

Abstract
The Nuclear Factor-kappa B (NF-κB) pathway is vital for immune system regulation and pro-inflammatory signaling. Many inflammatory disorders and diseases, including cancer, are linked to dysregulation of NF-κB signaling. When macrophages recognize the presence of a pathogen, the signaling pathway is activated, resulting in the nuclear translocation of the transcription factor, NF-κB, to turn on pro-inflammatory genes. Here, we demonstrate the effects of a novel microtubule depolymerizer, NT-07-16, a polysubstituted pyrrole compound, on this process. Treatment with NT-07-16 decreased the production of pro-inflammatory cytokines in RAW264.7 mouse macrophages. It appears that the reduction in pro-inflammatory mediators produced by the macrophages after exposure to NT-07-16 may be due to activities upstream of the translocation of NF-κB into the nucleus. NF-κB translocation occurs after its inhibitory protein, IκB-α is phosphorylated which signals for its degradation releasing NF-κB so it is free to move into the nucleus. Previous studies from other laboratories indicate that these processes are associated with the microtubule network. Our results show that exposure to the microtubule-depolymerizer, NT-07-16 reduces the phosphorylation of IκB-α and also decreases the association of NF-κB with tubulin which may affect the ability of NF-κB to translocate into the nucleus. Therefore, the anti-inflammatory activity of NT-07-16 may be explained, at least in part, by alterations in these steps in the NF-κB signaling pathway leading to less NF-κB entering the nucleus and reducing the production of pro-inflammatory mediators by the activated macrophages.
Keywords: NT-07-16 pyrrole compound, macrophage, inflammation, NF-κB, signaling, microtubule
1. Introduction
Inflammation is the body’s natural means of protection against infection and the restoration to homeostasis after cellular and tissue damage from injury or metabolic stress1–3. Macrophages play an important role in initiating the inflammatory response by recognizing damage-associated molecular patterns (DAMPs) released by necrotic cells or by recognizing pathogen-associated molecular patterns (PAMPs) found on pathogens, and then releasing cytokines and chemokines that are involved in recruiting other immune cells to the affected area4–6. Lipopolysaccharide (LPS) is a PAMP in gram-negative bacterial cell walls that elicits an inflammatory response by binding to the toll-like receptor 4 (TLR4) complex located on the cell surface of macrophages4,7. TLR4 binding triggers the NF-κB inflammatory pathway by activating IκB Kinase (IKK) which in turn phosphorylates the inhibitory protein IκB-α which is then ubiquitinated, targeting it for proteasomal degradation7–9. After being released by IκB-α, NF-κB translocates into the nucleus where it stimulates the transcription of pro-inflammatory genes7. The resulting mRNA is translated and the macrophages secrete these signals to mediate a local inflammatory response in the infected region7. These mediators include reactive oxygen and nitrogen species as well as cytokines such as interleukin-6 (IL-6), interleukin-1β (IL-1β), and tumor necrosis factor-alpha (TNF-α)7. Additionally, the activation of the NF-κB signaling pathway has been correlated to the dynamics of the microtubule cytoskeleton network10–15. Although inconsistencies have been found about how stabilization or depolymerization of the microtubules affect the pathway, most studies are in agreement that the translocation of NF-κB from the cytoplasm into the nucleus is influenced by microtubule dynamics10–17.
Acute inflammation is protective and usually self-limiting but under some conditions it may become chronic and lead to disease18–20. Therefore, the dysregulation of the pro-inflammatory NF-κB pathway may lead to chronic inflammation and trigger inflammatory diseases. It may also influence tumor development and the aggressiveness of the cancer by enhancing angiogenesis and tumor proliferation while repressing apoptosis21–23. Thus, NF-κB appears to serve as a link between inflammation and many disease states, and therefore, is an attractive therapeutic target.
Since microtubules play a critical role in cell physiology, compounds that target the microtubule network have been studied for their potential use as chemotherapeutic agents24–27. However, some of these compounds, such as colchicine, are too toxic to be used in a clinical setting25–27. Polysubstituted pyrroles like JG-03-14 (3,5-dibromo-4-(3,4-dimethoxyphenyl)-1H-pyrrole-2-carboxylic_acid ethyl ester) also bind microtubules at the “colchicine site” but have been shown to be less toxic while still inhibiting the proliferation of many cancer cell lines28–32. Previous studies from our laboratory with the polysubstituted pyrrole, JG-03-14, have also shown significant decreases in pro-inflammatory signaling in activated RAW264.7 macrophages exposed to the compound33. Treatment with JG-03-14 resulted in the depolymerization of microtubules and decreased the production of nitric oxide (NO) and the pro-inflammatory cytokine TNF-α in RAW264.7 mouse macrophages. These studies provide further evidence that pyrrole-containing compounds may exhibit anti-inflammatory properties while also suggesting that this activity may enhance the chemotherapeutic potential of this microtubule-depolymerizing compound34–39.
Although JG-03-14 clearly depolymerizes microtubules, an earlier study demonstrated that this compound also has some off-target effects40. Therefore, Rohena et al. used information from a diverse group of colchicine site-interacting compounds to design improved analogs of JG-03-1440. NT-07-16 (2,3,4-trimethoxyphenyl analog; Fig. 1), is a refined analog of JG-03-14 with an additional methoxy group at the 2-position of the phenyl ring, which enhances its ability to interact with tubulin and reduces its off-target effects29,40. Further, the addition of the methoxy group enhances the water solubility of NT-07-16 improving its ability to serve as a chemotherapeutic agent40.
Figure 1.
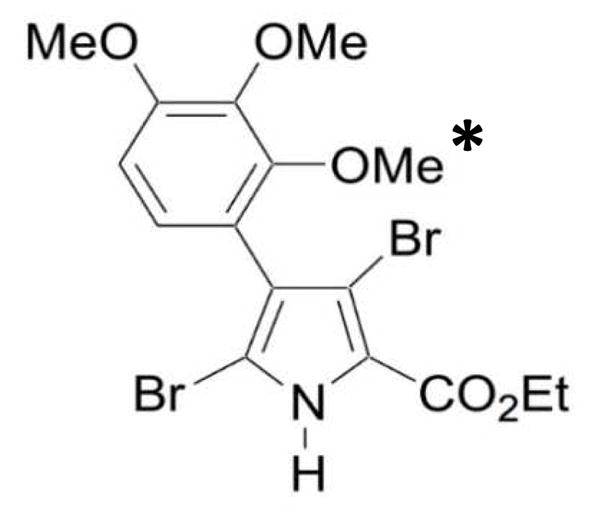
Structure of NT-07-16. An additional methoxy group (marked with an *) was added to the 2-position on the phenyl ring of the parent compound, JG-03-14 (not shown), in order to enhance its ability to interact with tubulin.
To further understand the effects of polysubstituted pyrroles, specifically NT-07-16, on macrophage pro-inflammatory activity, we used the widely accepted model, the RAW264.7 mouse macrophage cell line for our studies. Macrophages were exposed to NT-07-16 prior to activation with LPS and the production of pro-inflammatory cytokines was measured. Also, since the previous results from Ciemniecki et al. suggest that the anti-inflammatory properties of the parent compound, JG-03-14 may be due to a decrease in the ability of NF-κB to translocate into the nucleus we performed fluorescence microscopy and image anaylsis to directly assess this event. We also analyzed the phosphorylation of IκB-α and the interaction of NF-κB with microtubulues to better understand how the depolymerization of the microtubules by NT-07-16 may alter NF-κB nuclear translocation33.
2. Materials and Methods
2.1 Cell culture and reagents
RAW264.7 mouse macrophages were purchased from ATCC and plated in T75 filter-top flasks. Cells were maintained under sterile conditions in RPMI complete medium with 10% fetal calf serum and supplemented with -glutamine, penicillin/streptomycin, non-essential amino acids, and MEM vitamins. Cells incubated at 37 °C with 5% CO2 and grown to approximately 80–95% confluency prior to being plated for experiments. NT-07-16 (3,5-dibromo-4-(2,3,4-trimethoxyphenyl)-1H-pyrrole-2-carboxylic acid ethyl ester) was provided by Dr. John Gupton (Chemistry department, University of Richmond) and was solubilized in DMSO and then diluted in fresh RPMI media (<1% DMSO) prior to use in experiments. Lipopolysaccharide (E. coli 055:B5) was purchased from Sigma-Aldrich.
2.2 MTT cell viability assay
RAW264.7 macrophages were plated 5 x 103 cells/well in a 96-well plate in 100 μl RPMI complete medium and allowed to adhere for 2 hours at 37 °C with 5% CO2. Then the media was removed and replaced with fresh media. Cells were exposed to concentrations of NT-07-16 ranging from 0.0156 μM to 2 μM and incubated for an hour. LPS (500 ng/ml) was then introduced to designated wells so that each concentration of drug was observed with both activated and control macrophages. Cells were then incubated for an additional 20 hours. A volume of 10 μl of MTT (Sigma-Aldrich) solution was added to each well and allowed to sit for 4 hours in the dark. Then 100 μl of isopropanol in 0.04 μM HCl was added to each well and the absorbance at 570 nm was read using a Beckman-Coulter DTX 800 Multimode Detector.
2.3 ELISA for TNF-α and IL-6
Populations of 1.0 x 106 cells were cultured in 3 ml of complete RPMI in 6-well plates. Each culture was incubated for 1 hour in the presence or absence of various concentrations of NT-07-16 (0.25 μM, 0.5 μM, 1.0 μM). Then cells were activated with LPS (500 ng/ml) and incubated for an additional 20 hours. Supernatants were collected from each culture and diluted 1:100 in order to analyze them and quantify the amount of TNF-α and IL-6 present using an OptEIA™ ELISA Assay kit (BD Biociences). Assays were carried out according to the manufacturer’s protocol.
2.4 qPCR for TNFA, IL1B, and IL6
Populations of 3 x 105 cells were treated as described above but were only activated with 500 ng/ml of LPS for 4 hours. RNA from these cultures was harvested using the Qiagen RNEasy Mini-Kit. After RNA was collected, cDNA was created using Origene's First-Strand cDNA Synthesis Kit following Origene's standard protocols and using a Bio-Rad DNAEngine thermo-cycler. The gene of interest was amplified using Origene primers (qSTAR qPCR primer pairs against Mus musculus TNF-α, IL-6, IL-1β and actin) and Quanta Biosciences B-R SYBR Green SuperMix for iQ in 20 μl reactions. Reaction progress was monitored using a Bio-Rad CFX Connect Real-Time System and its associated software. Cq data were analyzed using a normalized expression method (κCq).
2.5 SDS-PAGE/Western blotting
Populations of 1.0 x 106 cells were cultured in 6-well plates and then incubated with NT-07-16 for 1 hour prior to LPS activation (100 ng/ml) for 2.5 minutes up to 2 hours. Then supernatants were removed and the adherent cells were washed with sterile PBS and lysed with lysis buffer (0.05 M Tris buffer pH 7, 0.3 M NaCl, 2 mM EDTA, 0.5% Triton-X 100, 2 μg/mL leupeptin, 1 μg/ml aprotinin, and 0.2 mM PMSF). Total protein concentration from each macrophage culture was determined using a Bio-Rad protein assay. Then 30 μg of protein from each sample was run on a 10% gel (Bio-Rad Mini-PROTEAN TGX Stain-Free Pre-Cast Gels) and transferred to a nitrocellulose membrane. After transferring, the membranes were blocked for 1 hour in a blocking solution of 5% non-fat dry milk solubilized in Tris-buffered saline (TBS). Primary antibodies were diluted in 5 ml TBS with 0.1% Tween-20 (TBST) and membranes were incubated overnight at 4 °C with gentle rotation. The following primary antibodies were used: mouse monoclonal anti-IκB-α antibody diluted 1:1000 (Cell Signaling Technology, Inc.), mouse monoclonal anti-phospho-IκB-α antibody diluted 1:1000 (Cell Signaling Technology, Inc.), 5 μg of a rabbit polyclonal anti-NF-κB antibody (1:200 dilution; Santa-Cruz Biotechnology, Inc.), and 0.2 μg of a mouse monoclonal anti-α-tubulin antibody diluted 1:5000 (Santa-Cruz Biotechnology, Inc.). Then after three 10 minute washes in TBST, blots were subsequently incubated with either 0.4 μg of goat anti-rabbit or goat anti-mouse secondary antibody (1:5,000 dilution; Santa-Cruz Biotechnology, Inc.) in 5 ml TBST for 1 hour at room temperature with gentle rotation. For immunoprecipitation samples, ImmunoCruz™ IP/WB Optima C Western Blot Detection Reagent (Santa-Cruz Biotechnology, Inc.) was diluted 1:5000 in place of the secondary antibody for NF-κB blots. Following incubation with secondary antibody, blots were washed four times for 5 minutes in TBST and then placed in Bio-Rad Clarity Western Substrate reagents. Results were visualized using a ChemiDoc Touch Imaging System (Bio-Rad Laboratories, Inc.) following the manufacturer’s instructions for using stain-free gels. Blots were stripped with Millipore ReBlot Plus Mild Antibody Stripping Solution (Millipore) and re-blocked with blocking solution according to the manufacturer’s protocol and then re-probed with control antibodies as described above.
2.6 Immunoprecipitation
Populations of 1.0 x 106 cells were cultured in 6-well plates and total cell lysates were prepared as described above. Immunoprecipitation was performed using an ImmunoCruz™ IP/WB Optima C System (Santa Cruz Biotechnology, Inc., sc-45040) with a Mouse IP Matrix and mouse α-tubulin IP antibody according to the manufacturer’s protocol. IP matrix was incubated with the IP antibody overnight at 4°C on a rotator. Then 100 μg of total cellular protein was incubated with the IP antibody-IP matrix complex overnight with rotation at 4 °C. Samples were pelleted and washed with PBS 3 times before a final aspiration. Pellets were resuspended in 2X reducing electrophoresis buffer and samples were boiled for 5 minutes prior to running them on a 10% gel (Bio-Rad Mini-PROTEAN TGX Pre-Cast Gels) as described above. Western blotting analysis was performed using anti-NF-κB antibodies and anti-tubulin antibodies as the control.
2.7 Fluorescence microscopy and image analysis
Acid-washed 22 mm2 #1.5 microscope coverslips were coated with 0.01% poly-lysine for 5 minutes, dried, UV sterilized, and placed into 6-well cell culture dishes. RAW264.7 cells (2.0 x 105) were cultured in 2 ml RPMI complete medium and allowed to adhere overnight under normal culture conditions (37 ºC, 5% CO2). Samples were then incubated for 1 hour in the presence or absence of 1μM NT-07-16 or 1μM nocodazole under normal culture conditions. Then designated cultures were activated with 500 ng/ml of LPS and allowed to incubate in the same conditions for an additional hour. After LPS treatment, cells were immediately fixed with 4% formaldehyde in PBS, permeabilized with 0.5% Triton X-100 in PBS, and then blocked in PBS with 5% donkey serum and 0.1% Triton X-100. The cells were then incubated with primary antibodies diluted in blocking buffer for 30 mintues: 0.2 μg/ml anti-α-tubulin (mouse monoclonal, Sigma DM1A) and 0.4 μg/ml anti-NF-κB p65 (rabbit polyclonal, Santa Cruz Biotechnology). Cells were then washed with PBS (4 times for 5 minutes) followed by a secondary antibody incubation for 30 minutes: 0.3 μg/ml ALEX647 donkey anti-mouse (Jackson Immunochemical), 0.3 μg/ml ALEXA488 donkey anti-rabbit (Jackson Immunochemical), 6.6 nM ALEXA568-phalloidin (Invitrogen), and 75 nM DAPI (Invitrogen). Coverslips were washed 4 times for 5 minutes each with PBS, and then mounted onto slides in PBS, 80% glycerol, and 0.5% n-propyl gallate. Images were acquired using an Olympus IX-83 microscope outfitted with a UPLFLN 40x/1.3NA DIC objective, an EXFO mixed gas light source, Sutter filter wheels and shutters, a Hamamatsu ORCA-Flash 4.0 V2 sCMOS camera, and Metamorph imaging software. For fixed cell imaging, z-stack images (0.5 μm steps) were captured sequentially using the Sedat Quad filter-set (Chroma), and exposure times maintained constant within an experimental day across all conditions.
Image analysis was performed using FIJI41,42. Briefly, the ratio of the average p65 signal pixel intensity for a region inside the nucleus was divided by the average p65 signal pixel intensity for a cytoplasmic region. This ratio was standardized against the nucleus/cytoplasm ratio for the unstimulated cells. In this way, a numerical value greater than 1 would indicate an increase in p65 within the nucleus.
3. Results
3.1 NT-07-16 does not affect the viability of RAW264.7 macrophages
In order to be certain that observed changes in macrophage inflammatory activity were not due to cytotoxic effects of NT-07-16, a cell viability assay was performed. No significant decrease in viability of RAW264.7 macrophages was observed following a 20-hour exposure to NT-07-16 in the presence (data not shown) or in the absence of LPS activation (Fig. 2).
Figure 2.

Effect of NT-07-16 on RAW264.7 cell viability. Cells were exposed to varying concentrations of NT-07-16 for 20 hours. Viability was assessed using an MTT assay and is reported as a percentage of the control sample (macrophages not exposed to NT-07-16). Results are representative of three independent experiments and error bars indicate the standard error of the mean within each treatment group.
3.2 NT-07-16 reduces the production of pro-inflammatory cytokines TNF-α and IL-6 by activated RAW264.7 macrophages
In order to investigate the effect of NT-07-16 on pro-inflammatory cytokine secretion, ELISAs were performed using supernatants from macrophage cultures to measure levels of extracellular TNF-α and IL-6. Treatment with NT-07-16 prior to LPS activation significantly decreased secretion of TNF-α (Fig. 3A) and IL-6 (Fig. 3B) from activated macrophages. The release of an additional pro-inflammatory cytokine, IL-1β, was not measured using these supernatants since RAW264.7 macrophages express little, if any, apopotosis-associated speck-like protein containing a C-terminal caspase-activating recruiting domain (ASC), so they lack the required caspase-1 inflammasome activity needed to process IL-1β into its mature, secreted form43.
Figure 3.

Effect of NT-07-16 on IL-6 and TNFα production by activated RAW264.7 macrophages. Cells were exposed to NT-07-16 for one hour prior to activation with LPS. Cells were incubated for 20 hours and then supernatants were collected. OptEIA™ ELISA Assay kits were used to quantify (A) TNF-α and (B) IL-6 released from the cells. Variation among treatments was determined by one-way ANOVA. The means of each sample were compared to the LPS control, **** p<0.0001; significant differences in expression were seen in independently treated samples, A= p<0.01, B= p<0.05 (Tukey-Kramer post-hoc test). Results are representative of three independent experiments and error bars indicate the standard error of the mean within each treatment group.
3.3 NT-07-16 downregulates transcription of pro-inflammatory cytokine genes in activated RAW264.7 macrophages
In order to measure the effect of NT-07-16 on the transcription of pro-inflammatory signaling genes, qPCR analysis of TNFA, IL1B, and IL6 in activated macrophages was performed. In concurrence with the previous experiments, pretreatment with NT-07-16 significantly decreased transcription of TNFA (Fig. 4A), IL1B (Fig. 4B), and IL6 (Fig. 4C) in the LPS activated macrophages. Transcription of the gene for inducible nitric oxide synthase (NOS2), the enzyme responsible for catalyzing the reaction that results in the production of the pro-inflammatory radical, nitric oxide, was also measured from these same RNA preparations, however, the mRNA expression of this gene was not consistently affected by NT-07-16 exposure (data not shown).
FIGURE 4.

Effect of NT-07-16 on TNFA, IL1B, and IL6 expression in RAW264.7 macrophages. Cells were exposed to NT-07-16 for one hour prior to activation with LPS. Cells were incubated for 4 hours before RNA isolation. Relative expression of (A) TNFA, (B) IL1B, and (C) IL6 was measured using qPCR with β-actin as the reference gene. Variation among treatments was determined by one-way ANOVA. The means of each sample were compared to the LPS, * p<0.05, **** p<0.0001; significant differences in expression were also seen between independently treated samples, A= p<0.01, B= p<0.05 (Tukey-Kramer post-hoc test). Results are representative of three independent experiments and error bars indicate the standard error of the mean within each treatment group.
3.4 NT-07-16 alters phosphorylation of IκB-α during activation of RAW264.7 macrophages
IκB-α phosphorylation in LPS activated macrophages not treated or pre-treated with NT-07-16 was measured over time. Levels of both IκB-α and phosphorylated IκB-α (p-IκB-α ) in cells not treated with NT-07-16 (Fig. 5A) and treated with NT-07-16 (Fig. 5B) were examined. These results suggest that exposure to NT-07-16 reduced the amount of IκB-α phosphorylation as well as the length of time that it remained phosphorylated in these macrophages (Fig. 5C)
Figure 5.

Effect of NT-07-16 on the phosphorylation of IκB-α over time. RAW264.7 macrophages were activated with LPS for the indicated length of time and cell lysates were prepared and analyzed by Western blotting using antibodies specific for IκB-α and p-IκB-α. Macrophages were either (A) not exposed to or (B) were exposed to 0.5 μM NT-07-16 for one hour prior to LPS activation. Normalization factors are below each blot. These values were determined by dividing the total adjusted protein volume (background subtracted) of each lane by the total adjusted protein volume of lane 1 on each blot. (C) Relative IκB-α phosphorylation was calculated by dividing the normalized densitometry values of p-IκB-α by the normalized densitometry values of IκB-α for each time period. Low values indicate little p-IκB-α compared to the unphosporylated form for each time period whereas high values indicate high levels of pIκB-α compared to the unphosporylated form. Results are representative of three independent experiments.
3.5 NT-07-16 reduces the association of NF-κB with α-tubulin
To further explore the mechanism by which NT-07-16 exerts its anti-inflammatory properties we examined the association of NF-κB with α-tubulin in order to more fully understand the role of microtubule dynamics in the translocation of NF-κB during the inflammatory response. Immunoprecipitation with western blotting indicated that there was an increase in the association of NF-κB with α-tubulin in RAW264.7 macrophages following activation of the cells with LPS. Additionally, results suggested that exposure of the macrophages to NT-07-16 prior to activation decreased the association between NF-κB and α-tubulin when compared to activated macrophages not exposed to NT-07-16 (Fig. 6).
Figure 6.


Effect of NT-07-16 on the association of NF-κB with tubulin in RAW264.7 macrophages. Cells were exposed to NT-07-16 for 1 hour prior to activation with LPS for 30 minutes. Total cell lysates were collected and immunoprecipitation was performed to isolate α-tubulin. The association of NF-κB with α-tubulin was analyzed by western blotting (A). Densitometry was performed to analyze the amount of NF-κB that co-immunoprecipitated with α-tubulin before and after exposure to NT-07-16 in control and LPS-activated macrophages (B). Data incorporate results from four independent experiments. * p<0.05 (paired t-test).
3.6 Confirmation of the microtubule depolymerization activity of NT-07-16
As confirmation of the work of Rohena et al. demonstrating that NT-07-16 depolymerizes microtubules, RAW264.7 macrophages and LLCPK1 kidney cells (data not shown) were exposed to 1 μM NT-07-16 or 1 μM nocodazole (a known microtubule-depolymerizer, positive control) and subsequently activated with LPS for 1 hour while still in the presence of the compounds (Fig. 7)40. Immunofluorescence microscopy showed the induction of the typical flattened and rounded phenotype, as well as an increase in microtubule stabilization exhibited by LPS activated macrophages44–46. Additionally, these images demonstrated that although the known microtubule-depolymerizer, nocodazole, consistently showed the highest degree of depolymerization, exposure to NT-07-16 also altered the microtubule network in multiple cell types.
Figure 7.

Pretreatment of RAW264.7 cells with microtubule depolymerizing compounds disrupts the typical microtubule networking phenotype. Qualitative image analysis demonstrates microtubule depolymerization of RAW264.7 cells treated with either nocodazole or NT-07-16. Arrows indicate the altered microtubule network in the nocodazole and NT-07-16 treated macrophages. Scale bar = 20 μm.
3.7 NT-07-16 decreases nuclear translocation of NF-κB
RAW264.7 cells were cultured in the presence or absence of 1 μM NT-07-16 or 1 μM nocodazole and prior to a 1 hour LPS activation. Immunofluoresence microscopy to visualize the p65 subunit of NF-κB revealed increased NF-κB localization in the nucleus upon LPS activation, compared to control cells. Treatment with either nocodazole or NT-07-16 abrogated NF-κB nuclear localization in LPS-activated cells (Fig. 8A). NF-κB nuclear localization was quantified by calculating the ratio of nuclear signal to cytoplasmic signal, and normalizing that ratio against control cells (Fig. 8B). Activated macrophages pre-treated with either of the two microtubule-depolymerizing compounds showed significantly less NF-κB nuclear translocation than those macrophages activated with LPS but not exposed to the microtubule-depolymerizers, indicating that the microtubule network is involved in NF-κB nuclear translocation. No difference was found between RAW264.7 cells that were not activated but exposed to NT-07-16 or nocodazole and the control macrophages. These data support our hypothesis that NT-07-16 anti-inflammatory activity is at least partially due to microtubule destabilization.
Figure 8.

Effect of microtubule depolymerization on nuclear translocation of NFκB in RAW264.7 macrophages. (A) Intensity-reversed representative images of p65 staining in control RAW264.7 macrophages, or cells activated with 500 ng/ml of LPS, in the absence or presences of 1μM nocodazole, or 1μM NT-07-16. Arrows represent position of nuclei. Scale bar = 20μm. (B) Normalized mean nuclear localization ratios were calculated from in vitro immunofluorescence imaging of the p65 subunit of NF-κB. LPS activation resulted in an increase in nuclear NF-κB compared to cells that were not activated. Exposure to either nocodazole or NT-07-16 for 1 hour prior to LPS activation decreased the response (*Dunnett’s Test compared to LPS treatment, p < 0.0001). Error bars represent standard error of the mean. n >190 cells for each condition across five independent experiments.
4. Discussion
While acute inflammation is important for combatting pathogens, chronic inflammation has been shown to contribute to many disease states such as inflammatory bowel disease, arthritis, heart disease, neurodegenerative diseases and even cancers21–23. Therefore, in order to develop new treatments for these diseases it is crucial to understand the signaling pathways that are responsible for triggering the inflammation. Due to the importance of the NF-κB signaling pathway in the inflammatory response it has recently become a target for many broad-sweeping drug therapies21–23. The results of our work contribute to the broader understanding of the activation of this pathway leading to the translocation of NF-κB into the nucleus to turn on pro-inflammatory genes, and provide further information about the anti-inflammatory properties of polysubstituted pyrrole compounds.
Treatment of RAW264.7 macrophages with the novel microtubule depolymerizer NT-07-16 yields a down-regulation of pro-inflammatory behavior in the macrophages without compromising cell viability. When exposed to the compound prior to activation, the macrophages secreted less pro-inflammatory cytokines than those not treated with NT-07-16. Additional experiments demonstrated that exposure to NT-07-16 also decreased transcription of pro-inflammatory genes suggesting that NT-07-16 acts upstream of NF-κB’s nuclear activities. Our studies suggest that microtubule depolymerization induced by NT-07-16 interferes with the phosphorylation of the IκB-α inhibitory protein as well as decreases the association of NF-κB with α-tubulin. These two events may be related although this has not yet been studied experimentally. Earlier work from other laboratories has shown that the phosphorylation of IκB-α is the signal for its degradation but that NF-κB remains bound to IκB-α until this inhibitory protein is ubiquitinated by the ubiquitin-activating enzyme, E147,48. Another study demonstrated that the E1 enzyme co-localizes with the cytoskeleton49. Therefore, the decrease in phosphorylation may reduce the signal required to degrade IκB-α and release NF-κB for nuclear translocation. In addition, the disruption of the microtubule network may reduce the ability of NF-κB to interact with the microtubules making it more difficult for the E1 enzyme to carry out its function. Together these effects may contribute to the observed reduction in the nuclear translocation of NF-κB which results in a decrease in the production of pro-inflammatory mediators by activated macrophages exposed to NT-07-16. Therefore, our data provide further evidence that the microtubule network is involved in the activation of the NF-κB transcription factor and its ability to move from the cytoplasm into the nucleus following its dissociation from the IκB-α inhibitory protein. Shrum et al. found that stimulated NF-κB translocation into the nucleus relies on the dynein molecular motor associated with minus-end movement on microtubules but this mechanism has yet to be studied in macrophages14.
It is important to note that although these data do not specifically exclude the possibility that depolymerization of the microtubules may also contribute to a cytokine secretion defect it is unlikely. Our findings demonstrated a significant reduction in the mRNA expression levels of the cytokines tested which suggests that the effects of NT-07-16 are pre-transcriptional. Also, an earlier study using several microtubule disrupting drugs found a similar decrease in TNF-α transcription but little effect on TNF-α secretion in rat peritoneal macrophages50. And more recently, a study showed that while anterograde transport of TNF-α was initially reduced after microtubule depolymerization it appears that these findings were due to a delay in the functional maturation of Golgi elements required for post-Golgi trafficking and not dependent on polymerized microtubules51. However, further studies are still needed to fully understand the role of microtubules in post-Golgi transport of cytokines, specifically in macrophages.
It is clear from our studies that NT-07-16 possesses anti-inflammatory activity. These data suggest that treatment with NT-07-16 may affect the activation of the pro-inflammatory transcription factor, NF-κB by reducing its ability to translocate into the nucleus and turn on pro-inflammatory genes in activated macrophages. As such, NT-07-16, which has already shown activity as an anti-proliferative agent towards cancer cells, may also be effective as an inhibitor of the NF-κB pro-inflammatory signaling pathway further enhancing its chemotherapeutic potential.
Supplementary Material
Highlights.
NT-07-16 is a microtubule-depolymerizing compound with antitumor activity
NT-07-16 decreases pro-inflammatory cytokine production by activated macrophages
NT-07-16 reduces IκB-α phosphorylation and NF-κB association with microtubules
Exposure to NT-07-16 decreases NF-κB nuclear translocation in macrophages
Less NF-κB nuclear translocation may explain anti-inflammatory activity of NT-07-16
Acknowledgments
Funding: This work was supported in part by the National Institutes of Health grant R15-CA67236 (JTG) and the NIGMS grant R15-GM119077 (OAQ), and the Floyd D. and Elisabeth S. Gottwald Endowment and the University of Richmond Faculty Research Grant program (JTG). The Howard Hughes Medical Institute (HHMI 52007567) provided a summer fellowship and research funds for ALKG, the University of Richmond Undergraduate Summer Fellowship and Research Grant program provided support for SPG, ALKG and ECL.
Abbreviations
- NF-κB
Nuclear factor-kappa B
- DAMP
Damage-associated molecular pattern
- PAMP
Pathogen-associated molecular pattern
- LPS
Lipolysaccharide
- TLR4
Toll-like receptor 4
- IKK
Inhibitor of nuclear factor kappa B kinase
- IκB-α
Inhibitor of nuclear factor kappa B alpha
- TNF-α
Tumor necrosis factor alpha
- IL-6
Interleukin-6; IL-1β
- NO
Nitric oxide
Footnotes
Author contributions: This manuscript was written with contributions from all authors. KFS conceived the idea for the project. JTG synthesized and provided NT-07-16 for the studies. OAQ designed the immunofluorescence experiments. SPG and KFS drafted the manuscript. SPG performed ELISAs and the immunofluorescence experiments. ALKG performed ELISAs, and the phosphorylation and immunoprecipitation experiments. ECL performed ELISAs and the qPCR experiments. SEDLR performed the immunofluorescence experiments. All authors analyzed data, read, edited and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Antonelli A, Kushner I. It’s time to redefine inflammation. FASEB J. 2017;31:1787–1791. doi: 10.1096/fj.201601326R. [DOI] [PubMed] [Google Scholar]
- 2.Kondylis V, Kumari S, Vlantis K, Pasparakis M. The interplay of IKK, NF- B and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol Rev. 2017;277:113–127. doi: 10.1111/imr.12550. [DOI] [PubMed] [Google Scholar]
- 3.Munn LL. Cancer and inflammation. WIREs Syst Biol Med. 2017 Mar-Apr; doi: 10.1002/wsbm.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell. 2002;111:927–930. doi: 10.1016/s0092-8674(02)01201-1. [DOI] [PubMed] [Google Scholar]
- 5.Komai K, Shichita T, Ito M, Kanamori M, Chikuma S. Role of scavenger receptors as damage-associated molecular pattern receptors in Toll-like receptor activation. Int Immunol. 2017;29:59–70. doi: 10.1093/intimm/dxx010. [DOI] [PubMed] [Google Scholar]
- 6.Martin SJ. Cell death and inflammation: the case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J. 2016;283:2599–2615. doi: 10.1111/febs.13775. [DOI] [PubMed] [Google Scholar]
- 7.Zhang G, Ghosh S. Toll-like receptor-mediated NF-κB activation: a phylogenetically conserved paradigm in innate immunity. J Clin Invest. 2001;107:13–19. doi: 10.1172/JCI11837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017;168:37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crépieux P, Kwon H, Leclerc N, Spencer W, Richard S, Lin R, Hiscott J. IκBα physically interacts with a cytoskeleton-associated protein through its signal response domain. Mol Cell Biol. 1997;17:7375–7384. doi: 10.1128/mcb.17.12.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackman RW, Rhoads MG, Cornwell E, Kandarian SC. Microtubule-mediated NF-κB activation in the TNF-α signaling pathway. Exp Cell Res. 2009;315:3242–3249. doi: 10.1016/j.yexcr.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rai A, Kapoor S, Singh S, Chatterji BP, Panda D. Transcription factor NF-κB associates with microtubules and stimulates apoptosis in response to suppression of microtubule dynamics in MCF-7 cells. Biochem Pharm. 2015;93:277–289. doi: 10.1016/j.bcp.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Rosette C, Karin M. Cytoskeletal control of gene expression: depolymerization of microtubules activates NF-κB. J Cell Biol. 1995;128:1111–1119. doi: 10.1083/jcb.128.6.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shrum CK, DeFrancisco D, Meffert MK. Stimulated nuclear translocation of NF-κB and shuttling differentially depend on dynein and the dynactin complex. Proc Nat Acad Sci USA. 2009;106:2647–2652. doi: 10.1073/pnas.0806677106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spencer W, Kwon H, Crépieux P, Leclerc N, Lin R, Hiscott J. Taxol selectively blocks microtubule dependent NF-κB activation by phorbol ester via inhibition of IκBα phosphorylation and degradation. Oncogene. 1999;18:495–505. doi: 10.1038/sj.onc.1202335. [DOI] [PubMed] [Google Scholar]
- 16.Manié S, Schmid-Alliana A, Kubar J, Ferrua B, Rossi B. Disruption of microtubule network in human monocytes induces expression of interleukin-1 but not that of interleukin-6 nor tumor necrosis factor-α. J Biol Chem. 1993;268:13675–13681. [PubMed] [Google Scholar]
- 17.Rao P, Falk LA, Dougherty SF, Sawada T, Pluznik DH. Colchicine down-regulates lipopolysaccharide-induced granulocyte-macrophage colony-stimulating factor production in murine macrophages. J Immunol. 1997;159:3531–3539. [PubMed] [Google Scholar]
- 18.Chai EZP, Siveen KS, Shanmugam MK, Arfuso F, Sethi G. Analysis of the intricate relationship between chronic inflammation and cancer. Biochem J. 2015;468:1–15. doi: 10.1042/BJ20141337. [DOI] [PubMed] [Google Scholar]
- 19.Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol. 2015;12:584–596. doi: 10.1038/nrclinonc.2015.105. [DOI] [PubMed] [Google Scholar]
- 20.Marelli G, Sica A, Vannucci L, Allavena P. Inflammation as target in cancer therapy. Curr Opin Pharmacol. 2017;35:57–65. doi: 10.1016/j.coph.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 21.Shen HM, Tergaonkar V. NFκB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis. 2009;14:348–363. doi: 10.1007/s10495-009-0315-0. [DOI] [PubMed] [Google Scholar]
- 22.Sun XF, Zhang H. NFKB and NFKBI polymorphisms in relation to susceptibility of tumour and other diseases. Histol Histopathol. 2007;22:1387–1398. doi: 10.14670/HH-22.1387. [DOI] [PubMed] [Google Scholar]
- 23.Verstrepen L, Beyaert R. Receptor proximal kinases in NF-κB signaling as potential therapeutic targets in cancer and inflammation. Biochem Pharmacol. 2014;92:519–529. doi: 10.1016/j.bcp.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 24.Avila J. Microtubule functions. Life Sci. 1991;50:327–334. doi: 10.1016/0024-3205(92)90433-p. [DOI] [PubMed] [Google Scholar]
- 25.Jordan A, Hadfield JA, Lawrence AT, McGown AT. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med Res Rev. 1998;18:259–296. doi: 10.1002/(sici)1098-1128(199807)18:4<259::aid-med3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 26.Owellen RJ, Owens AH, Jr, Donigian DW. The binding of vincristine, vinblastine and colchicine to tubulin. Biochem Biophys Res Comm. 1972;47:685–691. doi: 10.1016/0006-291x(72)90546-3. [DOI] [PubMed] [Google Scholar]
- 27.Stanton RA, Gernert KM, Nettles JH, Aneja R. Drugs that target dynamic microtubules: a new molecular perspective. Med Res Rev. 2011;31:443–481. doi: 10.1002/med.20242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mooberry SL, Weiderhold KN, Dakshanamurthy S, Hamel E, Banner EJ, Kharlamova A, Hempel J, Gupton JT, Brown ML. Identification and characterization of a new tubulin-binding tetrasubstituted brominated pyrrole. Mol Pharmacol. 2007;72:132–140. doi: 10.1124/mol.107.034876. [DOI] [PubMed] [Google Scholar]
- 29.Da C, Telang N, Barelli P, Jia X, Gupton JT, Mooberry SL, Kellog GE. Pyrrole-based antitubulin agents: two distinct binding modalities are predicted for C-2 analogs in the colchicine site. Med Chem Lett. 2012;3:53–57. doi: 10.1021/ml200217u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Da C, Telang N, Hall K, Kluball E, Barelli P, Finzel K, Jia X, Gupton JT, Mooberry SL, Kellogg GE. Developing novel C-4 analogues of pyrrole-based antitubulin agents: weak but critical hydrogen bonding in the colchicine site. Med Chem Comm. 2013;4:417–421. doi: 10.1039/C2MD20320K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arthur CR, Gupton JT, Kellogg GE, Yeudall WA, Cabot MC, Newsham IF, Gewirtz DA. Autophagic cell death, polyploidy and senescence induced in breast tumor cells by the substituted pyrrole JG-03–14, a novel microtubule poison. Biochem Pharmacol. 2007;74:981–991. doi: 10.1016/j.bcp.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biggers JW, Nguyen T, Di X, Gupton JT, Henderson SC, Emery SM, Alotaibi M, White KL, Brown R, Almenara J, Gewirtz DA. Autophagy, cell death and sustained senescence arrest in B16/F10 melanoma cells and HCT-116 colon carcinoma cells in response to the novel microtubule poison, JG-03–14. Cancer Chemother Pharmacol. 2013;71:441–455. doi: 10.1007/s00280-012-2024-6. [DOI] [PubMed] [Google Scholar]
- 33.Ciemniecki JA, Lewis CP, Gupton JT, Fischer-Stenger K. Effects of a pyrrole-based, microtubule-depolymerizing compound on RAW 264.7 macrophages. Chem Biol Interact. 2016;246:63–68. doi: 10.1016/j.cbi.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anzini M, Di Capua A, Valenti S, Brogi S, Rovini M, Giuliani G, Cappelli A, Vomero S, Chiasserini L, Sega A, Poce G, Giorgi G, Calderone V, Martelli A, Testai L, Sautebin L, Rossi A, Pace S, Ghelardini C, Di Cesare Mannelli L, Benetti V, Giordani A, Anzellotti P, Dovizio M, Patrignani P, Biava M. Novel analgesic/anti-inflammatory agents: 1,5-diarylpyrrole nitrooxyalkyl ethers and related compounds as cyclooxygenase-2 inhibiting nitric oxide donors. J Med Chem. 2013;56:3191–3206. doi: 10.1021/jm301370e. [DOI] [PubMed] [Google Scholar]
- 35.Battilocchio C, Poce G, Alfonso S, Porretta GC, Consalvi S, Sautebin L, Pace S, Rossi A, Ghelardini C, Di Cesare Mannelli L, Schenone S, Giordani A, Di Francesco L, Patrignani P, Biava M. A class of pyrrole derivatives endowed with analgesic/anti-inflammatory activity. Bioorg Med Chem. 2013;21:3695–701. doi: 10.1016/j.bmc.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 36.Hua KF, Chou JC, Lam Y, Tasi YL, Chen A, Ka SM, Fang Z, Liu ML, Yang FL, Yang YL, Chiu YC, Wu SH. Polyenylpyrrole derivatives inhibit NLRP3 inflammasome activation and inflammatory mediator expression by reducing reactive oxygen species production and mitogen-activated protein kinase activation. PLoS ONE. 2013 doi: 10.1371/journal.pone.0076754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim KJ, Choi MJ, Shin JS, Kim M, Choi HE, Kang SM, Jin JH, Lee KT, Lee JY. Synthesis, biological evaluation, and docking analysis of a novel family of 1-methyl-1H-pyrrole-2,5-diones as highly potent and selective cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med Chem Lett. 2014;24:1958–1962. doi: 10.1016/j.bmcl.2014.02.074. [DOI] [PubMed] [Google Scholar]
- 38.Maddila S, Gorle S, Sampath Ch, Lavanya P. Synthesis and anti-inflammatory activity of some new 1,3,4-thiadiazoles containing pyrazole and pyrrole nucleus. J Saudi Chem Soc. 2012;3:16–25. [Google Scholar]
- 39.Mohamed MS, Kamel R, Fathallah SS. Synthesis of new pyrroles of potential anti-inflammatory activity. Arch Pharm Chem Life Sci. 2011;344:830–39. doi: 10.1002/ardp.201100056. [DOI] [PubMed] [Google Scholar]
- 40.Rohena CC, Telang NS, Da C, Risinger AL, Sikorski JA, Kellog GE, Gupton JT, Mooberry SL. Biological characterization of an improved pyrrole-based colchicine site agent identified through structure-based design. Mol Pharmacol. 2016;89:287–296. doi: 10.1124/mol.115.101592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noursadeghi M, Tsang J, Haustein T, Miller RF, Chain BM, Katz DR. Quantitative imaging assay for NF-κB nuclear translocation in primary human macrophages. J Immunol Methods. 2008;329:194–200. doi: 10.1016/j.jim.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2X7 receptor differentially couples to distinct release pathways for IL-1β in mouse macrophages. J Immunol. 2008;180:7147–7157. doi: 10.4049/jimmunol.180.11.7147. [DOI] [PubMed] [Google Scholar]
- 44.McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci USA. 2013;110:17253–17258. doi: 10.1073/pnas.1308887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Binker MG, Zhao DY, Pang SJY, Harrison RE. Cytoplasmic linker protein-170 enhances spreading and phagocytosis in activated macrophages by stabilizing microtubules. J Immunol. 2007;179:3780–3791. doi: 10.4049/jimmunol.179.6.3780. [DOI] [PubMed] [Google Scholar]
- 46.Hanania R, Sun HS, Xu K, Pustylnik S, Jeganathan S, Harrison RE. Classically activated macrophages use stable microtubules for matrix metalloproteinase-9 (MMP-9) secretion. J Biol Chem. 2012;287:8468–8483. doi: 10.1074/jbc.M111.290676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin YC, Brown K, Siebenlist U. Activation of NF-κB requires proteolysis of the inhibitor IκB-α: signal-induced phosphorylation of IκB-α alone does not release active NF-κB. Proc Natl Acad Sci USA. 1995;92:552–556. doi: 10.1073/pnas.92.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyamoto S, Maki M, Schmitt MJ, Hatanaka M, Verma IM. Tumor necrosis factorα-induced phosphorylation of IκBα is a signal for its degradation but not dissociation from NF-κB. Proc Natl Acad Sci USA. 1994;91:12740–12744. doi: 10.1073/pnas.91.26.12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trausch JS, Grenfell SJ, Handley-Gearhart PM, Ciechanover A, Schwartz AL. Immunofluorescent localization of the ubiquitin-activating enzyme, E1, to the nucleus and cytoskeleton. Cell Phys. 1993;264:C93–C102. doi: 10.1152/ajpcell.1993.264.1.C93. [DOI] [PubMed] [Google Scholar]
- 50.Li Z, Davis GS, Mohr C, Nain M, Gemsa D. Inhibition of LPS-induced tumor necrosis factor-α production by colchicine and other microtubule disrupting drugs. Immunobiol. 1996;95:624–639. doi: 10.1016/s0171-2985(96)80027-1. [DOI] [PubMed] [Google Scholar]
- 51.Fourriere L, Divoux S, Roceri M, Perez F, Boncompain G. Microtubule-independent secretion requires functional maturation of Golgi elements. J Cell Sci. 2016;129:3238–3250. doi: 10.1242/jcs.188870. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.