

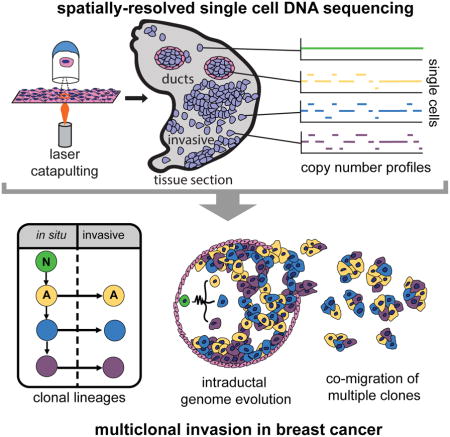
SUMMARY
Ductal Carcinoma in situ (DCIS) is an early-stage breast cancer that infrequently progresses to invasive ductal carcinoma (IDC). Genomic evolution has been difficult to delineate during invasion due to intratumor heterogeneity and the low number of tumor cells in the ducts. To overcome these challenges, we developed Topographic Single Cell Sequencing (TSCS) to measure genomic copy number profiles of single tumor cells while preserving their spatial context in tissue sections. We applied TSCS to 1293 single cells from 10 synchronous patients with both DCIS and IDC regions, in addition to exome sequencing. Our data reveal a direct genomic lineage between in situ and invasive tumor subpopulations, and further shows that most mutations and copy number aberrations evolved within the ducts, prior to invasion. These results support a multi-clonal invasion model, in which one or more clones escape the ducts and migrate into the adjacent tissues to establish the invasive carcinomas.
In-brief
Retaining spatial information in single cell analysis provides insight into clonal invasion patterns and disease progression in patients with DCIS-IDC breast cancer.
INTRODUCTION
Ductal carcinoma in situ (DCIS) is the most common form of early-stage breast cancer and is often detected during routine mammography. Only a small percentage of cases (10–30%) progress to invasive ductal carcinoma (IDC), resulting in a major clinical challenge in determining which patients to treat (Allred, 2010). The genomic and evolutionary basis of invasion remains poorly understood in DCIS due to several technical challenges in bulk tissue analysis, including the extensive intratumor heterogeneity in breast cancer (Gao et al., 2016; Shah et al., 2012; Yates et al., 2015), the limited number of tumor cells in the ducts (Allred et al., 2008), and the large amount of stromal cells in DCIS tumors (Virnig et al., 2010).
Different evolutionary models have been proposed for invasion. In the independent lineage model, different initiating cells in the breast tissues give rise to the in situ and invasive subpopulations, through distinct cell lineages that evolve in parallel and do not share any genomic aberrations (Sontag and Axelrod, 2005). This model is supported by targeted studies that have shown discordant molecular markers between the in situ and invasive regions (Gerlinger et al., 2014; Miron et al., 2010; Yates et al., 2015). In contrast, the evolutionary bottleneck model posits that a direct genomic lineage exists between the in situ cells in the ducts and the invasive cells in the adjacent tissues (Cowell et al., 2013). This model postulates that multiple clones evolve within the ducts, after which a single clone escapes the basement membrane and expands to form the invasive tumor mass (Cowell et al., 2013). The bottleneck model is supported by genomic studies that report concordant mutations between the ductal and invasion regions, in addition to many invasive-specific mutations and copy number aberrations that are selected during invasion (Kim et al., 2015; Kroigard et al., 2015; Newburger et al., 2013; Yates et al., 2015). However, precise clonal lineages have been difficult to distinguish in studies that have analyzed bulk tissue samples from DCIS patients.
Single cell DNA sequencing methods have emerged as powerful tools for resolving intratumor heterogeneity (Navin et al., 2011; Xu et al., 2012; Zong et al., 2012), delineating stromal cell types (Patel et al., 2014; Tirosh et al., 2016), and detecting rare subpopulations (Aceto et al., 2014; Lohr et al., 2014; Song et al., 2017). These methods can reconstruct evolutionary lineages in heterogeneous tumors from single time-point samples (McPherson et al., 2016; Wang et al., 2014). However, a major limitation is that most single cell isolation methods require the preparation of cell suspensions, including methods such as FACS (Baslan et al., 2012; Leung et al., 2015), micromanipulation (Grindberg et al., 2013), microdroplets (Macosko et al., 2015), or nanowells (Gao et al., 2017). These procedures inherently lose all spatial information, which is critical for studies of early stage cancers, such as DCIS, where histopathology is necessary to classify tumor cells as in situ or invasive.
To address this limitation, we developed Topographical Single Cell Sequencing (TSCS), an approach that combines laser-catapulting (Datta et al., 2015) and single cell DNA sequencing to measure genomic copy number profiles of single tumor cells while preserving their spatial information in tissue sections. We hypothesized that invasive cells share a direct genomic lineage with one (or more) single cells in the ducts. To investigate this question, we applied TSCS, along with deep-exome sequencing to trace clonal evolution during invasion in 10 high-grade frozen tumor samples from synchronous DCIS-IDC patients. Our results support a direct genomic lineage between the in situ and invasive tumor cell subpopulations and further shows that most mutations and copy number aberrations evolved within the ducts prior to invasion. These data suggest that multiple clones escaped from the ducts and co-migrated into the adjacent tissues to establish invasive carcinomas.
RESULTS
Spatially-Resolved Single Cell DNA Sequencing
To isolate single tumor cells from frozen tissue sections, while preserving their spatial positions and morphology in situ, we developed TSCS. This approach combines laser-capture-microdissection, laser-catapulting, whole-genome-amplification (WGA), and single cell DNA sequencing (Figure 1, Methods). First, frozen tissue sections were sectioned with a cryomicrotome and stained with H&E to identify in situ and invasive regions by histopathology. Whole-tissue imaging was performed to create a global tissue map of the tumor to identify ductal (in situ) and invasive regions prior to capture (Figure 1A). LCM was used to cut a circumference around each single cell with a 1-micron laser (Figure 1B) followed by laser-catapulting to transfer the cell into collection tubes (Figure 1C) using a high-throughput robotic stage (Figure 1D, Methods). Laser-catapulting is a touchless approach that transfers cells using UV energy, thereby mitigating bacterial contamination and adjacent cell contamination compared to standard LCM systems that require physical contact with the tissues (Vandewoestyne et al., 2013). We optimized the UV laser parameters in normal breast tissues to minimize DNA damage from the UV laser and refined the tissue section thickness to 14 microns to mitigate the probability of cutting through individual nuclei on the cryomicrotome during tissue sectioning (Methods).
Figure 1. Topographic Single Cell Sequencing of DCIS Tissues.

(A) Whole-tissue scanning is performed on H&E stained synchronous DCIS tissues at low 10X magnification. (B) UV laser-microdissection of a single cell at 63X magnification (C) laser-catapulting transfer of a single cell into a collection tube. (D) automated robotic depositing of single cells into 8-well strip tubes with lysis buffer into a 96-well manifold, followed by whole-genome-amplification using DOP-PCR. (E) Construction of barcoded single cell libraries for multiplexed pooling and sparse whole-genome sequencing on the Illumina platform. (F) Processing of brightfield images of single cells and spatial XY coordinates. (G) Mapping of spatial coordinates and genomic data in tissue sections, showing examples of genomic copy number profiles from a normal cell, in situ tumor cell, and an invasive tumor cell.
After isolation, the single cells were lysed and WGA was performed using Degenerative-Oligonucleotide-PCR (DOP-PCR) following the Single-Nucleus-Sequencing (SNS) protocol (Gao et al., 2016; Navin et al., 2011) (Figure 1D). Barcoded libraries were prepared and 48–96 single cells were pooled together for multiplexed next-generation sequencing (NGS) (Figure 1E). After NGS, the data was demultiplexed by cell library barcode and processed to calculate genomic copy number profiles from read depth at 220kb resolution (Methods). Under these optimized parameters, TSCS generated reproducible high-resolution single cell copy number profiles (Supplementary Figure 1). The spatial coordinates (X, Y) were extracted from each cell and transformed in cases where multiple sections were used (Figure 1F, Methods). The single cell genomic data was then mapped to the spatial coordinates to delineate the topographic organization of different clonal genotypes in the tissue sections (Figure 1G).
Cohort of Synchronous DCIS-IDC Patients
We selected a unique cohort of 10 DCIS-IDC patients with frozen tumors that had synchronous regions of both in situ and invasive tumor cells matched in the same tissue sections as defined by histopathology (Methods, Supplementary Table 1). Synchronous samples provide a number of advantages over ‘pure DCIS’ and recurrent IDC samples, that are often collected many years apart. Longitudinal samples may be confounded by intervening therapies and differences in spatial sampling, which can lead to additional mutations that are not associated with invasion. Previous work has highlighted the utility of using synchronous DCIS-IDC samples to study invasion in breast cancer, which are matched in space and time (Hernandez et al., 2012; Johnson et al., 2012; Kroigard et al., 2015; Martelotto et al., 2017). Our patient cohort consisted of 5 triple-negative (ER-, PR-, HER2-) and 5 estrogen-receptor (ER) positive breast cancer patients (Supplementary Table 1). Most of the patients had high tumor grade, with the exception of P3 and P4. Importantly, the frozen tumor samples were collected prior to any therapeutic intervention. For each patient, an average of 129 single cells were sequenced to quantify genome-wide copy number. In parallel, laser-capture-microdissection (LCM) was used to isolate thousands of tumor cells from the in situ and invasive regions. Exome libraries were prepared from each region (in situ, invasive, and matched normal) and sequenced at high coverage depth (mean=162.8X, SEM=18.9) to detect somatic point mutations (Supplementary Table 2). Matched normal breast tissues were sequenced in parallel at high coverage depth (mean=144.1X, SEM=20.3) to identify and filter germline variants.
Copy Number Evolution During Invasion in Patient 8
We investigated copy number evolution during invasion in P8 using TSCS to profile 85 in situ cells and 150 invasive cells from tissue sections from four different tumor regions (R1–R4) (Figure 2). Single-Nucleus-Sequencing (SNS) was performed and genome-wide copy number profiles were calculated at 220kb resolution (Methods). We performed 1-dimensional clustering, which revealed 1 major population of diploid cells (N) and 3 clonal aneuploid tumor subpopulations (A, B, C) (Figure 2A). Within each subpopulation (A, B, C) the copy number profiles were highly correlated (A=0.64, B=0.71, C=0.80, Pearson correlations), representing stable clonal expansions. Consensus profiles were calculated and compared from each tumor subpopulation, which identified shared amplifications on chromosome 2p (ALK), 8q (MYC), 14q (FOXA1) and 21q (RUNX1), in addition to many subpopulation-specific CNAs. Clone A had focal deletions in chromosome 4p (RHOH), 9p (CDKN2A), Xq (COL4A5) and focal amplifications on chromosome 17p (MAP2K3, NF1, BCAS3), 12p (ALG10B and ERBB3), and chromosome Xq (AR). Clone B had deletions on chromosome 3p (FHIT), 13 (RB1) 8p (DBC2), and amplifications on chromosomes 2q (GALNT13), 11p (WT1) and Xp (PDK3), while clone C shared many CNA events with clone B, including an amplification on 7p (EGFR).
Figure 2. Single Cell Copy Number Profiling in Patient P8.

(A) Clustered heatmap of single cell copy number profiles with headers indicating the major subpopulations and tissue regions (in situ or invasive) from which the cells were isolated. Lower panels show consensus profiles of the major clonal subpopulations, with known cancer gene annotations for common CNAs listed above and divergent CNAs listed below. (B) Clonal lineages of the major tumor subpopulations plotted with Timescape with inferred common ancestors indicated in grey, and clonal frequencies labelled. (C) MDS plot of single cell copy number profiles with in situ or invasive regions indicated. (D) Spatial maps of tissue sections from four different tumor regions, with single cells marked as in situ or invasive. Tumor cells are color coded by their clonal genotypes or by diploid genomes, and ducts are annotated with different colors.
To delineate clonal evolution during invasion, we inferred genomic lineages and plotted the data using Timescape (Smith et al., 2017) (Figure 2B, Methods). This analysis identified a common ancestor that evolved in the ducts with amplifications of ALK, MYC, FOXA1, and RUNX1 that subsequently diverged to form clones A and C. Clone B was a common ancestor of clone C, but diverged and evolved additional CNAs in RB1, FHIT and DBC2. This data showed that all 3 subclones evolved in the ducts from a common ancestor prior to invasion, and subsequently co-migrated into the surrounding tissues where they underwent stable clonal expansions. These data did not detect any new CNAs that were acquired in the clones during invasion, however, did reveal a decreased frequency of subclone A (40% to 5%) in the invasive regions.
To understand the relationship between the clonal genotypes and their spatial positions, we performed multi-dimensional scaling (MDS), which identified 4 discrete clusters corresponding to different subpopulations (1 normal cells and 3 tumor subpopulations). Each subpopulation consisted of single cells that were isolated from both the in situ and invasive cells, with no clonal genotype specifically associated with the in situ or invasive regions (Figure 2C). MDS showed that subpopulations C and B were adjacent in high-dimensional space, while subpopulation A was the most distant.
The clonal genotypes were mapped to their spatial coordinates in the four tissue sections (R1–R4) to delineate their topography, which showed that all three tumor clones were localized to both the ductal and the invasive regions, with no single genotype mapping exclusively to one region (Figure 2D). However, clone A was more restricted to the ductal regions (R3), while clones B and C were more frequent in the invasive regions. Consistent with the spatial distributions, we found that clones B and C had an amplification of EGFR which was previously shown to be associated with cell migration (Andl et al., 2004), while clone C had an additional deletion of FHIT, which has been shown to suppress EMT and cell migration (Suh et al., 2014).
Copy Number Evolution During Invasion in Patient 4
We investigated copy number evolution during invasion in P4 using TSCS to sequence 46 in situ cells and 58 invasive cells from two tumor regions (R1, R2) (Figure 3). Hierarchical clustering of single cell copy number profiles identified one subpopulation of diploid cells (N) and two aneuploid tumor subpopulations (A, B) (Figure 3A, upper panel). Within each subpopulation, the single cell copy number profiles showed high correlations (A= 0.89, B=0.60, Pearson correlations), representing stable clonal expansions. Consensus copy number profiles showed that both clones shared a common amplification of chromosome 1p (MDM4, ABL2), in addition to many subpopulation-specific CNAs (Figure 3A, lower panels). In clone A we identified many focal amplifications, including chromosome 3q (EVI1), 4p (CPEB2), 11q (CASP12), 13q (PCDH17) and an amplification of chromosome 12q (CDK2, MDM2). In contrast, clone B harbored many large hemizygous chromosomal deletions including 3p (SETD2, FHIT), 4 (FGFR3, NEK1), 5q (PIK3R1, APC), 14q (AKT1), 15q (NTRK3), 16q (CDH1), 17p (TP53, MAP2K4), 18 (SMAD4), and 22 (NF2).
Figure 3. Single Cell Copy Number Profiling in Patient P4.

(A) Clustered heatmap of single cell copy number profiles with headers indicating the major subpopulations and in situ or invasive regions from which the cells were isolated. Lower panels show consensus profiles of the major clonal subpopulations, with known cancer gene annotations for common CNAs listed above and divergent CNAs listed below. (B) Clonal lineages of the major tumor subpopulations plotted using Timescape with inferred common ancestors indicated in grey, and clonal frequencies labelled. (C) MDS plot of single cell copy number profiles with in situ or invasive regions indicated. (D) Spatial maps of tissue sections from two different tumor regions, with single cells marked as in situ or invasive. Tumor cells are colored by their clonal genotypes or by diploid genomes, and ducts are annotated with different colors.
Clonal lineages, inferred from the major subpopulations, identified a common ancestor with an amplification of chromosome 1q that gave rise to the two tumor subpopulations in the ducts: one that had many focal amplifications of cancer genes including MDM2 and CDK2 (clone A), and another that had many large hemizygous deletions, including CDH1, TP53, FHIT and SMAD4 (clone B) (Figure 3C). This data showed that genomic copy number evolution occurred within the ducts and gave rise to two major tumor subpopulations. During invasion, the frequency of clone B increased from 16% to 67%, while the frequency of clone A decreased from 84% to 33% in the invasive tissues.
MDS identified three distinct clusters that corresponded to the normal cells (N) and the two tumor clones (A, B). The MDS plot showed that each clonal genotype was composed of both in situ and invasive tumor cells, with no specific genotype associated with either region (Figure 3B). Next, we mapped the clonal genotypes to their spatial coordinates in the two tissue sections (R1, R2) which showed that both clones were located in the ductal and invasive regions (Figure 3D). This map also showed that in region 1 most of the normal diploid cells were localized to the invasive regions, which may reflect the difficulty in distinguishing stromal from tumor cells in these regions by histopathology. Furthermore, these data showed that clone A was highly localized to the four ducts (d1 – d4) in region 2, while clone B was more prevalent in the invasive regions. Consistent with the invasive spatial localization, we found that clone B had deletions in a number of cancer genes involved in cell migration, including AKT1, APC, FGFR3, CDH1 and SMAD4.
Copy Number Evolution During Invasion in the Patient Cohort
TSCS was applied to 8 additional synchronous DCIS-IDC patients to study copy number evolution during invasion. Whole-tissue scanning of H&E tissue sections from each patient was performed to identify in situ and invasive regions for single cell isolation. In total 425 in situ and 503 invasive cells were sequenced from the 10 patients, in addition to 365 stromal diploid cells. The data was analyzed to delineate clonal substructure and copy number evolution during invasion (Figure 4). Clustering of single cell CNA profiles showed that most patients harbored 1–5 major tumor subpopulations, and that these subpopulations were located in both the in situ and invasive regions (Figure 4A).
Figure 4. Copy Number Substructure and Clonal Evolution in 10 DCIS Patients.

(A) Bar plots of clonal frequencies calculated from single cell copy number profiles in the in situ (D) or invasive (I) regions. (B) Shannon Diversity indexes calculated from single cell copy number profiles from the in situ and invasive regions of each patient with confidence intervals. (C) MDS plots of single cell copy number profiles from each DCIS patient with clonal subpopulations and normal cells indicated by color, and in situ or invasive regions indicated by shape. (D) Clonal lineages of the major tumor subpopulations plotted using Timescape, with common ancestors indicated in grey and clonal frequencies labeled for the in situ and invasive regions.
Four tumors were found to be monoclonal (P2, P3, P7, P9), while six tumors were polyclonal (P1, P4, P5, P6, P8, P10), harboring multiple clonal subclones in both the in situ and invasive regions (Figures 2, 3 and Supplementary Figures 2–5). Shannon Diversity indexes were calculated from the single cell CNA profiles ad showed that the amount of clonal diversity did not show major changes during invasion in most patients (Figure 4B, Methods). These data showed that the amount of genomic diversity correlated with the number of subpopulations detected in the in situ or invasive regions and was inconsistent with a population bottleneck, in which a decreased in clonal diversity is expected, due to the selection of a specific clonal genotype. MDS analysis of all 10 DCIS patients identified 1–6 major clusters in each patient, including the normal cells (N) and 1–5 major tumor subpopulations (A–E) that were often separated in high-dimensional space (Figure 4C). Moreover, the MDS plots show that within each genotype cluster, the tumor cells were localized to both the in situ and invasive regions.
Clonal lineages were inferred in the 6 polyclonal DCIS patients and plotted with Timescape (Smith et al., 2017) (Figure 4D, Supplementary Figures 2–5). These data showed that in all patients, the subpopulations shared a common evolutionary origin with shared truncal CNAs, suggesting that the tumors had evolved from a single cell in the duct. These data are inconsistent with an independent lineage model, in which different initiating cells give rise to the in situ and invasive subpopulations separately. In every patient, we found that the same clonal subpopulations present in the ducts and invasive regions. However, we did observe shifts in clonal frequencies in some patients (P4, P6, P8) suggesting that some genotypes may be more invasive than others. For example, in P4, clone B increased from 16% to 67% during invasion, while in P6, clone C increased from 19% to 49%. This data suggests that genome evolution initiated from a single cell in the ducts and gave rise to one or more clonal subpopulations that migrated into the adjacent tissues to establish the invasive tumor mass.
Mapping of Spatial Topography and Clonal Genotypes
To understand the distribution of clonal genotypes and their spatial organization in the polyclonal tumors, we constructed tanglegrams (Scornavacca et al., 2011). Genetic distance trees were calculated from single cell copy number profiles and mapped to spatial trees (X, Y coordinates) with minimal overlapping connections (Methods, Figure 5). In patient P4, clone A (81.5%) localized mainly to the ducts, with only a few cells (N=7) in the invasive regions, while clone B showed a higher frequency in the invasive regions. In patient P5, the two major clones (A, B) mapped to all three ducts and the invasive regions; however, clone B was restricted more to ducts 2 and 3. In patient P6 we identified 5 clonal subpopulations, in which clones A, B and C mapped more frequently to the invasive regions, while clones D and E, were found mainly in the ductal regions (ducts 1, 2, and 5). In patient P8, we identified 3 clonal subpopulations, in which clones B and C each mapped to 8 of the 10 ducts, while clone A was localized mainly to two ducts (d1 and d2). In other cases (P10 and P1) we found that the clones were equally distributed to the in situ and invasive regions. These data show that while all clones were detected in both the in situ and invasive regions, specific subclones were more restricted to the ducts, while others were more prevalent in the invasive regions, suggesting that they may have had a more invasive or migratory phenotype.
Figure 5. Mapping of Spatial Coordinates and Clonal Genotypes.

Genomic copy number trees were mapped to spatial coordinate trees using tanglegrams in the 6 polyclonal patients. Genotype trees are located on the left side for each patient, with clonal subpopulations indicated by color. Spatial trees are located on the right side with different ducts indicated by colors, and the invasive regions colored in grey. Mapping of cells coordinates and genotypes were performed by minimizing overlapping connections.
Mutational Evolution During Invasion
To investigate mutational evolution during invasion, we performed LCM to microdissect thousands of tumor cells from the in situ and invasive regions for deep-exome sequencing (mean=162.8X, SEM=18.9, Figure 6). Matched normal breast tissue (mean=144.1X, SEM=20.3) was sequenced in parallel to distinguish germline variants from somatic mutations. From this data we detected point mutations, which showed that the total number of exonic mutations (mean=23, SEM=3.3) were highly consistent between the in situ and invasive regions (t-test, p=0.868) (Figure 6A). To identify specific mutations that were discordant, we constructed oncomaps using nonsynonymous mutations (Figure 6B). Most nonsynonymous mutations (mean 87.4%) were concordant in the ducts and invasive regions, including mutations in known breast cancer genes such as TP53, PIK3CA, NCOA2, ABL2, PDE4DIP, AHNAK and RUNX1, suggesting that they were acquired in the ducts prior to invasion. However, a few mutations were in situ-specific (N=12) or invasive-specific (N=11) in 4 patients (P3, P4, P7, P8) and were not recurrent among the patients (Supplemental Table 3).
Figure 6. Exome Mutations in the in situ and Invasive Regions.

Exome sequencing of laser-capture microdissected in situ and invasive regions. (A) Bar plot of exonic mutation frequencies detected in the in situ and invasive regions of 10 DCIS-IDC patients. (B) Oncomap of nonsynonymous mutations in the in situ and invasive regions from each patient. The presence or absence of mutations has been updated based on the results from amplicon deep-sequencing validation data. Known breast cancer genes are indicated in bold, while mutations that were validated by deep-amplicon sequencing are in italics. Inset panels show examples of brightfield images of in situ or invasive regions isolated by laser-capture-microdissection. (C) Amplicon targeted deep-sequencing of in situ-specific mutations. (D) Amplicon targeted deep-sequencing of the invasive-specific mutations.
The invasive-specific mutations may have occurred at low frequencies in the ducts prior to invasion (below the exome sensitivity), or alternatively after invasion, during the expansion of the invasive tumor mass. Another possibility is that they were sampled from different geographical regions; however, this is unlikely in synchronous DCIS-IDC tissue since the cells were collected from adjacent regions in the same tissue sections. To determine if the invasive-specific mutations were acquired in the ducts or after invasion, we performed targeted deep-amplicon sequencing at high coverage depth (mean=453, 446X) for a subset of the in situ-specific (Figure 6C) and invasive-specific mutations (Figure 6D). In parallel, we performed targeted deep-amplicon sequencing of matched normal breast tissues to establish site-specific background error rates and identified significant mutations using DeepSNV (Gerstung et al., 2014).
The amplicon data showed that at higher coverage depth (226,000X) many of the in situ-specific mutations were present at low frequencies in the invasive regions. However, in contrast, most of the invasive-specific mutations (8/12) were found to be exclusive to the invasive tissues in patients P3 (DRD1, CRY1), P4 (TECRL), P7 (SCNA4, PCDHA5) and P8 (SORBS2, LAMTOR1, AKAP6) at higher coverage depths (Supplementary Table 5). These mutations are unlikely to play an important role in invasion, since they were acquired after the tumor cells escaped the basement membrane, during the expansion of the invasive carcinoma. However, in one patient (P8) we identified a few mutations (NCOA2, MMP8, RNF182, LTBP2) that were pre-existing at low frequencies and increased in frequency during invasion (Supplementary Table 4). These mutations included MMP8, which is a matrix metallopeptidase that plays a role in breaking down the extracellular matrix (Sarper et al., 2017), and LTBP2 that interacts with TGF-beta and to regulate cell adhesion (Vehvilainen et al., 2003).
We further investigated whether any of the concordant mutations showed large changes in mutation frequencies by constructing tumor-purity normalized line plots (Figure 7). This analysis showed that there were only minor changes in mutation frequencies during invasion in five patients (Figure 7A), while the other five patients had at least one mutation with a large (>0.5) frequency change (Figure 7B). From these data 7 mutations were identified that underwent large (>0.5) mutation frequency changes during invasion, including MEGF9 in P1 (19% to 100%), NPY4R in P3 (48% to 100%), AHDC1 in P5 (33% to 100%) and 4 mutations in P8 (Supplementary Table 4). However, most patients (P1, P3, P5) had only a single concordant mutation that underwent a large frequency shift during invasion, with the exception of P8.
Figure 7. Mutation Frequencies and Clonal Dynamics During Invasion.

Purity adjusted mutation frequencies and clonal subpopulations and frequencies inferred from exome data. (A) Patients with minor changes in mutation and clonal frequencies. (B) Patients with large mutation or clonal frequency changes during invasion. The left panels show purity-adjusted nonsynonymous mutation frequencies for the in situ and invasive regions. Lines in grey indicate mutations with minor frequency changes, while lines in pink show large frequency changes (>0.5) between the in situ and invasive regions. Mutations in dark grey indicate driver mutations, while mutations in blue are in-situ and red are invasive-specific. Right panels indicate clonal subpopulations and frequencies inferred by PyClone2 and CITUP, with lines indicating different clonal subpopulations.
To infer clonal dynamics during invasion, we applied PyClone2 (Roth et al., 2014) and CITUP (Malikic et al., 2015) to cluster mutation frequencies and estimated clonal subpopulations after purity and copy number normalization (Figure 7). This analysis identified 2–5 major subpopulations in each patient, which was higher than the number of subpopulations detected by single cell copy number profiling. Several tumors that were found to be monoclonal by single cell copy number profiling (P2, P3, P7 and P9) showed 2–5 subpopulations based on the inferred mutation clusters. This data suggests there was ongoing mutational evolution in the ducts after copy number evolution, leading to further subclonal diversification that occurred prior to invasion into the adjacent tissues. While some of the clonal frequencies shifted during invasion (Figure 7B), the total number of subpopulations estimated from exome mutations remained consistent in most patients.
Discussion
In this study we developed a spatially-resolved single cell DNA sequencing method and applied it to study genome evolution during invasion in 10 early stage breast cancer patients. Our study reports three important findings. First, we show that genome evolution occurs within the ducts, before the tumor cells escape the basement membrane. Second, our data suggest that all subclones in the ducts arise from a single initiating cell, as evidenced by many shared truncal mutations and CNAs. Third, our data shows that one or more clones co-migrated through the basement membrane into the adjacent tissues to establish the invasive tumor mass. We refer to this model as multiclonal invasion, to distinguish it from the evolutionary bottleneck or independent lineage models that have previously been proposed for invasion in DCIS (Supplementary Figure 6). Consistent with our model, a study using flow-sorting and single cell copy number profiling in a single DCIS patient also reported evidence that multiple clones crossed the basement membrane (Martelotto et al., 2017).
Our model contrasts with previous work that has posited that genomic evolution during invasion occurs via a population bottleneck (Hernandez et al., 2012; Heselmeyer-Haddad et al., 2012; Sakr et al., 2014), or through independent cell lineages (Foschini et al., 2013). In our data we show that the same subclones were present in both the in situ and invasive regions in all 10 patients, with no additional CNA events that were acquired during invasion and few invasive-specific mutations. These data suggest that a single clone was not selected during invasion through an evolutionary bottleneck. Furthermore, our data does not support an independent lineage model (Miron et al., 2010; Sontag and Axelrod, 2005), since we identified a large number of shared truncal mutations and CNAs in all tumor cells, suggesting that a field effect did not give rise to two different clones that formed the in situ and invasive regions independently.
The total number of clonal subpopulations we identified is similar to previous reports in invasive ductal carcinomas (Gao et al., 2016; Navin et al., 2010; Shah et al., 2012; Yates et al., 2015) and is consistent with a punctuated model of copy number evolution, in which short bursts of genome instability give rise to multiple clones that stably expand to form the tumor mass (Gao et al., 2016; Navin et al., 2011). However, previous studies could not resolve whether the punctuated bursts of genomic instability occurred within the ducts, or subsequently, during the expansion of the invasive tumor mass, the former of which our data strongly support. Although the mechanisms of punctuated copy number evolution remain unknown, we speculate that telomere crisis (Chin et al., 2004) is a plausible model. Interestingly, our data also show that most somatic mutations, including driver mutations in TP53 and PIK3CA, were acquired in the ducts prior to invasion, at the earliest stages of tumor progression.
The co-migration of multiple clones into the invasive regions raises interesting questions, by suggesting that invasion occurs either through: 1) the complete breakdown of the basement membrane and random escape of tumor clones into the adjacent tissues, or alternatively through 2) the cooperation of tumor clones that collectively breakdown the basement membrane. The latter may occur through mutualistic interactions between tumor clones, or through commensalism, in which a single leader clone breaks through the basement membrane and clears the path for subsequent follower clones to escape. Understanding these clonal interactions during invasion will require further functional studies using in vivo systems.
This study has a few notable limitations. First, the cohort size was limited to 10 patients, and thus we cannot exclude the possibility that some early breast cancer patients follow alternative evolutionary models, particularly in low-grade tumors. Second, we profiled a limited number of cells in each patient, which may lead to sampling bias. To investigate this question, we calculated posterior saturation curves (Gao et al., 2016) which suggest that we sampled sufficient cells to detect the major tumor subpopulations in most patients (Supplementary Figure 7). Third, our study did not investigate epigenetic modifications or stromal cell types, the latter of which may also modulate the ability of the tumor cells to invade the surrounding tissues (Hu et al., 2008; Sharma et al., 2010). These represent important future directions that can be addressed with single cell RNA and epigenomic profiling methods.
TSCS and other spatially-resolved single cell sequencing methods hold great potential for opening up new avenues of investigation in early stage cancers. Of particular interest will be the analysis of tumor initiation and invasion in early stage cancers with well-defined histopathologies, such as colorectal adenomas, lobular carcinoma in situ (LCIS), prostatic intraepithelial neoplasias (PIN) and pancreatic intraepithelial neoplasias (PanINs). In these cancers, spatial resolution can provide new insights into the context, organization and migration of tumor clones, as they escape the basement membranes and invade the surrounding tissues. These studies will begin to shed light onto the enigmatic question of why some premalignant cancers remain indolent for the lifetime of the patient, while others progress to invasive disease and ultimately cause morbidity in patients.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Nicholas Navin (nnavin@mdanderson.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ten frozen synchronous DCIS-IDC tissue from women with paired normal adjacent breast tissues were obtained from the UT MD Anderson Cancer Center Breast Tissue Bank. The patient ages ranged from 36–77 years and is indicated in Supplementary Table 1. Frozen tissues were selected based on the presence of both in situ and invasive legions, or classification as synchronous DCIS-IDC and availability of paired normal adjacent breast tissues. ER and PR status of <1% was determined by IHC; while, HER2 status was defined through FISH analysis using a CEP-17 centromere control probe (ratio of Her2/CEP17 < 2.2) and were scored separately for the in situ and invasive regions. Five of the ten samples were classified as triple-negative based on negative staining for ER, PR and HER2. Receptor status and clinical parameters such as age, stage, grade, and number of cell collected per region are provided in Supplementary Table 1. The study was approved by the IRB at the MD Anderson Cancer Center.
METHOD DETAILS
Frozen Tissue Section Staining
Frozen tumors were divided into 1–2mm tissue blocks and mounted on OCT Compound (Tissue-Tek, Cat# 25608-930) without embedding the tissue that was used for sectioning. Mounted tumor tissue was sectioned using Thermo Scientific CryoStar (NX70) or Leica Cryostat (cm3050S) at −23°C to −27°C. Sections 12 and 14μm thick were placed on untreated PEN-membrane slides (Carl Zeiss Microscopy, Cat# 415190-9041-001). Standard procedures for Harris’ Alum Hematoxylin (VWR Cat#638A-71) and Eosin Y (VWR Cat#588X-75) staining were used for staining. Tissue sections were scanned using the AxioCam iCC 1 on the PALM MicroBeam at 10x magnification prior to collection.
Single Cell Isolation by Laser-Catapulting
Single cells were identified in the in situ and invasive regions of the tissue sections and selected by histology (size, shape, location to nearest duct). PALM Robo wizard (Carl Zeiss) was used to optimize the UV cutting parameters. The optimal energy for laser-catapulting single cells was set between 20–25 delta in order to reduce DNA fragmentation and increase collection efficiency. Delta settings below 15 resulted in frequent cell transfer failures by laser catapulting. Brightfield images were collected before and after capture of each single cell, along with parameters used for UV cutting and catapulting.
Single Cell Whole Genome Amplification
Single cells were laser-catapulted into 8-strips of 0.2 ml PCR tube caps containing 10μl of lysis solution from Sigma-Aldrich GenomePlex© WGA4 kit (cat# WGA4-50RXN) in a 96-cell manifold with robotic automation. After capture, the cells and lysis buffer were spun down at 12,000 rpm. Single cell DNA was amplified using Degenerative-Oligonucleotide-Primer PCR (DOP-PCR) following the Single Nucleus Sequencing (SNS) protocol as previously described (Baslan et al., 2012; Navin et al., 2011). For quality control (QC), WGA DNA size distributions were determined through electrophoresis and only samples with fragment sizes >300bp were selected and purified (Genesee Cat # 11-303). Purified WGA DNA was measured on a Qubit 2.0 Fluorometer (Fisher Cat#Q32854) and samples containing > 200ng of DNA were selected for library construction and next-generation sequencing.
Single Cell Barcoded Library Construction
Single cell amplified DOP-PCR products that passed QC were sonicated to 250bp using the S220 acoustic sonicator (Covaris). Following sonication, TA-ligation based Illumina libraries were prepared as previously described (Gao et al., 2016a). Alterations to this protocol included increased ligation time at 20°C for 30 minutes and PCR amplification cycles were adjusted according to input DNA (8 cycles for 1ug, 9 cycles for 500ng, and 10 cycles for 200ng). The insert size distributions of pooled multiplexed libraries were measured using the Bioanalyzer 2100 or Tape Station (Agilent). Multiplexed libraries were sequenced for 76 cycles using single-end or paired-end flow cells lanes on the HiSeq2000 or HiSeq4000 systems (Illumina, Inc.).
Bulk Frozen Tissue Microdissection
Frozen tissue sections were fixed and H&E stained as described above. Whole-tissue sections on slides were scanned and marked as in situ, invasive, or stroma. Tissue was collected using a Laser-Capture-Microdissection (LCM) on the PALM System (Carl Zeiss). The pathologist reviewed adjacent 6μm H&E stained tissue sections to verify the in situ and invasive regions prior to LCM. Tissue regions containing thousands of cells were cut by a UV laser (settings of 72–81 delta) and catapulted (setting of 50–100 delta) into 0.2 mL adhesive PCR tube caps (Item #: Zeiss 415190-9181-000 or 415190-9191-000). DNA was isolated using the QIAamp DNA Micro Kit (QIAGEN Cat# 56304) according to manufacturer’s instructions, with one modification: the samples were incubated at 56°C overnight. DNA concentration was measured on Qubit 2.0. Fresh frozen adjacent normal tissue was also processed in parallel; DNA was isolated using the DNeasy Blood & Tissue Kit (QIAGEN Cat# 69506). DNA concentrations were quantified by Qubit 2.0.–Normal DNA was isolated using the QIAGEN DNeasy protocol (Cat # 69506).
Exome Library Construction and Sequencing
Exome libraries were constructed from DNA isolated by LCM from in situ and invasive regions, in addition to matched normal tissues. DNA was fragmented to 250 bp using the Covaris Sonicator and purified by Zymo DNA Clean & Concentrator Column Kit (Genesee Cat # 11-303 or 11-306) according to manufacturer’s instructions. Barcoded next-generation sequencing libraries were constructed using the NEBNext end repair (NEB, E6050L), dA-tailing module (NEB, E6053L) and quick ligation module (NEB, E6056L). Libraries were PCR amplified with NEBNext HiFi 2x PCRmix (NEB, M0541L). Capture reactions were quantified using Qubit 2.0 Fluorometer and measured by quantitative PCR using the KAPA Library Quantification Kit (KAPA Biosystems, KK4835). Exome captures were performed using Nimblegen’s SeqCap EZ Exome V2 kit (Roche, 05860482001) and sequenced on a 100 paired-end flowcell on the Illumina HiSeq4000 system.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single Cell Copy Number Calculations
Multiplexed single-cell FASTQ files corresponding to the single cell samples were deconvoluted using 1 mismatch of the 6pb barcodes. The deconvoluted FASTQ files were aligned to hg19 (NCBS build 36) using Bowtie 2 (2.1.0) alignment software. The aligned reads were converted from SAM files to BAM files, then sorted using SAMtools (0.1.16). PCR duplicates were marked and removed using SAMtools. The sequencing data was processed following the ‘variable binning’ pipeline (Baslan et al., 2012; Baslan et al., 2015). Briefly, reads were aligned to the human genome HG19 using Bowtie2 and counted in variable bins at a genomic resolution of 220kb. Unique normalized read counts were segmented using the circular binary segmentation (CBS) method from R Bioconductor ‘DNAcopy’ package (Shah et al., 2006) followed by MergeLevels to join adjacent segments with non-significant differences in segmented ratios. The parameters used for CBS segmentation were alpha=0.0001 and undo.prune=0.05. Default parameters were used for MergeLevels, which removed erroneous chromosome breakpoints. Data was filtering with more than 100 break points or identified as noise with the R package for Density-based spatial clustering of applications (Dudik et al., 2015; Martin Ester, 1996; Yue et al., 2004) with noise ‘dbscan’ (v1.1-1) (Piekenbrock, 2017). We used this package to determine technical noise within the copy number profiles. We examined the plots to find the elbow and recorded this value for selecting the eps to filter data using the ‘dbscan’ package. Using this number, dbscan determined which single cell samples exhibited to much technical noise and filtered approximately 20% of the total datasets for each patient.
Identification of Subclones from CNA Data
The optimal ‘k’ (number of clusters between 1–15) was determined using the R-package ‘cluster’, clusGap function (K.max=15, B = 100, FUNcluster = kmeans). The maxSE (method=“firstSEmax”) was used to select the best number of clusters using k-means clustering. The original method (Maechler and Hornik, 2012) used the location of the first f() values which is less than or equal to the first local maximum minus the standard error factor times the standard error function and within the range of the function of standard error maximum 1 S.E (Tibshirani, 2001). After this, a k-means matrix was calculated using 250 original start sites for k+1. The matrix was sampled by start sites. We used ward.D2 clustering to generate the genetic trees based on the k-means matrix. The tree was cut into k clusters to determine the number of clones. The internal Pearson and Spearman correlation of the samples within each cluster was calculated. Most of cells with technical noise were removed in the previous filtering steps, however in a few cases, we identified additional cells with an internal correlation of Pearson and Spearman of less than 0.2, which were excluded from further analysis.
Multi-dimensional-Scaling Analysis
MDS plots were constructed in R using the single cell genotype binary matrix with columns as single cells and rows as mutations. Multidimensional Scaling was performed using the following command: cmdscale(x, eig=TRUE, k=2).
Calculation of the Subclonal Diversity Index
To calculate the subpopulation diversity index for each tumor, we performed hierarchical clustering of copy number data to cluster the aneuploid tumor cells into 1–5 major groups (‘species’) based on Euclidean distances. From this grouping we calculated the proportion (p) of cells that belong to each distinct group. The subpopulation diversity index is then calculated as Shannon Index: Dc = −Σi(pi×lnpi), where larger values representing higher subclonal diversity within the tumor.
Spatial Image Data Processing
The XY spatial coordinates of each cell from the LCM tissue maps were extracted from the elements in the whole tissue scan at 10X magnification from each tissue section. Since multiple tissue sections were often used for collection we estimated the Z-axis by sequential sections that were cut at 12–14 microns. In cases where tissue sections had different orientations, we rotated the spatial coordinates and transposed the coordinate values. We also collected brightfield images at 63X magnification before and after laser-catapulting, to confirm that single cells were isolated without adjacent material from neighboring cells. Additionally, these images were used to validate that the individual cells were collected from in situ and invasive regions, or stroma. Following the genomic analysis of clonal subpopulations, single cells were colored by clonal genotypes in the whole-tissue scanned images, in addition to ducts that were false-colored and enumerated.
Mapping Spatial Coordinates and Genomic Data
Spatial trees were constructed using Euclidean distance between cell coordinates and clustered with the hclust function using “ward.D2” linkage in R. Cells of the same tumor from different sample vials were given an artificial distance to buffer samples in which the distance between the regions of the tissue sections was unknown. The genetic trees from copy number profiles were constructed as described above. The genetic and spatial trees were mapped using Tanglegram version 1.5.2 of the dendextend package in R. Tanglegrams were untangled to minimize artificial branch crossing by first testing 100 random shuffles then by local stepwise untangling.
Exome Data Processing and Analysis
Sequence reads in FASTQ files corresponding to the regional and normal samples were aligned to the hg19 using the Bowtie 2 alignment software. Samtools (0.1.16) converted SAM files to compressed BAM files and sorted BAM files by coordinate. Duplicate PCR reads were removed with Picard. GATK was used to detect variants and generate a multi-cell VCF file. GATK was also used to recalibrate variant quality scores. We ran GATK with default parameters for depth (maximum read coverage = 250x). Germline SNPs were filtered out that were identified in the matched normal tissue samples. Mutations with less than 20X depth or less than 5 variant reads were filtered from the VCF4 file. Variant annotation was performed on the VCF4 file using ANNOVAR (Wang et al., 2010). Cancer genes were annotated using the 413 genes compiled from multiple databases including the Cancer Gene Census (Futreal et al., 2004), The Cancer Gene Atlas Project (TCGA), and the NCI cancer gene index (Sophic Systems Alliance Inc., Biomax Informatics A.G) used in previous publications (Gao et al., 2016b; Wang et al., 2014).
Targeted Deep Sequencing of PCR Amplicons
The invasive specific mutations were validated by targeted deep-amplicon sequencing. The primers were designed using Primer 3 (bioinfo.ut.ee/primer3-0.4.0/), with five base pairs upstream and downstream from the SNP location used as a target. The amplicon size range was limited to 100–225bp (Supplemental Table 6). DNA isolated by LCM from invasive or in situ regions was used for PCR. The amplicons from different regions were pooled in equimolar amounts and sequencing libraries were constructed using NEBNext® DNA library Prep enzymes (NEB, #E6050L, E6053L, E6056L/M0202L, and M0541 for end-repair, 3′ adenylation, ligation and PCR amplification) according to manufacturer’s instructions. Following ligation, DNA underwent a negative and positive selection with Ampure XP beads (Beckman Coulter, #A63881), 0.7× and 0.15× respectively, prior to PCR amplification. Final library concentrations were measured using the Qubit 2.0 Fluorometer. Samples were diluted to 10nM and then sequenced on the MiSeq system (Illumina, 150 paired-end) to obtain a target coverage depth of >100,000X.
Detection of Rare Mutations in Amplicon Data
Statistical significance of observed variants was calculated using deepSNV version 1.16.0, which detects variants assuming a beta-binomial model (Gerstung et al., 2012). To estimate the over dispersion parameter of the model, data from the targeted sites plus flanking regions of 20bp on either side were used. DeepSNV was used to calculate p values for the null hypothesis that the targeted variant was equally frequent in primary tumor and control using separate one-tailed likelihood ratio tests for each strand orientation, and combining the p-values using Fisher’s method.
Saturation Analysis to Estimate Cell Numbers
To estimate whether we sequenced sufficient numbers of cells to discover the major tumor subpopulations in both in situ and invasive regions, we performed a post hoc saturation analysis as previously described (Gao et al., 2016). We defined the total number of subpopulations and the fractions of each subpopulation in both the in situ and invasive regions using the experimental single cell copy number data for each patient. We then calculated the accumulative multinomial distribution probability of observing at least 2 tumor cells in each subpopulation, given the numbers of cells sequenced in our experiments. Since we only consider the tumor cells subpopulations, we did not restrict the number of normal cells to be observed. The same calculations were performed for both the in situ and invasive regions and then pooled together with weighted averages using the post hoc fractions to obtain the total number of cells per patient. Only one monoclonal tumor, MP7 was excluded from this analysis since there was only a single clone detected with no normal diploid cells.
DATA AND SOFTWARE AVAILABILITY
Single cell copy number and exome LCM data are deposited in NCBI Sequence Read Archive under accession SRP116771.
Supplementary Material
Highlight.
Development of a spatially-resolved single cell genome sequencing method.
In DCIS-IDC breast cancer, genomic evolution occurred prior to invasion.
Invasion involved the co-migration of multiple clones into the adjacent tissues.
Acknowledgments
This work was supported by grants to N.N from the Lefkofsky Family Foundation, NCI (1RO1CA169244-01), American Cancer Society (129098-RSG-16-092-01-TBG) and Chan-Zuckerberg Initiative (HCA-A-1704-01668). N.N. is a T.C. Hsu Endowed Scholar, AAAS Wachtel Scholar, Andrew Sabin Family Fellow and Jack & Beverly Randall Innovator. AKC is supported by a Rosalie B. Hite Fellowship in Cancer Research. This work was also supported by the MD Anderson Sequencing and Microarray Core Facility (CA016672). We thank Hank Adams, Louis Ramagli, Erika Thompson, Hongli Tang, Alexander Davis and Jake Leighton for their help. We are also grateful to Sohrab Shah and his group for support using the Timescape software.
Footnotes
AUTHOR CONTRIBUTIONS
AKC performed experiments, data analysis and wrote the manuscript. AS, TC, TM and RG performed data analysis. AL, WP and ES performed experiments. FMB and MEE provided clinical samples and interpreted the data. NN designed the study, performed data analysis and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110–1122. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allred DC. Ductal carcinoma in situ: terminology, classification, and natural history. J Natl Cancer Inst Monogr. 2010;2010:134–138. doi: 10.1093/jncimonographs/lgq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allred DC, Wu Y, Mao S, Nagtegaal ID, Lee S, Perou CM, Mohsin SK, O’Connell P, Tsimelzon A, Medina D. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin Cancer Res. 2008;14:370–378. doi: 10.1158/1078-0432.CCR-07-1127. [DOI] [PubMed] [Google Scholar]
- Andl CD, Mizushima T, Oyama K, Bowser M, Nakagawa H, Rustgi AK. EGFR-induced cell migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1227–1237. doi: 10.1152/ajpgi.00253.2004. [DOI] [PubMed] [Google Scholar]
- Baslan T, Kendall J, Rodgers L, Cox H, Riggs M, Stepansky A, Troge J, Ravi K, Esposito D, Lakshmi B, et al. Genome-wide copy number analysis of single cells. Nat Protoc. 2012;7:1024–1041. doi: 10.1038/nprot.2012.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K, de Solorzano CO, Knowles D, Jones A, Chou W, Rodriguez EG, Kuo WL, Ljung BM, Chew K, Myambo K, et al. In situ analyses of genome instability in breast cancer. Nat Genet. 2004;36:984–988. doi: 10.1038/ng1409. [DOI] [PubMed] [Google Scholar]
- Cowell CF, Weigelt B, Sakr RA, Ng CK, Hicks J, King TA, Reis-Filho JS. Progression from ductal carcinoma in situ to invasive breast cancer: revisited. Mol Oncol. 2013;7:859–869. doi: 10.1016/j.molonc.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Malhotra L, Dickerson R, Chaffee S, Sen CK, Roy S. Laser capture microdissection: Big data from small samples. Histol Histopathol. 2015;30:1255–1269. doi: 10.14670/HH-11-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foschini MP, Morandi L, Leonardi E, Flamminio F, Ishikawa Y, Masetti R, Eusebi V. Genetic clonal mapping of in situ and invasive ductal carcinoma indicates the field cancerization phenomenon in the breast. Hum Pathol. 2013;44:1310–1319. doi: 10.1016/j.humpath.2012.09.022. [DOI] [PubMed] [Google Scholar]
- Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, Tsai PC, Casasent A, Waters J, Zhang H, et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet. 2016 doi: 10.1038/ng.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Kim C, Sei E, Foukakis T, Crosetto N, Chan LK, Srinivasan M, Zhang H, Meric-Bernstam F, Navin N. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nature communications. 2017;8:228. doi: 10.1038/s41467-017-00244-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–233. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstung M, Papaemmanuil E, Campbell PJ. Subclonal variant calling with multiple samples and prior knowledge. Bioinformatics. 2014;30:1198–1204. doi: 10.1093/bioinformatics/btt750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindberg RV, Yee-Greenbaum JL, McConnell MJ, Novotny M, O’Shaughnessy AL, Lambert GM, Arauzo-Bravo MJ, Lee J, Fishman M, Robbins GE, et al. RNA-sequencing from single nuclei. Proc Natl Acad Sci U S A. 2013;110:19802–19807. doi: 10.1073/pnas.1319700110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L, Wilkerson PM, Lambros MB, Campion-Flora A, Rodrigues DN, Gauthier A, Cabral C, Pawar V, Mackay A, A’Hern R, et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J Pathol. 2012;227:42–52. doi: 10.1002/path.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heselmeyer-Haddad K, Berroa Garcia LY, Bradley A, Ortiz-Melendez C, Lee WJ, Christensen R, Prindiville SA, Calzone KA, Soballe PW, Hu Y, et al. Single-cell genetic analysis of ductal carcinoma in situ and invasive breast cancer reveals enormous tumor heterogeneity yet conserved genomic imbalances and gain of MYC during progression. Am J Pathol. 2012;181:1807–1822. doi: 10.1016/j.ajpath.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y, et al. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13:394–406. doi: 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CE, Gorringe KL, Thompson ER, Opeskin K, Boyle SE, Wang Y, Hill P, Mann GB, Campbell IG. Identification of copy number alterations associated with the progression of DCIS to invasive ductal carcinoma. Breast Cancer Res Treat. 2012;133:889–898. doi: 10.1007/s10549-011-1835-1. [DOI] [PubMed] [Google Scholar]
- Kim SY, Jung SH, Kim MS, Baek IP, Lee SH, Kim TM, Chung YJ, Lee SH. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget. 2015;6:7597–7607. doi: 10.18632/oncotarget.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroigard AB, Larsen MJ, Laenkholm AV, Knoop AS, Jensen JD, Bak M, Mollenhauer J, Kruse TA, Thomassen M. Clonal expansion and linear genome evolution through breast cancer progression from pre-invasive stages to asynchronous metastasis. Oncotarget. 2015;6:5634–5649. doi: 10.18632/oncotarget.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung ML, Wang Y, Waters J, Navin NE. SNES: single nucleus exome sequencing. Genome Biol. 2015;16:55. doi: 10.1186/s13059-015-0616-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz-Gordillo P, Francis JM, Zhang CZ, Shalek AK, Satija R, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 2014;32:479–484. doi: 10.1038/nbt.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikic S, McPherson AW, Donmez N, Sahinalp CS. Clonality inference in multiple tumor samples using phylogeny. Bioinformatics. 2015;31:1349–1356. doi: 10.1093/bioinformatics/btv003. [DOI] [PubMed] [Google Scholar]
- Martelotto LG, Baslan T, Kendall J, Geyer FC, Burke KA, Spraggon L, Piscuoglio S, Chadalavada K, Nanjangud G, Ng CK, et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nat Med. 2017;23:376–385. doi: 10.1038/nm.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson A, Roth A, Laks E, Masud T, Bashashati A, Zhang AW, Ha G, Biele J, Yap D, Wan A, et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat Genet. 2016 doi: 10.1038/ng.3573. [DOI] [PubMed] [Google Scholar]
- Miron A, Varadi M, Carrasco D, Li H, Luongo L, Kim HJ, Park SY, Cho EY, Lewis G, Kehoe S, et al. PIK3CA mutations in in situ and invasive breast carcinomas. Cancer Res. 2010;70:5674–5678. doi: 10.1158/0008-5472.CAN-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Krasnitz A, Rodgers L, Cook K, Meth J, Kendall J, Riggs M, Eberling Y, Troge J, Grubor V, et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburger DE, Kashef-Haghighi D, Weng Z, Salari R, Sweeney RT, Brunner AL, Zhu SX, Guo X, Varma S, Troxell ML, et al. Genome evolution during progression to breast cancer. Genome Res. 2013;23:1097–1108. doi: 10.1101/gr.151670.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard-Cote A, Shah SP. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11:396–398. doi: 10.1038/nmeth.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakr RA, Weigelt B, Chandarlapaty S, Andrade VP, Guerini-Rocco E, Giri D, Ng CK, Cowell CF, Rosen N, Reis-Filho JS, et al. PI3K pathway activation in high-grade ductal carcinoma in situ--implications for progression to invasive breast carcinoma. Clin Cancer Res. 2014;20:2326–2337. doi: 10.1158/1078-0432.CCR-13-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarper M, Allen MD, Gomm J, Haywood L, Decock J, Thirkettle S, Ustaoglu A, Sarker SJ, Marshall J, Edwards DR, et al. Loss of MMP-8 in ductal carcinoma in situ (DCIS)-associated myoepithelial cells contributes to tumour promotion through altered adhesive and proteolytic function. Breast Cancer Res. 2017;19:33. doi: 10.1186/s13058-017-0822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scornavacca C, Zickmann F, Huson DH. Tanglegrams for rooted phylogenetic trees and networks. Bioinformatics. 2011;27:i248–256. doi: 10.1093/bioinformatics/btr210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Beck AH, Webster JA, Espinosa I, Montgomery K, Varma S, van de Rijn M, Jensen KC, West RB. Analysis of stromal signatures in the tumor microenvironment of ductal carcinoma in situ. Breast Cancer Res Treat. 2010;123:397–404. doi: 10.1007/s10549-009-0654-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Nielsen CB, Chan FC, McPherson A, Roth A, Farahani H, Machev D, Steif A, Shah SP. E-scape: interactive visualization of single-cell phylogenetics and cancer evolution. Nat Methods. 2017;14:549–550. doi: 10.1038/nmeth.4303. [DOI] [PubMed] [Google Scholar]
- Song Y, Tian T, Shi Y, Liu W, Zou Y, Khajvand T, Wang S, Zhu Z, Yang C. Enrichment and single-cell analysis of circulating tumor cells. Chem Sci. 2017;8:1736–1751. doi: 10.1039/c6sc04671a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag L, Axelrod DE. Evaluation of pathways for progression of heterogeneous breast tumors. J Theor Biol. 2005;232:179–189. doi: 10.1016/j.jtbi.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Suh SS, Yoo JY, Cui R, Kaur B, Huebner K, Lee TK, Aqeilan RI, Croce CM. FHIT suppresses epithelial-mesenchymal transition (EMT) and metastasis in lung cancer through modulation of microRNAs. PLoS genetics. 2014;10:e1004652. doi: 10.1371/journal.pgen.1004652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandewoestyne M, Goossens K, Burvenich C, Van Soom A, Peelman L, Deforce D. Laser capture microdissection: should an ultraviolet or infrared laser be used? Anal Biochem. 2013;439:88–98. doi: 10.1016/j.ab.2013.04.023. [DOI] [PubMed] [Google Scholar]
- Vehvilainen P, Hyytiainen M, Keski-Oja J. Latent transforming growth factor-beta-binding protein 2 is an adhesion protein for melanoma cells. J Biol Chem. 2003;278:24705–24713. doi: 10.1074/jbc.M212953200. [DOI] [PubMed] [Google Scholar]
- Virnig BA, Tuttle TM, Shamliyan T, Kane RL. Ductal carcinoma in situ of the breast: a systematic review of incidence, treatment, and outcomes. J Natl Cancer Inst. 2010;102:170–178. doi: 10.1093/jnci/djp482. [DOI] [PubMed] [Google Scholar]
- Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148:886–895. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, Aas T, Alexandrov LB, Larsimont D, Davies H, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338:1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.