

Abstract
Tissue plasminogen activator (tPA) is used in fewer than 4% patients after ischemic stroke because of its narrow therapeutic time window. We tested whether pyrrolidine dithiocarbamate (PDTC), a drug with multiple mechanisms to provide neuroprotection, can be used to extend the therapeutic time window of tPA. Three month old male Sprague-Dawley rats were subjected to embolic stroke in the area supplied by right middle cerebral artery. tPA at 10 mg/kg was given intravenously 4 h after the onset of stroke. PDTC at 50 mg/kg was given via gastric gavage at 30 min or 4 h after the onset of stroke. Two days after the stroke, neurological outcome was evaluated and the right frontal cortex area 1 (Fr1), an ischemic penumbral region, was harvested for analysis. PDTC given at 30 min after the stroke reduced infarct volumes and improved neurological functions no matter whether the rats received tPA. PDTC also reduced tPA-increased hemorrhagic volumes. Consistent with these results, PDTC in the presence or absence of tPA treatment attenuated the increase of proinflammatory cytokines, oxidative stress and matrix metalloprotease 2 activity in the right Fr1. However, PDTC given at 4 h after the onset of stroke did not improve the neurological outcome of rats treated with or without tPA. Our results suggest that PDTC given at an early time point but not in a delayed phase provides neuroprotection against embolic stroke and may be used to extend the therapeutic time window of tPA.
Keywords: hemorrhagic transformation, neuroprotection, pyrrolidine dithiocarbamate, embolic stroke, tissue plasminogen activator
Graphical Abstract
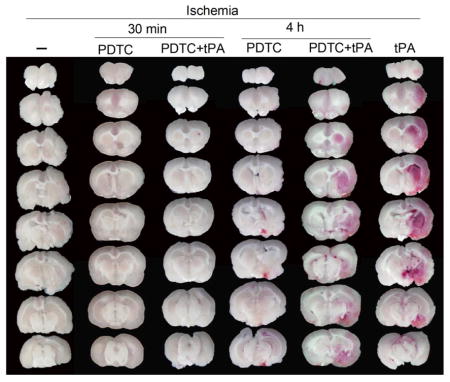
Rats treated with tPA 4 h after the onset of embolic stroke had increased hemorrhagic volumes compared to rats only with embolic stroke. The increased hemorrhagic volumes were attenuated by PDTC applied 30 min after the onset of embolic stroke but not by PDTC applied 4 h after the stroke.
Introduction
Stroke is a leading cause of mortality and morbidity. A fundamental pathophysiological process of stroke is ischemic brain injury (Lipton 1999; Martin et al. 1999). Despite intensive research, clinically effective and safe methods for reducing ischemic brain injury have yet to be established. Reopening the occluded cerebral vessels within a certain time frame represents an effective way to improve neurological outcome after the onset of ischemic stroke (Berkhemer et al. 2015; Goyal et al. 2015).
Tissue plasminogen activator (tPA) is the only drug therapy approved by Food and Drug Administration for ischemic stroke. It has a narrow therapeutic time window because its side effects will outweigh its benefits if it is used at a time point that is more than 3 h after the onset of brain ischemia (Adams et al. 2007), although this therapeutic time window may be extended to 4.5 h in selective cases (Del Zoppo et al. 2009). The major side effects of tPA include hemorrhagic transformation (HT), an effect that may be mediated by activation of matrix metalloprotease (MMP) (Montaner et al. 2003), and direct neurovascular toxicity (Liu et al. 2004; NINDS 1997; Wang et al. 2012; Wang et al. 1998). Due to this narrow therapeutic time window, intravenous tPA is currently used only in fewer than 4% patients with ischemic stroke (Kleindorfer et al. 2008). Methods to extend the therapeutic time window of tPA are highly sought to improve the outcome of patients with ischemic stroke.
Pyrrolidine dithiocarbamate (PDTC) is a small molecule with anti-oxidant and anti-inflammatory property (Liu et al. 1999). PDTC has been shown to activate Akt, protein that is considered to be pro-survival. PDTC can reduce focal brain ischemic injury in young adult rats (Nurmi et al. 2004a; Nurmi et al. 2004b). This protective effect occurs even if the application of PDTC is at 6 h after the onset of transient focal brain ischemia in those rats (Nurmi et al. 2004b). Our recent study showed that oral PDTC started at 10 min after brain ischemia improved neurological outcome assessed at 1 or 2 months later in young adult rats (Li et al. 2012). PDTC also reduces brain injury after ischemia and hypoxia in neonatal rats (Nurmi et al. 2006; Wang et al. 2013). Thus, we hypothesize that PDTC can be used to extend the therapeutic time window of tPA. To test this hypothesis, rats were subjected to embolic stroke and then treated with tPA in the presence or absence of PDTC. Their neurological outcome was assessed.
Methods and Materials
The animal protocol was approved by the Institutional Animal Care and Use Committee of the University of Virginia (Charlottesville, VA, USA). All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publications number 80-23) revised in 2011. Efforts were made to minimize the used number and suffering of animals. Our manuscript was written up in accordance with the Animal Research: Reporting In Vivo Experiments.
Animal groups
Male Sprague-Dawley rats (RRID: RGD_5508379) weighing from 280 g to 310 g (Charles River Laboratories Inc., Wilmington, MA) were randomly assigned to the following groups: 1) embolic stroke, 2) embolic stroke plus tPA treatment, 3) embolic stroke plus PDTC treatment at 30 min after the onset of stroke, 4) embolic stroke plus PDTC treatment at 30 min after the onset of stroke plus tPA treatment, 5) embolic stroke plus PDTC treatment at 4 h after the onset of stroke, and 6) embolic stroke plus PDTC treatment at 4 h after the onset of stroke plus tPA treatment. Each group had 8 rats. tPA was given intravenously at 4 h after the onset of stroke. PDTC was given via gastric gavage. Normal saline (NS) at equal volume was applied intravenously or via gastric gavage at the corresponding times in animals that did not receive PDTC or tPA. These rats were used to evaluate the neurological outcome at 2 days after the stroke.
A second cohort of rats were randomly assigned to the following groups: 1) sham, 2) embolic stroke, 3) embolic stroke plus tPA treatment, 4) embolic stroke plus PDTC treatment at 30 min after the onset of stroke, 5) embolic stroke plus PDTC treatment at 30 min after the onset of stroke plus tPA treatment. Each group also had 8 rats. tPA was also given intravenously at 4 h after the onset of stroke. These rats were used to harvest the right frontal cortex area 1 (Fr1), an ischemic penumbral region (Li et al. 2013; Li and Zuo 2011a; Li and Zuo 2011b), at 2 days after the stroke.
Preparation of the blood embolus
The method used to prepare a white embolus was modified from a previous study (Overgaard et al. 1992). Briefly, blood was withdrawn from a femoral artery of a donor rat under isoflurane anesthesia and then filled into a 20 cm long PE-50 tube. The blood was allowed to clot in the tube for 2 h at room temperature and then kept for 22 h at 4°C.
Five centimeter long PE-50 tube containing the clot was cut and attached at both ends to a 20 cm long PE-10 tube that was connected to a syringe via a 30 gauge needle. The PE-10 tube and syringes were filled with saline. The clot was shifted by alternating movement from one syringe to the other for 5 min. A single clot of 25 mm was cut and transferred to a PE-0402 tube (Anilab Software & Instruments Co., Ltd., Ningbo, China) filled with saline. The PE-0402 tube had a 0.38 mm outer diameter and 0.2 mm inner diameter.
Animal model
The embolic stroke model was modified from a previous study (Zhang et al. 1997). Rats were anesthetized with 2% isoflurane. Rectal temperature was maintained at 37°C with a feedback regulated heating pad. The right common carotid artery (CCA), external carotid artery (ECA) and internal carotid artery (ICA) were isolated under a microscope via a midline incision. A 6-0 silk suture was loosely tied at the origin of the ECA. Another suture was used to ligate the distal end of the ECA. The right CCA and ICA were temporarily clamped using a curved microvascular clip. The PE-0402 tube containing a 25 mm long clot was attached to a 100 μl Hamilton syringe and introduced into the ECA lumen through a small puncture. The suture around the origin of the ECA was tightened around the intraluminal catheter to prevent bleeding and the microvascular clip was removed. About 20 mm length of catheter was gently advanced from the ECA into the lumen of the ICA. The clot along with 50 μl of saline was injected into the ICA over 10 s. The catheter was removed from the right ECA 5 min after the injection. The right ECA was ligated. The duration of the entire surgical procedure was within 30 min. Heparin was not administered to any animals.
Drug application
tPA (Genentech, South San Francisco, CA) solution was prepared by diluting with sterile water to 3 mg/ml and kept at −80°C. tPA at 10 mg/kg was injected intravenously through a tail vein as reported before (Ding et al. 2005).
PDTC solution was prepared by dissolving PDTC powder in NS to 10 mg/ml just before use. PDTC at 50 mg/kg was administered to rats via gastric gavage (Li et al. 2012).
Evaluation of motor coordination, neurological deficit scores, infarct volumes and hemorrhagic volumes
Motor coordination was evaluated by using an accelerating rotarod as we described before (Li et al. 2013). Each rat was tested for three times. The latency and speed of rat’s falling off the rotarod were recorded. The speed–latency index (latency in seconds x speed in rpm) of each of the three tests was calculated and averaged for reporting. Animals were tested before the embolic stroke and immediately before they were sacrificed for assessment of brain infarct volume. The ratio of speed-latency index after the stroke/speed-latency index before the stroke was used to reflect the motor co-ordination.
Neurological deficit scores were evaluated based on an eight-point scale by a person who was blind to the group assignment as we described before (Li et al. 2013; Li and Zuo 2011b).
Infarct volumes were measured after staining of the 2-mm thick brain slices with 2% 2,3,5-triphenyltetrazolium chloride as we described before (Li et al. 2013; Li and Zuo 2011b). The percentage of brain infarct volume in the ipsilateral hemisphere volume was calculated to account for cerebral edema and differential shrinkage from brain ischemia and tissue processing and to correct for individual differences in brain volumes (Li and Zuo 2011a). The hemorrhagic volumes were measured in the same way as measuring infarct volumes before the slices were stained by 2,3,5-triphenyltetrazolium chloride (Deng et al. 2014).
Brain tissue harvesting
Rats with or without brain ischemia were deeply anesthetized by isoflurane and transcardially perfused with NS at 2 days after the embolic stroke. The right Fr1 between bregma +2 and bregma 0 mm, and between 1 to 3 mm from the midline was harvested as we described before (Li et al. 2013; Li and Zuo 2011a; Li and Zuo 2011b) for evaluation of oxidative stress, MMP activity and ELISA of cytokines.
ELISA assay of cytokines in the brain tissues
IL-1β, IL-6 and TNF-α levels in the Fr1 were determined with Quantikine ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions as we described before (Cao et al. 2012; Lin and Zuo 2011). The quantity of IL-1β, TNF-α and IL-6 in each brain sample was normalized to its protein contents.
Measurement of malondialdehyde (MDA) level, superoxide dismutase (SOD) activity and 4-hydroxy-2-nonenal (HNE) content
The levels of MDA, SOD activity and HNE in the brain tissues were assessed using the OxiSelect™ MDA Adduct Competitive ELISA Kit, OxiSelect™ Superoxide Dismutase Activity Assay and OxiSelect™ HNE Adduct Competitive ELISA Kit (Cell Bio labs Inc., San Diego, CA), respectively, according to the manufacture’s instruction.
MMP zymography
The MMP activity was measured by gelatin zymography as we reported before (Ding et al. 2005). Forty five micrograms of cytoplasmic protein prepared from Fr1 of each animal were loaded per lane onto 10% polyacrylamide gel containing 0.1% gelatin and then subjected to electrophoresis. Gelatinolytic activity in the gel was revealed with a series of incubations with re-naturing buffer, developing buffer and de-staining solution. MMP activity was reflected by the density of clear bands in the gel.
Statistical Analysis
Parametrical data are presented as means ± S.D. Neurological deficit scores were analyzed by one-way analysis of variance on ranks followed by Tukey’s post-hoc pair-wise comparison testing. All other results were analyzed by one-way analysis of variance followed by Tukey’s post-hoc pair-wise comparison testing if the data were normally distributed, or one-way analysis of variance on the ranks followed by the Tukey’s post-hoc pairwise comparison testing if the data were not normally distributed. A p ≤ 0.050 decision rule was utilized as the null hypothesis rejection criterion for all statistical comparisons. All statistical analyses were performed using the SigmaStat program (SYSTAT Software Inc., Point Richmond, CA, USA; RRID: SCR_010285).
Results
No rats had an episode of hypoxia during the surgery to create the embolic stroke. No rats died during the perioperative period (within one hour after the surgery).
PDTC gave at an early time point extended the therapeutic time window of tPA
Use of tPA at 4 h after the onset of stroke did not affect the infarct brain volume and the severity of brain edema as reflected by brain edema index (Fig. 1). It also did not affect the neurological functions as reflected by neurological deficit scores and performance on rotarod (Fig. 2). However, this use significantly increased the hemorrhagic volumes. Consistent with this increase, MMP2 (at ~64 kDa) activity was increased by tPA use (Fig. 3). We could not resolve a clear band with a molecular size corresponding to MMP9 or any other bands on the zymogram. These results suggest potential harm of tPA application at a delayed time point. However, PDTC applied 30 min after the onset of stroke decreased infarct brain volume (overall test with 6 groups by one-way analysis of variance: F(5, 42) = 8.184, n = 8 per group, P < 0.001; infarct volume was decreased from 38.6 ± 21.7% to 9.4 ± 9% in the absence of tPA, n = 8, P = 0.020 and from 45.2 ± 17.2% to 6.5 ± 9.1% in the presence of tPA, n = 8, P < 0.001) and severity of brain edema and improved neurological functions no matter whether the rats were treated with or without tPA (Figs. 1 and 2). This PDTC application also attenuated the increased hemorrhagic volume induced by tPA (from 25.0 ± 20.3% to 0.5 ± 1.0%, n = 8, P < 0.001; overall test with 6 groups by one-way analysis of variance: F(5, 42) = 8.387, n = 8 per group, P < 0.001) (Fig. 3). The rate of hemorrhage in the brain was also decreased (from 100% in the brain ischemia plus tPA group to 37.5% in the brain ischemia plus PDTC plus tPA group, n = 8, P = 0.031 by Z test). However, application of PDTC 4 h after the onset of stroke did not affect infarct brain volume, severity of edema, neurological functions and hemorrhagic volume in the presence or absence of tPA (Figs. 1 to 3).
Fig. 1. The effects of tPA and PDTC on brain infarct volume and edema index.

Rats were subjected to an embolic stroke. They received intravenous tPA at 4 h after the onset of stroke or PDTC via gastric gavage at 30 min or 4 h after the onset of stroke. Rats were evaluated 2 days after the stroke. A: representative brain slices stained with 2,3,5-triphenyl-tetrazolium chloride. B: brain infarct volume. C: edema index. Results are in box plot. Inside boxes: 25–75% interval including the median of the data (n = 8). * P < 0.050 compared with ischemia alone group. # P < 0.050 compared with ischemia plus tPA group.
Fig. 2. The effects of tPA and PDTC on neurological function.

Rats were subjected to an embolic stroke. They received intravenous tPA at 4 h after the onset of stroke or PDTC via gastric gavage at 30 min or 4 h after the onset of stroke. Rats were evaluated 2 days after the stroke. A: neurological deficit scores. B: performance in rotarod. Results are in box plot. Inside boxes: 25–75% interval including the median of the data (n = 8). * P < 0.050 compared with ischemia alone group. # P < 0.050 compared with ischemia plus tPA group.
Fig. 3. The effects of tPA and PDTC on hemorrhagic volume and MMP2 activity.

Rats were subjected to an embolic stroke. They received intravenous tPA at 4 h after the onset of stroke or PDTC via gastric gavage at 30 min or 4 h after the onset of stroke. Rats were evaluated 2 days after the stroke. A: representative images of fresh brain slices. B: quantitative data of hemorrhagic volume. C: representative MMP zymogram with samples prepared from the right frontal cortex area 1 in rats treated with or without PDTC at 30 min after the stroke. D: quantitative data of MMP2 activity in the samples prepared from the right frontal cortex area 1 from rats treated with or without PDCT at 30 min after the stroke. Results are in box plot. Inside boxes: 25–75% interval including the median of the data (n = 7 – 8). * P < 0.050 compared with ischemia alone group. # P < 0.050 compared with ischemia plus tPA group. ^ P < 0.050 compared with sham group.
PDTC gave at an early time point reduced inflammatory and oxidative response in the ischemic brain tissues
There was a significant increase in the expression of TNF-α, IL-1β, and IL-6, proinflammatory cytokines, in the ischemic penumbral Fr1. This increase in the presence or absence of tPA was attenuated by PDTC gave at 30 min after the onset of stroke (from 208 ± 103 pg/mg protein to 52 ± 17 pg/mg protein for IL-1β in the absence of tPA, n = 8, P = 0.001; overall test with 5 groups by one-way analysis of variance: F(4, 35) = 25.807, n = 8 per group, P < 0.001) (Fig. 4). The activity of SOD, an anti-oxidative enzyme, was decreased in the ischemic Fr1. This decrease was reversed by PDTC. Consistent with this change of SOD activity, the levels of 4-HNE and MDA, two indices of oxidative stress, were increased in the ischemic Fr1 but this increase was attenuated by PDTC (Fig. 5).
Fig. 4. The effects of tPA and PDTC on proinflammatory cytokine expression.

Rats were subjected to an embolic stroke. They received intravenous tPA at 4 h after the onset of stroke or PDTC via gastric gavage at 30 min after the onset of stroke. The right frontal cortex area 1 was evaluated 2 days after the stroke. A: IL-1β. B: IL-6. C: TNF-α. Results are in box plot. Inside boxes: 25–75% interval including the median of the data (n = 8). * P < 0.050 compared with sham group. ^ P < 0.050 compared with ischemia alone group. # P < 0.050 compared with ischemia plus tPA group.
Fig. 5. The effects of tPA and PDTC on oxidative stress.

Rats were subjected to an embolic stroke. They received intravenous tPA at 4 h after the onset of stroke or PDTC via gastric gavage at 30 min after the onset of stroke. The right frontal cortex area 1 was evaluated 2 days after the stroke. A: SOD activity. B: 4-HNE. C: MDA. Results are in box plot. Inside boxes: 25–75% interval including the median of the data (n = 8). * P < 0.050 compared with sham group. ^ P < 0.050 compared with ischemia alone group. # P < 0.050 compared with ischemia plus tPA group.
Discussions
Our results clearly showed that PDTC applied 30 min after the onset of brain ischemia induced neuroprotection. More importantly, application of PDTC at this time point improved the outcome of rats treated with tPA that was given 4 h after the onset of brain ischemia, suggesting that PDTC can be used to extend the therapeutic time window of tPA.
Reopening the occluded blood vessels as soon as possible after the onset of ischemic stroke is obviously an effective intervention (Berkhemer et al. 2015; Goyal et al. 2015). Use of tPA for this purpose within 3 h after the onset of stroke is considered beneficial (Adams et al. 2007). Multiple factors may contribute to the loss of benefits with the delayed use of tPA. First, tPA causes direct neurovascular injury and HT. tPA knockout mice have better neurological outcome after brain ischemia. tPA also induces apoptosis to endothelial cells and neurons (Liu et al. 2004; NINDS 1997; Wang et al. 2012; Wang et al. 1998). Second, brain tissues distal to the occluded artery may be severely injured or suffer from irreversible injury when tPA is used at a delayed time point. Third, the reperfusion after tPA use can cause significant reperfusion injury (Lipton 1999). The harmful effects caused by these factors outweigh the beneficial effects induced by blood reflowing to the ischemic tissues with a delayed use of tPA. In our study, rats treated with tPA at 4 h after the onset of embolic stroke had a degree of infarct brain volume, brain edema and neurological deficit similar to rats with embolic stroke alone. However, the hemorrhagic volumes in rats treated with tPA was increased compared to rats with embolic stroke alone. These results suggest that tPA use at a delayed time point does not improve neurological outcome but increases hemorrhagic volume. It is not clear whether the failure to improve neurological outcome is due to the failure to save any cells by blood reflowing to the ischemic area or the count balance between the beneficial effects of blood reflowing and the detrimental effects of tPA, reperfusion and increased hemorrhage. However, application of PDTC at 30 min after the onset of stroke improved neurological outcome and attenuated hemorrhagic volume increase caused by tPA, strongly suggesting its neuroprotective effects in the presence of tPA.
We thought to use an agent that has multiple mechanisms to provide neuroprotection for extending the therapeutic time window of tPA because of the complex etiology of ischemia-reperfusion injury (Lipton 1999) and tPA-induced damage (Liu et al. 2004; NINDS 1997; Wang et al. 2012; Wang et al. 1998). PDTC is neuroprotective in many brain ischemia models (Li et al. 2012; Nurmi et al. 2006; Nurmi et al. 2004a; Nurmi et al. 2004b; Wang et al. 2013) and has multiple targets for neuroprotection. First, PDTC is an anti-oxidant that scavenges free radicals (Hayakawa et al. 2003). Second, PDTC is an anti-inflammatory agent. It inhibits the nuclear factor κB (NF-κB) (Liu et al. 1999; Schreck et al. 1992), a transcription factor that is necessary for the expression of proinflammatory mediators. PDTC reduces ischemia-reperfusion-induced IL-1β and TNFα production in the brain of young adult rat (Nurmi et al. 2004b). Third, it has been shown that PDTC can activate Akt, which then activates downstream proteins to support cell survival (Nurmi et al. 2006). Apart from these three mechanisms, we showed that PDTC reduced ischemia-reperfusion-induced amyloid β peptide (Aβ) production (Li et al. 2012). Aβ can cause oxidative injury to cells (Agostinho et al. 2010). Thus, reduced Aβ level by PDTC also may contribute to its neuroprotection. Consistent with the previous studies (Li et al. 2012; Nurmi et al. 2006; Nurmi et al. 2004a; Nurmi et al. 2004b; Wang et al. 2013), our results showed that PDTC given 30 min after the embolic stroke improved neurological outcome no matter whether the rats were treated with tPA at 4 h after the onset of stroke. PDTC also reduced the increase of hemorrhagic volume in rats treated with tPA. As expected, PDTC reduced proinflammatory cytokines, such as TNF-α, IL-1β and IL-6, and oxidative stress as reflected by the decreased 4-HNE, a lipid oxidative stress index (Li and Zuo 2011a), and MDA, a highly reactive compound, in the ischemic penumbral region. PDTC also preserved SOD activity in the ischemic penumbral region. SOD is an important antioxidative enzyme (McCord and Fridovich 1988). The preservation of SOD activity may facilitate the reduction of oxidative stress in the ischemic tissues.
There are at least two mechanisms for the hemorrhage to occur after ischemic stroke: direct neurovascular injury and activation of MMPs. Use of tPA can enhance hemorrhage because tPA can cause direct neurovascular injury (Liu et al. 2004; NINDS 1997; Wang et al. 2012; Wang et al. 1998) and enhance MMP activation (Montaner et al. 2003). In addition, blood reflowing to the ischemic area may facilitate the formation of hemorrhage. Consistent with these ideas, tPA use at 4 h after the onset of embolic stroke increased the hemorrhagic volume and MMP2 activity and these effects were attenuated by PDTC. These results suggest a role of MMP2 in the HT after tPA treatment. Previous studies have shown a role of MMP9 in ischemic brain injury and the brain damage after tPA treatment (Montaner et al. 2003; Turner and Sharp 2016). It is unclear why we could not detect a band corresponding to MMP9 in our study.
A previous study showed that PDTC use started at 6 h after the onset of ischemic stroke induced by an intravascular suture was neuroprotective in rats (Nurmi et al. 2004b). PDTC was given every 12 h and brain ischemia was for 90 min in that study. In our study, PDTC given at 4 h after the onset of embolic stroke was not protective. Application of PDTC at this time did not provide any benefits to the rats treated with tPA at 4 h after the onset of the stroke. We gave only one dose of PDTC. Multiple differences in the experimental design between our study and the previous study (Nurmi et al. 2004b) may explain the discrepancy in findings of PDTC effects when it is applied in a delayed phase. Giving multiple doses may be useful but our previous study showed that one dose of PDTC is as protective as multiple doses of PDTC (Wang et al. 2013). In addition, one dose of PDTC given at 30 min after the onset of stroke improved neurological outcome in the current study.
As discussed above, tPA can induce neurovascular injury (Liu et al. 2004; NINDS 1997; Wang et al. 2012; Wang et al. 1998). It has been shown that tPA can cause remote parenchymal hemorrhage, especially in patients with microbleeds (Charidimou et al. 2015; Kimura et al. 2013; Prats-Sanchez et al. 2016). Interestingly, recombinant activated protein C has been shown to provide neuroprotective effects and reduce parenchymal hemorrhage by blocking the damaging pathways induced by tPA (Cheng et al. 2006; Liu et al. 2004; Wang et al. 2012). The safety and effectiveness of intravenous recombinant activated protein C in patients receives tPA are tested in a phase 2 clinical trial (NCT02222714) (Amar et al. 2015). Of note, our current study showed that PDTC via gastric gavage, similar to intravenous activated protein C, reduced cerebral hemorrhage volume caused by tPA.
To reduce the interference of anesthesia/anesthetics in the neurological outcome after a stroke, we did not monitor cerebral blood flow to determine the number of rats that had recanalization to the ischemic brain region after tPA application. A previous study using a similar embolic stroke model showed that up to 50% rats had recanalization after 10 mg/kg tPA compared with 20% rats with recanalization in the control group at 24 h after the embolic stroke (Ding et al. 2005). Of note, the effects of PDTC on cerebral blood flow to the ischemic brain region are not known. However, PDTC via its inhibition on inflammation can attenuate cerebral vasospasm caused by subarachnoid hemorrhage in rabbits (Zhou et al. 2007). Thus, it is conceivable that PDTC may improve blood flow to the ischemic brain regions, which may be a mechanism for its neuroprotection.
Our findings may have significant implications. PDTC was given via gastric gavage. This route of application can be achieved in human by mouth, which is very convenient. Patients who cannot swallow may receive intranasal spray as this method of application is neuroprotective in our previous study (Wang et al. 2013). In addition, PDTC is a safe drug (Chabicovsky et al. 2010). It is our vision that patients can take PDTC by mouth or via intranasal spray at home soon after the onset of stroke symptoms if the neuroprotective effects of PDTC are confirmed in human. Patients can then go to hospital to have further treatment including tPA therapy. However, PDTC can cause sedation, central nervous excitation, gastrointestinal lesions and kidney injury in rodents in a pre-clinical safety evaluation study (Chabicovsky et al. 2010). Nevertheless, the doses to induce these toxic effects were much higher than the dose used in this current study. The intravenous maximum tolerance dose was defined at 100 mg/kg in rats and the oral median lethal dose was > 2000 mg/kg in male rats. There were no significant kidney and liver injury if PDTC dose was < 100 mg/kg via intranasal application once a day for 28 days in rats (Chabicovsky et al. 2010). We did not observe side effects after brain ischemia in rats that were treated with oral PDTC at 50 mg/kg once a day for 2 months (Li et al. 2012). Intranasal application of PDTC at 100 mg/kg once a day for 3 days provided neuroprotection without noticeable side effects in neonatal rats (Wang et al. 2013). Thus, current studies suggest the safety of using PDTC to provide neuroprotection. However, additional pre-clinical studies are needed to understand the safety profile of PDTC to determine its translational potential for providing neuroprotection.
Our study has limitation. We used male rats only in this initial study to avoid the interference of sex difference in the results. Future pre-clinical studies using animals of both sexes will determine whether there is any sex difference in the neuroprotective effects of PDTC.
In summary, our results suggest that PDTC is neuroprotective in embolic stroke. PDTC applied early after the onset of embolic stroke can extend the therapeutic time window of tPA. These neuroprotective effects may be mediated by its anti-inflammatory and anti-oxidative properties.
Significance statement.
Stroke is a leading cause of mortality and morbidity. About 4% patients benefit from using tissue plasminogen activator (tPA) that can open the blocked blood vessels. Most patients arrive hospital too late, i.e. not within the therapeutic time window of tPA. We showed that pyrrolidine dithiocarbamate (PDTC) improved neurological outcome after a delayed use of tPA in rats with stroke. PDTC reduced bleeding in the brain of animals treated with tPA. Bleeding is a side-effect of tPA. Thus, our study identified a small molecule that potentially can be used with tPA to improve the outcome of patients with stroke.
Acknowledgments
Grant support: This study was supported by a grant (R01 GM098308 to Z Zuo) from the National Institutes of Health, Bethesda, MD, by a grant from the International Anesthesia Research Society (2007 Frontiers in Anesthesia Research Award to Z Zuo), Cleveland, OH, by a Grant-in-Aid from the American Heart Association Mid-Atlantic Affiliate (10GRNT3900019 to Z Zuo), Baltimore, MD, and the Robert M. Epstein Professorship endowment, University of Virginia, Charlottesville, VA.
Footnotes
The research work was performed in and should be attributed to the Department of Anesthesiology, University of Virginia, Charlottesville, VA 22908, U.S.A.
Disclosure/conflict of interest: The authors declare no competing financial interests.
Author contributions: ZW: research design, performance of experiments, data analysis and drafting of the methods and materials section; WS: performance of experiments and data analysis; JC: Performance of experiments; MW: research design; WH: research design; ZZ: project concept inception, research design, data analysis and manuscript writing.
References
- Adams HP, Jr, del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, Grubb RL, Higashida RT, Jauch EC, Kidwell C, Lyden PD, Morgenstern LB, Qureshi AI, Rosenwasser RH, Scott PA, Wijdicks EF. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38:1655–1711. doi: 10.1161/STROKEAHA.107.181486. [DOI] [PubMed] [Google Scholar]
- Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharm Des. 2010;16:2766–2778. doi: 10.2174/138161210793176572. [DOI] [PubMed] [Google Scholar]
- Amar AP, Griffin JH, Zlokovic BV. Combined neurothrombectomy or thrombolysis with adjunctive delivery of 3K3A-activated protein C in acute ischemic stroke. Front Cell Neurosci. 2015;9:344. doi: 10.3389/fncel.2015.00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhemer OA, Fransen PS, Beumer D, van den Berg LA, Lingsma HF, Yoo AJ, Schonewille WJ, Vos JA, Nederkoorn PJ, Wermer MJ, van Walderveen MA, Staals J, Hofmeijer J, van Oostayen JA, Lycklama a Nijeholt GJ, Boiten J, Brouwer PA, Emmer BJ, de Bruijn SF, van Dijk LC, Kappelle LJ, Lo RH, van Dijk EJ, de Vries J, de Kort PL, van Rooij WJ, van den Berg JS, van Hasselt BA, Aerden LA, Dallinga RJ, Visser MC, Bot JC, Vroomen PC, Eshghi O, Schreuder TH, Heijboer RJ, Keizer K, Tielbeek AV, den Hertog HM, Gerrits DG, van den Berg-Vos RM, Karas GB, Steyerberg EW, Flach HZ, Marquering HA, Sprengers ME, Jenniskens SF, Beenen LF, van den Berg R, Koudstaal PJ, van Zwam WH, Roos YB, van der Lugt A, van Oostenbrugge RJ, Majoie CB, Dippel DW Investigators MC. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372:11–20. doi: 10.1056/NEJMoa1411587. [DOI] [PubMed] [Google Scholar]
- Cao L, Li L, Lin D, Zuo Z. Isoflurane induces learning impairment that is mediated by interleukin 1beta in rodents. PLoS ONE. 2012;7:e51431. doi: 10.1371/journal.pone.0051431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabicovsky M, Prieschl-Grassauer E, Seipelt J, Muster T, Szolar OH, Hebar A, Doblhoff-Dier O. Pre-clinical safety evaluation of pyrrolidine dithiocarbamate. Basic Clin Pharmacol Toxicol. 2010;107:758–767. doi: 10.1111/j.1742-7843.2010.00573.x. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Shoamanesh A, Wilson D, Gang Q, Fox Z, Jager HR, Benavente OR, Werring DJ. Cerebral microbleeds and postthrombolysis intracerebral hemorrhage risk Updated meta-analysis. Neurology. 2015;85:927–924. doi: 10.1212/WNL.0000000000001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernandez JA, LaRue B, Griffin JH, Chopp M, Zlokovic BV. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12:1278–1285. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- Del Zoppo GJ, Saver JL, Jauch EC, Adams HP., Jr Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: a science advisory from the American Heart Association/American Stroke Association. Stroke. 2009;40:2945–2948. doi: 10.1161/STROKEAHA.109.192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Zhang J, Feng C, Xiong L, Zuo Z. Critical role of matrix metalloprotease-9 in chronic high fat diet-induced cerebral vascular remodelling and increase of ischaemic brain injury in mice. Cardiovasc Res. 2014;103:473–484. doi: 10.1093/cvr/cvu154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding G, Jiang Q, Zhang L, Zhang ZG, Li L, Knight RA, Ewing JR, Wang Y, Chopp M. Analysis of combined treatment of embolic stroke in rat with r-tPA and a GPIIb/IIIa inhibitor. J Cereb Blood Flow Metab. 2005;25:87–97. doi: 10.1038/sj.jcbfm.9600010. [DOI] [PubMed] [Google Scholar]
- Goyal M, Pradhan G, Gupta S, Kapoor S. Hypohidrotic ectodermal dysplasia with ankylosis of temporomandibular joint and cleft palate: A rare presentation. Contemp Clin Dent. 2015;6:110–112. doi: 10.4103/0976-237X.149304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K. Evidence that reactive oxygen species do not mediate NF-kappaB activation. Embo J. 2003;22:3356–3366. doi: 10.1093/emboj/cdg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Aoki J, Shibazaki K, Saji N, Uemura J, Sakamoto Y. New appearance of extraischemic microbleeds on T2*-weighted magnetic resonance imaging 24 hours after tissue-type plasminogen activator administration. Stroke. 2013;44:2776–2781. doi: 10.1161/STROKEAHA.113.001778. [DOI] [PubMed] [Google Scholar]
- Kleindorfer D, Lindsell CJ, Brass L, Koroshetz W, Broderick JP. National US estimates of recombinant tissue plasminogen activator use: ICD-9 codes substantially underestimate. Stroke. 2008;39:924–928. doi: 10.1161/STROKEAHA.107.490375. [DOI] [PubMed] [Google Scholar]
- Li H, Yin J, Li L, Deng J, Feng C, Zuo Z. Isoflurane postconditioning reduces ischemia-induced nuclear factor-kappaB activation and interleukin 1beta production to provide neuroprotection in rats and mice. Neurobiol Dis. 2013;54:216–224. doi: 10.1016/j.nbd.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Sheng W, Feng C, Zuo Z. Pyrrolidine dithiocarbamate attenuates brain Abeta increase and improves long-term neurological outcome in rats after transient focal brain ischemia. Neurobiol Dis. 2012;45:564–572. doi: 10.1016/j.nbd.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zuo Z. Glutamate transporter type 3 knockout reduces brain tolerance to focal brain ischemia in mice. J Cereb Blood Flow Metab. 2011a;31:1283–1292. doi: 10.1038/jcbfm.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zuo Z. Isoflurane postconditioning induces neuroprotection via Akt activation and attenuation of increased mitochondrial membrane permeability. Neurosci. 2011b;199:44–50. doi: 10.1016/j.neuroscience.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Zuo Z. Isoflurane induces hippocampal cell injury and cognitive impairments in adult rats. Neuropharmacology. 2011;61:1354–1359. doi: 10.1016/j.neuropharm.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiological Reviews. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu D, Cheng T, Guo H, Fernandez JA, Griffin JH, Song X, Zlokovic BV. Tissue plasminogen activator neurovascular toxicity is controlled by activated protein C. Nat Med. 2004;10:1379–1383. doi: 10.1038/nm1122. [DOI] [PubMed] [Google Scholar]
- Liu SF, Ye X, Malik AB. Inhibition of NF-kappaB activation by pyrrolidine dithiocarbamate prevents In vivo expression of proinflammatory genes. Circulation. 1999;100:1330–1337. doi: 10.1161/01.cir.100.12.1330. [DOI] [PubMed] [Google Scholar]
- Martin JA, Smith BL, Matthews TJ, Ventura SJ. Births and Deaths: Preliminary Data for 1998. National Vital Statistics Reports. 1999;47:1–45. [PubMed] [Google Scholar]
- McCord JM, Fridovich I. Superoxide dismutase: the first twenty years (1968–1988) Free Radic Biol Med. 1988;5:363–369. doi: 10.1016/0891-5849(88)90109-8. [DOI] [PubMed] [Google Scholar]
- Montaner J, Molina CA, Monasterio J, Abilleira S, Arenillas JF, Ribo M, Quintana M, Alvarez-Sabin J. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation. 2003;107:598–603. doi: 10.1161/01.cir.0000046451.38849.90. [DOI] [PubMed] [Google Scholar]
- NINDS. Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. The NINDS t-PA Stroke Study Group. Stroke. 1997;28:2109–2118. doi: 10.1161/01.str.28.11.2109. [DOI] [PubMed] [Google Scholar]
- Nurmi A, Goldsteins G, Narvainen J, Pihlaja R, Ahtoniemi T, Grohn O, Koistinaho J. Antioxidant pyrrolidine dithiocarbamate activates Akt-GSK signaling and is neuroprotective in neonatal hypoxia-ischemia. Free Radic Biol Med. 2006;40:1776–1784. doi: 10.1016/j.freeradbiomed.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, Weih F, Frank N, Schwaninger M, Koistinaho J. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004a;35:987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- Nurmi A, Vartiainen N, Pihlaja R, Goldsteins G, Yrjanheikki J, Koistinaho J. Pyrrolidine dithiocarbamate inhibits translocation of nuclear factor kappa-B in neurons and protects against brain ischaemia with a wide therapeutic time window. J Neurochem. 2004b;91:755–765. doi: 10.1111/j.1471-4159.2004.02756.x. [DOI] [PubMed] [Google Scholar]
- Overgaard K, Sereghy T, Boysen G, Pedersen H, Hoyer S, Diemer NH. A rat model of reproducible cerebral infarction using thrombotic blood clot emboli. J Cereb Blood Flow Metab. 1992;12:484–490. doi: 10.1038/jcbfm.1992.66. [DOI] [PubMed] [Google Scholar]
- Prats-Sanchez L, Camps-Renom P, Sotoca-Fernandez J, Delgado-Mederos R, Martinez-Domeno A, Marin R, Almendrote M, Dorado L, Gomis M, Codas J, Llull L, Gomez Gonzalez A, Roquer J, Purroy F, Gomez-Choco M, Canovas D, Cocho D, Garces M, Abilleira S, Marti-Fabregas J Catalan Stroke, C, Reperfusion C. Remote Intracerebral Hemorrhage After Intravenous Thrombolysis: Results From a Multicenter Study. Stroke. 2016;47:2003–2009. doi: 10.1161/STROKEAHA.116.013952. [DOI] [PubMed] [Google Scholar]
- Schreck R, Meier B, Mannel DN, Droge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med. 1992;175:1181–1194. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RJ, Sharp FR. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front Cell Neurosci. 2016;10:56. doi: 10.3389/fncel.2016.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Z, Chow N, Davis TP, Griffin JH, Chopp M, Zlokovic BV. An activated protein C analog with reduced anticoagulant activity extends the therapeutic window of tissue plasminogen activator for ischemic stroke in rodents. Stroke. 2012;43:2444–2449. doi: 10.1161/STROKEAHA.112.658997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhao H, Peng S, Zuo Z. Intranasal pyrrolidine dithiocarbamate decreases brain inflammatory mediators and provides neuroprotection after brain hypoxia-ischemia in neonatal rats. Exp Neurol. 2013;249:74–82. doi: 10.1016/j.expneurol.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RL, Chopp M, Zhang ZG, Jiang Q, Ewing JR. A rat model of focal embolic cerebral ischemia. Brain Res. 1997;766:83–92. doi: 10.1016/s0006-8993(97)00580-5. [DOI] [PubMed] [Google Scholar]
- Zhou ML, Shi JX, Hang CH, Cheng HL, Qi XP, Mao L, Chen KF, Yin HX. Potential contribution of nuclear factor-kappaB to cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. J Cereb Blood Flow Metab. 2007;27:1583–1592. doi: 10.1038/sj.jcbfm.9600456. [DOI] [PubMed] [Google Scholar]