

Abstract
The HIV-1 RNA genome contains complex structures with many structural elements playing regulatory roles during viral replication. A recent study has identified multiple RNA structures with unknown functions that are conserved among HIV-1 and two simian immunodeficiency viruses. To explore the roles of these conserved RNA structures, we introduced synonymous mutations into the HIV-1 genome to disrupt each structure. These mutants exhibited similar particle production, viral infectivity, and replication kinetics relative to the parent NL4-3 virus. However, when replicating in direct competition with the wild-type NL4-3 virus, mutations of RNA structures at inter-protein domain junctions can cause fitness defects. These findings reveal the ability of HIV-1 to tolerate changes in its sequences, even in apparently highly conserved structures, which permits high genetic diversity in HIV-1 population. Our results also suggest that some conserved RNA structures may function to fine-tune viral replication.
Keywords: HIV, lentivirus, RNA structure, viral replication, replication fitness
Introduction
The HIV-1 full-length RNA (referred to hereafter as HIV-1 RNA) plays at least two major roles during viral replication: it encodes viral proteins and serves as the virion genome (Sundquist and Krausslich, 2012). HIV-1 RNA is translated to generate Gag/Gag-Pol polyproteins, the viral structural proteins and enzymes required for replication. Furthermore, two copies of HIV-1 RNA are packaged into each particle as virion genomes (Chen et al., 2009); hence, all the genetic information that needs to be passed down to the progeny must reside within the HIV-1 RNA. In addition to the primary sequence, HIV-1 RNA also contains complex structures, many of which serve important functional roles. Together with viral and host proteins, several RNA elements regulate different steps of HIV-1 gene expression. For example, the trans-activation response (TAR) element has an important regulatory role in transcription elongation; the Rev response element (RRE) is important for the nuclear export of intron-containing HIV-1 RNA (Cochrane et al., 1990; Heaphy et al., 1990; Malim et al., 1989; Malim et al., 1990; Zapp and Green, 1989); and the Gag-Pol ribosomal frameshift signal dictates the proportion of Gag-Pol polyprotein translated (Bidou et al., 1997; Parkin et al., 1992; Wilson et al., 1988). RNA elements residing in the 5′ untranslated region (UTR) mediate and regulate HIV-1 RNA dimerization and packaging during virus assembly. For example, the dimerization initiation site located in the loop of stem loop 1 plays an important role in partner selection of co-packaged RNA (Chen et al., 2009; Chin et al., 2005; Moore et al., 2007); in addition, multiple unpaired elements serve as specific Gag-binding sites to mediate RNA packaging (Abd El-Wahab et al., 2014; Keane et al., 2015; Rye-McCurdy et al., 2016; Wilkinson et al., 2008). Therefore, high-order RNA structure serves as an additional layer of information encoded beyond that encoded in the primary sequence.
The structure of the 9-kb HIV-1 RNA in virion has been extensively examined using the SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) chemical probing strategy (Merino et al., 2005; Siegfried et al., 2014; Watts et al., 2009; Wilkinson et al., 2008). These studies revealed complex structures form along the length of HIV-1 RNA. In addition to RNA elements with known functions, there are other regions that form distinct RNA structures with unknown functions, many of which are as structurally complex as the known functional elements. The virion RNA structures of three primate lentiviruses have been compared (Lavender et al., 2015): those of HIV-1 and those from two simian immunodeficiency viruses (SIVs) isolated from rhesus macaque (SIVmac) and chimpanzee (SIVcpz). The nucleotide sequences of HIV-1 (NL4-3 molecular clone) and SIVmac239 contain ~55% sequence identity. Several regions of these three viral RNAs exhibited structures conserved at the level of their per-nucleotide SHAPE reactivities. These elements comprised most of the strongly validated functional elements including motifs in the 5′ UTR, the Gag-Pol ribsosomal frameshift signal, and the RRE. In addition, five RNA structures without known function showed apparent structural conservation (Lavender et al., 2015). Three of these conserved structures were located between domains of the Gag or Gag-Pol polyproteins. It was hypothesized that these RNA structures may slow down the translation machinery to allow proper protein folding (Lavender et al., 2015; Watts et al., 2009). Intriguingly, one of the RNA structures is located downstream from the Gag stop codon and overlaps the junction of protease (PR) and reverse transcriptase (RT). The location of this element is reminiscent of a recently identified RNA stability element in Rous sarcoma virus (RSV) (Ge et al., 2016; Quek and Beemon, 2014; Withers and Beemon, 2011), which is located downstream from the Gag-Pro stop codon and affects the stability of the full-length viral RNA. Thus, it was possible these three conserved HIV-1 RNA structures might have more than one function. In addition, the comparisons of HIV-1 and SIV RNA structures also identified two regions with unknown function containing long helical elements with highly conserved base-pairing, located in capsid (CA) and nef.
In this report, we sought to explore the potential functions of these five conserved RNA structures in the HIV-1 genome. We generated five mutants in which each RNA structure was disrupted by synonymous mutations such that the RNA structure, but not protein-coding capacity, was affected. We examined the properties of these mutants by virion production, viral infectivity, and replication kinetics. We then further characterized the replication fitness of these mutants using a competition assay in which a mixture of mutant and wild-type NL4-3 virus were used to infect cultured T cells or peripheral blood mononuclear cells (PBMCs), followed by monitoring the ratios of wild-type and mutant viruses. Our studies demonstrate that these five conserved RNA structures are not essential for viral replication but can affect replication fitness in modest but reproducible manners.
Results
Strategy to study the roles of the conserved RNA structures in HIV-1 replication
To explore the potential functions of the RNA structures conserved between HIV-1, SIVcpz, and SIVmac, we introduced synonymous mutations in the five regions in the context of HIV-1 and examined their effects on viral replication (Fig. 1). The locations of these RNA structures in the HIV-1 genome are shown in Fig. 1A. Three of the structures – termed A1, A2, and A3 – overlap regions between domains of the Gag/Gag-Pol polyproteins (Fig. 1A). RNA structures A1, A2, and A3 encompass sequences encoding the matrix (MA) and capsid (CA) junction, the protease (PR) and reverse transcriptase (RT) junction, and the RT and integrase (IN) junction, respectively.
Fig. 1.
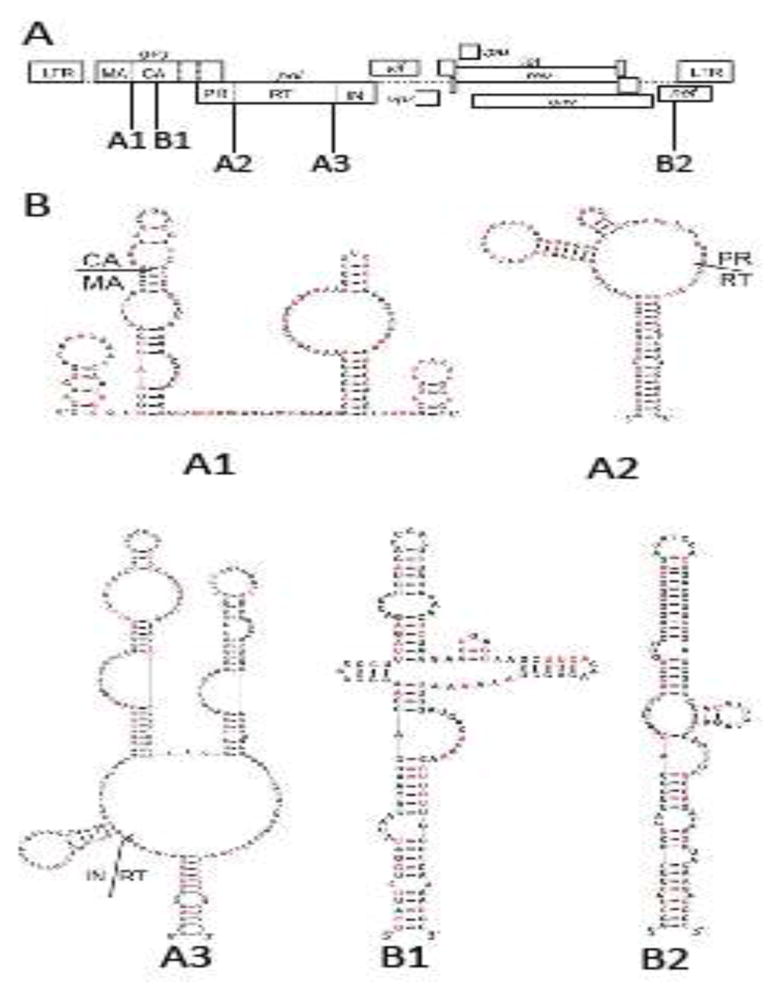
Five conserved RNA structures in HIV-1 genome. (A) Locations of the structures in HIV-1 genome. Three elements correspond to conserved structures encoding the junctions of polyprotein domains: MA and CA (A1), PR and RT (A2), RT and IN (A3). Two structures contain extended base-pairings are located in the regions encoding CA (B1) and nef (B2). (B) Nucleotide sequence and the predicted structures of the five RNA structures in the NL4-3 molecular clone. Nucleotides shown in red are replaced by synonymous mutations in mutant constructs.
The RNA structures as modeled in the SHAPE studies (Lavender et al., 2015) were altered by introducing synonymous mutations (Fig. 1B in red; nucleotide sequences and alignments are shown in Supplemental Figure 1). As virion RNA derived from NL4-3 was used in the SHAPE studies, we introduced mutations into the NL4-3 molecular clone. When possible, synonymous mutations were introduced to disrupt the base-paired nucleotides described in the predicted structures from the SHAPE studies. Consequently, 18 of the 36 described nucleotide pairs are disrupted in the A1 mutant, and 11 of the 28, and 26 of the 54 described pairs are disrupted in A2 and A3 mutants, respectively. RNA structure modeling of the mutant sequences suggests that these mutations substantially disrupted the A1, A2, and A3 structures (Supplemental Fig. 2). We have examined the abundance of tRNAs that correspond to the synonymous mutations in these regions and found that on average, these mutations slightly improved the codon usage of the three regions.
Mutations of conserved RNA structures do not affect viral particle production or replication
It was hypothesized that RNA structures between domains of polyproteins induce pauses in translation, thereby allowing proper protein folding. Therefore, disrupting the A1, A2, or A3 structure could affect proper folding of the Gag/Gag-Pol proteins and lead to lower viral production or infectivity compared with that of the wild-type virus. Additionally, the location of the A2 RNA structure is reminiscent of the RSV RNA stability element; synonymous mutations in the A2 mutant could disrupt this element, thereby leading to reduction of full-length RNA and viral particle production compared with wild-type virus.
To characterize the effects of these mutations, we transfected plasmids encoding wild-type or mutant NL4-3 into 293T cells. Viruses were harvested from transfected cells, quantified by the amount of CA (p24) proteins, and equal amounts of viruses were used to infect TZM-bl indicator cells (Fig. 2A). The viral particle production was measured by p24 CA ELISA and the level of viral infection was determined by measuring the luciferase activity of the TZM-bl cells; mutations in A1, A2, and A3 do not affect virus production or virus infectivity (Fig. 2B and 2C).
Fig. 2.

Characterization of HIV-1 mutants of conserved RNA structures. (A) Outline of the experimental protocol. (B) Relative virus production. Viruses were harvested 36 hours post-transfection and were quantified by p24 CA ELISA. (C) Relative infectivity of mutant viruses. Equal amounts of viruses (quantified by p24 CA) were used to infect TZM-bl cell and luciferase activities were measured. Measurements from wild-type NL4-3 (WT) were defined as 100%. Results from three independent experiments are summarized; error bars indicate standard deviations.
We then examined the replication kinetics of the mutant viruses in T cells by infecting CEM cells with equal amounts of wild-type or mutant virus and monitoring virions released into the supernatant using the p24 assay. Representative kinetics are shown in Fig. 3. For each mutant, the kinetics of the viral replication were compared to the kinetics of the wild-type virus performed in parallel. The p24 production of the wild-type and mutant viruses peaked at the same time point, although the absolute values of the p24 amount varied slightly between samples, indicating that these mutants have replication kinetics similar to NL4-3. Results from a total of three different sets of experiments all showed that the replication kinetics of the A1, A2, and A3 mutants were similar to that of the wild-type NL4-3.
Fig. 3.
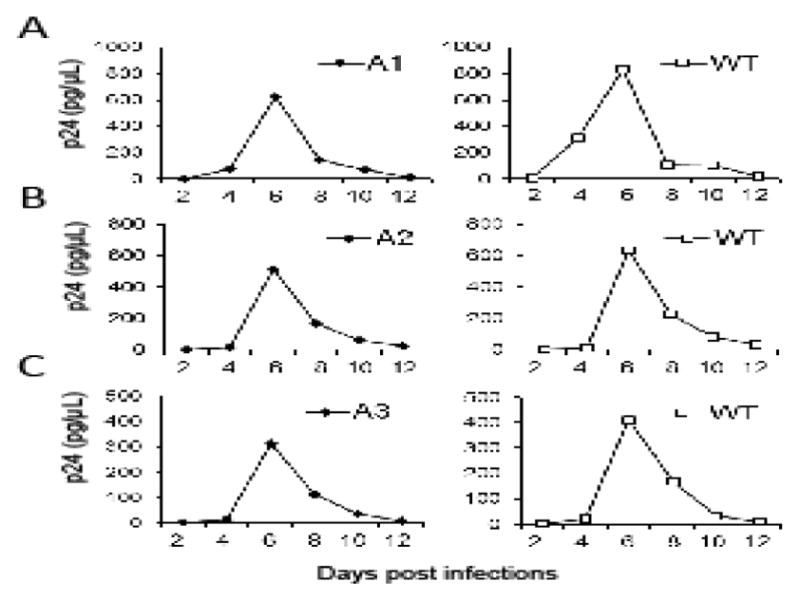
Replication kinetics of the wild-type and the mutant viruses. Representative replication kinetics of mutant viruses (A1, A2, A3) are shown on the left whereas those from NL4-3 virus performed in parallel are shown on the right. Equal amounts of viruses were used to infect CEM cells, supernatants were harvested at 2, 4, 6, 8, 10 or 12 days post-infection, and virus production was measured by p24 ELISA. Three independent experiments were performed for each mutant; one representative replication curve for each mutant virus is shown.
Mutations in the conserved RNA structures can cause HIV-1 fitness defects
To determine whether there are fitness costs associated with mutations in A1, A2, or A3 RNA structures, we performed competition experiments between each mutant and wild-type NL4-3 (Fig. 4A). Briefly, based on p24 CA measurements, a stock of the wild-type virus and a stock of mutant virus were mixed together at either a 1:1 or a 1:9 ratio; these virus mixtures were then used to infect T cells. Aliquots of the supernatant were removed from the culture as a function of time, and the proportions of wild-type and mutant viruses were determined by a DNA sequencing method (Nikolaitchik et al., 2006). Briefly, RNA was reverse transcribed into cDNA, a region of the viral genome was amplified and subjected to DNA sequencing using primers flanking the mutated sequences. At the mutated positions, there are two signals in the chromatogram, corresponding to the mutant and the wild-type viruses, from which the ratio of these viruses are calculated. Standard curves were generated using known proportion of mutant and wild-type plasmid mixtures; only signals that were in the linear detection range of the assay were used. The accuracy of this assay is comparable to allelic-specific PCR when used to detect viruses above 10% of the total population (Nikolaitchik et al., 2006).
Fig. 4.

Replication fitness of mutant viruses measured by competition assay. (A) Experimental design of the competition assay. (B–D) Results from competition assay between NL4-3 (WT) virus and mutant virus A1 (B), A2 (C), and A3 (D). In each set of competition assays, wild-type and mutant viruses were mixed at ratios of 1:9 (top panels) and 1:1 (bottom panels). Competition assays were performed using CEM cells (left panels) and PBMCs (right panels). Results from two independent experiments using CEM cells are summarized in the left panels, with error bars indicating standard deviation. Results from one representative experiment using PBMCs are shown in the right panels. PBMCs from three donors were used in three independent experiments. Day zero samples are the results from the input virus of each infection. X-axis: Days post infection. Y-axis: Proportions of wild-type and mutant virion RNA released in the supernatant.
We examined the replication fitness of the A1 mutant in two independent experiments using CEM T cells. Results from these experiments are summarized on the left panels in Fig. 4B. Additionally, three sets of competition experiments were also performed using PBMCs from three different donors. Results from one representative experiment are shown in the right panels of Fig. 4B. In all these experiments, the proportion of the wild-type virus increased while the proportion of the A1 virus decreased. Therefore, when in competition with the wild-type NL4-3, A1 mutant displays a fitness defect in both CEM T cells and PBMCs. We have also examined the replication fitness of the A2 mutant (Fig. 4C) and found that A2 had little fitness defect compared with wild-type NL4-3 virus in CEM T cells and in PBMCs. On the other hand, A3 mutant demonstrated a detectable replication fitness defect in CEM T cells and in PBMCs.
We calculated the absolute fitness (replication) difference between wild-type NL4-3 and mutant viruses (Bonhoeffer et al., 2002; Maree et al., 2000). The average and standard deviation of the absolute fitness difference values for A1, A2, and A3 mutants in CEM cells were −0.27 ± 0.06, −0.06 ± 0.03, −0.24 ± 0.06, respectively. Using the absolute fitness difference and replication rate of wild-type virus calculated from the amounts of viruses measured at various time points, we estimated the relative fitness of the A1, A2, and A3 mutant viruses to be ~0.82, 0.96, and 0.86, respectively, when the wild-type virus fitness is defined as 1. Therefore, A1 and A3 mutants have mild fitness defects whereas A2 mutant does not have observable fitness defects.
Disrupting the long conserved B1 and B2 RNA helices does not affect viral replication
In addition to the A1, A2, and A3 structures, two other conserved RNA structures were identified in SHAPE studies (Lavender et al., 2015). These structures, referred to as B1 and B2, appear to contain long irregular helices conserved between HIV-1 and the two SIVs. B1 is located in the CA-coding region whereas B2 is located in the Nef-encoding region of the HIV-1 genome (Fig. 1). We generated B1 and B2 mutants containing synonymous mutations in these structures. The B1 mutant disrupted 22 of the 43 base pairs whereas the B2 mutant disrupted 14 of the 36 base pairs in the predicted structures.
We examined the virus production and infectivity of the B1 and B2 mutants and, when compared to wild-type NL4-3 virus, these mutants do not have observable defects in these assays (Fig. 5A and 5B). Similarly, B1 and B2 mutants have replication kinetics comparable to that of wild-type NL4-3 (Fig. 5C and 5D). Furthermore, B1 and B2 mutants replicated with fitness comparable to that of the wild-type virus even in the highly sensitive competition assays in CEM T cells and in PBMCs (Fig. 5E and 5F). Therefore, disrupting the B1 or B2 structure thus does not result in detectable defects in viral production, infectivity, replication kinetics, or fitness of the NL4-3 virus.
Fig. 5.

Replication properties of the B1 and B2 mutant viruses. Relative virus production (A) and infectivity (B) of the mutant viruses. Viruses were harvested 36 hours post-transfection and quantified by p24 CA ELISA; equal amounts of viruses were used to infect TZM-bl cell and luciferase activities were measured. Measurements from wild-type NL4-3 (WT) were defined as 100%. Results from three independent experiments are summarized; error bars indicate standard deviations. Replication of the wild-type and the B1 (C) and B2 (D) mutant viruses. Representative replication kinetics of the mutant virus are shown on the left whereas those from NL4-3 virus performed in parallel are shown on the right. Equal amounts of viruses were used to infect CEM cells, supernatants were harvested at 2, 4, 6, 8, 10 or 12 days post-infection, and virus production was measured by p24 ELISA. Three independent experiments were performed for each mutant; one representative replication curve for each mutant virus is shown. Replication fitness of the B1 (E) and B2 (F) mutant virus measured by competition assay. Wild-type and mutant viruses were mixed at the ratios of 1:9 (top panels) and 1:1 (bottom panels). Results from two independent experiments using CEM cells are summarized in the left panels with error bars indicating standard deviation, whereas results from one representative experiment using PBMCs are shown in the right panels. PBMCs from three donors were used in three independent experiments. Day zero samples are the results from the input virus of each infection. X-axis: Days post infections. Y-axis: Proportions of wild-type and mutant virion RNA released in the supernatant.
Discussion
Multiple RNA structures play regulatory roles in various steps of HIV-1 replication. In this report, we investigated the putative role(s) of five RNA structures conserved in several lentiviruses. These mutants contain synonymous mutations in the RNA structures such that in each mutant, 40% to 50% of the predicted base pairs in the structure were disrupted. We examined two classes of RNA motifs. In B1 and B2, we disrupted two RNA elements with apparent strong structural conservation among HIV-1, SIVcpz and SIVmac but without known functional roles (Lavender et al., 2015). As the structures do not have known functions, we could not predict the anticipated phenotypes of such mutants. However, neither of these mutants showed any significant effect in viral replication or competition assay, despite their apparent conservation among different lentiviruses. We have also disrupted three RNA structures located between domains of polyproteins (A1, A2, and A3), which were hypothesized to promote folding of protein domains during the translation of polyproteins (Siegfried et al., 2014; Watts et al., 2009). This model has recently garnered additional support based on genome-wide analysis of the correlation between RNA and protein structure (Tang et al., 2016). Our studies indicate that these RNA structures are not absolutely required to generate functional polyproteins as they display little effects on viral production, one-round infectivity, or overall replication kinetics (Fig. 2 and 3). However, in A1 and A3, replacing these structures with mutant sequences lead to clear and reproducible, albeit mild, replication fitness defects in a competition assay (Fig. 4); such defects could come from losing the original structure, introducing the new sequence, or both. It is unclear whether the mutations in these elements cause defects in one particular step or multiple steps of the viral replication. The mild fitness defects make it difficult to further dissect the step(s) in which the mutants have decreased functions given the limited sensitivities of current biochemical assays. Nonetheless, the magnitude of these effects is such that individual genomes containing mutations in these regions would likely be readily out-competed in the viral swarms generated after a few rounds of infection.
Retroviral full-length RNAs have long 3′ UTRs, which correlate with susceptibility to nonsense-mediated mRNA decay (Hogg and Goff, 2010). The full-length RSV RNA contains an RNA stability element, which interacts with host proteins, including polypyrimidine track binding protein, to prevent activation of nonsense-mediated decay pathway (Ge et al., 2016; Quek and Beemon, 2014; Withers and Beemon, 2011). We hypothesized that the A2 structure may be the HIV-1 stability element based on its location and structure. However, mutations destroying this structure did not affect viral production, infectivity, or replication kinetics; these results do not support the hypothesis that A2 is an HIV-1 RNA stability element. Future studies are needed to determine how HIV-1 RNA avoids the detection of the host surveillance system and nonsense mediated RNA decay despite the presence of a long 3′ UTR.
The magnitude of effects obtained in this work is comparable to prior work in which the effects of disrupting pseudoknot structures in HIV-1 were examined (Siegfried et al., 2014). In all cases, HIV RNA genome structures, identified de novo by either the ability to form stable RNA structures (Siegfried et al., 2014) or by apparent conservation across diverse strains (Lavender et al., 2015) yielded clear, but modest, effects in direct competition experiments. First, these results illustrate the flexibility of the HIV-1 genome and the ability of HIV-1 to tolerate changes in its sequences. The ability to tolerate different primary sequences provides the foundation for high genetic diversity in HIV populations. This work also supports a second conclusion that stable conserved RNA motifs may play functional, but modest roles in viral replication. We hypothesize that such stable RNA elements might function in a manner analogous to many semi-specific RNA-protein interactions or micro RNA interactions and serve to fine tune expression and regulation of viral genes.
Materials and Methods
Constructs, Cell culture, DNA transfection, luciferase assay and RNA structure modeling
All of the HIV-1 constructs described in this report were derived from NL4-3 molecular clone. Synonymous mutations were introduced into NL4-3 by replacing HIV-1 sequences with synthesized DNA fragments containing synonymous mutations using standard molecular cloning techniques. Constructs were verified using restriction enzyme mapping and DNA sequencing.
Human 293T and TZM-bl cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (HyClone), penicillin (50 U/ml; Gibco) and streptomycin (50 μg/ml; Gibco). CEM cell line and PBMCs were grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum, penicillin (50 U/ml) and streptomycin (50 μg/ml). Mononuclear cells were collected after apheresis from healthy donors and separated through a Ficoll-Hypaque gradient (Sigma). T cell activation was performed by culturing cells with medium containing phytohemagglutinin-P (2 μg/ml) for three days. Interleukin-2 (20 U/ml) was added to the PBMC culture prior to infection. Cells were maintained in humidified 37°C incubators with 5% CO2. DNA transfection was performed using FuGENE® HD transfection reagent (Promega) according to manufacturer’s recommendations. Cell culture supernatants were harvested 36-h post-transfection, clarified through a 0.45-μm-pore-size filter, and either stored at −80°C or used immediately. Firefly luciferase activities were determined at 36-h post-transfection using Britelite Plus Reporter Gene Assay System (PerkinElmer). Structure modeling of the mutant RNA structures was performed using Superfold (Reuter and Mathews, 2010; Siegfried et al., 2014)
Viral infection and replication fitness assay
The production of HIV-1 viral particles was quantified using HIV-1 p24 ELISA kit (XpressBio). For viral infectivity and replication kinetics experiments, viruses were first quantified by the p24 CA measurement and the same amount of the wild-type or mutant virus was used to infect TZM-bl cells and CEM cells, respectively. Multi-round competition assays were performed between the mutant virus and the wild-type NL4-3 virus in CEM cells or PBMCs. The wild-type and mutant viruses were first quantified by p24 amounts and then mixed together at a ratio of 1:1 or 1:9. In the competition assay, one million CEM cells and ten million PBMCs were infected with 5 pg and 1 μg of p24 measurement of HIV-1, respectively. All the infected cells were maintained in 3 ml of media in 25-cm flasks; at various time points, 1 ml of supernatant was collected and replaced by 1 ml of fresh medium. Viral RNAs were isolated using QIAamp® Viral RNA Mini Kit (QIAGEN) and used for cDNA synthesis using sequence-specific primer that annealed to a sequence 350 bp downstream of the mutated RNA sequences and Transcriptor First Strand cDNA Synthesis Kit (Roche). The resulting cDNAs were amplified using primers flanking the mutated region, sequenced using Sanger sequencing (Macrogen), and the resulting chromatograms were analyzed. To ensure the measurement of wild-type and mutant ratio is accurate, in each experiment a standard curve was generated by mixing the wild-type NL4-3 plasmid and a mutant plasmid at various ratios, the DNA mixtures were subjected to sequencing reactions and the wild-type and mutated nucleotide signals in the chromatogram were determined. Measurements within the linear dynamic range of the assays were used.
Viral fitness was quantified as r – r′ = r – [(1 + s)r] = rs; the absolute fitness difference between wild-type and mutant is r – r′, where r and r′ are the wild type and mutant virus replication rates, respectively, s is coefficient of selection, the relative fitness of the mutant virus is 1+ s (Bonhoeffer et al., 2002; Maree et al., 2000). The rs is defined by the slope of the logarithmic time plot of mutant to wild-type ratio; virus ratios used for the calculations were summarized in Fig. 4 and described in Supplemental Table 1; graphs that defined rs values are shown in Supplemental Fig. 3. The net replication rate is defined by the slope of the logarithmic time plot of the virus production measured by p24 released into the supernatant and is the sum of replication rate minus death rate, which is assumed to be 0.5 (Bonhoeffer et al., 2002). Therefore, the coefficient of selection is calculated as s = [rs/(net replication rate + 0.5)].
Supplementary Material
Supplemental Fig. 1. Nucleotide sequence alignments of NL4-3 (wt) and mutants containing synonymous mutations. Sequence identities are marked by grey highlight.
Supplemental Fig. 2. Comparison of the NL4-3 structures (wild-type) and the predicted mutant RNA structures. To illustrate the change of RNA structures, elements in the NL4-3 structures are color-coded and the corresponding mutated sequences are coded in the same colors.
Supplemental Fig. 3. The natural log of the mutant to WT virus ratios were plotted over times and the slope the fitted linear line represents the absolute replication differences of each mutant compared to WT virus. Red, blue, gold and grey lines correspond to Experiment 1A, 2A, 1B, and 2B listed in supplemental Table 1, respectively.
Supplemental Table 1. Mutant and wild-type (NL4-3) virus ratios from various time points of the competition assay. These results were summarized in the CEM portion of Fig. 4B, 4C, and 4D.
Research Highlight.
We analyzed three conserved HIV-1 RNA structures in inter-protein domain junctions
These conserved RNA structures are not essential for HIV-1 replication
Altering some RNA structures with synonymous mutations caused fitness defects
The conserved RNA structures can be altered, illustrating HIV-1 genome flexibility
We suggest that conserved RNA structures may play a role in fine-tuning replication
Acknowledgments
We thank Jonathan Rawson for discussions and critical review of the manuscript; Krista Delviks-Frankenberry for advice on primary cell infection, and Frank Maldarelli for discussions on viral fitness calculation. This work is supported in part by Intramural Research Program, Center for Cancer Research, National Cancer Institute; IATAP grants to W.S.H. and to V.K.P.; and NIH grant AI068462 to K.M.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abd El-Wahab EW, Smyth RP, Mailler E, Bernacchi S, Vivet-Boudou V, Hijnen M, Jossinet F, Mak J, Paillart JC, Marquet R. Specific recognition of the HIV-1 genomic RNA by the Gag precursor. Nat Commun. 2014;5:4304. doi: 10.1038/ncomms5304. [DOI] [PubMed] [Google Scholar]
- Bidou L, Stahl G, Grima B, Liu H, Cassan M, Rousset JP. In vivo HIV-1 frameshifting efficiency is directly related to the stability of the stem-loop stimulatory signal. RNA (New York, NY. 1997;3:1153–1158. [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer S, Barbour AD, De Boer RJ. Procedures for reliable estimation of viral fitness from time-series data. Proceedings Biological sciences. 2002;269:1887–1893. doi: 10.1098/rspb.2002.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Nikolaitchik O, Singh J, Wright A, Bencsics CE, Coffin JM, Ni N, Lockett S, Pathak VK, Hu WS. High efficiency of HIV-1 genomic RNA packaging and heterozygote formation revealed by single virion analysis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13535–13540. doi: 10.1073/pnas.0906822106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin MP, Rhodes TD, Chen J, Fu W, Hu WS. Identification of a major restriction in HIV-1 intersubtype recombination. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9002–9007. doi: 10.1073/pnas.0502522102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane AW, Chen CH, Rosen CA. Specific interaction of the human immunodeficiency virus Rev protein with a structured region in the env mRNA. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:1198–1202. doi: 10.1073/pnas.87.3.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Z, Quek BL, Beemon KL, Hogg JR. Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway. eLife. 2016:5. doi: 10.7554/eLife.11155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy S, Dingwall C, Ernberg I, Gait MJ, Green SM, Karn J, Lowe AD, Singh M, Skinner MA. HIV-1 regulator of virion expression (Rev) protein binds to an RNA stem-loop structure located within the Rev response element region. Cell. 1990;60:685–693. doi: 10.1016/0092-8674(90)90671-z. [DOI] [PubMed] [Google Scholar]
- Hogg JR, Goff SP. Upf1 senses 3′UTR length to potentiate mRNA decay. Cell. 2010;143:379–389. doi: 10.1016/j.cell.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane SC, Heng X, Lu K, Kharytonchyk S, Ramakrishnan V, Carter G, Barton S, Hosic A, Florwick A, Santos J, Bolden NC, McCowin S, Case DA, Johnson BA, Salemi M, Telesnitsky A, Summers MF. Structure of the HIV-1 RNA packaging signal. Science (New York, NY. 2015;348:917–921. doi: 10.1126/science.aaa9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavender CA, Gorelick RJ, Weeks KM. Structure-Based Alignment and Consensus Secondary Structures for Three HIV-Related RNA Genomes. PLoS computational biology. 2015;11:e1004230. doi: 10.1371/journal.pcbi.1004230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature. 1989;338:254–257. doi: 10.1038/338254a0. [DOI] [PubMed] [Google Scholar]
- Malim MH, Tiley LS, McCarn DF, Rusche JR, Hauber J, Cullen BR. HIV-1 structural gene expression requires binding of the Rev trans-activator to its RNA target sequence. Cell. 1990;60:675–683. doi: 10.1016/0092-8674(90)90670-a. [DOI] [PubMed] [Google Scholar]
- Maree AF, Keulen W, Boucher CA, De Boer RJ. Estimating relative fitness in viral competition experiments. Journal of virology. 2000;74:11067–11072. doi: 10.1128/jvi.74.23.11067-11072.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- Moore MD, Fu W, Nikolaitchik O, Chen J, Ptak RG, Hu WS. Dimer initiation signal of human immunodeficiency virus type 1: its role in partner selection during RNA copackaging and its effects on recombination. Journal of virology. 2007;81:4002–4011. doi: 10.1128/JVI.02589-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaitchik O, Rhodes TD, Ott D, Hu WS. Effects of mutations in the human immunodeficiency virus type 1 Gag gene on RNA packaging and recombination. Journal of virology. 2006;80:4691–4697. doi: 10.1128/JVI.80.10.4691-4697.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin NT, Chamorro M, Varmus HE. Human immunodeficiency virus type 1 gag-pol frameshifting is dependent on downstream mRNA secondary structure: demonstration by expression in vivo. Journal of virology. 1992;66:5147–5151. doi: 10.1128/jvi.66.8.5147-5151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quek BL, Beemon K. Retroviral strategy to stabilize viral RNA. Current opinion in microbiology. 2014;18:78–82. doi: 10.1016/j.mib.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye-McCurdy T, Olson ED, Liu S, Binkley C, Reyes JP, Thompson BR, Flanagan JM, Parent LJ, Musier-Forsyth K. Functional Equivalence of Retroviral MA Domains in Facilitating Psi RNA Binding Specificity by Gag. Viruses. 2016:8. doi: 10.3390/v8090256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried NA, Busan S, Rice GM, Nelson JA, Weeks KM. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP) Nat Methods. 2014;11:959–965. doi: 10.1038/nmeth.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundquist WI, Krausslich HG. HIV-1 assembly, budding, and maturation. Cold Spring Harbor perspectives in medicine. 2012;2:a006924. doi: 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Assmann SM, Bevilacqua PC. Protein Structure Is Related to RNA Structural Reactivity In Vivo. Journal of molecular biology. 2016;428:758–766. doi: 10.1016/j.jmb.2015.11.012. [DOI] [PubMed] [Google Scholar]
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS biology. 2008;6:e96. doi: 10.1371/journal.pbio.0060096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson W, Braddock M, Adams SE, Rathjen PD, Kingsman SM, Kingsman AJ. HIV expression strategies: ribosomal frameshifting is directed by a short sequence in both mammalian and yeast systems. Cell. 1988;55:1159–1169. doi: 10.1016/0092-8674(88)90260-7. [DOI] [PubMed] [Google Scholar]
- Withers JB, Beemon KL. The structure and function of the rous sarcoma virus RNA stability element. Journal of cellular biochemistry. 2011;112:3085–3092. doi: 10.1002/jcb.23272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapp ML, Green MR. Sequence-specific RNA binding by the HIV-1 Rev protein. Nature. 1989;342:714–716. doi: 10.1038/342714a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. Nucleotide sequence alignments of NL4-3 (wt) and mutants containing synonymous mutations. Sequence identities are marked by grey highlight.
Supplemental Fig. 2. Comparison of the NL4-3 structures (wild-type) and the predicted mutant RNA structures. To illustrate the change of RNA structures, elements in the NL4-3 structures are color-coded and the corresponding mutated sequences are coded in the same colors.
Supplemental Fig. 3. The natural log of the mutant to WT virus ratios were plotted over times and the slope the fitted linear line represents the absolute replication differences of each mutant compared to WT virus. Red, blue, gold and grey lines correspond to Experiment 1A, 2A, 1B, and 2B listed in supplemental Table 1, respectively.
Supplemental Table 1. Mutant and wild-type (NL4-3) virus ratios from various time points of the competition assay. These results were summarized in the CEM portion of Fig. 4B, 4C, and 4D.