

Abstract
Epidemiology studies suggest associations between schizophrenia and cancer. However, the underlying genetic mechanisms are not well understood, and difficult to identify from epidemiological data. We investigated if there is a shared genetic architecture between schizophrenia and cancer, with the aim to identify specific overlapping genetic loci. First, we performed genome-wide enrichment analysis and second, we analyzed specific loci jointly associated with schizophrenia and cancer by the conjunction false discovery rate. We analyzed the largest genome-wide association studies of schizophrenia and lung, breast, prostate, ovary, and colon-rectum cancer including more than 220,000 subjects, and included genetic association with smoking behavior. Polygenic enrichment of associations with lung cancer was observed in schizophrenia, and weak enrichment for the remaining cancer sites. After excluding the major histocompatibility complex region, we identified three independent loci jointly associated with schizophrenia and lung cancer. The strongest association included nicotinic acetylcholine receptors and is an established pleiotropic locus shared between lung cancer and smoking. The two other loci were independent of genetic association with smoking. Functional analysis identified downstream pleiotropic effects on epigenetics and gene-expression in lung and brain tissue. These findings suggest that genetic factors may explain partly the observed epidemiological association of lung cancer and schizophrenia.
Introduction
Schizophrenia (SCZ) is a mental disorder that greatly impacts the life of the affected individuals and ranks globally among the leading causes of disability. Genetic factors are important for development of SCZ, and heritability estimates range up to 0.81. Large genome-wide association studies (GWAS) suggest that SCZ is a polygenic disease with many genetic variants associated, each with a small effect2. Recently, several lines of evidence indicate genetic overlap between SCZ and other brain disorders3 as well as cardiovascular risk factors4. Due to the polygenic nature of SCZ, it is possible that shared genetic factors may also underlie other diseases or traits associated with SCZ.
Epidemiological studies report both inverse and direct co-morbidity between SCZ and some cancer types. For example, a meta-analysis of cancer incidence in more than 500,000 participants showed an increased risk for breast cancer and decreased risk for melanoma and lung cancer5. Similarly, a prospective cohort study found increased risk of breast cancer for women and lung cancer for men6. Additional support for comorbidity between SCZ and lung cancer was given by a Danish nation-wide registry study7. In contrast, a large UK cohort study did not show any significant difference in incidence of colorectal cancer, breast cancer and lung cancer between SCZ and controls8. Another study investigating parents of patients with SCZ did not find any significantly reduced risk for overall cancer types, although it reported an increased risk for lung cancer in mothers of patients with SCZ9. Furthermore, first-degree relatives of patients with SCZ showed significantly reduced overall cancer risk10. In summary, the literature seems to provide inconsistent results. This can be due to study design, as well as confounders including lifestyle factors, such as smoking or diet, antipsychotic medication, and different approaches to cancer screening and treatment. Additionally, cancer is a disease of the older ages, while patients with SCZ have a decreased life expectancy of 10–25 years.
Combining GWAS from multiple disorders provides insights into genetic pleiotropy, a single genetic variant associated with more than one distinct phenotype, and could elucidate shared pathophysiology. We used a genetic epidemiology framework based on the conjunction false discovery rate (FDR), which enables identification of specific loci of cross-phenotype association independent of direction, thus making it particularly useful to test overlap between different diseases where directions of effects are unknown4. Since the FDR framework requires only summary statistics we were able to integrate GWAS data from SCZ and cancer sites from more than 220,000 subjects (Supplementary Table 1). Our first aim was to visualize polygenic overlap between SCZ and cancer in a genome-wide enrichment analysis and if this varies depending on cancer sites. Secondly, we aimed at identifying specific loci sharing association between SCZ and cancer using conjunction FDR, a two-dimensional extension of the FDR. Finally, we functionally characterized the shared loci using epigenetic and expression data in relevant tissue types to better understand joint disease etiologies.
Results
Enrichment pattern between schizophrenia (SCZ) and cancer
A stratified quantile-quantile (Q-Q) plot showed a strong enrichment pattern for SCZ given lung cancer (Fig. 1). While the blue line shows the standard enrichment of the main trait of interest (SCZ) including all SNPs irrespective of their association with the secondary trait (lung cancer), we observe a stronger leftward deflection from the dashed line of no association with increasingly stronger association with lung cancer. We did not see any similar enrichment pattern for any other cancer sites. Breast cancer showed weak enrichment (Supplementary Figure 1A), i.e. strata conditional on association with breast cancer did not diverge from the line of all SNPs. Conditioning on prostate cancer did not result in any deflection (Supplementary Figure 1B) from the Q-Q line of all SNPs. Furthermore, there was no substantial enrichment given strata defined by ovarian cancer (Supplementary Figure 1C) or colon cancer (Supplementary Figure 1D), which might be due to the comparatively small sample sizes of these GWAS.
Figure 1.
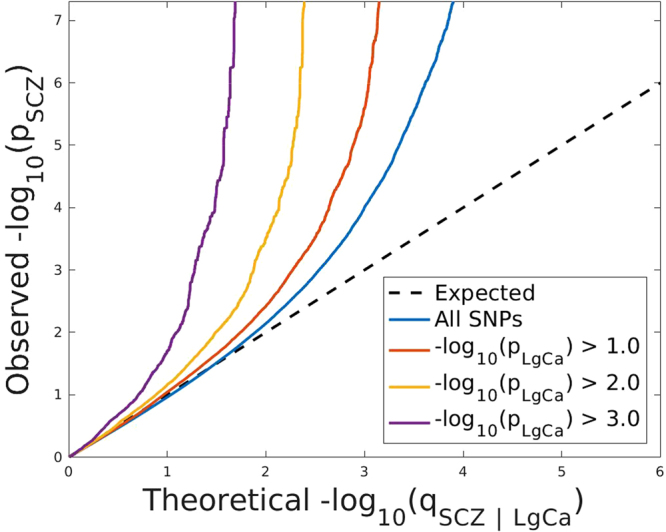
Stratified Q-Q plot for schizophrenia (SCZ) given lung cancer (LgCa). Stratified Q-Q plot of theoretical vs empirical −log10 p-values (corrected for genomic control) in schizophrenia (SCZ) below the standard GWAS threshold of -log10 p-values equal to 7.3 (equals p-values above 5 × 10−8) as a function of significance of association with lung cancer (LgCa) at the level of p < 1 (all SNPs), p < 0.1, p < 0.01, p < 0.001 respectively. Dotted lines indicate the theoretical line in case of no association. Prior to this analysis single nucleotide polymorphisms (SNPs) in the major histocompatibility complex (MHC) have been excluded.
To test for statistical significance of enrichment for the Q-Q plot strata we used LD-score regression11. After adjusting for multiple testing (four cancer traits and three strata) we detected an increase in the enrichment parameter for SCZ given lung cancer ranging from 1.424 (−log10pval >1) to 2.190 (−log10pval >2), and 6.512 (-log10pval >3) of which the first stratum is significantly enriched after multiple testing correction, and the second stratum is nominally significant (Supplementary Table 2). None of the other traits showed significant enrichment of any strata. The prostate cancer study was excluded from the enrichment analysis since its coverage (211,155 SNPs) using a customised genotyping platform was too low. All analysis was performed after excluding SNPs mapping to the major histocompatibility complex (MHC, genomic position (hg 19): chr6:29,528,318- 33,373,64912) since the MHC has been shown to be one of the key driving factors for enrichment of genetic association in SCZ13. In order to check involvement of the MHC region, we repeated the stratified Q-Q plot for SCZ given lung cancer (Supplementary Figure 2) including all SNPs mapping to the MHC, but we did not find substantial changes in enrichment as seen in the stratified Q-Q plots between analysis including the MHC (Supplementary Figure 2) and excluding the MHC (Fig. 1). Further we note the symmetry of the observed enrichment and show the stratified Q-Q plot for lung cancer given SCZ in Supplementary Figure 3.
Shared risk loci between schizophrenia (SCZ) and lung cancer
Three independent (r 2 < 0.2) loci shared between SCZ and lung cancer passed the conjunctional FDR < 0.01 threshold. See Table 1 for p-values and effect directions and Fig. 2 for the conjunctional FDR Manhattan plot. Variants mapping to the MHC have been removed prior to fitting the conjunction FDR.
Table 1.
Independent (r 2 < 0.2) loci associated with both schizophrenia (SCZ) and lung cancer (LgCa) as defined by conjunction false discovery rates (ConjFDR < 0.01).
| SNP | Gene | Band | A1 | A2 | p-value | p-value | p-value | z-score | z-score | z-score | ConjFDR | ConjFDR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SCZ | LgCa | CPD | SCZ | LgCa | CPD | SCZ_LgCa | SCZ_CPD | |||||
| rs7749305 | ZNF184 | 6p22.1 | T | C | 2.385e-17 | 5.084e-06 | NaN | 8.47 | −4.56 | NaN | 2.340e-04 | NaN |
| rs2081361 | AK096335 | 11q12.1 | C | T | 2.525e-04 | 1.667e-05 | 2.517e-01 | −3.66 | −4.31 | −1.15 | 5.891e-03 | 1 |
| rs8042374 | CHRNA3 | 15q25.1 | A | G | 2.056e-08 | 5.302e-32 | 5.067e-23 | 5.61 | 11.77 | 9.88 | 9.317e-07 | 1.960e-05 |
In addition, we include cross-phenotype association of SCZ and smoking status (measured by number of cigarettes per day (CPD)). For each locus we report the lead single nucleotide polymorphism (SNP), closest annotated gene (Gene), genomic position (Band), p-values and z-scores with A1 (reference allele) and A2 (effect allele) for the specific traits. The major histocompatibility complex (MHC) was excluded from the analyses. The SNP rs7749305 on band 6p22.1 has the genomic position (hg19) chr6:27,446,566 and is thus outside the physical boundaries of the MHC. Still, it is an eQTL with a MHC-related gene (BTN3A2, Supplementary Table 3B). Not available number (NaN) if not available in the summary data file.
Figure 2.
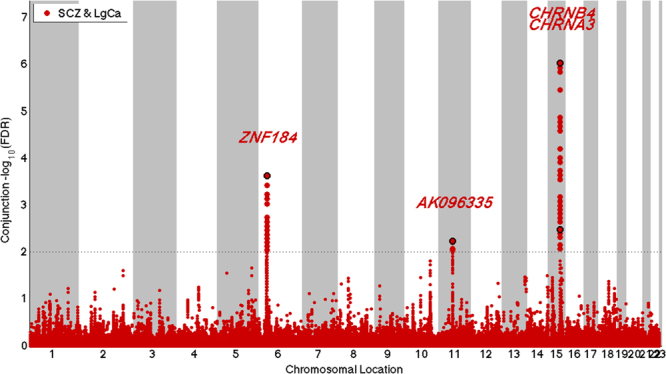
Manhattan plot for independent (r 2 < 0.2) loci associated with both schizophrenia (SCZ) and lung cancer (LgCa) as defined by conjunction false discovery rates (ConjFDR) < 0.01 after excluding single nucleotide polymorphisms in the major histocompatibility complex.
The three loci with joint association between SCZ and lung cancer were explored with functional follow-up studies. The strongest association was found for the locus on 15q25.1 mapping to genes of the nicotinic acetylcholine receptors, which has been previously implicated for cross-phenotype association between lung cancer and smoking14. The 15q25.1 locus showed a concordant effect direction between SCZ and lung cancer. There was one LD proxy (rs2904130, r 2 = 0.89) of the lead SNP rs8042374, which is an expression quantitative trait loci (eQTL) with neuronal acetylcholine receptor subunit alpha-5 (CHRNA5) and in both in lung and brain (caudate) tissue (Genotype-Tissue Expression (GTEx)15 Supplementary Table 3A). The locus on 6p22.1, has been identified and replicated as a cross-phenotype association between lung cancer and blood triglycerides16. This locus harbors two SNPs (rs28360634 and rs72839477) in strong LD (r 2 = 1) with the lead SNP rs7749305, which are eQTL (GTEx15) in both brain and lung tissue with the same gene butyrophilin subfamily 3 member A2 (BTN3A2). The eQTL in brain tissue was confirmed in the independent Brain eQTL dataset (Braineac17, Supplementary Table 3B). The lead SNP rs7749305 is outside of the physical boundaries of the MHC, but it is an eQTL with BTN3A2, a MHC-related gene, underscoring the complicated and extensive LD structure in this region. The third association was on 11q12.1 and included the lead SNP rs2081361, which was an eQTL (GTEx) in lung tissue with the gene translocase of inner mitochondrial membrane 10 homolog (TIMM10) and with the leucine-rich repeat-containing protein 55 (LRRCP55) which is an auxiliary protein of the large-conductance, voltage and calcium-activated potassium channel. Further we found evidence for rs2081361 to be a moderate eQTL with TIMM10 in brain tissue in the Braineac database (Supplementary Table 3B). We found epigenetic evidence for rs2081361 in lung tissue (normal human lung fibroblast (NHLF) and adenocarcinomic human alveolar basal epithelial cells (A549)), and Henrietta Lacks (HeLa) S3 cells (Supplementary Figure 4). In particular, for A549 and HeLa S3 we found CCCTC-binding factor (CTCF) binding and for NHLF open chromatin as characterized by DNase1 was discovered. A summary of the eQTL data is given in Supplementary Table 3 (A. GTEx15, B. Braineac17).
Genetic overlap and shared risk loci between schizophrenia (SCZ) and smoking
Smoking is the main risk factor for lung cancer, and there is a higher prevalence of smoking among patients with SCZ than controls. There is also one reported cross-phenotype association between lung cancer and smoking14,18. Thus, we investigated if association with smoking behavior measured by cigarettes per day (CPD), correlated with the polygenic overlap between SCZ and lung cancer. As shown in the stratified Q-Q plot (Supplementary Figure 5A.), there is an enrichment of SCZ association given CPD (after removing MHC region). After removing SNPs mapping to the nicotinic acetylcholine receptors (genomic position (hg 19) chr15: 78,686,690-79,231,478) the enrichment of SCZ given CPD disappears (Supplementary Figure 5B) which suggests that the shared signal between SCZ and CPD is driven by genetic variation within the nicotinic acetylcholine receptors.
To detect cross-phenotype association between SCZ and smoking behavior we computed the conjunction FDR for joint association between SCZ and CPD. There is only one locus, 15q25.1, with conjunction FDR < 0.01 between SCZ and CPD. This has a concordant association between lung cancer and smoking, as reported earlier14,18, and the effect direction is also concordant for SCZ. Remarkably, the other two loci shared by SCZ and lung cancer had a conjunction FDR for SCZ and CPD close to one, which indicates no association between SCZ and CPD apart from the locus on 15q25.1 (Table 1). We included further smoking traits such as onset, cessation, and initiation into the analysis, but except for the locus on 15q25.1 none of the cross-phenotype associated SNPs shows any association with any other smoking trait (Supplementary Table 4).
Shared risk loci between schizophrenia (SCZ) and squamous cell carcinoma type of lung cancer
Furthermore, we refined the definition of shared genetic variants between SCZ and lung cancer to subtypes of lung cancer, adenocarcinoma (ADENO) and squamous cell carcinoma (SQUAM). We analyzed the two subtypes and presented stratified Q-Q plots for SCZ given SQUAM (Supplementary Figure 6A) and for SCZ given ADENO (Supplementary Figure 6B). Noteworthy, we observed a strong enrichment for SCZ for SQUAM, and a weaker enrichment for ADENO. This is in line with previous findings of a different genetic architecture of the two cancer sub-types19.
We found three independent loci with conjunction FDR < 0.01 for SCZ&SQUAM, and one locus with conjunction FDR < 0.01 for SCZ&ADENO (Supplementary Table 5). The locus shared between SCZ, SQUAM and ADENO is the locus on 15q25.1, which was the strongest association in the general lung cancer analysis.
Discussion
We report polygenic enrichment between SCZ and lung cancer, but not for any other cancer site. This suggests that shared genetic risk factors may underlie the association between SCZ and lung cancer shown in epidemiological studies. Smoking is strongly associated with both SCZ and lung cancer, and here we show that variants mapping to the nicotinic acetylcholine receptors may contribute to this overlap. The current findings of shared variants associated with these three phenotypes have implications for the underlying pathophysiological processes, and interpretation of epidemiological findings. In particular, the finding of partly genetic causes for the high smoking prevalence in SCZ are of clinical relevance. It underscores the importance of preventive measures against smoking initiation and smoking cessation programs in mental health care, and suggests evaluation of lung cancer screening programs in SCZ.
The conjunction FDR is a genome-wide approach and it is possible that inclusion of larger LD blocks such as the MHC can impact the model fit and confound the results. Therefore, the main results are based on the analysis after excluding the MHC and re-fitting the FDR estimate, which showed associations of three loci (6p22.1, 11q22.1, and 15q25.1). The statistical framework we used has the advantage of pinpointing loci of cross-phenotype associations even when the effect directions are mixed as it is the case for the three loci we identified here (Table 1). In contrast, LD score regression11, a useful approach for genome-wide co-heritability analysis as presented in Supplementary Table 2, is neither able to identify specific genetic regions nor pleiotropic traits with mixed effect direction20.
The locus on chromosome 15q25.1, including the nicotinic acetylcholine receptors CHRNA3, CHRNA5 and CHRNB4, showed concordant effect direction between SCZ, lung cancer, and smoking behavior. When the two lung cancer sub-types were analyzed, the associations with SCZ were in same direction. The locus on chromosome 11q12.1 showed concordant effect direction for SCZ and lung cancer. It harbors several variants that are moderate eQTL in both lung and brain tissue with the gene translocate of inner mitochondrial membrane 10 (TIMM10). The protein encoded by TIMM10 functions as a preprotein translocase for the import of proteins into inner and outer membranes, particularly inner membrane metabolite carriers21. Under-expression of genes of the TIMM family has been associated with neurodegenerative diseases22. Additionally TIMM10 transcript was recently identified as significantly down-regulated in dorsolateral prefrontal cortex layer 3 pyramidal cells isolated from tissue from SCZ patients23. However, the present evidence for involvement of TIMM10 is moderate and replication and further investigations are needed.
We found associations between SCZ and both histological types of lung cancer, squamous cell carcinoma and adenocarcinoma. The enrichment was stronger and more extensive in the squamous cell type, which had three loci associated with SCZ, and only one with adenocarcinoma (the CHRNA3/CHRNA5/CHRNB4 cluster on chromosome 15q25.1). It was reported that more than 90% of patients with squamous cell carcinoma were or had been smokers, as compared to about 55% of those suffering from adenocarcinoma24.
We expect that the present findings will form the basis for future studies of the role of the 15q25.1 in smoking behavior, lung cancer, and SCZ. Our approach aimed at identifying cross-phenotype associations and cannot distinguish between biological and mediated pleiotropy25. The present findings demonstrate the importance of further functional follow-up studies and further investigations using other approaches such as Mendelian Randomisation, which can help to distinguish between biological and mediated pleiotropy. Recent epidemiological studies, including a Mendelian Randomisation study26 and a prospective co-relative control study27 have found evidence for smoking initiation as putative risk factor for SCZ.
The present findings suggest pleiotropic downstream effects of the cross-phenotype associations. Especially eQTL studies in relevant tissue types provide important insights how genetic variants exert downstream effects on gene-expression28. The evidence that all three cross-phenotype associations from our pleiotropic analysis are eQTL with the same gene (nicotinic acetylcholine receptors, BTN3A2, TIMM10) in relevant tissue types including lung and brain further support the claim of downstream pleiotropy between SCZ and lung cancer and complement observed associations from epidemiology studies. Further analyses of the molecular downstream consequences of these genetic variants are beyond the scope of this manuscript. One should be cautious with interpretation, as the relationship between SCZ and lung cancer is complex. Cancer risk in SCZ seems to vary with age, with higher than expected frequencies during young ages and lower than expected frequencies later in life29. Also lung cancer followed this pattern, with higher standardized incidence ratios at ages less than 60 years, and lower incidences at higher ages29. We do not have data stratified for age in the present study.
In conclusion, we identified shared genetic variation between SCZ and lung cancer in the CHRNA3/CHRNA5/CHRNB4 cluster on chromosome 15q25.1, and two other loci (6p22.1, 11q12.1) show cross-phenotype association and downstream pleiotropic effects on gene-expression in relevant tissue types for lung cancer and SCZ. The genetic effects are however complex, giving rise to both increased and decreased risk of the disorders. Further efforts into fine-mapping, causal analysis, and functional annotation are needed to clarify how these cross-phenotype associations exert their pleiotropic effects. Especially of interest is the role of the nicotinic acetylcholine receptors in the synthesis of smoking behavior, lung cancer and SCZ.
Methods
Genome-wide association studies (GWAS) Samples
GWAS summary statistics on SCZ were provided by the Psychiatric Genomic Consortium (PGC) and comprised association analyses of 32,405 cases and 42,221 controls2. The summary statistics on five cancer sites were obtained from the Genetic Associations and Mechanisms in Oncology (GAME-ON) consortium and included lung cancer (13,373 cases and 26,014 controls)19 (including sub-types referred to as adenocarcinoma (ADENO) and squamous cell carcinoma (SQUAM)), breast cancer (15,863 cases and 40,022 controls)30, prostate cancer (25,074 cases and 24,272 controls)31, colon cancer (5,100 cases and 7,529 controls)32, and ovarian cancer (3,995 cases and 3,277 controls)33. Additionally, we included GWAS data on smoking behavior measured by cigarettes per day (CPD) (74,503 individuals)34. For more details see Supplementary Table 1.
Pre-processing
As a first pre-processing step we aligned all summary statistics to a common set of reference single nucleotide polymorphisms (SNPs) (of size d = 2,558,411) generated from the 1000 genomes project. As summary statistics we saved for each reference SNP and each trait one p-value and one z-score. Next we performed genomic control35, and finally we adjusted for overlap between samples36. There were overlaps between controls of the PGC study on SCZ and controls of the cancer studies, i.e. n = 3,179 individuals for lung cancer, n = 4,834 for breast cancer, and n = 713 for colon cancer. All p-values reported are adjusted for genomic control, all false discovery rates reported are adjusted for genomic control and sample overlap. As reference panel for the computation of the linkage disequilibrium (LD) structure between SNPs we use the European populations from the 1000 genomes project. The European population best reflects the mainly European composition of the PGC study on SCZ and the lung cancer GWAS.
Quantile-Quantile (Q-Q) plots
Q-Q plots are standard tools in genomics to visualize the distribution of the observed p-values with the expected distribution of p-values under the null hypothesis, or in other words under no association of the tagged SNPs with the phenotype of interest. Q-Q plots depict the quantiles of the observed p-values on the y-axis against the theoretical quantiles under no association on the x-axis. In order to focus on the tails, Q-Q plots are often displayed on the −log10 scale. In case of no association a Q-Q plot follows a straight line. Deflection from this null line describes enrichment, i.e. the presence of lower p-values as expected by chance. Stratified Q-Q plots investigate differential enrichment between pre-specified strata of SNPs37,38. When investigating polygenic shared architecture between two traits we focused on the p-values of trait 1 (SCZ), and defined the strata based on trait 2 (cancer). More specifically we plotted the p-values of trait 1 given or conditional on different strength of association with trait 2 (i.e. p-value > −log10 p-values of 1, 2, or 3). Thus, we were able to visualize if conditioning on a secondary trait leads to stronger enrichment in the primary trait of interest. A strong enrichment increasing with association on the secondary trait is an indicator of a shared polygenic architecture between the two traits.
Large blocks of linkage disequilibrium (LD) may confound the results. To account for this we applied a random pruning approach, where one random SNP per LD block (defined by an r 2 of 0.8) were used and averaged over 100 random pruning runs. The impact of differing correlation parameters (from 0.7 to 0.3) on the Q-Q plots is displayed in Supplementary Figure 7. Further we focus the Q-Q plots on the region below genome-wide significance (−log10 p-values < 7.3) in order to highlight the polygenic component of the cross-phenotype association.
In order to test for differential enrichment of the Q-Q plot strata we use LD score regression11 to test for fold enrichment. We assess the fold enrichment of each of the three strata (i.e. p-value > −log10 p-values of 1, 2, or 3) represented in the stratified Q-Q plots with the total LD score as covariate. The prostate cancer study was excluded from the analysis since its coverage (211,155 SNPs) using a customised genotyping platform was too low. Multiple-testing correction is performed for four cancer traits and for the three strata (p_adjusted = p–value × 4 cancer types × 3 strata).
Conditional and conjunction false discovery rate (FDR)
The second part of our genetic epidemiology framework aimed at pinpointing shared cross-phenotype associations using the conjunction false discovery rate (FDR). The basic FDR framework is based on the assumption that the distribution of p-values follows a mixture distribution where SNPs are either associated (non-null) or not associated (null) with the phenotype39. The (tail-area based) FDR is defined as the probability that a given SNP is null given that its p-value is as small as or smaller than the observed one. Note that in context of the FDR all modeling is done on the summary statistic level, and no access to genotype data is needed. The conditional FDR is a simple extension of the standard FDR that allows including additional information on the association of a SNP with a secondary trait or more precisely, with the p-value of the same SNP in a secondary trait. It is defined as the probability that a specific SNP is null given that the p-values for both, trait 1 and trait 2, are as small as or smaller than the observed ones37,38. Low values of conditional FDR can be driven by the first trait only. To detect SNPs associated jointly with both traits at the same time we employed the conjunction FDR. It is defined as the probability of being null for either trait, or for both traits simultaneously given that the p-values for the two traits are as small as or smaller than the observed ones. Thus, a true discovery is only the case when a SNP is non-null for both traits jointly. This symmetric behavior of the conjunction FDR weights both traits equal. Low values in conjunction FDR can only be found when a SNP is associated with both traits jointly. For example, for lung cancer and SCZ this symmetric behavior is best demonstrated by a stratified Q-Q plot of SCZ given lung cancer and then vice-versa lung cancer given SCZ (Supplementary Figure 3).
For more information on conditional and conjunction FDR we refer to40. We set a conservative FDR level of 0.01 per pair-wise comparison, which relates to one expected false positive finding within 100 reported findings. The conjunction FDR provides a genome-wide unbiased scan and is thus a suitable technique to discover novel associations that are not detected by a univariate conservative Bonferroni threshold.
Functional follow up
To investigate downstream effects of the cross-phenotype associated genetic loci we looked up expression quantitative trait loci (eQTL) in relevant tissue types (especially lung and brain) in the Genotype-Tissue Expression (GTEx) database15, and the UK Brain Expression Consortium (Braineac)17.
Electronic supplementary material
Acknowledgements
The work was supported by the Research Council of Norway, KG Jebsen Stiftelsen, and South East Norway Health Authority. The authors would like to thank the study participants and the members of the different consortia contributing summary statistics data. This work would not have been possible without the Psychiatric Genomics Consortium (PGC) Schizophrenia Work Groups, the Tobacco and Genetics (TAG) Consortium, and the Colon Cancer Family Registries (Colon CFR), and ColoRectal Transdisciplinary Study (CORECT) consortia, the Discovery, Biology, and Risk of Inherited Variants in Breast Cancer (DRIVE) consortium, the Follow-up of Ovarian Cancer Genetic Association and Interaction Studies (FOCI) Research Team, the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) Consortium, Transdisciplinary Research in Cancer of the Lung (TRICL) Research Team. In particular, the Discovery, Biology, and Risk of Inherited Variants in Breast Cancer (DRIVE) breast cancer genome-wide association study meta-analysis was supported by U19 CA148065. The following genome-wide association studies and investigators that shared genome-wide summary data as part of the DRIVE meta-analysis: the Australian Breast Cancer Family Study (ABCFS) (John L Hopper, Melissa C. Southey, Enes Makalic, Daniel F. Schmidt), the British Breast Cancer Study (BBCS) (Olivia Fletcher, Julian Peto, Lorna Gibson, Isabel dos Santos Silva), the Breast and Prostate Cancer Cohort Consortium (BPC3) (David J. Hunter, Sara Lindström, Peter Kraft), the Breast Cancer Family Registries (BCFR) (Habib Ahsan, Alice Whittemore), the Dutch Familial Bilateral Breast Cancer Study (DFBBCS) (Quinten Waisfisz, Hanne Meijers-Heijboer, Muriel Adank, Rob B van der Luijt, Andre G Uitterlinden, Albert Hofman), German Consortium for Hereditary Breast and Ovarian Cancer (GC-HBOC) (Alfons Meindl, Rita K. Schmutzler, Bertram Müller-Myhsok, Peter Lichtner), the Helsinki breast cancer family Study (HEBCS) (Heli Nevanlinna, Taru A Muranen, Kristiina Aittomäki, Carl Blomqvist), the Mammary Carcinoma Risk factor Investigation (MARIE) (Jenny Chang-Claude, Rebecca Hein, Norbert Dahmen, Lars Beckman), SardiNIA (Laura Crisponi), the Singapore and Sweden Breast Cancer Study (SASBAC) (Per Hall, Kamila Czene, Astrid Irwanto, Jianjun Liu), and the UK2 GWAS (Douglas F Easton, Clare Turnbull, Nazneen Rahman). Furthermore, the COGS study would not have been possible without the contributions of the following: Per Hall (COGS); Douglas F. Easton, Paul Pharoah, Kyriaki Michailidou, Manjeet K. Bolla, Qin Wang (BCAC), Andrew Berchuck (OCAC), Rosalind A. Eeles, Douglas F. Easton, Ali Amin Al Olama, Zsofia Kote-Jarai, Sara Benlloch (PRACTICAL), Georgia Chenevix-Trench, Antonis Antoniou, Lesley McGuffog, Fergus Couch and Ken Offit (CIMBA), Joe Dennis, Alison M. Dunning, Andrew Lee, and Ed Dicks, Craig Luccarini and the staff of the Centre for Genetic Epidemiology Laboratory, Javier Benitez, Anna Gonzalez-Neira and the staff of the CNIO genotyping unit, Jacques Simard and Daniel C. Tessier, Francois Bacot, Daniel Vincent, Sylvie LaBoissière and Frederic Robidoux and the staff of the McGill University and Génome Québec Innovation Centre, Stig E. Bojesen, Sune F. Nielsen, Borge G. Nordestgaard, and the staff of the Copenhagen DNA laboratory, and Julie M. Cunningham, Sharon A. Windebank, Christopher A. Hilker, Jeffrey Meyer and the staff of Mayo Clinic Genotyping Core Facility. Funding for the iCOGS infrastructure came from: the European Community’s Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A 10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112 - the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. A full list of consortium members appears in the Supplementary Information.
Author Contributions
V.Z., E.G.J., I.G.M., and O.A.A. have conceived and designed the work and drafted the manuscript. V.Z. has performed the analysis with support from O.F., A.W., F.B., W.K.T., A.S., Y.W., O.B.S. S.D., I.D., A.Y., R.S.D., A.M.D. have contributed to the interpretation of the results. S.K. and S.B. have provided critical feedback and help with the interpretation. All authors have read and approved the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-16481-4.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–1192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 2.Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature511, 421–427, 10.1038/nature13595 (2014). [DOI] [PMC free article] [PubMed]
- 3.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet381, 1371–1379, 10.1016/S0140-6736(12)62129-1 (2013). [DOI] [PMC free article] [PubMed]
- 4.Andreassen OA, et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. American journal of human genetics. 2013;92:197–209. doi: 10.1016/j.ajhg.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catala-Lopez F, et al. Inverse and direct cancer comorbidity in people with central nervous system disorders: a meta-analysis of cancer incidence in 577,013 participants of 50 observational studies. Psychother Psychosom. 2014;83:89–105. doi: 10.1159/000356498. [DOI] [PubMed] [Google Scholar]
- 6.Tran E, et al. Cancer mortality in patients with schizophrenia: an 11-year prospective cohort study. Cancer. 2009;115:3555–3562. doi: 10.1002/cncr.24383. [DOI] [PubMed] [Google Scholar]
- 7.Benros ME, Laursen TM, Dalton SO, Nordentoft M, Mortensen PB. The risk of schizophrenia and child psychiatric disorders in offspring of mothers with lung cancer and other types of cancer: a Danish nationwide register study. PLoS One. 2013;8:e79031. doi: 10.1371/journal.pone.0079031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osborn DP, et al. Relative incidence of common cancers in people with severe mental illness. Cohort study in the United Kingdom THIN primary care database. Schizophr Res. 2013;143:44–49. doi: 10.1016/j.schres.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Dalton SO, Laursen TM, Mellemkjaer L, Johansen C, Mortensen PB. Risk for cancer in parents of patients with schizophrenia. Am J Psychiatry. 2004;161:903–908. doi: 10.1176/appi.ajp.161.5.903. [DOI] [PubMed] [Google Scholar]
- 10.Catts VS, Catts SV, O’Toole BI, Frost AD. Cancer incidence in patients with schizophrenia and their first-degree relatives - a meta-analysis. Acta Psychiatr Scand. 2008;117:323–336. doi: 10.1111/j.1600-0447.2008.01163.x. [DOI] [PubMed] [Google Scholar]
- 11.Finucane HK, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet. 2009;54:15–39. doi: 10.1038/jhg.2008.5. [DOI] [PubMed] [Google Scholar]
- 13.Andreassen OA, et al. Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: differential involvement of immune-related gene loci. Mol Psychiatry. 2015;20:207–214. doi: 10.1038/mp.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hung RJ, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 15.Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuber, V. et al. Pleiotropic Analysis of Lung Cancer and Blood Triglycerides. J Natl Cancer Inst108, 10.1093/jnci/djw167 (2016). [DOI] [PMC free article] [PubMed]
- 17.Ramasamy A, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci. 2014;17:1418–1428. doi: 10.1038/nn.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thorgeirsson TE, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Timofeeva MN, et al. Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Hum Mol Genet. 2012;21:4980–4995. doi: 10.1093/hmg/dds334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schork AJ, Wang Y, Thompson WK, Dale AM, Andreassen OA. New statistical approaches exploit the polygenic architecture of schizophrenia–implications for the underlying neurobiology. Curr Opin Neurobiol. 2016;36:89–98. doi: 10.1016/j.conb.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sokol AM, Sztolsztener ME, Wasilewski M, Heinz E, Chacinska A. Mitochondrial protein translocases for survival and wellbeing. FEBS Lett. 2014;588:2484–2495. doi: 10.1016/j.febslet.2014.05.028. [DOI] [PubMed] [Google Scholar]
- 22.Liang WS, et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci USA. 2008;105:4441–4446. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arion D, et al. Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol Psychiatry. 2015;20:1397–1405. doi: 10.1038/mp.2014.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toh CK, et al. Never-smokers with lung cancer: epidemiologic evidence of a distinct disease entity. J Clin Oncol. 2006;24:2245–2251. doi: 10.1200/JCO.2005.04.8033. [DOI] [PubMed] [Google Scholar]
- 25.Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet. 2013;14:483–495. doi: 10.1038/nrg3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gage SH, et al. Investigating causality in associations between smoking initiation and schizophrenia using Mendelian randomization. Sci Rep. 2017;7:40653. doi: 10.1038/srep40653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kendler KS, Lonn SL, Sundquist J, Sundquist K. Smoking and schizophrenia in population cohorts of Swedish women and men: a prospective co-relative control study. Am J Psychiatry. 2015;172:1092–1100. doi: 10.1176/appi.ajp.2015.15010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet. 2015;16:197–212. doi: 10.1038/nrg3891. [DOI] [PubMed] [Google Scholar]
- 29.Lin CY, et al. Inverse association between cancer risks and age in schizophrenic patients: a 12-year nationwide cohort study. Cancer Sci. 2013;104:383–390. doi: 10.1111/cas.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michailidou, K. et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet45, 353–361, 361e351–352, 10.1038/ng.2563 (2013). [DOI] [PMC free article] [PubMed]
- 31.Eeles, R. A. et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet45, 385–391, 391e381–382, 10.1038/ng.2560 (2013). [DOI] [PMC free article] [PubMed]
- 32.Schumacher FR, et al. Genome-wide association study of colorectal cancer identifies six new susceptibility loci. Nature communications. 2015;6:7138. doi: 10.1038/ncomms8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pharoah, P. D. et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet45, 362–370, 370e361–362, 10.1038/ng.2564 (2013). [DOI] [PMC free article] [PubMed]
- 34.Tobacco & Genetics, C Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet42, 441–447, 10.1038/ng.571 (2010). [DOI] [PMC free article] [PubMed]
- 35.Schork AJ, et al. All SNPs Are Not Created Equal: Genome-Wide Association Studies Reveal a Consistent Pattern of Enrichment among Functionally Annotated SNPs. PLoS Genet. 2013;9:e1003449. doi: 10.1371/journal.pgen.1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin DY, Sullivan PF. Meta-analysis of genome-wide association studies with overlapping subjects. Am J Hum Genet. 2009;85:862–872. doi: 10.1016/j.ajhg.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andreassen OA, et al. Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional False Discovery Rate. PLoS Genet. 2013;9:e1003455. doi: 10.1371/journal.pgen.1003455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu JZ, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nature Genetics. 2013;45:670. doi: 10.1038/ng.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Efron BS. power and false discovery rates. Annals of Statistics. 2007;35:1351–1377. doi: 10.1214/009053606000001460. [DOI] [Google Scholar]
- 40.Andreassen, O. A. et al. Shared common variants in prostate cancer and blood lipids. Int J Epidemiol, 10.1093/ije/dyu090 (2014). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.