca.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 65.Rosslenbroich V, Dai L, Franken S, Gehrke M, Junghans U, Gieselmann V, Kappler J. Subcellular localization of collapsin response mediator proteins to lipid rafts. Biochem Biophys Res Commun. 2003;305:392–9. doi: 10.1016/s0006-291x(03)00754-x. [DOI] [PubMed] [Google Scholar]
- 66.Rowbotham MC, Twilling L, Davies PS, Reisner L, Taylor K, Mohr D. Oral opioid therapy for chronic peripheral and central neuropathic pain. N Engl J Med. 2003;348:1223–32. doi: 10.1056/NEJMoa021420. [DOI] [PubMed] [Google Scholar]
- 67.Sankaram MB. Membrane interaction of small N-myristoylated peptides: implications for membrane anchoring and protein-protein association. Biophys J. 1994;67:105–12. doi: 10.1016/S0006-3495(94)80459-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmidt EF, Strittmatter SM. The CRMP family of proteins and their role in Sema3A signaling. Adv Exp Med Biol. 2007;600:1–11. doi: 10.1007/978-0-387-70956-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scholz A, Gruss M, Vogel W. Properties and functions of calcium-activated K+ channels in small neurones of rat dorsal root ganglion studied in a thin slice preparation. J Physiol. 1998;513:55–69. doi: 10.1111/j.1469-7793.1998.055by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scott DA, Wright CE, Angus JA. Actions of intrathecal omega-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur J Pharmacol. 2002;451:279–86. doi: 10.1016/s0014-2999(02)02247-1. [DOI] [PubMed] [Google Scholar]
- 71.Sezgin E, Kaiser HJ, Baumgart T, Schwille P, Simons K, Levental I. Elucidating membrane structure and protein behavior using giant plasma membrane vesicles. Nat Protoc. 2012;7:1042–51. doi: 10.1038/nprot.2012.059. [DOI] [PubMed] [Google Scholar]
- 72.Stenmark P, Ogg D, Flodin S, Flores A, Kotenyova T, Nyman T, Nordlund P, Kursula P. The structure of human collapsin response mediator protein 2, a regulator of axonal growth. J Neurochem. 2007;101:906–17. doi: 10.1111/j.1471-4159.2006.04401.x. [DOI] [PubMed] [Google Scholar]
- 73.Sutton KG, Martin DJ, Pinnock RD, Lee K, Scott RH. Gabapentin inhibits high-threshold calcium channel currents in cultured rat dorsal root ganglion neurones. Br J Pharmacol. 2002;135:257–65. doi: 10.1038/sj.bjp.0704439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tillu DV, Melemedjian OK, Asiedu MN, Qu N, De Felice M, Dussor G, Price TJ. Resveratrol engages AMPK to attenuate ERK and mTOR signaling in sensory neurons and inhibits incision-induced acute and chronic pain. Mol Pain. 2012;8:5. doi: 10.1186/1744-8069-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Todorovic SM, Jevtovic-Todorovic V. T-type voltage-gated calcium channels as targets for the development of novel pain therapies. Br J Pharmacol. 2011;163:484–95. doi: 10.1111/j.1476-5381.2011.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K, Kolattukudy P, Honnorat J, Goshima Y. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells. 2005;10:165–79. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- 77.Utreras E, Terse A, Keller J, Iadarola MJ, Kulkarni AB. Resveratrol inhibits Cdk5 activity through regulation of p35 expression. Mol Pain. 2011;7:49. doi: 10.1186/1744-8069-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Viard P, Butcher AJ, Halet G, Davies A, Nurnberg B, Heblich F, Dolphin AC. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat Neurosci. 2004;7:939–46. doi: 10.1038/nn1300. [DOI] [PubMed] [Google Scholar]
- 79.Wang LH, Strittmatter SM. Brain CRMP forms heterotetramers similar to liver dihydropyrimidinase. J Neurochem. 1997;69:2261–9. doi: 10.1046/j.1471-4159.1997.69062261.x. [DOI] [PubMed] [Google Scholar]
- 80.Wang YX, Pettus M, Gao D, Phillips C, Scott Bowersox S. Effects of intrathecal administration of ziconotide, a selective neuronal N-type calcium channel blocker, on mechanical allodynia and heat hyperalgesia in a rat model of postoperative pain. PAIN. 2000;84:151–8. doi: 10.1016/s0304-3959(99)00197-9. [DOI] [PubMed] [Google Scholar]
- 81.Ward NE, O’Brian CA. Inhibition of protein kinase C by N-myristoylated peptide substrate analogs. Biochemistry. 1993;32:11903–9. doi: 10.1021/bi00095a020. [DOI] [PubMed] [Google Scholar]
- 82.Weisner CM, Campbell CI, Ray GT, Saunders K, Merrill JO, Banta-Green C, Sullivan MD, Silverberg MJ, Mertens JR, Boudreau D, Von Korff M. Trends in prescribed opioid therapy for non-cancer pain for individuals with prior substance use disorders. PAIN. 2009;145:287–93. doi: 10.1016/j.pain.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9:1099–115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- 84.Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW. Ca (V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell. 2012;149:1112–24. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilson SM, Brittain JM, Piekarz AD, Ballard CJ, Ripsch MS, Cummins TR, Hurley JH, Khanna M, Hammes NM, Samuels BC, White FA, Khanna R. Further insights into the antinociceptive potential of a peptide disrupting the N-type calcium channel-CRMP-2 signaling complex. Channels (Austin) 2011;5:449–56. doi: 10.4161/chan.5.5.17363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilson SM, Moutal A, Melemedjian OK, Wang Y, Ju W, Francois-Moutal L, Khanna M, Khanna R. The functionalized amino acid (S)-Lacosamide subverts CRMP2-mediated tubulin polymerization to prevent constitutive and activity-dependent increase in neurite outgrowth. Front Cell Neurosci. 2014 Jul 24;8:196. doi: 10.3389/fncel.2014.00196. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wilson SM, Schmutzler BS, Brittain JM, Dustrude ET, Ripsch MS, Pellman JJ, Yeum TS, Hurley JH, Hingtgen CM, White FA, Khanna R. Inhibition of transmitter release and attenuation of aids therapy-induced and tibial nerve injury-related painful peripheral neuropathy by novel synthetic Ca2+ channel peptides. J Biol Chem. 2012;287:35065–77. doi: 10.1074/jbc.M112.378695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu G, Felsted RL. Effect of myristoylation on p27 nef subcellular distribution and suppression of HIV-LTR transcription. Virology. 1992;187:46–55. doi: 10.1016/0042-6822(92)90293-x. [DOI] [PubMed] [Google Scholar]
- 89.Zhang XL, Mok LP, Katz EJ, Gold MS. BKCa currents are enriched in a subpopulation of adult rat cutaneous nociceptive dorsal root ganglion neurons. Eur J Neurosci. 2010;31:450–62. doi: 10.1111/j.1460-9568.2009.07060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.