

Abstract
The mevalonate pathway produces isopentenyl diphosphate (IPP), a building block for polyisoprenoid synthesis, and is a crucial pathway for growth of the human bacterial pathogen Enterococcus faecalis. The final enzyme in this pathway, mevalonate diphosphate decarboxylase (MDD), acts on mevalonate diphosphate (MVAPP) to produce IPP while consuming ATP. This essential enzyme has been suggested as a therapeutic target for the treatment of drug-resistant bacterial infections. Here, we report functional and structural studies on the mevalonate diphosphate decarboxylase from E. faecalis (MDDEF). The MDDEF crystal structure in complex with ATP (MDDEF–ATP) revealed that the phosphate-binding loop (amino acids 97–105) is not involved in ATP binding and that the phosphate tail of ATP in this structure is in an outward-facing position pointing away from the active site. This suggested that binding of MDDEF to MVAPP is necessary to guide ATP into a catalytically favorable position. Enzymology experiments show that the MDDEF performs a sequential ordered bi-substrate reaction with MVAPP as the first substrate, consistent with the isothermal titration calorimetry (ITC) experiments. On the basis of ITC results, we propose that this initial prerequisite binding of MVAPP enhances ATP binding. In summary, our findings reveal a substrate-induced substrate-binding event that occurs during the MDDEF-catalyzed reaction. The disengagement of the phosphate-binding loop concomitant with the alternative ATP-binding configuration may provide the structural basis for antimicrobial design against these pathogenic enterococci.
Keywords: crystal structure, decarboxylase, drug resistance, Enterococcus, enzyme kinetics, enzyme mechanism, isothermal titration calorimetry (ITC), mevalonate pathway
Introduction
Multidrug-resistant microorganisms have been found in humans and food animals due to the overuse of antibiotics (1). In the United States, a surveillance of antibacterial resistance from the Centers for Disease Control and Prevention reports the deaths and the cost resulting from infectious diseases and emphasizes the urgency to control drug-resistant bacteria (2). This critical list includes vancomycin-resistant enterococci (VRE),2 which cause a range of enterococcal infections such as bacteremia, urinary tract infections, intra-abdominal and pelvic infections, central nervous system infections, skin and skin structure infections, and infective enterocarditis (2–4).
Enterococci are Gram-positive and facultative anaerobes colonized in the gastrointestinal tract. The first clinical isolates of vancomycin-resistant enterococci were reported in Europe in 1988 (5, 6). The VRE bacteria are intrinsically insensitive to detergents and antibiotics (clindamycin, cephalosporins, and aminoglycosides) and keep gaining drug resistance to other newly invented antibiotics, such as linezolid and daptomycin (7–11). The emergence of drug-resistant enterococci results in difficulties of treatment of VRE infections (12–14). For the protection of patients in health care settings, new approaches and new antimicrobial agents against enterococcal infections are urgently needed.
In 2000, Wilding et al. (15) reported that the mevalonate pathway is pivotal in the Gram-positive bacterial pathogens, enterococci, staphylococci, and streptococci. This pathway includes six enzymes, acetyl-CoA acetyltransferase, HMG-CoA synthase, HMG-CoA reductase, mevalonate kinase, phosphomevalonate kinase, and mevalonate diphosphate decarboxylase (EC 4.1.1.33), that ultimately produce IPP. IPP then serves as a building block for polyisoprenoid synthesis in living organisms ranging from bacteria to humans (16–18). In bacteria, isoprenoid production is also involved in the biosynthesis of bacterial cell wall and electron carriers in the respiratory chain (16, 19–21).
The five enzymes in the mevalonate pathway (except acetyl-CoA acetyltransferase) have been identified to be critical for bacterial growth (15). Experiments with the mammalian enzyme have shown that the ATP-dependent decarboxylation of MVAPP catalyzed by MDD is a rate-limiting step in IPP synthesis (22), implicating MDD as a suitable drug target for the disease treatment. Interestingly, in Streptococcus pneumoniae, an in vitro experiment has indicated that a higher level of MVAPP can also inhibit mevalonate kinase (23). This implies that the mevalonate pathway is sophisticatedly regulated by its downstream products and strengthens the case for MDD as a promising drug target for treatments of enterococcal infections.
In this research, we have studied MDD from Enterococcus faecalis (MDDEF) from functional, biophysical, and structural points of view to decipher the substrate-binding mechanism of MDDEF. MDD proteins trigger the irreversible ATP-dependent decarboxylation of MVAPP to produce IPP in the last step of the mevalonate pathway (Fig. 1) (24). Several residues (aspartate, lysine, serine and arginine) in the active site of MDD proteins have been identified as being involved in substrate binding and enzyme catalysis (25–27). In 2012, Barta et al. (27) reported several crystal structures of the wild-type and mutant forms of MDD from Staphylococcus epidermidis (MDDSE) with or without substrates/analogues binding to the active-site cleft. From their structural models, they proposed that MVAPP is the first substrate binding to the deeper pocket in the active site of MDDSE, where Ser-106, Arg-144, Ser-192, and Arg-193 hold MVAPP in position. Next, ATP sits in the ATP-binding pocket interacting with Ser-94 and Asn-96, and the phosphate tail is clamped by Ser-106, Ser-107, and the phosphate-binding loop (Ala-101, Gly-103, and Leu-104), followed by enzyme catalysis (27). The substrate-binding order of MDD isolated from chicken liver was examined with results that suggest avian MDD performs a sequential ordered bi-substrate mechanism with MVAPP as the first substrate (28, 29). However, no kinetic evidence was presented that prokaryotic MDDs act on substrates in the same manner,. A case in point is the enzyme mechanism of proteins in the GHMP kinase family (galactokinases, homoserine kinases, mevalonate kinases, and phosphomevalonate kinases), which vary due to different biological sources (27). In this work, we aimed to obtain a crystal structure of MDDEF bound with ATP alone to investigate structural differences between the MDDEF–ATP structure and those complex structures of MDDSE (supplemental Table S1). With that, we could better understand how the two substrates initially bind to the active site, which may aid in our rational drug design in vitro and in silico.
Figure 1.

Decarboxylation reaction of MVAPP to IPP by MDD. MVAPP and MgATP are the two substrates of MDD enzymes. MgADP, CO2, phosphate, and IPP are the products.
Results
MDDEF protein expression and purification
Protein expression and purification conditions are detailed under “Experimental procedures.” Overexpressed His-tagged MDDEF in Escherichia coli (BL21) was purified via nickel-affinity chromatography (Fig. 2A). Eluted protein fractions were examined by SDS-PAGE (Fig. 2B). Fractions containing MDDEF were collected for TEV treatment to remove the fusion N-terminal His tag from MDDEF (Fig. 2C) (“Experimental procedures”). Finally, we obtained a His tag-removed MDDEF (less than 5% impurities, Fig. 2C, flow-through) with a protein yield of 80–120 mg/liter of culture and adjusted the protein concentration to 8–10 mg/ml.
Figure 2.

Protein purification and structure determination of MDDEF. A, purification profile of MDDEF from a nickel affinity column. The detailed procedures of protein purification are discussed under “Experimental procedures.” mAU, milli-absorbance unit. B, SDS-polyacrylamide gel for the MDDEF protein purification. M, protein marker; Pellet, sample in the pellet after French press; Flow through, an aliquot of flow-through containing proteins that cannot be trapped on the Ni-NTA column; fractions from 12 to 18 were combined. C, SDS-polyacrylamide gel for the TEV-protease treatment profile of MDDEF. The lane 0 hr shows the His-tagged MDDEF without TEV treatment. The TEV treatment process is done in 2 days (Day1 and Day2; 4 and *16 hrs indicate the TEV treatment at different time courses; *, dialysis against buffer without DTT and EDTA; RT, treatment at room temperature; Flow through, an aliquot containing MDDEF proteins without a His tag which cannot be trapped on a Ni-NTA column; Strip, an aliquot after 1 column volume of strip buffer (containing 50 mm EDTA) flowing through the column. D, superposition of MDDEF and the MDDEF–ATP complex. Two structures of MDDEF (gray) and MDDEF–ATP (pink) were Cα-aligned (r.m.s.d. = 0.174 Å). The position of the phosphate-binding loop (97–104) can be determined in MDDEF–ATP but not MDDEF. The missing residues (dashed line) in MDDEF range from 183 to 192; in MDDEF–ATP, residues from 184 to 189 are missing.
Crystal structure of MDDEF-ATP and the apo form of MDDEF
The detailed procedures for obtaining co-crystals of MDDEF bound with ATP (MDDEF–ATP, PDB accession number 5V2L) and apo-MDDEF (MDDEF, PDB accession number 5V2M), data collection, and structure refinement are summarized under “Experimental procedures.” The data collection and refinement statistics of both structures are listed in Table 1. According to the CATH protein structure classification database analysis, the overall structure of the MDD family of proteins contains α and β secondary structure elements (α and β class) (Fig. 2D), and the folding architecture is characterized as a two-layer sandwich (30). The protein family annotation (Pfam) (31) indicates MDD proteins belong to the GHMP kinase (galactokinase, homoserine kinase, mevalonate kinase, and phosphomevalonate kinase) superfamily, which catalyzes ATP-dependent reactions. The overall structures of MDDEF and MDDEF–ATP superimpose well with an r.m.s.d of 0.174 Å. In MDDEF, the phosphate-binding loop (97–104) and residues from 183 to 192 are missing. Similarly, in MDDEF–ATP, residues from 184 to 189 cannot be determined. However, the phosphate-binding loop can be observed (Fig. 2D). In the literature, the phosphate-binding loop in MDD is known for ATP binding (27). In our crystal structure of MDDEF–ATP, the phosphate-binding loop does not interact with the ATP molecule (Fig. 3A), although ATP binds to the ATP-binding pocket via the interactions between ATP and the conserved residues (Ser-93, Asn-95, and Ser-105 (Fig. 3B).
Table 1.
Data collection and refinement statistics of MDDEF-ATP and MDDEF
r.m.s.d. is root mean square deviation.
| MDDEF–ATP | MDDEF | |
|---|---|---|
| Data collection | ||
| Space group | P21212 | P21212 |
| Cell dimensions: | ||
| a, b, c (Å) | 82.0, 97.7, 46.3 | 81.9, 98.1, 46.1 |
| α = β = γ (°) | 90.0 | 90.0 |
| Wavelength (Å) | 1.0332 | 0.9794 |
| Resolution range (Å) | 40.0–2.1 (2.18–2.10) | 20.0–2.0 (2.07–2.00) |
| Rmerge (%) | 7.5 (40.9) | 6.4 (12.9) |
| 〈I〉/〈σI〉 | 24.1(4.3) | 17.4 (10.6) |
| Completeness (%) | 99.8 (99.8) | 95.4 (86.7) |
| Redundancy | 4.4 | 5.9 |
| Refinement | ||
| Resolution range (Å) | 31.4–2.1 (2.17–2.09) | 19.7–2.0 (2.07–1.99) |
| No. reflections in working set | 21,102 | 23,223 |
| Rworka/Rfreeb | 0.172/0.192 | 0.179/0.205 |
| No. of atoms | ||
| Protein | 2483 | 2624 |
| Solvent | 112 | 154 |
| Ligand | 36 | 10 |
| Average B-factor (Å2) | 28.1 | 25.5 |
| Protein | 27.5 | 25.2 |
| Solvent | 30.0 | 30.9 |
| Ligand | 58.8 | 24.7 |
| r.m.s.d. from ideal geometry | ||
| Bond lengths (Å) | 0.012 | 0.012 |
| Bond angles (degrees) | 1.07 | 1.10 |
| Ramachandran plot | ||
| Most favored (%) | 98.7 | 98.7 |
| Allowed (%) | 1.3 | 1.3 |
| Outliers (%) | 0.0 | 0.0 |
| Occupancy of ligandc | ATP(0.90), PO43−(0.80) | SO42−(0.61, 0.39) |
a Rwork = Σ‖Fo| − |Fc‖/Σ|Fo|.
b Rfree was calculated against 5–5.3% of the reflections removed at random.
c Phosphate and sulfate ions are in the individual crystallization conditions.
Figure 3.

Superimposition of complex structures of MDDEF–ATP and MDD from S. epidermidis bound with FMVAPP and ATPγS (MDDS.E.–FMVAPP–ATPγS). A, ribbon model of the crystal structure of MDDEF–ATP is shown in pink with the bound ATP molecule as a stick model. B, ATP molecule is surrounded by the SA-omit map (mFo − DFc at a contour of 3σ, cropped at 5 Å from ATP). Residues (Gln-68, Ser-93, Asn-95, and Ser-105) that form hydrogen bonds with ATP are shown as stick models. The hydrogen bonding partners (Gln-68–O and ATP–O2′, 3.3 Å; Ser-93–Oγ and ATP–N6, 3.0 Å; Asn-95–Oδ and ATP–N6, 2.9 Å; Asn-95–Nδ and ATP–N7, 3.3 Å; Ser-105–N and ATP–Oα, 2.9 Å) are connected by dashed lines. C, overlay of the models of MDDSE–FMVAPP–ATPγS (PDB code 4DPT, green) and MDDEF–ATP (pink) (r.m.s.d. = 0.66 Å) is depicted with the phosphate-binding loops emphasized. D, ligands from two structures are shown as stick models (FMVAPP and ATPγS from MDDSE–FMVAPP–ATPγS; ATP from MDDEF–ATP). The arrows in black indicate the distinct orientations of the phosphate tails of ATPγS and ATP from these two structures.
Structural differences between MDDEF-ATP and MDDSE–FMVAPP–ATPγS infer the pivotal role of MVAPP in ATP binding
By comparing our crystal structure of MDDEF–ATP with the tertiary crystal structure of MDDSE (MDDSE–FMVAPP–ATPγS, PDB code 4DPT) (27), three major differences were found in the phosphate-binding loop (Fig. 3C) and the ATP/ATPγS configurations (Fig. 3D). First, the phosphate-binding loop in MDDEF–ATP remains in an open conformation and does not bend down to form contacts with ATP, if compared with the phosphate-binding loop in MDDSE–FMVAPP–ATPγS (Fig. 3C). The largest distance moved by the backbone atoms in the phosphate-binding loop is 11.1 Å (Cα of Ala-100 in MDDEF–ATP and Cα of Ala-101 in the MDDSE complex structure). Second, the γ-thiophosphate group of ATPγS in MDDSE–FMVAPP–ATPγS is reported to form contacts (hydrogen bonding) with the backbone amide of two residues, Ser-107 and Ala-108 in Helix α2 of MDDSE(106–121) (supplemental Fig. S1A), which are located in the active site; instead, the γ-phosphate of ATP in our structure of MDDEF–ATP is facing out and does not interact with any residues in the molecule (Fig. 3D and supplemental Fig. S1B). Third, the α- and β-phosphates in these two structures are located and oriented differently and thus result in the distinct orientations of the phosphate tails in these structures (Fig. 3D). The adenosine group of ATP in our MDDEF–ATP binds to the ATP-binding pocket with a temperature factor similar to the average B-factor of the protein (Table 1). However, the phosphate tail of ATP does not interact with any residues, consistent with its higher temperature factor (B-factor >60), suggesting ATP does not initially bind to MDDEF in its favored position. These findings lead us to hypothesize the prerequisite binding of MVAPP to MDDEF may be important for forming the ATP-binding pocket. To test the hypothesis, we utilized enzymatic and thermodynamic approaches to determine the substrate-binding order of MDDEF and to examine whether the binding of MVAPP enhances binding of ATP.
Kinetics reveals a compulsory ordered bi-substrate mechanism of MDDEF with MVAPP binding first
The crystal structure of MDDEF–ATP described above implied that the binding of MVAPP may affect ATP binding. The previous work on avian MDD suggests the substrate-binding order of MDD from chickens belongs to an ordered bi-substrate mechanism with MVAPP binding first (28). From our crystal structure of MDDEF–ATP, ATP can bind to the ATP-binding pocket by itself, even if in an unproductive form (Fig. 3, B and D). Therefore, the question arises whether the substrate-binding order is conserved in both eukaryotic and prokaryotic MDD proteins. To address this issue, we performed kinetics studies introducing ATPγS as a competitive inhibitor of ATP to determine the enzyme mechanism of MDDEF. We utilized the coupled enzyme method (pyruvate kinase/lactate dehydrogenase) to determine the enzymatic activity of MDDEF at varying concentrations of the two substrates and inhibitors (25, 29, 32). The detailed procedures are described under “Experimental procedures.”
MDD triggers MVAPP decarboxylation coupled with ATP consumption. MVAPP and ATP are its two substrates. To differentiate whether the reaction belongs to either the ping-pong or the sequential bi-substrate mechanism, we have performed enzymatic reactions at varying concentrations of both substrates of MDDEF, MVAPP, and MgATP (“Experimental procedures”) (Fig. 4, A and C), and we analyzed the kinetic data by the Lineweaver-Burk method (Fig. 4, B and 4D). Each 1/v (reciprocal reaction velocity) was plotted as a function of either 1/[MVAPP] (reciprocal of varying concentrations of MVAPP) (Fig. 4B) or 1/[MgATP] (reciprocal of varying concentrations of MgATP) (Fig. 4D). The data were best represented by a sequential bi-substrate model using the general equation for a bi-substrate enzyme catalysis system as represented in Equation 1,
| (Eq. 1) |
where Vmax is the maximum velocity; [A] is the varying concentration of MVAPP; [B] is the varying concentration of ATP; KIA is KdMVAPP, KB is KmMgATP; and KA is KmMVAPP (33).
Figure 4.

Kinetic analysis of MDDEF. A, set of kinetic data at varying concentrations of MVAPP and several fixed concentrations of MgATP ((●) 50, (○) 100, (▾) 200, (▿) 400, (■) 600, (□) 800, and (♦) 1000 μm) is shown as a Michaelis-Menten plot. B, same data set from A is shown as a Lineweaver-Burk plot. C, data set at varying concentrations of MgATP and several fixed concentrations of MVAPP ((●) 10, (○) 15, (▾) 25, (▿) 50, (■) 100, (□)150, (♦) 200, and (♢) 300 μm) is shown as a Michaelis-Menten plot. D, same data set from C is shown as a Lineweaver-Burk plot. Kinetic data are represented by Equation 1 and used for a sequential bi-substrate model. Each data point represents independent triplicate results, and the error bar for each point indicates standard deviations.
The patterns in Fig. 4, B and D, show that the kinetic curves in the double-reciprocal plots converge to the x axis. We could then confirm that the enzyme mechanism of MDDEF belongs to a sequential bi-substrate mechanism instead of a ping-pong mechanism or a rapid-equilibrium sequential ordered mechanism. From the curve fitting, we obtain the following kinetic parameters: Vmax = 16.1 ± 0.3 μmol/min/mg; kcat = 9.8 ± 0.2 s−1; KmMVAPP = 39.7 ± 2.8 μm; and KmMgATP = 166 ± 12 μm (Table 2).
Table 2.
Enzyme kinetic parameters
The means and the standard deviation of each data point were obtained from a triplicate test (means ± S.D.).
| Vmax | Vmax | kcat | Km, MVAPP | K>m, MgATP | KI, ATPγS | Metal | Inhibition pattern |
|---|---|---|---|---|---|---|---|
| μmol/min/mg | μm/s | s−1 | μm | μm | μm | ||
| 16.1 ± 0.3 | 0.75 ± 0.02 | 9.8 ± 0.2 | 39.7 ± 2.8 | 166 ± 12 | Mg2+ | ||
| 20.4 ± 0.8 | 0.94 ± 0.04 | 12.4 ± 0.6 | 42.6 ± 3.4 | 173 ± 16 | Mg2+ | Uncompetitive | |
| 24.0 ± 0.6 | 1.12 ± 0.2 | 14.6 ± 0.4 | 165 ± 9 | 199 ± 13 | Mg2+ | Competitive |
To determine whether substrates bind to MDDEF in an ordered or random manner, we introduced ATPγS into the reaction buffer as a dead-end inhibitor of MDDEF and then determined the substrate-binding order based on the inhibitory patterns of ATPγS (supplemental Table S2) (28, 33). Two sets of inhibitory assays were conducted. The first set of experiments was performed at a constant MVAPP concentration of 40 μm and varying concentrations of MgATP and ATPγS (see under “Experimental procedures”). The kinetic data from the inhibitory assays were best represented by a modified competitive inhibition model as shown in Equation 2,
| (Eq. 2) |
where Vmax is the maximum velocity; [B] is the varying concentrations of ATP; KB is KmMgATP; KIA is KdMVAPP; KA is KmMVAPP; KIA/KA is 0.8, which was derived from the enzymatic reactions described above; [I] is the varying concentration of ATPγS, and KI is KIAATPγS (33).
The kinetics data are shown as a Michaelis-Menten plot (Fig. 5A) and a Lineweaver-Burk plot (Fig. 5B). The values of Vmax, KmMgATP, and KIATPγS were derived from Equation 2 and are summarized in Table 2.
Figure 5.

Inhibitory assays of MDDEF. A, different fixed concentrations of ATPγS ((●) 0, (○) 100, (▾) 200, or (▿) 400 μm) were added to the reactions at a fixed concentration of MVAPP (40 μm) and varying concentrations of MgATP (100, 150, 200, or 400 μm). The set of inhibitory data is shown as a Michaelis-Menten plot. B, same set of data from A is shown as a Lineweaver-Burk plot. C, different fixed concentrations of ATPγS ((●) 0, (○) 100, (▾) 200, and (▿) 400 μm) were added to the reactions at a fixed concentration of MgATP (200 μm) and varying concentrations of MVAPP (10, 20, 40, and 80 μm). The set of inhibitory data is shown as a Michaelis-Menten plot. D, same set of data from C is shown as a Lineweaver-Burk plot. A and B can be analyzed as a competitive inhibition model; C and D can be analyzed as an uncompetitive inhibition model. The means and the standard deviation of each data point were obtained from a triplicate test. Each data point represents independent triplicate results, and the error bar for each point indicates standard deviations.
The second set of experiments was performed at a constant MgATP concentration of 200 μm and varying concentrations of MVAPP and ATPγS (“Experimental procedures”). The data set was best represented by a modified uncompetitive inhibition model as shown in Equation 3,
| (Eq. 3) |
where Vmax is the maximum velocity; [A] is the varying concentrations of MVAPP; KA is KmMVAPP; KIA is KdMVAPP; KIA/KA is 0.8, which was derived from the enzymatic reactions described above; [I] is the varying concentration of ATPγS, and KI is KIATPγS (33).
The kinetics data are shown as a Michaelis-Menten plot (Fig. 5C) and a Lineweaver-Burk plot (Fig. 5D). The values of Vmax, KmMVAPP, and KIATPγS were also derived from Equation 3 and are summarized in Table 2. According to the inhibitory patterns with the addition of ATPγS in the enzymatic reactions under the designed conditions, we suggest that the enzymatic reaction of MDDEF belongs to a sequential ordered bi-substrate mechanism with MVAPP binding first, and we find the substrate-binding mechanism of MDD to be conserved between the eukaryotic and prokaryotic species in the family of MDD proteins.
Isothermal titration calorimetry (ITC) results suggest an MVAPP-induced ATP-binding mechanism of MDDEF
From the kinetics study on MDDEF, MVAPP is identified as the first substrate in the reaction mechanism. Our complex structure of MDDEF–ATP shows that in the absence of MVAPP, ATP binds the ATP-binding pocket in an unusual configuration (Fig. 3, B and D), and the phosphate-binding loop does not bend down to interact with the ATP molecule (Fig. 3C). These results imply the binding of MVAPP may help form a binding site for ATP in its catalytically favored configuration. In other words, the binding of ATP might be enhanced by the prerequisite binding of MVAPP. To test this, we employed thermodynamic approaches and utilized ITC (34) to determine the dissociation constants of the first substrate, MVAPP, the second substrate, MgATP, and the ATP analogues, ATPγS and AMPPCP.
We have determined the Kd values of substrates or ligands (MVAPP, MgATP, AMPPCP, and ATPγS). The raw data for titrations of MDDEF are shown in Fig. 6. All the derived thermodynamic parameters, Kd (1/Ka), are listed in Table 3. The KdMVAPP value is 20.4 ± 9.3 μm (Fig. 6A); the KdATP value is 288 ± 36 μm (Fig. 6B); and the KdATPγS value is 215 ± 8 μm (Fig. 6C). Next, we determined KdATPγS under the condition in which MDDEF was pre-incubated with MVAPP. MDDEF was incubated with 1 mm MVAPP for 30 min to ensure that the MVAPP-binding pocket of MDDEF was nearly fully occupied (about 97.8%), according to the dissociation constant at equilibrium as shown in Equation 4, where [MDDEF]total is the total concentration of MDDEF; [MVAPP]total is the total concentration of MVAPP; and x is the concentration of the MDDEF–MVAPP complex.
| (Eq. 4) |
Figure 6.

Original titration curves from ITC experiments with MDDEF. A, MDDEF (100 μm) titrated with MVAPP (2 mm). B, MDDEF titrated with ATP (3 mm). C, MDDEF titrated with ATPγS (3 mm). D, MDDEF pre-incubated with MVAPP (1 mm) and then titrated with ATPγS (2 mm). The protein concentration is adjusted to 100 μm, and all the protein and titrants are dissolved in the buffer containing 100 mm HEPES, pH 7, 100 mm KCl, and 10 mm MgCl2.
Table 3.
Thermodynamic parameters
Titration experiments were done at 25 °C. NBD indicates no detectable binding.
| Species | Substrate | Kd | ΔGa | ΔH | TΔSb | ΔS | |
|---|---|---|---|---|---|---|---|
| μm | kcal/mol | kcal/mol | kcal/mol | cal/mol·K | |||
| a | MDDEF (100 μm) | MVAPP (2 mm) | 20.4 ± 9.3 | −6.5 ± 0.3 | −0.7 ± 0.1 | 5.8 ± 0.2 | 19.5 ± 0.5 |
| b | MDDEF (100 μm) | ATP (3 mm) | 288 ± 36 | −4.8 ± 0.1 | −3.8 ± 0.5 | 1.0 ± 0.3 | 3.5 ± 1.0 |
| c | MDDEF (100 μm) | ATPγS (3 mm) | 215 ± 8 | −5.00 ± 0.02 | −6.4 ± 0.3 | −1.4 ± 0.2 | −4.7 ± 0.6 |
| d | MDDEF (100 μm) + MVAPP (1 mm) | ATPγS (2 mm) | 25.4 ± 5.5 | −6.3 ± 0.1 | −4.8 ± 1.1 | 1.5 ± 0.6 | 5.0 ± 2.1 |
| e | MDDEF (100 μm) | AMPPCP (3 mm) | 271 ± 43 | −4.87 ± 0.09 | −3.20 ± 0.50 | 1.67 ± 0.36 | 5.61 ± 1.21 |
| f | MDDEF (100 μm) + MVAPP (1 mm) | AMPPCP (5 mm) | NDB | NDB | NDB | NDB | NDB |
| g | MDDEF (100 μm) +AMPPCP (1 mm) | MVAPP (2 mm) | 18.7 ± 7.4 | −6.50 ± 0.23 | 0.91 ± 0.18 | 7.41 ± 0.20 | 24.88 ± 0.69 |
a The mean of ΔG (μ) is derived from the equation: μ = −RTln(m/√1 + (v/m2)) and the standard deviation of ΔG (σ) is calculated from the equation: σ = RT√ln(1 + (v/m2)), where m is the mean of the association constant (Ka), and v is the variance of Ka derived from each ITC experiment.
b The mean of TΔS (μ1–2) is derived from the difference between the means of ΔG (μ1) and ΔH (μ2) and the standard deviation (σ1–2) of TΔS is derived from the equation: σ1–2 = √(σ12/n + σ22/n), where σ1 is the standard deviation of ΔG and σ2 is the standard deviation of ΔH.
Under these conditions, the Kd value between ATPγS and MDDEF–MVAPP was determined to be 25.4 ± 5.5 μm (Fig. 6D), which is 10-fold less than the Kd value between ATPγS and MDDEF alone, supporting our proposed model in which the pre-binding of MVAPP would strengthen ATPγS binding. Our results show that the binding of ATPγS is enhanced by the prerequisite binding of MVAPP to MDDEF, suggesting the binding of MVAPP would trigger conformational changes of MDDEF to accommodate the ATP molecule in its catalytically favored position for the subsequent chemical steps in the reaction.
Discussion
GHMP family kinases employ a variety of mechanisms and enzyme order. Previously, the substrate-binding mechanism of MDD from chickens was determined to belong to a sequential bi-substrate mechanism with MVAPP as the first substrate (28); from our study, we determined prokaryotic MDD from E. faecalis also binds MVAPP first during the enzymatic reaction. MDD is characterized as a member GHMK of the kinase family (26). For the galactokinase from pig liver, MgATP is the first substrate of the galactokinase, followed by the binding of galactose (35); however, in E. coli, the galactokinase enzyme performs a random bi-substrate mechanism (36). For the homoserine kinase in E. coli, MgATP is preferred to bind to the enzyme first (37). For the mevalonate kinase from hog liver, a sequential ordered bi-substrate mechanism is performed with mevalonate binding first (38). The enzyme mechanism of the phosphomevalonate kinase from pigs belongs to a sequential ordered bi-substrate mechanism with phosphomevalonate binding first (39). In the case of the MDD, the enzyme mechanism may be conserved among the family of proteins; however, in other cases in the GHMP kinase family, the enzyme mechanisms may vary in species. Although in this study we aim at elucidating the intriguing observations from the structure of MDDEF–ATP and the structural basis of MVAPP for assisting ATP binding to MDDEF, alternative substrates or product inhibition strategies at both saturated and unsaturated concentrations of MVAPP and MgATP (40) are also the ways to comprehensively investigate the overall enzyme mechanism of MDDEF.
MDD proteins have been known to trigger the ATP-dependent decarboxylation of MVAPP, but with a requirement for metal ions (Mg2+) to perform catalysis under physiological conditions (29). In our crystal structure of MDDEF–ATP, the ATP molecule does not have magnesium binding to the α- and β-phosphate group. In the crystallization condition, we have 5 mm magnesium chloride and 10 mm ATP in the solution. The buffer condition is 50 mm sodium acetate at pH 4.6, and the protonation state of ATP is mainly HATP3− under this condition (41). The dissociation constant between HATP3− and magnesium is about 1.58 mm (log Ka = 2.79 ± 0.15) (42), and the MgATP concentration under this condition is about 4 mm, suggesting the concentration of free HATP3− is about 6 mm. The reason for not seeing magnesium-bound ATP in the crystal structure of MDDEF–ATP could be due to the competitive binding of free HATP3− to the ATP-binding pocket of MDDEF. It is also possible that a higher average temperature factor of the phosphate tail of ATP results in poorly defined electron density in this region so that we cannot identify a magnesium ion in this structure. In 2012, Barta et al. (27) reported experimental findings on the substrate-binding order of MDDSE. However, they do not identify any metal-binding sites in their crystal structures of MDDSE with substrate analogues. Based on the observations above, we suggest the current structure models of MDD proteins in the PDB do not necessarily represent the final conformation of MDD proteins upon substrate binding and enzyme catalysis. This could also be a problem for structure-based drug design, which is heavily dependent on the structural details in atomic models of macromolecules. To study the structure and function of the MDD family of proteins remains an important but challenging field.
The open conformation of the phosphate-binding loop and the unusual configuration of ATP in our crystal structure of MDDEF–ATP suggest the importance of MVAPP binding in forming the productive ATP-binding site. We proposed that MVAPP could induce ATP binding to MDDEF, and the thermodynamic results are in agreement with our proposal. In addition to the examination of changes of KdATPγS in the presence of excess amounts of MVAPP in the solution, we also tested whether this enhanced binding occurs in the titration experiments using AMPPCP. The KdAMPPCP value was 271 ± 43 μm (Table 3, row e); however, in the presence of MVAPP, we could not detect heat changes and so could not obtain the thermodynamic parameters in this case (Table 3, row f). If instead MDDEF is pre-incubated with excess amounts of AMPPCP (10-fold, MDDEF–AMPPCP complex formation rate: 77.4%) and then the mixture is titrated with MVAPP (Table 3, row g), the KdMVAPP value in this case is 18.7 ± 7.4 μm, which is a similar value as from the MDDEF–MVAPP titration (Table 3, row a), indicating AMPPCP may have little effect on MVAPP binding. We have also done a preliminary test on ATPγS. MDDEF was pre-incubated with excess amounts of ATPγS (10-fold, MDDEF–ATPγS complex formation rate: 81.1%) and then the mixture was titrated with MVAPP (data not shown). The thermogram shows an unusual two-process binding event and reaches saturation with no further heat change after the 6th injection, suggesting a tight binding event occurs. KdMVAPP was roughly determined to be 76 nm (data not shown). Although there are no other experimental results to explain the two-process binding, a quick saturation after the 6th injection indicates that MVAPP binds MDDEF–ATPγS much tighter than MVAPP binds MDDEF alone (Table 3, row a). It also implies that AMPPCP might not be able to access a catalytically-favored conformation for “locking” MVAPP in the active site, which might need, for example, an aid from the conformational changes of MDDEF upon substrate binding. The conserved phosphate-binding loop of the MDD family of proteins functions to interact with the phosphate tail of ATP. It has been shown that AMPPCP has poor inhibitory activity (Ki > 1 mm) against MDD from chickens (28) and MDDEF in our preliminary enzymatic study (data not shown). AMPPCP has a replacement of the bridging oxygen with the methyl group (CH2–) between the β- and γ-phosphorus atoms. We therefore propose that the β-,γ-bridging oxygen of ATP serves as a key checkpoint for the molecular recognition of ATP by MDDEF during the enzymatic reaction.
To summarize our results, we suggest that an induced substrate-binding mechanism of MDD proteins occurs during the enzymatic reaction (Fig. 7), in which the binding of the first substrate, MVAPP, to MDD will trigger the conformational changes of the enzyme to accommodate ATP binding to the ATP-binding pocket in its catalytically favored position, followed by enzyme catalysis and product release. The observation that ATP can bind by itself but in a catalytically unfavorable position may also provide an alternative configuration of the MDDEF enzyme that provides a new structural basis for antibacterial design for pathogenic enterococci.
Figure 7.
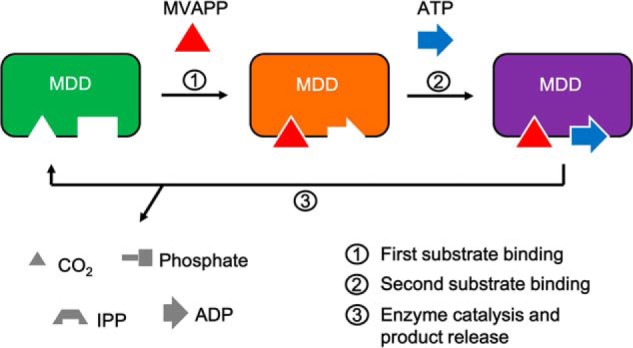
Induced substrate-binding mechanism of MDD proteins. Left panel (green), apo-form of MDD in which two pockets for substrate binding are empty; middle panel (orange), the MVAPP-bound MDD in which the binding of MVAPP (shown in a red triangle) triggers conformational changes of the enzyme and reshapes the ATP-binding pocket, which allows the binding of ATP (shown in a blue arrow) to its catalytically favored position; right panel (purple), two-substrate-bound MDD. Step 1, the binding of MVAPP; step 2, the binding of ATP; step 3, enzyme catalysis and product release. Products in different shapes are shown in gray.
Experimental procedures
Cloning, overexpression, and purification of the recombinant form of MDDEF
A gene fragment encoding MDD from E. faecalis (MDDEF) was amplified via PCR and subcloned into the expression plasmid pET30a (43). Upon confirmation of the DNA sequence, the construct was transformed into E. coli BL21 (DE3). Transformed cells were cultured in LB broth supplemented with kanamycin (50 mg/ml) at 37 °C to an A600 nm of 1.0. Protein expression of MDDEF was induced by addition of isopropyl 1-thio-β-d-galactopyranoside (0.1 mm) for another 4 h at 37 °C.
Cells were harvested by centrifugation at 8000 rpm, resuspended in binding buffer (50 mm sodium phosphate, pH 7.4, 300 mm NaCl, and 10 mm imidazole), and lysed to homogeneity by a French press. His-tagged MDDEF protein was soluble in the supernatant after centrifugation and trapped on a Ni2+-NTA column followed by the elution with increasing concentrations of elution buffer (50 mm sodium phosphate, pH 7.4, 300 mm NaCl, and 300 mm imidazole). Eluted fractions were pooled and desalted against dialysis buffer (25 mm Tris-HCl, pH 8.0, 100 mm NaCl, and 10 mm MgSO4) twice, the first time with β-mercaptoethanol (2-ME) (20 mm) and the second time without 2-ME. The N-terminal His tag was removed from MDDEF by treatment with recombinant TEV protease in dialysis buffer containing 1 mm DTT and 0.5 mm EDTA overnight at room temperature for 4 h followed by a final dialysis procedure at 4 °C in the dialysis buffer without DTT and EDTA. His-tagged TEV and residual His-tagged MDDEF were removed by passing the protein mixture through a nickel affinity resin. The artificial sequence NA remains at the N terminus of MDDEF after the TEV treatment. The purified MDDEF protein solution was concentrated to 8–10 mg/ml by ultrafiltration and stored at −20 °C.
Protein crystallization
The co-crystal of MDDEF in complex with ATP (MDDEF–ATP) grew under the crystallization condition (10 mm ATP, 26% PEG 3350, 50 mm sodium acetate, pH 4.6, and 5 mm MgCl2) by sitting drop vapor diffusion. ATP was added to the protein solution to a final concentration of 10 mm. 1 μl of MDDEF–ATP solution was mixed with 1 μl of the reservoir. Crystals formed after a 2-day equilibrium period. Cryo-protectant (26% PEG 3350, 17% PEG 400, 50 mm sodium acetate, pH 4.6, and 5 mm MgCl2) was prepared and gradually added into the protein/reservoir mixture to prevent ice formation when flash-freezing the crystal in liquid nitrogen.
A stable condition (1.6 m ammonium sulfate, 50 mm sodium acetate, pH 4.6) for growing crystals of the apo-form of MDDEF was obtained and optimized from a commercialized crystallization screening kit (Qiagen, class II, condition A2: 0.1 m sodium acetate, pH 4.5, 2 m ammonium sulfate). Protein solution (8 to 9 mg/ml) was mixed with the reservoir solution at a 1:1 ratio and equilibrated by vapor diffusion of sitting drops at 20 °C for 2 days. In a 96-well plate, there were about 20 wells containing single cuboid crystals with a size of 0.2 to 0.3 mm. Cryo-protectant (25% glycerol, 1.6 m ammonium sulfate, 50 mm sodium acetate, pH 4.6) was prepared and gradually added into the protein/reservoir mixture to prevent ice formation when flash-freezing the crystal in liquid nitrogen.
Data collection and structure determination, refinement analysis, and refine statistics
The diffraction data from crystals of MDDEF co-crystallized with ATP (MDDEF–ATP) and an apo-form of MDDEF (MDDEF) were collected at the 23-ID-D and 19-ID-BM beamlines at the Advanced Photon Source (APS) at Argonne National Laboratory in Chicago. The HKL2000 software was used for data integration, data reduction, and data scaling (44), generating a scale pack reflection file. The space group of the crystal was determined as P21212 (MDDEF–ATP: a, b, c = 82.0, 97.7, 46.3 Å; α, β, γ = 90°; MDDEF: a, b, c = 81.9, 98.1, 46.1 Å; α, β, γ = 90°) by the software Pointless in CCP4 and phenix.phaser (45, 46). The software in CCP4, scalepack2mtz, was then used to convert the scale pack reflection file (.sca) to an MTZ format (.mtz) with R-free flag assigned (5–5.5%) (47).
One molecule was expected in an asymmetric unit by analyzing the Matthews coefficient and the water content in each case (MDDEF–ATP: cell volume,: 370710.5 Å3, molecular mass: 36,489.5 Da, Matthews coefficient: 2.54, % solvent: 51.6; MDDEF: cell volume: 370,279.3 Å3, molecular mass: 36489.5 Da, Matthews coefficient: 2.54, % solvent: 51.6). Phenix.phaser was used to solve the phases and determine the structure of MDDEF. MDDEF and MDDSE share 60% sequence identity. A modified search model (polyalanine) generated from MDD from S. epidermidis (MDDSE, PDB code 3QT5) using CHAINSAW in CCP4 (48) was used to produce initial phases for the co-crystal structure of MDDEF–ATP in molecular replacement with phenix.phaser (45). In each step of molecular replacement, only one solution was found. The rotation function with log-likelihood gain greater than 0, the translation function with Z-score greater than 8, and no violations after the packing analysis also suggest the success of the molecular replacement approach. The phases for determining the crystal structure of the apo-form of MDDEF were produced by molecular replacement with Phenix.phaser (45) using a polyalanine-modified MDDEF–ATP structure (described above) as a search model.
After phase determination, the structure refinement was performed in phenix.refine (45). During structure refinement, strategies for refining the model geometry (XYZ coordinates), atomic positions (Real space), and atomic B-factors (individual B-factors) were chosen, and after the refinement, the missing side chains of the residues in the structural model were manually rebuilt in the graphics program Coot (49) based on observation of the electron density map (2Fo − Fc) and the difference map (Fo − Fc). The simulated-annealing (Cartesian) option was also employed in the first few runs of structure refinement. In phenix, we used the phenix.composit_omit_map software to generate the omit map and used the maps to evaluate the refined structural models. Target function optimization was also chosen for refining the weight between X-ray data and the structural model (optimize X-ray/stereochemistry weight, optimize X-ray/ADP weight). Water molecules were built by either phenix.refine or Coot and inspected in the Coot interface.
The crystallographic information file (.cif) and the PDB format file (.pdb) of ATP were generated using phenix. eLBOW (45). After a few runs of structure refinement without ligands, ATP was manually placed and fitted into the weighted difference electron density maps in Coot (49). Geometry and rotamer outliers were inspected and evaluated after each structure refinement. Finally, ATP was omitted by setting the ligand occupancy to zero or simply removing it from the structure for calculating a simulated-annealing-ligand-omit map in phenix.composite_omit_map, and the ligand-omit map and its corresponding structure were examined in PyMOL (50).
Enzyme kinetics of wild-type MDDEF
Enzymatic activity of MDDEF was determined using an ATP/NADH enzyme-coupled assay. In this assay, ADP produced from the MDD reaction is converted back to ATP by pyruvate kinase, coupled with the conversion of phosphoenolpyruvate to pyruvate, and the pyruvate is then converted to lactate by lactate dehydrogenase, coupled with the oxidation of NADH to NAD+. The oxidation of NADH to NAD+ results in an absorbance decrease at 340 nm. Thus, the ADP production can be detected by measuring the decrease in A340 nm. The MDD enzymatic activity would be proportional to the slope of the continuously declining value of A340 nm. We have utilized this method to determine the enzymatic activity of MDDEF under different conditions for obtaining kinetic parameters of MDDEF. Each reaction was performed at 30 °C under the buffer condition (100 mm HEPES, pH 7.0, 100 mm KCl, 10 mm MgCl2, 0.2 mm NADH, 0.4 mm phosphoenolpyruvate, 4 units of pyruvate kinase, 4 units of lactate dehydrogenase, and 100 nm MDDEF) (27). Initial velocity of each reaction was determined at a range of concentrations of MgATP (50, 100, 150, 200, 400, 600, 800, and 1000 μm) and MVAPP (10, 15, 25, 50, 100, 150, 200, and 300 μm). Final enzymatic parameters, Km and Vmax, were determined by fitting kinetic data to a sequential bi-substrate mechanism model using SigmaPlot12.5/Enzyme Kinetic Module 1.3 (Systat Software, Inc.).
Inhibition assays
ATPγS was used as a competitive inhibitor of ATP for determining the inhibition kinetics of MDDEF. Different fixed concentrations of ATPγS (0, 100, 200 and 400 μm) were added into the reactions versus varying MgATP (50, 100, 200, and 400 μm) and fixed MVAPP (40 μm) or varying MVAPP (10, 20, 40, 80 μm) and fixed MgATP (200 μm). Assays were performed under the conditions as described above. The kinetic data with a fixed MVAPP concentration were fit with a competitive inhibition model; the kinetic data with a fixed MgATP concentration were fit with an uncompetitive inhibition model using SigmaPlot as described previously.
ITC experiments
The preparation of TEV-treated MDDEF was described above. The protein solution was dialyzed against the same buffer as used in the enzymatic reactions described previously (100 mm HEPES, pH 7.0, 100 mm KCl and 10 mm MgCl2). All the buffer solutions in ITC experiments were filtered through an 0.45-μm filter and degassed for 1 h at room temperature. The protein concentration was adjusted to 100 μm (260 μl). Each ligand (MVAPP, ATP, AMPPCP, and ATPγS) was prepared in the same dialysis buffer to avoid buffer mismatch. The concentration of each titrant was optimized in different experiments based on the experimental designs simulated using the MicroCal Origin 7.0 software package, and the final concentration of each ligand was adjusted to 2 or 3 mm. The ITC instrument, MicroCal iTC200, was employed for isothermal titrations in this study, and the reference cell was filled with double-distilled H2O containing 0.01% sodium azide. The experimental temperature was set at 25 °C. Each experimental profile was composed of the addition of an initial aliquot of 0.4 μl, followed by 22 aliquots of 1.8 μl of the substrate or ligand solution. The time interval between two consecutive injections was 180 s. The data were further processed with NITPIC (51) and analyzed using an one-site model in SEDPHAT (52). Figures were generated using GUSSI in SEDPHAT.
Author contributions
C. C.-L. and C. V. S. designed the experiments. J. C. M. cloned the MDD protein. C. C.-L. purified the MDDEF protein for the study on enzymology, thermodynamics, and crystallization. C. C.-L. analyzed data and determined the substrate-binding order and the crystal structures of the target protein. L. N. P. designed and performed the ITC experiments. C. N. S. assisted in solving the MDD X-ray crystal structure. C. C.-L. and C. V. S. collaborated in writing the article.
Supplementary Material
Acknowledgments
We thank Tim Schmidt (Macromolecule Crystallography X-ray Diffraction Lab, Purdue University) for help on the home source X-ray data collection and the Advanced Photon Source (APS) at Argonne National Laboratory in Chicago for access to the beam lines 19-BM-D and 23-ID-D that contributed to the results presented here.
This work was supported by the Purdue Center for Cancer Research and National Institutes of Health Grant P30 CA 023168. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Fig. S1 and Tables S1 and S2.
The atomic coordinates and structure factors (codes 5V2L and 5V2M) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- VRE
- vancomycin-resistant enterococcus
- MVAPP
- mevalonate diphosphate
- FMVAPP
- 6-fluoromevalonate diphosphate
- IPP
- isopentenyl diphosphate
- ATPγS
- adenosine 5′-(γ-thio)triphosphate
- TEV
- tobacco etch virus
- MDD
- mevalonate diphosphate decarboxylase
- MDDEF
- MDD from E. faecalis
- MDDSE
- MDD from S. epidermidis
- ITC
- isothermal titration calorimetry
- AMPPCP
- adenosine 5′-(β,γ-methylenetriphosphate)
- Ni-NTA
- nickel-nitrilotriacetic acid
- 2-ME
- β-mercaptoethanol
- r.m.s.d.
- root mean square deviation.
References
- 1. World Health Organization (2014) Antimicrobial Resistance: Global Report on Surveillance 2014
- 2. Centers for Disease Control and Prevention (2013) Antibiotic Resistance Threats in the United States, Report 2013
- 3. Paulsen I. T., Banerjei L., Myers G. S., Nelson K. E., Seshadri R., Read T. D., Fouts D. E., Eisen J. A., Gill S. R., Heidelberg J. F., Tettelin H., Dodson R. J., Umayam L., Brinkac L., Beanan M., et al. (2003) Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299, 2071–2074 [DOI] [PubMed] [Google Scholar]
- 4. O'Driscoll T., and Crank C. W. (2015) Vancomycin-resistant enterococcal infections: epidemiology, clinical manifestations, and optimal management. Infect. Drug Resist. 8, 217–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Uttley A. H., Collins C. H., Naidoo J., and George R. C. (1988) Vancomycin-resistant enterococci. Lancet 1, 57–58 [DOI] [PubMed] [Google Scholar]
- 6. Leclercq R., Derlot E., Duval J., and Courvalin P. (1988) Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N. Engl. J. Med. 319, 157–161 [DOI] [PubMed] [Google Scholar]
- 7. Barrett J. F. (2000) Linezolid Pharmacia Corp. Curr. Opin. Invest. Drugs 1, 181–187 [PubMed] [Google Scholar]
- 8. Vilhena C., and Bettencourt A. (2012) Daptomycin: a review of properties, clinical use, drug delivery and resistance. Mini Rev. Med. Chem. 12, 202–209 [DOI] [PubMed] [Google Scholar]
- 9. Johnson A. P., Tysall L., Stockdale M. V., Woodford N., Kaufmann M. E., Warner M., Livermore D. M., Asboth F., and Allerberger F. J. (2002) Emerging linezolid-resistant Enterococcus faecalis and Enterococcus faecium isolated from two Austrian patients in the same intensive care unit. Eur. J. Clin. Microbiol. Infect. Dis. 21, 751–754 [DOI] [PubMed] [Google Scholar]
- 10. Burleson B. S., Ritchie D. J., Micek S. T., and Dunne W. M. (2004) Enterococcus faecalis resistant to linezolid: case series and review of the literature. Pharmacotherapy 24, 1225–1231 [DOI] [PubMed] [Google Scholar]
- 11. Munoz-Price L. S., Lolans K., and Quinn J. P. (2005) Emergence of resistance to daptomycin during treatment of vancomycin-resistant Enterococcus faecalis infection. Clin. Infect. Dis. 41, 565–566 [DOI] [PubMed] [Google Scholar]
- 12. Weigel L. M., Clewell D. B., Gill S. R., Clark N. C., McDougal L. K., Flannagan S. E., Kolonay J. F., Shetty J., Killgore G. E., and Tenover F. C. (2003) Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 302, 1569–1571 [DOI] [PubMed] [Google Scholar]
- 13. Sundsfjord A., and Willems R. (2010) Enterococcus research: recent developments and clinical challenges. Clin. Microbiol. Infect. 16, 525–526 [DOI] [PubMed] [Google Scholar]
- 14. Arias C. A., and Murray B. E. (2012) The rise of the Enterococcus: beyond vancomycin resistance. Nat. Rev. Microbiol. 10, 266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilding E. I., Brown J. R., Bryant A. P., Chalker A. F., Holmes D. J., Ingraham K. A., Iordanescu S., So C. Y., Rosenberg M., and Gwynn M. N. (2000) Identification, evolution, and essentiality of the mevalonate pathway for isopentenyl diphosphate biosynthesis in Gram-positive cocci. J. Bacteriol. 182, 4319–4327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goldstein J. L., and Brown M. S. (1990) Regulation of the mevalonate pathway. Nature 343, 425–430 [DOI] [PubMed] [Google Scholar]
- 17. Boronat A., and Rodríguez-Concepción M. (2015) Terpenoid biosynthesis in prokaryotes. Adv. Biochem. Eng. Biotechnol. 148, 3–18 [DOI] [PubMed] [Google Scholar]
- 18. Sacchettini J. C., and Poulter C. D. (1997) Creating isoprenoid diversity. Science 277, 1788–1789 [DOI] [PubMed] [Google Scholar]
- 19. Buhaescu I., and Izzedine H. (2007) Mevalonate pathway: a review of clinical and therapeutical implications. Clin. Biochem. 40, 575–584 [DOI] [PubMed] [Google Scholar]
- 20. Ruiz N. (2015) Lipid flippases for bacterial peptidoglycan biosynthesis. Lipid Insights 8, 21–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Inokoshi J., Nakamura Y., Komada S., Komatsu K., Umeyama H., and Tomoda H. (2016) Inhibition of bacterial undecaprenyl pyrophosphate synthase by small fungal molecules. J. Antibiot. 69, 798–805 [DOI] [PubMed] [Google Scholar]
- 22. Ramachandran C. K., and Shah S. N. (1976) Decarboxylation of mevalonate pyrophosphate is one rate-limiting step in hepatic cholesterol synthesis in suckling and weaned rats. Biochem. Biophys. Res. Commun. 69, 42–47 [DOI] [PubMed] [Google Scholar]
- 23. Andreassi J. L. 2nd., Dabovic K., and Leyh T. S. (2004) Streptococcus pneumoniae isoprenoid biosynthesis is downregulated by diphosphomevalonate: an antimicrobial target. Biochemistry 43, 16461–16466 [DOI] [PubMed] [Google Scholar]
- 24. Bloch K., Chaykin S., Phillips A. H., and De Waard A. (1959) Mevalonic acid pyrophosphate and isopentenylpyrophosphate. J. Biol. Chem. 234, 2595–2604 [PubMed] [Google Scholar]
- 25. Qiu Y., Gao J., Guo F., Qiao Y., and Li D. (2007) Mutation and inhibition studies of mevalonate 5-diphosphate decarboxylase. Bioorg. Med. Chem. Lett. 17, 6164–6168 [DOI] [PubMed] [Google Scholar]
- 26. Krepkiy D., and Miziorko H. M. (2004) Identification of active site residues in mevalonate diphosphate decarboxylase: implications for a family of phosphotransferases. Protein Sci. 13, 1875–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barta M. L., McWhorter W. J., Miziorko H. M., and Geisbrecht B. V. (2012) Structural basis for nucleotide binding and reaction catalysis in mevalonate diphosphate decarboxylase. Biochemistry 51, 5611–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jabalquinto A. M., and Cardemil E. (1989) Substrate binding order in mevalonate 5-diphosphate decarboxylase from chicken liver. Biochim. Biophys. Acta 996, 257–259 [DOI] [PubMed] [Google Scholar]
- 29. Jabalquinto A. M., and Cardemil E. (1987) Kinetic effects of ATP, divalent metal-ions and pH on chicken liver mevalonate 5-diphosphate decarboxylase. Biochim. Biophys. Acta 916, 172–178 [DOI] [PubMed] [Google Scholar]
- 30. Sillitoe I., Lewis T. E., Cuff A., Das S., Ashford P., Dawson N. L., Furnham N., Laskowski R. A., Lee D., Lees J. G., Lehtinen S., Studer R. A., Thornton J., and Orengo C. A. (2015) CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 43, D376–D381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Finn R. D., Coggill P., Eberhardt R. Y., Eddy S. R., Mistry J., Mitchell A. L., Potter S. C., Punta M., Qureshi M., Sangrador-Vegas A., Salazar G. A., Tate J., and Bateman A. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barta M. L., Skaff D. A., McWhorter W. J., Herdendorf T. J., Miziorko H. M., and Geisbrecht B. V. (2011) Crystal structures of Staphylococcus epidermidis mevalonate diphosphate decarboxylase bound to inhibitory analogs reveal new insight into substrate binding and catalysis. J. Biol. Chem. 286, 23900–23910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism and Data Analysis, 2nd Ed., pp. 352–359, John Wiley & Sons, Inc., New York [Google Scholar]
- 34. Jelesarov I., and Bosshard H. R. (1999) Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J. Mol. Recognit. 12, 3–18 [DOI] [PubMed] [Google Scholar]
- 35. Ballard F. J. (1966) Kinetic studies with liver galactokinase. Biochem. J. 101, 70–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gulbinsky J. S., and Cleland W. W. (1968) Kinetic studies of Escherichia coli galactokinase. Biochemistry 7, 566–575 [DOI] [PubMed] [Google Scholar]
- 37. Shames S. L., and Wedler F. C. (1984) Homoserine kinase of Escherichia coli: kinetic mechanism and inhibition by l-aspartate semialdehyde. Arch. Biochem. Biophys. 235, 359–370 [DOI] [PubMed] [Google Scholar]
- 38. Soler M., Jabalquinto A. M., and Beytía E. (1979) Hog liver mevalonate kinase: inactivation by pyridoxal-5′-phosphate and evidence of dead-end inhibition by one of the substrates. Int. J. Biochem. 10, 931–935 [DOI] [PubMed] [Google Scholar]
- 39. Eyzaguirre J., Valdebenito D., and Cardemil E. (2006) Pig liver phosphomevalonate kinase: kinetic mechanism. Arch. Biochem. Biophys 454, 189–196 [DOI] [PubMed] [Google Scholar]
- 40. Leskovac V. (2003) Comprehensive Enzyme Kinetics, pp. 171–182, Kluwer Academic/Plenum Publishers, New York [Google Scholar]
- 41. Alberty R. A., Smith R. M., and Bock R. M. (1951) The apparent ionization constants of the adenosine phosphates and related compounds. J. Biol. Chem. 193, 425–434 [PubMed] [Google Scholar]
- 42. Pecoraro V. L., Hermes J. D., and Cleland W. W. (1984) Stability constants of Mg2+ and Cd2+ complexes of adenine nucleotides and thionucleotides and rate constants for formation and dissociation of MgATP and MgADP. Biochemistry 23, 5262–5271 [DOI] [PubMed] [Google Scholar]
- 43. Mermoud J., Stauffacher C., V., Charbonneau H., and Friedman A. (2009) Characterization of Mevalonate Diphosphate Decarboxylase and Isopentenyl Diphosphate Isomerase in Enterococcus Faecalis. M.Sc. thesis, Purdue University, West Lafayette, IN [Google Scholar]
- 44. Otwinowski Z., and Minor W. (1997) 20 Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 45. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 47. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stein N. (2008) CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643 [Google Scholar]
- 49. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 50. Schrödinger, LLC (2015) The PyMOL Molecular Graphics System. Version 1.5.0.4, Schrödinger, LLC, New York [Google Scholar]
- 51. Keller S., Vargas C., Zhao H., Piszczek G., Brautigam C. A., and Schuck P. (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84, 5066–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Houtman J. C., Brown P. H., Bowden B., Yamaguchi H., Appella E., Samelson L. E., and Schuck P. (2007) Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: Application to adaptor protein complexes in cell signaling. Protein Science 16, 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.