

Abstract
Epigenetic regulation of activity-induced gene expression involves multiple levels of molecular interaction, including histone and DNA modifications, as well as mechanisms of DNA repair. Here we demonstrate that the genome-wide deposition of Inhibitor of growth family member 1 (ING1), which is a central epigenetic regulatory protein, is dynamically regulated in response to activity in primary cortical neurons. ING1 knockdown leads to decreased expression of genes related to synaptic plasticity, including the regulatory subunit of calcineurin, Ppp3r1. In addition, ING1 binding at a site upstream of the transcription start site (TSS) of Ppp3r1 depends on yet another group of neuroepigenetic regulatory proteins, the Piwi-like family, which are also involved in DNA repair. These findings provide new insight into a novel mode of activity-induced gene expression, which involves the interaction between different epigenetic regulatory mechanisms traditionally associated with gene repression and DNA repair.
Keywords: Epigenetics, Gene regulation, Neuronal activation, ING1, Piwi-like proteins, DNA repair
Introduction
Epigenetic mechanisms, including DNA methylation, ATP-dependent chromatin remodelling factors, post-transcriptional modification of histone proteins, and various non-coding RNA pathways, are now recognised as fundamental for the regulation of activity-induced neuronal gene expression. While some of these epigenetic pathways are relatively well known, others remain poorly characterised, and ongoing research continues to reveal novel epigenetic regulators in post-mitotic neurons that were previously believed to respond only to stress, DNA damage, or replication error. One such mechanism is the recent discovery that DNA double-strand breaks are induced in an experience-dependent manner, and associated with transcriptional activity (Suberbielle et al., 2013; Bunch et al., 2015; Madabhushi et al., 2015); however, the precise mechanism by which this process interacts with canonical epigenetic mechanisms remains to be determined.
Inhibitor of growth family member 1 (ING1), initially characterised as a tumour suppressor gene involved in cell cycle arrest and apoptosis and associated with several forms of cancer (Nouman et al., 2003; Ythier et al., 2008), has recently been identified as a central epigenetic regulator. ING1 acts as both a transcriptional activator and repressor through interactions with DNA and a multitude of epigenetic regulatory proteins, including histone modifications, by modulating the activity of the mSIN3A/HDAC1 transcriptional repression complex (Skowyra et al., 2001; Binda et al., 2008). It is also a reader for the histone mark H3K4me3, and promotes active DNA demethylation at H3K4me3 sites by recruiting the DNA damage response factor GADD45α (Schäfer et al., 2013). In addition, ING1 contributes to transcriptional regulation of the miRNA-processing gene Dgcr8, and loss of ING1 has been shown to lead to the dysregulation of miRNA expression in mouse fibroblasts (Gómez- Cabello et al., 2010). Finally, there is substantial evidence for a fundamental role for ING1 in DNA repair. ING1 is upregulated in response to DNA damage (Cheung Jr et al., 2000; Cheung et al., 2001; Ceruti et al., 2013) and mutant forms of ING1 derived from tumours are unable to promote DNA repair (Ceruti et al., 2013). Moreover, overexpression of ING1 enhances the repair of UV-damaged DNA in a p53-dependent manner (Cheung et al., 2001). Whether ING1 is also involved in DNA repair in differentiated neurons, and represents an underlying mechanism of activity-induced gene expression, is currently unknown.
Another epigenetic pathway that is also involved in DNA repair and transcriptional regulation is the Piwi pathway. Piwi-like proteins are an evolutionarily conserved subset of the Argonaute family with similarity to Drosophila PIWI (Juliano et al., 2011; Iwasaki et al., 2015), and are required for the biogenesis and activity of Piwi-interacting RNAs (piRNAs). piRNAs comprise a distinct class of small non-coding RNAs which have several characteristic features, including occurring in genomic clusters, a tendency to have uracil as the first base, a length of 26–31nt, and 2’-O-methylation on the terminal nucleotide (Iwasaki et al., 2015). To date, it is thought that the dominant function of piRNAs is to protect the genome by controlling the activity of transposons (Juliano et al., 2011; Iwasaki et al., 2015), and although they have a broad role in stem cell maintenance and reproduction in many species, their activity in mammals occurs predominantly within the male germline (Iwasaki et al., 2015). As indicated, Piwi proteins have been linked to DNA repair; Yin et al. have reported that Piwil2 lies upstream of the DNA damage response pathway, and is also activated by DNA damage in fibroblasts. Preventing Piwil2 activation blocks histone H3 acetylation and subsequent chromatin relaxation that is necessary for DNA repair, resulting in a DNA repair defect (Yin et al., 2011). Furthermore, the Piwi pathway has recently been implicated in neuronal function and brain organisation (Rajasethupathy et al., 2012; Zhao et al., 2015; Nandi et al., 2016; Viljetic et al., 2017) with numerous reports of piRNA expression in the mammalian brain (Lee et al., 2011; Saxena et al., 2012; Ghosheh et al., 2016; Nandi et al., 2016).
Given the potential overlap between ING1 and the Piwi pathway as epigenetic regulators, we investigated whether ING1 is involved in activity-induced gene expression and whether Piwi plays a role in this process. To model neuronal activation, we cultured primary neurons from embryonic mouse cortex and used potassium chloride to induce depolarisation. This well-established tissue culture model activates all of the neuronal subtypes present in primary cortical neuronal cultures, and recapitulates many of the cellular and transcriptional programs that occur in neurons in response to physiological stimulation occurring in vivo (Martinowich et al., 2003; Flavell et al., 2008; Greer & Greenberg, 2008; Moon et al., 2009; Bouvier et al., 2010; Connolly et al., 2010; Grubb et al., 2010; Kim et al., 2010; Day et al., 2013; Malik et al., 2014; Chand et al., 2015; Chen et al., 2015, Liang et al., 2015; Ataman et al., 2016; Grassi et al., 2017).
Our findings indicate that ING1 accumulates at regulatory elements of genes involved in synaptic function in response to KCl stimulation in primary neurons. We show that ING1 accumulation is involved in activity-dependent regulation of several genes relevant to neuronal plasticity and function, and that the function of ING1 in regulating a specific target gene, Ppp3r1, can be modulated by the Piwi pathway. This is the first evidence of an interaction between these two conserved epigenetic pathways, revealing a novel mode of activity-induced epigenetic regulation of gene expression in primary cortical neurons.
Experimental Procedures
Adeno-associated virus construction and packaging
For knockdown of Piwil1 and Piwil2, pAAV2-mu6-shRNAs were constructed as described previously (Hommel et al., 2003), with slight modifications. In brief, siRNAs were designed using siDirect version 2.0 (Naito et al., 2004), which were synthesised along with their antisense strand separated by a loop sequence to form a hairpin. Forward and reverse strands of shRNAs were annealed and cloned into the pAAV-MCS vector, containing the mU6 promoter and eGFP, in between XbaI and SapI restriction sites.
HEK293T cells were grown to 80% confluence in 150mm dishes. Lipofectamine was used to transfect cells with the plasmids AAV1, AAV2, pFdelta6, and pAAV-MCS. Transfected cells were cultured for a further 4 days, then collected and lysed by 3 freeze-thaw cycles. The cell lysate was filtered, treated with Benzonase and AAV particles were concentrated by ultracentrifugation. Concentrated viral pellets were resuspended in PBS and snap-frozen. Three AAVs were produced, one carrying a scrambled control shRNA with no specificity to any known mouse transcript and one targeting each of Piwil1 and Piwil2. The combination of packaging plasmids AAV1 and AAV2 in a 1:1 ratio produced approximately a 1:1 mixture of AAV serotypes 1 and 2, which have a complementary transduction pattern in neurons.
Lentivirus construction and packaging
Lentivirus was packaged according to our previously published protocol (Lin et al., 2011). Briefly, HEK293T cells were grown to 70% confluence in triple-layer flasks. Lipofectamine was used to transfect cells with the plasmids pMDG, pRSV-rev and pMDLg/pRRE and the transfer vector (gene-specific shRNA cloned into FG12). Transfected cells were cultured for 48 hours, after which the culture medium was collected, clarified, filtered, and lentivirus particles concentrated by ultracentrifugation. Concentrated viral pellets were resuspended in PBS and snap-frozen. Four lentiviruses were produced: one carrying a control shRNA with no specificity to any known mouse transcript, and the other three targeting ING1 (ING1 shRNA 1, 2, 3).
Culture of primary mouse cortical neurons
Experiments involving animals were approved by the Animal Ethics Committee of the University of Queensland. Pregnant female C57BL/6 mice were euthanised at E16 and embryos immediately collected into ice-cold PBS. Embryonic cortices were dissected and enzymatically dissociated with 5U/mL papain at 37°C for 20 minutes. Papain was washed out with neuronal culture medium (Neurobasal medium supplemented with 1% GlutaMAX and 2% B-27). The cells were then mechanically dissociated, counted, and plated at a density of 750–1000 cells/mm2 in culture dishes coated with poly-D-ornithine. Neuronal cultures were maintained in an incubator at 37°C with 5% CO2. The culture medium was replaced 24–48 hours after preparation of the cultures, and thereafter cultures were fed every third day by replacement of 50% of the culture medium. To knock down expression of genes of interest, cultured neurons at 2–3 days in vitro were exposed to lentivirus or AAV for 12 hours, followed by replacement of the culture medium. To investigate activity-dependent gene regulation, cultured neurons at 7–10 days in vitro were depolarised by the addition of 20mM KCl to the culture medium for three hours. The cells were collected immediately for molecular analysis following KCl exposure.
Culture of primary mouse embryonic fibroblasts
Pregnant female C57BL/6 mice were euthanised at E16 and embryos immediately collected into ice-cold PBS. Embryos were decapitated and the thoracic and abdominal organs discarded; remaining tissue was minced and stored at 4°C overnight in 3 volumes of 0.05% trypsin. Following incubation at 37°C for 45 minutes, tissue was washed and cells were mechanically dissociated, counted, and plated into uncoated T75 flasks at 25% density in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum. Cells were maintained in an incubator at 37°C with 5% CO2; cultures were passaged 3 times before use, at which time cultures were free from cells of non-fibroblast morphology.
To induce ING1 activity, MEFs at passage 3 were treated with ultraviolet light to damage genomic DNA. Growth medium was removed and cells washed once with Hank’s buffered salt solution, then placed without liquid into a Stratalinker (Stratagene) and irradiated with 40 J/m2 (0.87 J/m2/s at 254nm). Following irradiation, growth medium was replaced and cells were returned to the incubator for approximately 1–3 hours before collection for molecular analysis.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed according to our previously published protocol (Wei et al., 2012). Briefly, neurons were fixed in 1% formaldehyde to establish protein-DNA crosslinks. Crosslinked neuronal lysates were sheared by sonication in ChIP lysis buffer to generate chromatin fragments with an average length of 300–500 bp. Immunoprecipitation was carried out using an antibody specific to ING1 (Cell Signaling Technologies #14625) overnight at 4°C; antibody-protein- DNA complexes were isolated by incubation with Protein G Dynabeads (Thermo) for 1h at 4°C, followed by three washes in low-salt buffer, and three washes in high-salt buffer. The precipitated protein-DNA complexes were resuspended in ChIP dilution buffer, then incubated for 4 hours at 65°C to reverse formaldehyde cross-linking. Protein was removed by Proteinase K digestion and the ING1-associated DNA fragments were recovered by phenol-chloroform extraction and ethanol precipitation.
ChIP-Seq and sequencing data analysis
ING1-associated DNA isolated by ChIP was used to generate paired-end (PE) sequencing libraries following the protocol of the Illumina DNA kit. Libraries were sequenced using the Illumina HiSeq 2500 sequencing platform, with the read length of 126bp*2. Image processing and sequence data extraction were performed using the standard Illumina Genome Analyzer software and CASAVA (v1.8.2) software. Cutadapt (v1.8.1) was used to cut the adaptor sequences as well as low quality nucleotides at both ends (with the option of “-q 20,20”). Reads were aligned to the mouse reference genome (mm9) using BWA (v0.6.2) (Li & Durbin, 2009). Samtools (v0.1.17) (Li et al., 2009) was then used to convert “SAM” files to “BAM” files, sort and index the “BAM” files, and remove duplicate reads. Reads with low mapping quality (<20) or which were not properly paired-end aligned to the reference genome were excluded from the downstream peak calling analysis. MACS (v1.4.2) (Zhang et al., 2008) was used to call peaks for each sample with the parameter setting “-f BAM –keep-dup= all –nomodel –shiftsize 100 –g mm –p 1e-5 –bdg”. Peak summits identified by MACS from all samples were collected to generate a list of potential binding sites. Peak summits located within 600bp of each other were grouped together using a custom PERL script as these nearby peaks may represent the same binding site. Peaks that were detected by MACS in at least 2 biological replicates of either KCl+ or KCl- samples were defined as potential ING1 binding sites and only these sites were used for downstream analysis in this study. Genes near the ING1 binding sites (i.e. +/−10 kb regions from the peak summits) in KCl+ samples were extracted for GO enrichment analysis using DAVID (version 6.8) (Huang et al., 2009a, 2009b). We then categorised all potential binding sites into six groups, including promoter (defined as 2kb upstream of transcription start sites in known genes), 5’ UTR, CDS, intron, 3’ UTR, and intergenic. To determine the ING1 occupancy between KCl+ and KClsamples, a custom PERL script was applied to count the number of fragments (hereafter referred to as ‘counts’) that cover the peak summit in each sample. Each pair of properly aligned PE reads covering the peak summit represents one count. The total counts in each sample were normalised to 10 million for the normalised coverage plot between conditions.
RNA isolation and reverse transcription
To extract RNA, cells were lysed with Nucleozol (Macherey-Nagel) directly in the culture dish and RNA was isolated following the manufacturer’s instructions. Reverse transcription was carried out using the QuantiTect kit (QIAGEN) using the provided RT Primer Mix and following the manufacturer’s instructions.
Quantitative PCR for ChIP samples
Quantitative PCR reactions were prepared in duplicate, using 2x SYBR master mix (Qiagen), 500μM of each primer, and 1μL of template DNA purified as described previously after chromatin immunoprecipitation. Reactions were run on the Rotor-Gene Q platform and results were analysed using percent input method. Data was analysed using Student’s t-test or one-way ANOVA, as appropriate. Primer sequences for loci of interest are shown in Table 1. Control primer sets were obtained from Active Motif: the positive control primer set (Actb-2; #71017) amplifies a region of the beta-actin gene and the negative control (#71011) is located within a gene desert.
Table 1.
List of primer and shRNA sequences used in this study.
| shRNA sequences | |
| ING1-sh1 | CCGCAATTTAGAAACTACAA |
| ING1-sh2 | CGGCTGTGACAACGACGAA |
| ING1-sh3 | AACCATGTTGAGTCCTGCCAA |
| Universal control | GCGCGATAGCGCTAATAAT |
| Piwil1-sh | TGAAGTCTAGGACAGTCTCGC |
| Piwil2-sh | AGTTGTTTCAGCGGAATATCC |
| Scramble control | GGAATTTAGAAACCCGGCTCCAC |
| qPCR primers | |
| qING1 F | CGAATAACAAGCGGTCCAGG |
| qING1 R | GTTGCACAGACAGTACGTGG |
| qPPP3r1 F | GAAGGAGTGTCTCAGTTCAGTG |
| qPPP3r1 R | ACGAAAAGCAAACCTCAACTTCT |
| qPiwil1 F | CAGCAACCTGGGTACATCCC |
| qPiwil1 R | CCAAGGTCATGGAAGTCTCGG |
| qPiwil2 F | TTGGCCTCAAGCTCCTAGAC |
| qPiwil2 R | GAACATGGACACCAAACCTACA |
| qPGK F | TGCACGCTTCAAAAGCGCACG |
| qPGK R | AAGTCCACCCTCATCACGACCC |
| qPlcb F | GGACAAGTGCTAGAATGTTCCC |
| qPlcb R | GAAGCCGATATTCACCAGATCC |
| qCdk19 F | GCACCTACGGGCATGTCTAC |
| qCdk19 R | TCCTGTGCCTTCGATTTGCTT |
| qCacnb2 F | GCCCATCCGATTCAGATGTGT |
| qCacnb2 R | GGTCCGAACTGCAAATGCAA |
| qLrrc7 F | CAAGCTCTACGGAAACTAAGCA |
| qLrrc7 R | ACACCGTTTTTACTGATGTCGAG |
| ChIP primers | |
| ING1-Ppp3r1 ChIP F | GCCAGGAAGAACACCACAGA |
| ING1-Ppp3r1 ChIP R | TCCTTAAGAGGGACGGGGTT |
| ING1-Plcb4 ChIP F | TCTTGCAGAGGAGGGATGGA |
| ING1-Plcb4 ChIP R | CCCAATCCCACCACAGTTGT |
| ING1-Cdk19 ChIP F | CCTCCCTCTGCTGTGTGAAG |
| ING1-Cdk19 ChIP R | GGCCTACACCAAGAGCCTTT |
| ING1-Cacnb2 ChIP F | CTACGTGGAGCCAAAGGAAG |
| ING1-Cacnb2 ChIP R | TTAGATTTATGCCGGCTTCG |
| ING1-Lrrc7 ChIP F | CATTTTGGGCTCCAGACATC |
| ING1-Lrrc7 ChIP R | GAGCCTGAAACAGGTTCTGC |
Quantitative PCR for gene expression
Quantitative PCR reactions were prepared in duplicate, in a 10μL reaction volume, using 2X SYBR master mix (Qiagen), 500μM of each primer, and 1μL per reaction of a cDNA sample (the cDNA dilution factor varied according to target abundance). Reactions were run on the Rotor-Gene Q platform and results were analysed using the delta-delta-CT method, normalised to the reference gene Pgk (phosphoglycerate kinase). Data was analysed using Student’s t-test or one-way ANOVA as appropriate.
Results
Neural activity leads to a pronounced change in the genome-wide deposition of ING1
To determine whether ING1 functions as an activity-dependent epigenetic regulator in neurons, we first validated an antibody (Cell Signaling Technologies #14625) for chromatin immunoprecipitation (ChIP). ING1 ChIP was performed on mouse embryonic fibroblasts with and without ING1 induction via UV irradiation, and qPCR detected successful enrichment of the known ING1 target Mageb2 (Schäfer et al., 2013) from the UV-treated MEFs using this antibody (Figure 1A).
Figure 1. Genome-wide profiling reveals the dynamic landscape of ING1 binding sites in mouse primary cortical neurons following KCl-induced depolarisation.

The antibody detects accumulation of ING1 at the promoter of the known ING1 target Mageb1 in UV-irradiated MEFs (n=4) compared to untreated MEFs (n=4) and was therefore considered suitable for ChIP (A). Distribution analysis of ING1 ChIP-seq peaks identified in untreated (KCl-) (B) or 3 hours KCl-stimulated (KCl+) (C) primary cortical neurons, shown as a percentage of reads corresponding to each genomic region. (D) ING1 ChIP-seq peak counts in KCl- (n=3) and KCl+ (n=3) groups. Blue and red represent KCl- and KCl+, respectively. (E) Shown are the top 10 enriched GO terms associated with genes within ING1 binding regions in KCl+ samples (FDR<0.05).
We next performed ChIP-seq, using the same antibody, to examine the genome-wide distribution of ING1 in primary cortical neurons in response to KCl-induced depolarisation (n=3 per group). A total of 275 binding sites for ING1 were identified and found to be predominantly localised to intergenic regions (59.86% and 59.03% in KCl+/-, respectively) (Figure 1B and 1C). Of the 275 ING1 binding sites, 116 sites (42.18%) showed specific KCl-induced binding (Figure 1D). Therefore, although the global distribution pattern of ING1 binding did not change in terms of broad genomic regions where ING1 is deposited, the qualitative interaction with the genome was altered, with significantly more and different genomic loci targeted following KCl-induced depolarisation (Figure 1B, 1C and 1D; Appendix A). Intriguingly, in response to neuronal stimulation, ING1 binding was predominantly located (~53%) within 10kb upstream of the TSS of the nearest gene, representing a regulatory region including the promoter and potentially a cis-acting enhancer. Another ~39% of ING1 binding sites overlapped with coding genes (including binding sites in the exon, intron, 5’ or 3’ UTR); only 8% of binding sites were located more than 10kb away from any protein coding gene. This strongly suggests a role for ING1 binding in the regulation of gene expression. We performed a gene ontology analysis on the genes associated with ING1 occupancy, which revealed that most of the candidates are highly associated with synaptic functions, including synaptic transmission, calcium channel activity, ion binding and transport (Figure 1E).
ING1 binding is associated with gene induction by KCl stimulation
To validate our ChIP-seq data, we selected 5 genomic loci that showed an increase in ING1 occupancy subsequent to neuronal activation (Figure 2). We plotted raw ING1 ChIP-seq data (read counts) to demonstrate an increase in sequencing coverage (corresponding to increased ING1 occupancy) at these five loci: Protein phosphatase 3 regulatory subunit B, alpha (Ppp3r1), Phospholipase C Beta 4 (Plcb4), Cyclin Dependent Kinase 19 (Cdk19), Calcium Voltage-Gated Channel Auxiliary Subunit Beta 2 (Cacnb2) and Leucine Rich Repeat Containing 7 (Lrrc7) (Figure 2A, 2D, 2G, 2J and 2M). These binding profiles show that ING1 potentially interacts with intron, promoter, coding sequence (CDS) and intergenic regions separately. The KCl-dependent increase in ING1 occupancy at these loci was validated using ING1 ChIP-qPCR (Figure 2B, 2E, 2H, 2K and 2N). Given that the pattern of ING1 deposition was strongly suggestive of a role for ING1 in gene regulation, we used qPCR to measure mRNA expression of these five genes with and without KCl treatment. Expression of all five genes associated with ING1 binding sites was significantly induced by 3 hours of KCl treatment, indicating that ING1 binding is associated with nearby gene activation during stimulation with KCl (Figure 2C, 2F, 2I, 2L and 2O). Taken together, these findings show that ING1 binding is activity-induced, and support the hypothesis that ING1 is acting as a dynamic epigenetic regulator involved in the activity-dependent induction of gene expression in post-mitotic neurons.
Figure 2. ING1 ChIP-seq plots and validation of selected genes highlighting dynamic peaks in the presence (KCl+) or absence (KCl-) of KCl-induced depolarisation.

Dynamic peaks were identified within the URE of Ppp3r1 (A), the intron of Plcb4 (D), the promoter of Cdk19 (G), the CDS of Cacnb2 (J), and the intergenic region in front of Lrrc7 (M) respectively; the arrows indicate the peak regions. Y-axis represents mean of normalised coverage. Increased ING1 occupancy at each locus was confirmed by ChIP-qPCR (B, E, H, K and N) under KCl depolarisation (20mM, 3h) (n=5, Student’s t-test, *p <0.05; **p<0.01). mRNA expression of Ppp3r1 (C), Plcb4 (F), Cdk19 (I), Cacnb2 (L) and Lrrc7 (O) are inducible by KCl-induced depolarisation in primary cortical neurons (20mM, 3h) (n=5, Student’s t-test, *p <0.05, **p<0.01). Data represent mean ± SEM.
Knockdown of Ing1 blocks induction of Ppp3r1 by KCl stimulation
Ppp3r1 encodes a regulatory subunit of calcineurin, a protein phosphatase that is involved in calcium signalling in neurons (Baumgärtel & Mansuy, 2012). Disruption of calcineurin activity has severe effects on memory in animal models (Malleret et al., 2001; Zeng et al., 2001; Cottrell et al., 2013), making Ppp3r1 an interesting candidate gene in the context of neuronal function. We therefore selected this gene for further investigation. The binding of ING1 at the Ppp3r1 locus in primary cortical neurons occurs at a site 6580bp upstream of the TSS (upstream regulatory element/URE). This binding site is specific to KCl treated neurons and is one of the predominantly intergenic ING1 binding sites identified by ChIP-Seq. ING1 occupancy at this site shows an obvious increase in response to KCl stimulation, with higher normalised read coverage in KCl+ samples than that in KCl-samples (Figure 2A).
To investigate the functional role of ING1 in the epigenetic regulation of gene expression at this locus, we designed lentiviral short-hairpin RNA (shRNA) constructs (Figure 3A) based on our previously published protocols (Lin et al., 2011). Three different Ing1 shRNAs (Table 1) were validated by Western blot and all produced a significant knockdown of ING1 at the protein level, compared to a universal shRNA control which has no specificity to any known mouse transcripts. Furthermore, treatment of primary cortical neurons with a cocktail of all 3 Ing1 shRNAs blocked neural activity-induced ING1 binding at the Ppp3r1 locus (Figure 3B).
Figure 3. Activity-dependent ING1 occupancy is associated with increased Ppp3r1 mRNA expression in primary cortical neurons.

(A) shRNA targeting Ing1 inhibits ING1 protein expression in primary cortical neurons. Quantitative Western blot analysis confirms that each of the three Ing1 shRNAs tested (sh1, sh2, sh3) knocks down expression of ING1 compared to the universal shRNA control (Con) (n=3, Student’s t-test, *p <0.05). (B) Ing1 knockdown eliminated activity-induced ING1 occupancy at the Ppp3r1 upstream regulatory region after 3 hours of KCl-induced depolarisation (n=4, one-way ANOVA, F (3, 12) = 11.09, ***p<0.001). (C) Expression of Ppp3r1 mRNA is inhibited by Ing1 knockdown (n=4–5, one-way ANOVA, F(3, 9) = 4.446, *p <0.05). ‘Ing1 shRNA’ = cocktail of sh1, sh2 and sh3-expressing lentiviruses. Data represent mean ± SEM.
Stimulation of primary cortical neurons with KCl results in a robust upregulation of Ppp3r1 mRNA, which reached approximately 160% of its baseline expression (Figure 2C). In neurons treated with lentivirus carrying shRNAs targeting Ing1, the induction of Ppp3r1 by KCl-induced depolarisation was entirely abolished, revealing the importance of ING1 deposition at the Ppp3r1 locus for the activity-dependent induction of the Ppp3r1 gene (Figure 3C). Taken together, these observations indicate that the KCl-mediated increase in Ppp3r1 mRNA expression is due, at least in part, to increased ING1 binding at the Ppp3r1 URE region.
Piwi proteins are expressed in primary cortical neurons
As part of our exploration of the crosstalk between potential epigenetic regulatory mechanisms and neuronal gene expression, we also examined the expression of the Piwi-like genes Piwil1 and Piwil2 in primary cortical neurons. We found that Piwil1 and Piwil2 are both expressed in primary cortical neurons (Figure 4A, B). This is consistent with the observation of neuronal expression of piRNAs in several previously published studies (Lee et al., 2011; Saxena et al., 2012; Ghosheh et al., 2016; Nandi et al., 2016; Roy et al., 2017). In addition, we found that Piwil1 and Piwil2 are both induced by KCl-induced depolarisation. After 3 hours of KCl exposure, Piwil1 was upregulated threefold, while Piwil2 expression doubled (Figure 3A, B). These data indicate the presence of low-level, but potentially biologically relevant, expression of components of the Piwi pathway in mouse neurons, and support a link between the Piwi pathway and neuronal activity.
Figure 4. Regulation of Ppp3r1 mRNA expression by ING1 is mediated by the Piwi pathway.

Piwil1 (A) and Piwil2 (B) mRNA is detected in primary cortical neurons at baseline; mRNA of both Piwil1 and Piwil2 significantly increases following 3 hours of KCl stimulation (n=4–6, Student’s t-test, *p<0.05, **p <0.01). Expression of Piwil1 (C) and Piwil2 (D) is significantly reduced in primary cortical neurons 4 days after treatment with the respective shRNA-expressing AAVs, compared to neurons treated with a non-targeting control. (n=4, Student’s t-test, **p <0.01, ***p <0.001). (E) Ppp3r1 mRNA induction by KCl-induced depolarisation (20mM, 3h) in primary cortical neurons is inhibited by simultaneous knockdown of Piwil1 and Piwil2 (n=3–5, one way ANOVA, F (3, 10) = 8.101, **p <0.01). (F) Simultaneous knockdown of Piwil1 and Piwil2 eliminates activity-induced ING1 occupancy at the Ppp3r1 upstream regulatory region under KCl-induced depolarisation (n=3–4, one way ANOVA, F (3, 9) = 4.556, *p <0.05). Con = scrambled non-targeting shRNA. Data represent mean ± SEM.
Knockdown of Piwil1 and Piwil2 blocks Ppp3r1 mRNA expression and prevents ING1 binding to the Ppp3r1 upstream regulatory region
Given the evidence that the Piwi pathway lies upstream of DNA repair, in which ING1 is involved as an epigenetic regulator, we next asked whether the effect of disruption of the Piwi pathway on Ppp3r1 expression was similar to that of Ing1 knockdown. Given that little is known about the function of either Piwil1 or Piwil2 in neurons, we chose to investigate the function of the pathway as a whole by simultaneously knocking down both Piwil1 and Piwil2. We used adeno-associated viral vectors (AAVs) carrying shRNA sequences directed against Piwil1, Piwil2, and as a negative control, a scrambled sequence with no targets in the mouse genome (Table 1.) We confirmed that AAVs carrying shRNA against Piwil1 and Piwil2 significantly knocked down expression of target mRNAs in primary cortical neurons four days after application (Figure 4C, 4D.)
We next examined the expression of Ppp3r1 mRNA in primary cortical neurons treated with both Piwil1-shRNA and Piwil2-shRNA AAVs. Simultaneous knockdown of Piwil1 and Piwil2 blocked the neural activity-induced upregulation of Ppp3r1 mRNA expression (Figure 4E). Surprisingly, we also found that disruption of the Piwi pathway abolishes the activity-dependent Ppp3r1 induction by modulating ING1 binding dynamics. ChIP-qPCR revealed that knockdown of Piwil1 and Piwil2 eliminated the KCl-dependent increase in ING1 occupancy at the Ppp3r1 URE (Figure 4F), thereby blocking the ING1-mediated upregulation of this gene in response to neuronal activation.
Activating histone marks accumulate at the Ppp3r1 locus in response to neuronal activation
After discovering the activity-induced increase in ING1 binding at the Ppp3r1 URE, we looked for evidence of change in the local chromatin landscape that is associated with ING1 binding and Ppp3r1 mRNA expression. Given the previously identified interaction between ING1 and histone modification (Skowyra et al., 2001; Feng et al., 2002; Binda et al., 2008), we reasoned that ING1 may interact with the local chromatin landscape in an activity-induced manner, thereby providing a further link between ING1 binding and the epigenetic regulation of gene expression. To explore this possibility, we used ChIP-qPCR to interrogate the chromatin environment surrounding the ING1 binding site upstream of the Ppp3r1 gene for increases in four permissive histone marks: H3K4me3, H3K4me1, H3K9ac and H3K14ac.
To begin, we validated and optimised antibodies for specific chromatin marks (Fig 5A and 5B). Subsequently, using ChIP-qPCR, we found that there was no effect of KCl stimulation on the occupancy of H3K4me1 or H3K14ac at the Ppp3r1 URE (Figure 6A and 6B); these two histone marks are typically present within active enhancer regions (Wang et al., 2008; Heintzman et al., 2009). However, ChIP-qPCR revealed increased occupancy (~2.5-fold) of H3K4me3 at the Ppp3r1 URE after 3 hours of KCl stimulation (Figure 6C). H3K4me3 is primarily associated with active promoters and supports the initiation of transcription (Guenther et al., 2007; Wang et al., 2008), but also occurs within active enhancer regions (Chen et al., 2015). Additionally, we detected approximately 1.5-fold enrichment of H3K9ac at the Ppp3r1 URE in the KCl-stimulated cells (Figure 6D); this histone mark is broadly associated with active promoters (Wang et al., 2008; Heintzman et al., 2009) and is associated with euchromatin at enhancers and other sites (Schebesta et al., 2007). The increase in occupancy of the activating marks H3K4me3 and H3K9ac at the Ppp3r1 URE following KCl stimulation indicates that the chromatin state at this site has become more permissive for transcription, and for binding of transcription factors and regulatory elements. This finding is consistent with both the increased ING1 binding and enhanced Ppp3r1 expression observed under depolarising conditions.
Figure 5. Positive and negative controls for H3K4me3 and H3K9ac ChIP assays.
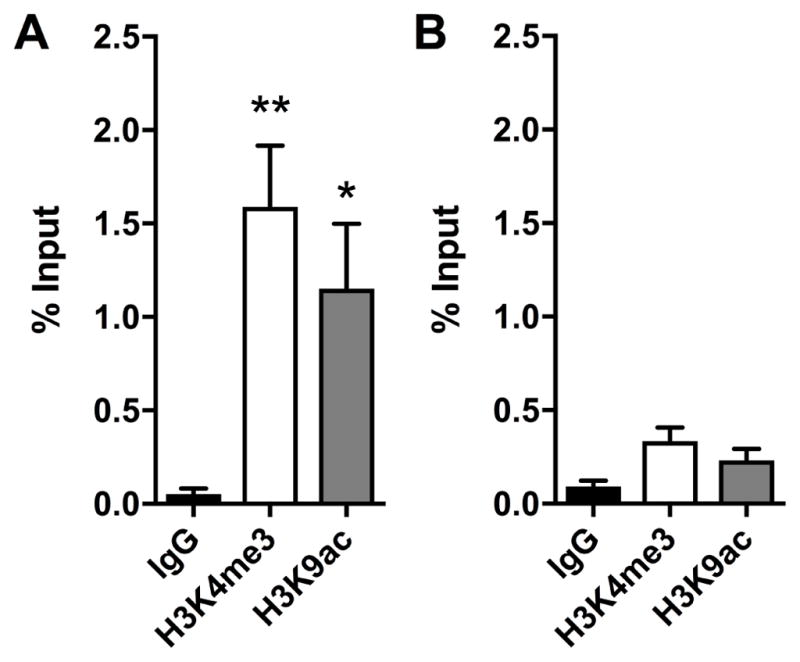
H3K4me3 and H3K9ac occupancy at a known binding locus Actb (A), and at a gene desert locus (B) at basal condition in primary cortical neurons. (n=3–4, one way ANOVA, F (2, 9) = 8.178, *p <0.05, **p <0.01). Data represent mean ± SEM.
Figure 6. ING1 binding at the Ppp3r1 locus is associated with activating histone marks, which are abolished following knockdown of Piwil1 and Piwil2.

There is a significant increase in H3K3me3 (A) and H3K9ac (B) occupancy at the Ppp3r1 upstream regulatory region under KCl-induced depolarisation (20mM, 3h), but no significant difference in H3K4me1 (C) and H3K14ac (D) occupancy. Simultaneous knockdown of Piwil1 and Piwil2 prevents accumulation of both H3K4me3 (A) and H3K9ac (B) at this locus. (n=3–4, Student’s t-test, *p <0.05, ***p <0.001). Data represent mean ±SEM.
We next asked whether disruption of the Piwi pathway, which is known to have a role in chromatin remodelling, could perturb the development of this permissive chromatin state in response to neuronal activity. Interestingly, we observed that simultaneous knockdown of Piwil1 and Piwil2 prevented accumulation of either H3K4me3 or H3K9ac at the Ppp3r1 URE in response to KCl stimulation (Figure 6a, 6b), indicating that the Piwi pathway is upstream of histone modification at the Ppp3r1 locus in this system.
Discussion
In this study, we have demonstrated that 1) ING1, a protein with a wide range of epigenetic and regulatory functions, exhibits altered binding dynamics in response to neuronal activity in primary cortical neurons in vitro, 2) increased occupancy of ING1 at an upstream regulatory region near the Ppp3r1 gene is necessary for the activity-induced upregulation of Ppp3r1 mRNA expression, and 3) disruption of the Piwi pathway blocks the upregulation of Ppp3r1 by preventing ING1 binding at the Ppp3r1 URE. ING1 is a multifaceted epigenetic regulator, and many of the epigenetic processes to which it contributes are known to be important for neuronal function. Our results represent the first specific evidence that ING1 is involved in regulating activity-dependent gene expression in normal neurons.
The abundance of ING1 mRNA and protein is unchanged by KCl stimulation (data not shown), and the overall distribution of ING1 genomic binding sites is similar (Figure 1b and 1c). However, ING1 occupancy was markedly increased at specific sites, many of which are expected to regulate plasticity-related genes (Figure 1e), and all five candidate ING1 targets that we tested were upregulated by KCl stimulation (Figure 2). These data strongly imply a nuanced activity of ING1 in the transcriptional regulation of plasticity-related genes during neuronal activity. Also of note is that background ING1 binding is present at the candidate loci, including Ppp3r1, under basal conditions (Figure 2). However, while knockdown of Ing1 prevents upregulation of Ppp3r1 by KCl, it does not reduce the baseline expression of Ppp3r1 (Figure 3c). This suggests that the depleted pool of ING1 (~30%) remaining in the cells after Ing1 knockdown is sufficient to support at least some of the baseline functions of ING1, but insufficient to respond to neuronal activation. The unchanged abundance of ING1 during stimulation, and the normal transcription of Ppp3r1 in unstimulated cells despite Ing1 knockdown by ~70%, may imply that much of the ING1 protein present in the cell is not bound to DNA in the baseline state. This hypothesis could be investigated by genetic deletion of Ing1 to remove it from the cells completely.
ING1 is known to be involved in chromatin remodelling (Skowyra et al., 2001; Binda et al., 2008, Schäfer et al., 2013), and we considered the possibility that ING1 either causes or responds to an altered chromatin environment. We did not find an obvious enrichment of ING1 binding sites within a specific chromatin context when we overlaid the ING1 binding sites identified by this study with ENCODE datasets (Sloan et al., 2016) including H3K4me1, H3K4me3, H3K9ac, H3K27ac and DNase I. This is probably due to the different cell types of origin and state dependence of samples analysed as part of the ENCODE datasets, which are dissimilar at the regulatory and transcriptomic level from primary cortical neurons under activity-dependent conditions. To clarify the nature of interactions between ING1 and chromatin modifications, genome-wide histone modification profiles from basal and stimulated neurons are warranted.
Consistent with its role as a tumour suppressor, Ing1 is a DNA damage response gene (Cheung Jr et al., 2000; Cheung et al., 2001; Ceruti et al., 2013) that is involved in DNA repair through several pathways (Ceruti et al., 2013). It has recently been reported that DNA double-strand breaks occur in neurons in an experience-dependent manner (Suberbielle et al., 2013; Bellesi et al., 2016), and that DNA breaks occur at the promoters of neuronal early-response genes (Madabhushi et al., 2015) and other stimulus-inducible genes (Bunch et al., 2015) and are required for their transcription. These findings raise the possibility that proteins involved in the recognition of DNA damage and subsequent DNA repair could act as a mechanism to target epigenetic or transcriptional machinery to specific loci in response to experience, such as learning. This possibility led us to ask whether disruption of the Piwi pathway affects activity-dependent gene regulation in a similar way to knockdown of Ing1. In addition to the mounting evidence that the Piwi pathway is functional in neurons, other unexpected roles for this pathway in a range of somatic cell types have recently been revealed (Ross et al., 2014). For example, Yin et al. showed that Piwil2, which is normally silenced in fibroblasts, is transiently activated in response to DNA damage (Yin et al., 2011). This is normally followed by histone H3 acetylation (H3K9ac and H3K14ac) which results in chromatin relaxation, allowing DNA repair enzymes to access the DNA. In cells deficient in PIWIL2, histone acetylation and chromatin relaxation do not occur in response to DNA damage, resulting in defective DNA repair and revealing that PIWIL2 is necessary at the first stage of the DNA damage response.
We speculate that the Piwi pathway is upstream of ING1 during activity-dependent regulation of Ppp3r1 in primary cortical neurons. Specifically, it is known that DNA breakage occurs in response to neuronal activity (Suberbielle et al., 2013; Bellesi et al., 2016) and Piwi proteins are known to respond to DNA breakage by promoting chromatin relaxation (Yin et al., 2011). We observed that simultaneous knockdown of Piwil1 and Piwil2 prevents the accumulation of the permissive histone marks H3K4me3 and H3K9ac at the Ppp3r1 URE in response to KCl stimulation. One of these histone marks, H3K9ac, was previously found to increase in response to DNA damage in fibroblasts; this increase did not occur in PIWIL2−/− cells (Yin et al., 2011). Taken together, these observations strongly suggest that knockdown of Piwil1 and Piwil2 results in inappropriate chromatin structure which impedes access of ING1 at the Ppp3r1 promoter, resulting in the loss of both ING1 binding and Ppp3r1 induction that is observed in the Piwil1/Piwil2 knockdown neurons. The Piwi pathway is primarily involved in transcriptional and epigenetic silencing, and it is not currently clear how Piwilike proteins affect histone modifications. However, the Piwi pathway is known to be involved in chromatin remodelling in Drosophila (Pal-Bhadra et al., 2004; Brower-Toland et al., 2007; Huang et al., 2013), including one case where a specific piRNA drives a euchromatic state at a complementary locus (Yin & Lin, 2007).
It is also possible that Piwil1/Piwil2 knockdown has an effect on ING1-mediated Ppp3r1 expression through a less direct mechanism. Given that H3K4me3 is able to recruit ING1, it is possible that ING1 binding at the Ppp3r1 URE is driven by the accumulation of this epigenetic mark. If this is the case, any activity of the Piwi pathway which promotes accumulation of H3K4me3 at the Ppp3r1 URE could contribute to the loss of ING1 binding observed in neurons where Piwil1 and Piwil2 are knocked down. Several studies have detected expression of piRNAs in neurons and observed altered piRNA expression in response to various stimuli (Lee et al., 2011; Rajasethupathy et al., 2012; Saxena et al., 2012; Ghosheh et al., 2016; Nandi et al., 2016). The targets of virtually all of these small regulatory RNAs remain unidentified, but could certainly include chromatin-modifying enzymes, and other genes which may affect ING1 binding or Ppp3r1 expression in other ways. Another possibility is that Piwil1 and/or Piwil2 may be acting independently of piRNAs; this has been suggested by Viljetic et al. who found that although Piwil1 knockdown disrupts mouse corticogenesis, it has little effect on cortical piRNA expression (Viljetic et al., 2017).
The downstream target identified by this study, Ppp3r1, encodes a regulatory subunit of calcineurin, a calcium and calmodulin-dependent serine/threonine protein phosphatase. Calcineurin is one of the most abundant protein phosphatases in the brain, and modulates the function of other proteins in response to calcium signalling (Baumgärtel & Mansuy, 2012). Calcineurin has been extensively implicated in learning and memory (Malleret et al., 2001; Zeng et al., 2001; Lin et al., 2003; Havekes et al., 2006; Baumgärtel & Mansuy, 2012; Cottrell et al., 2013), schizophrenia (Gerber et al., 2003; Miyakawa et al., 2003; Eastwood et al., 2005) and depression (Seimandi et al., 2013; Yu et al., 2013). Altered expression or regulation of one of its subunits, as we observed in the present study, could therefore be expected to have broad implications for neuronal function. Calcineurin has also been implicated in Alzheimer’s disease due to its effects on Tau metabolism (Gong et al., 1994; Luo et al., 2008; Karch et al., 2013), with a genetic variant of Ppp3r1 being strongly associated with the rapid progression of Alzheimer’s disease in humans (Cruchaga et al., 2010; Peterson et al., 2014). This provides strong justification for further investigation of Ppp3r1 as a gene of potential relevance to learning, memory and human health.
In conclusion, we report that ING1 binding is dynamically regulated in neurons and is involved in the epigenetic regulation of neuronal gene expression. We also show that ING1 binding to at least one target regulatory region, a site upstream of the TSS of Ppp3r1, depends on neuronal Piwi-like proteins. These findings provide important new insight into the epigenetic regulation of activity-induced gene expression in neurons, which has relevance for understanding neural plasticity, learning and memory.
Highlights.
Neuronal activation alters genome-wide deposition of the epigenetic regulator ING1.
ING1 promotes gene expression in post-mitotic neurons.
PIWIL1 and PIWIL2 are upstream of ING1 in neuronal gene regulation.
Acknowledgments
This work was supported by grants from the National Institutes of Mental Health (1R01MH109588 to T.W.B.) and Australian National Health and Medical Research Council (GNT1062570 to T.W.B.) and postgraduate scholarships from the Westpac Bicentennial Foundation and the Australian Government Research Training Program (administered by The University of Queensland) to L.J.L. We thank Ms. Rowan Tweedale for helpful editing of this paper.
Footnotes
Author contributions: L.J.L., Q.Z., T.W.B and W.W. designed the experiments. N.K., A.K., X.L., C.D., S.L. and W.W. designed and assembled shRNA constructs. L.J.L., W.W., X.L., C.D., P.R.M., E.Z., and S.L. conducted experiments. Q.Z. and Y.W. analysed ChIP-seq data. L.J.L., Q.Z., and W.W. wrote the paper. All authors reviewed and edited the manuscript.
Conflicts of interest: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ataman B, Boulting GL, Harmin DA, Yang MG, Baker-Salisbury M, Yap EL, Malik AN, Mei K, Rubin AA, Spiegel I. Evolution of Osteocrin as an activity-regulated factor in the primate brain. Nature. 2016;539:242–247. doi: 10.1038/nature20111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgärtel K, Mansuy IM. Neural functions of calcineurin in synaptic plasticity and memory. Learning & Memory. 2012;19:375–384. doi: 10.1101/lm.027201.112. [DOI] [PubMed] [Google Scholar]
- Bartel DP, Sheng M, Lau LF, Greenberg ME. Growth factors and membrane depolarization activate distinct programs of early response gene expression: dissociation of fos and jun induction. Genes & Development. 1989;3:304–313. doi: 10.1101/gad.3.3.304. [DOI] [PubMed] [Google Scholar]
- Bellesi M, Bushey D, Chini M, Tononi G, Cirelli C. Contribution of sleep to the repair of neuronal DNA double-strand breaks: evidence from flies and mice. Scientific Reports. 2016;6 doi: 10.1038/srep36804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda O, Nassif C, Branton P. SIRT1 negatively regulates HDAC1-dependent transcriptional repression by the RBP1 family of proteins. Oncogene. 2008;27:3384–3392. doi: 10.1038/sj.onc.1211014. [DOI] [PubMed] [Google Scholar]
- Bouvier D, Tremblay ME, Riad M, Corera AT, Gingras D, Horn KE, Fotouhi M, Girard M, Murai KK, Kennedy TE, McPherson PS, Pasquale EB, Fon EA, Doucet G. EphA4 is localized in clathrin-coated and synaptic vesicles in adult mouse brain. Journal of Neurochemistry. 2010;113(1):153–165. doi: 10.1111/j.1471-4159.2010.06582.x. [DOI] [PubMed] [Google Scholar]
- Brower-Toland B, Findley SD, Jiang L, Liu L, Yin H, Dus M, Zhou P, Elgin SC, Lin H. Drosophila PIWI associates with chromatin and interacts directly with HP1a. Genes & development. 2007;21:2300–2311. doi: 10.1101/gad.1564307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch H, Lawney BP, Lin Y-F, Asaithamby A, Murshid A, Wang YE, Chen BP, Calderwood SK. Transcriptional elongation requires DNA break-induced signalling. Nature communications. 2015;6 doi: 10.1038/ncomms10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceruti JM, Ogara MF, Menéndez C, Palmero I, Cánepa ET. Inhibitor of growth 1 (ING1) acts at early steps of multiple DNA repair pathways. Molecular and cellular biochemistry. 2013;378:117–126. doi: 10.1007/s11010-013-1601-2. [DOI] [PubMed] [Google Scholar]
- Chand AN, Galliano E, Chesters RA, Grubb MS. A Distinct Subtype of Dopaminergic Interneuron Displays Inverted Structural Plasticity at the Axon Initial Segment. Journal of Neuroscience. 2015;35(4):1573–1590. doi: 10.1523/JNEUROSCI.3515-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nature genetics. 2015;47(10):1149–57. doi: 10.1038/ng.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhao X, Zhang M, Wei S. Nuclear respiratory factor-2α and adenosine triphosphate synapses in rat primary cortical neuron cultures: The key role of adenosine monophosphate-activated protein kinase. Molecular Medicine Reports. 2015;35(4):6323–6329. doi: 10.3892/mmr.2015.4140. [DOI] [PubMed] [Google Scholar]
- Cheung KJ, Jr, Bush J, Jia W, Li G. Expression of the novel tumour suppressor p33ING1 is independent of p53. British journal of cancer. 2000;83:1468–1472. doi: 10.1054/bjoc.2000.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Mitchell D, Lin P, Li G. The tumor suppressor candidate p33ING1 mediates repair of UV-damaged DNA. Cancer Research. 2001;61:4974–4977. [PubMed] [Google Scholar]
- Connolly S, Kingsbury TJ. Caffeine modulates CREB-dependent gene expression in developing cortical neurons. Biochemical and Biophysical Research Communication. 2010;397(2):152–156. doi: 10.1016/j.bbrc.2010.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell JR, Levenson JM, Kim SH, Gibson HE, Richardson KA, Sivula M, Li B, Ashford CJ, Heindl KA, Babcock RJ. Working memory impairment in calcineurin knock-out mice is associated with alterations in synaptic vesicle cycling and disruption of high-frequency synaptic and network activity in prefrontal cortex. Journal of Neuroscience. 2013;33:10938–10949. doi: 10.1523/JNEUROSCI.5362-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Kauwe JS, Mayo K, Spiegel N, Bertelsen S, Nowotny P, Shah AR, Abraham R, Hollingworth P, Harold D. SNPs associated with cerebrospinal fluid phospho-tau levels influence rate of decline in Alzheimer's disease. PLoS Genet. 2010;6:e1001101. doi: 10.1371/journal.pgen.1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Childs D, Guzman-Karlsson MC, Kibe M, Moulden J, Song E, Tahir A, Sweatt JD. DNA methylation regulates associative reward learning. Nature Neuroscience. 2013;16:1445–1452. doi: 10.1038/nn.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood SL, Burnet PW, Harrison PJ. Decreased hippocampal expression of the susceptibility gene PPP3CC and other calcineurin subunits in schizophrenia. Biological psychiatry. 2005;57:702–710. doi: 10.1016/j.biopsych.2004.12.029. [DOI] [PubMed] [Google Scholar]
- Feng X, Hara Y, Riabowol K. Different HATS of the ING1 gene family. Trends in cell biology. 2002;12:532–538. doi: 10.1016/s0962-8924(02)02391-7. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME. Genome-Wide Analysis of MEF2 Transcriptional Program Reveals Synaptic Target Genes and Neuronal Activity-Dependent Polyadenylation Site Selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber DJ, Hall D, Miyakawa T, Demars S, Gogos JA, Karayiorgou M, Tonegawa S. Evidence for association of schizophrenia with genetic variation in the 8p21. 3 gene, PPP3CC, encoding the calcineurin gamma subunit. Proceedings of the National Academy of Sciences. 2003;100:8993–8998. doi: 10.1073/pnas.1432927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosheh Y, Seridi L, Ryu T, Takahashi H, Orlando V, Carninci P, Ravasi T. Characterization of piRNAs across postnatal development in mouse brain. Scientific reports. 2016;6 doi: 10.1038/srep25039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Cabello D, Callejas S, Benguría A, Moreno A, Alonso J, Palmero I. Regulation of the MicroRNA Processor DGCR8 by the Tumor Suppressor ING1. Cancer Research. 2010;70:1866–1874. doi: 10.1158/0008-5472.CAN-09-2088. [DOI] [PubMed] [Google Scholar]
- Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Alzheimer's Disease Abnormally Phosphorylated τ Is Dephosphorylated by Protein Phosphatase-2B (Calcineurin) Journal of neurochemistry. 1994;62:803–806. doi: 10.1046/j.1471-4159.1994.62020803.x. [DOI] [PubMed] [Google Scholar]
- Grassi D, Franz H, Vezzali R, Bovio P, Heidrich S, Dehghanian F, Lagunas N, Belzung C, Krieglstein K, Vogel T. Neuronal Activity, TGFβ-Signaling and Unpredictable Chronic Stress Modulate Transcription of Gadd45 Family Members and DNA Methylation in the Hippocampus. Cerebral Cortex. 2017;27(8):4166–4181. doi: 10.1093/cercor/bhx095. [DOI] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465(7301):1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59:846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havekes R, Nijholt IM, Luiten PG, Van der Zee EA. Differential involvement of hippocampal calcineurin during learning and reversal learning in a Y-maze task. Learning & Memory. 2006;13:753–759. doi: 10.1101/lm.323606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommel JD, Sears RM, Georgescu D, Simmons DL, DiLeone RJ. Local gene knockdown in the brain using viral-mediated RNA interference. Nature medicine. 2003;9:1539–1544. doi: 10.1038/nm964. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009a;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009b;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XA, Yin H, Sweeney S, Raha D, Snyder M, Lin H. A major epigenetic programming mechanism guided by piRNAs. Developmental cell. 2013;24:502–516. doi: 10.1016/j.devcel.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki YW, Siomi MC, Siomi H. PIWI-interacting RNA: its biogenesis and functions. Annual review of biochemistry. 2015;84:405–433. doi: 10.1146/annurev-biochem-060614-034258. [DOI] [PubMed] [Google Scholar]
- Juliano C, Wang J, Lin H. Uniting germline and stem cells: the function of Piwi proteins and the piRNA pathway in diverse organisms. Annual review of genetics. 2011;45:447–469. doi: 10.1146/annurev-genet-110410-132541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo JY, Schaukowitch K, Farbiak L, Kilaru G, Kim TK. Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nature Neuroscience. 2016;19(1):75–83. doi: 10.1038/nn.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Jeng AT, Goate AM. Calcium phosphatase calcineurin influences tau metabolism. Neurobiology of aging. 2013;34:374–386. doi: 10.1016/j.neurobiolaging.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS, Haddad C, Xia Z, Greenberg ME. CREB Transcriptional Activity in Neurons Is Regulated by Multiple, Calcium-Specific Phosphorylation Events. Neuron. 2002;34(2):221–33. doi: 10.1016/s0896-6273(02)00655-4. [DOI] [PubMed] [Google Scholar]
- Lee EJ, Banerjee S, Zhou H, Jammalamadaka A, Arcila M, Manjunath B, Kosik KS. Identification of piRNAs in the central nervous system. Rna. 2011;17:1090–1099. doi: 10.1261/rna.2565011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Ye T, Zhou X, Lai KO, Fu AK, Ip NY. Cdk5 regulates activity-dependent gene expression and dendrite development. Journal of Neuroscience. 2015;35(45):15127–15134. doi: 10.1523/JNEUROSCI.1443-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Leu TH, Chang WC, Wang ST, Gean PW. Identification of calcineurin as a key signal in the extinction of fear memory. Journal of Neuroscience. 2003;23:1574–1579. doi: 10.1523/JNEUROSCI.23-05-01574.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Wei W, Coelho CM, Li X, Baker-Andresen D, Dudley K, Ratnu VS, Boskovic Z, Kobor MS, Sun YE, Bredy TW. The brain-specific microRNA miR-128b regulates the formation of fear-extinction memory. Nature Neuroscience. 2011;14:1115–1117. doi: 10.1038/nn.2891. [DOI] [PubMed] [Google Scholar]
- Luo J, Ma J, Yu DY, Bu F, Zhang W, Tu LH, Wei Q. Infusion of FK506, a specific inhibitor of calcineurin, induces potent tau hyperphosphorylation in mouse brain. Brain research bulletin. 2008;76:464–468. doi: 10.1016/j.brainresbull.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell. 2015;161:1592–1605. doi: 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, Greenberg ME. Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nature Neuroscience. 2014;17(10):1330–1339. doi: 10.1038/nn.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302(5646):890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell. 2001;104:675–686. doi: 10.1016/s0092-8674(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Leiter LM, Gerber DJ, Gainetdinov RR, Sotnikova TD, Zeng H, Caron MG, Tonegawa S. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proceedings of the National Academy of Sciences. 2003;100:8987–8992. doi: 10.1073/pnas.1432926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon IS, Cho SJ, Seog DH, Walikonis R. Neuronal activation increases the density of eukaryotic translation initiation factor 4E mRNA clusters in dendrites of cultured hippocampal neurons. Experimental & Molecular Medicine. 2009;41(8):601–610. doi: 10.3858/emm.2009.41.8.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y, Yamada T, Ui-Tei K, Morishita S, Saigo K. siDirect: highly effective, target-specific siRNA design software for mammalian RNA interference. Nucleic Acids Research. 2004;32:W124–W129. doi: 10.1093/nar/gkh442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi S, Chandramohan D, Fioriti L, Melnick AM, Hébert JM, Mason CE, Rajasethupathy P, Kandel ER. Roles for small noncoding RNAs in silencing of retrotransposons in the mammalian brain. Proceedings of the National Academy of Sciences. 2016;113:12697–12702. doi: 10.1073/pnas.1609287113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouman G, Anderson J, Lunec J, Angus B. The role of the tumour suppressor p33ING1b in human neoplasia. Journal of clinical pathology. 2003;56:491–496. doi: 10.1136/jcp.56.7.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal-Bhadra M, Leibovitch BA, Gandhi SG, Rao M, Bhadra U, Birchler JA, Elgin SC. Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science. 2004;303:669–672. doi: 10.1126/science.1092653. [DOI] [PubMed] [Google Scholar]
- Peterson D, Munger C, Crowley J, Corcoran C, Cruchaga C, Goate AM, Norton MC, Green RC, Munger RG, Breitner JC. Variants in PPP3R1 and MAPT are associated with more rapid functional decline in Alzheimer's disease: The Cache County Dementia Progression Study. Alzheimer's & Dementia. 2014;10:366–371. doi: 10.1016/j.jalz.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasethupathy P, Antonov I, Sheridan R, Frey S, Sander C, Tuschl T, Kandel ER. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell. 2012;149:693–707. doi: 10.1016/j.cell.2012.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RJ, Weiner MM, Lin H. PIWI proteins and PIWI-interacting RNAs in the soma. Nature. 2014;505:353–359. doi: 10.1038/nature12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy J, Sarkar A, Parida S, Ghosh Z, Mallick B. Small RNA sequencing revealed dysregulated piRNAs in Alzheimer's disease and their probable role in pathogenesis. Molecular BioSystems. 2017;13:565–576. doi: 10.1039/c6mb00699j. [DOI] [PubMed] [Google Scholar]
- Saxena A, Tang D, Carninci P. piRNAs warrant investigation in Rett Syndrome: an omics perspective. Disease markers. 2012;33:261–275. doi: 10.3233/DMA-2012-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A, Karaulanov E, Stapf U, Döderlein G, Niehrs C. Ing1 functions in DNA demethylation by directing Gadd45a to H3K4me3. Genes & development. 2013;27:261–273. doi: 10.1101/gad.186916.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schebesta A, McManus S, Salvagiotto G, Delogu A, Busslinger GA, Busslinger M. Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity. 2007;27:49–63. doi: 10.1016/j.immuni.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Seimandi M, Seyer P, Park CS, Vandermoere F, Chanrion B, Bockaert J, Mansuy IM, Marin P. Calcineurin interacts with the serotonin transporter C-terminus to modulate its plasma membrane expression and serotonin uptake. Journal of Neuroscience. 2013;33:16189–16199. doi: 10.1523/JNEUROSCI.0076-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra D, Zeremski M, Neznanov N, Li M, Choi Y, Uesugi M, Hauser CA, Gu W, Gudkov AV, Qin J. Differential association of products of alternative transcripts of the candidate tumor suppressor ING1 with the mSin3/HDAC1 transcriptional corepressor complex. Journal of Biological Chemistry. 2001;276:8734–8739. doi: 10.1074/jbc.M007664200. [DOI] [PubMed] [Google Scholar]
- Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, Rowe LD, Dreszer TR, Roe G, Podduturi NR, Tanaka F, Hong EL, Cherry JM. ENCODE data at the ENCODE portal. Nucleic Acids Research. 2016;44(D1):D726–D732. doi: 10.1093/nar/gkv1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC, Mucke L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-[beta] Nature neuroscience. 2013;16:613–621. doi: 10.1038/nn.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Mortimer N, Bellini S, Molinos I, Sanfeliu A, Shovlin S, McAllister D, Gill M, Mitchell K, Corvin A. Expression of nuclear Methyl-CpG binding protein 2 (Mecp2) is dependent on neuronal stimulation and application of Insulin-like growth factor 1. Neuroscience Letter. 2016;16(621):111–116. doi: 10.1016/j.neulet.2016.04.024. [DOI] [PubMed] [Google Scholar]
- Viljetic B, Diao L, Liu J, Krsnik Z, Wijeratne SHR, Kristopovich R, Dutre-Clarke M, Kraushar ML, Song J, Xing J, Chen K, Rasin M-R. Multiple roles of PIWIL1 in mouse neocorticogenesis. bioRxiv 2017 [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ. Combinatorial patterns of histone acetylations and methylations in the human genome. Nature genetics. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Coelho CM, Li X, Marek R, Yan S, Anderson S, Meyers D, Mukherjee C, Sbardella G, Castellano S. p300/CBP-associated factor selectively regulates the extinction of conditioned fear. Journal of Neuroscience. 2012;32:11930–11941. doi: 10.1523/JNEUROSCI.0178-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin DT, Wang Q, Chen L, Liu MY, Han C, Yan Q, Shen R, He G, Duan W, Li JJ. Germline stem cell gene PIWIL2 mediates DNA repair through relaxation of chromatin. PLoS One. 2011;6:e27154. doi: 10.1371/journal.pone.0027154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Lin H. An epigenetic activation role of Piwi and a Piwi-associated piRNA in Drosophila melanogaster. Nature. 2007;450:304–308. doi: 10.1038/nature06263. [DOI] [PubMed] [Google Scholar]
- Ythier D, Larrieu D, Brambilla C, Brambilla E, Pedeux R. The new tumor suppressor genes ING: genomic structure and status in cancer. International journal of cancer. 2008;123:1483–1490. doi: 10.1002/ijc.23790. [DOI] [PubMed] [Google Scholar]
- Yu JJ, Zhang Y, Wang Y, Wen ZY, Liu XH, Qin J, Yang JL. Inhibition of calcineurin in the prefrontal cortex induced depressive-like behavior through mTOR signaling pathway. Psychopharmacology. 2013;225:361–372. doi: 10.1007/s00213-012-2823-9. [DOI] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, Bear MF, Tonegawa S. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–629. doi: 10.1016/s0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W. Model-based analysis of ChIP-Seq (MACS) Genome biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P-p, Yao M-j, Chang S-y, Gou L-t, Liu M-f, Qiu Z-l, Yuan X-b. Novel function of PIWIL1 in neuronal polarization and migration via regulation of microtubule-associated proteins. Molecular brain. 2015;8:39. doi: 10.1186/s13041-015-0131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]