

Abstract
Histone deacetylases (HDACs) catalyze deacetylation of acetyl-lysine residues within proteins. To date, HDAC substrate specificity and selectivity have been largely estimated using peptide substrates. However, it is unclear whether peptide substrates accurately reflect the substrate selectivity of HDAC8 toward full-length proteins. Here, we compare HDAC8 substrate selectivity in the context of peptides, full-length proteins, and protein–nucleic acid complexes. We demonstrate that HDAC8 catalyzes deacetylation of tetrameric histone (H3/H4) substrates with catalytic efficiencies that are 40–300-fold higher than those for corresponding peptide substrates. Thus, we conclude that additional contacts with protein substrates enhance catalytic efficiency. However, the catalytic efficiency decreases for larger multiprotein complexes. These differences in HDAC8 substrate selectivity for peptides and full-length proteins suggest that HDAC8 substrate preference is based on a combination of short- and long-range interactions. In summary, this work presents detailed kinetics for HDAC8-catalyzed deacetylation of singly-acetylated, full-length protein substrates, revealing that HDAC8 substrate selectivity is determined by multiple factors. These insights provide a foundation for understanding recognition of full-length proteins by HDACs.
Keywords: enzyme kinetics, enzyme turnover, histone, histone acetylation, histone deacetylase (HDAC), nucleosome, peptides, protein complex, non-natural amino acid incorporation
Introduction
The histone deacetylase (HDAC)4 family of enzymes includes 18 proteins that catalyze the hydrolysis of the acetyl moiety from acetyl-lysine residues within substrate proteins (1, 2). Protein acetylation, catalyzed by lysine acetyltransferases (KATs), alters various properties of the modified protein (e.g. protein–protein interactions) (3). These alterations can in turn affect downstream cellular events (4, 5). As a result, regulation of acetylation by the respective activities of KATs and HDACs is important for effective cellular signaling and homeostasis; aberrant acetylation/deacetylation is implicated in pathologies ranging from neurological diseases (6, 7) to cancers (8, 9). HDAC inhibitors have been approved by the Food and Drug Administration for the treatment of T-cell lymphomas and multiple myeloma (10–12). Identifying the specific substrate set for each HDAC isozyme is important for understanding the role of HDACs in disease progression and therapeutic development.
To date, testing HDAC substrate specificity has remained a challenge, due in part to HDAC isozyme interchangeability and promiscuity and the inherent difficulty in measuring the disappearance of signal from a previously modified substrate. To mitigate these difficulties, the HDAC field has sought to identify sequence motifs that define the substrate selectivity of each isozyme (13–16). Most of these studies have used peptide substrates to determine HDAC recognition motifs. However, the use of peptides to mimic recognition of protein substrates has not been sufficiently validated. Gurard-Levin and Mrksich (15) proposed an exosite model, in which HDAC8 contains one or more substrate-binding surfaces away from the active site, after observing HDAC8 sequence selectivity distal to the acetyl-lysine in H4-based peptides longer than 20 amino acids. However, outside of qualitative experiments (17, 18), there has been no kinetic characterization of HDAC-catalyzed deacetylation of protein substrates. HDAC8 is the best-characterized HDAC, with numerous crystal structures (19–27), kinetic studies (28–30), and peptide substrate specificity measurements (13–16), providing an important background for the investigation of HDAC activity with protein substrates. Although several putative HDAC8 substrates have been identified by cellular studies, including overexpression (e.g. estrogen-related receptor α (31)), genetic mutation (e.g. structural maintenance of chromosome 3 (SMC3) (32, 33)), and proteomic studies (e.g. AT-rich interaction domain 1A (ARID1A) (34)), the complete protein substrate set for HDAC8 is largely undefined (31, 32, 35–40).
Histones are putative substrates for HDAC8. In HEK293 cells, H3 and H4 acetylation levels decrease upon overexpression of HDAC8 (41). Moreover, treatment with the HDAC8-specific suberanilohydroxamic acid-pyrrole-imidazole polyamide derivative inhibitor Jδ increased acetylated H3 levels and expression of HDAC8-regulated transcription factors in mouse embryonic fibroblasts (42). Furthermore, HDAC8 catalyzes deacetylation of core histones and H3-based peptides in vitro (43, 44); however, detailed kinetics have not previously been determined.
Recently, substrate specificity for the Saccharomyces cerevisiae lysine acetyltransferase piccolo NuA4 has been measured using histone protein substrates, demonstrating acetylation of multiple lysine residues (45). However, HDAC substrate specificity studies to date have utilized acetylated peptides (13–15, 46–50), predicated on the assumption that HDAC8 uses similar interactions to recognize peptide and full-length protein substrates. To test the validity of this assumption, we determined the deacetylation kinetics of peptides corresponding to three biologically relevant acetylation sites on the putative HDAC8 substrate histone H3 (H3 Lys-9, Lys-14, and Lys-56 (51, 52)), and we compared the values to HDAC8-catalyzed deacetylation of full-length histone substrates.
To elucidate HDAC8 substrate specificity and recognition of protein substrates, we present the first detailed kinetic study of HDAC-catalyzed deacetylation of singly-acetylated, full-length protein substrates. Single acetyl-lysine side chains are inserted into H3 using non-natural amino acid incorporation (53, 54). We directly compare HDAC8 activity toward peptide substrates and toward protein substrates with the same local primary sequences. Furthermore, we analyze the effect of large histone complexes (histone core octamer and mononucleosome) on HDAC8-catalyzed deacetylation of acetylated H3. We demonstrate that deacetylation of acetylated full-length H3 tetramer and octamer complexes catalyzed by HDAC8 is significantly faster (>8-fold) than that of acetylated peptides. However, the addition of DNA to form mononucleosomes decreases reactivity with HDAC8. These results demonstrate that HDAC8 specificity for H3 peptide tetramer substrates is not determined solely based on the six amino acids proximal to the acetyl-lysine; substrate specificity of HDAC8 is modulated by both long-range and short-range contacts for H3 substrates.
Results
Local sequence governs HDAC8 peptide specificity
We focused on the activity of HDAC8 with three acetylated lysine sites within histone H3, a histone known to be amenable to non-natural acetyl-lysine incorporation (54). Two H3 acetylation sites (H3K9ac and H3K14ac) are located within proximity to each other on the N-terminal tail and share an unfolded secondary structure. Because these sites differ only in amino acid sequence, the role of primary sequence in HDAC8 substrate specificity can be probed. A third site (H3K56ac), located on an α-helix within the globular structure of H3 (Fig. 1), allows the role of secondary structure in HDAC8 substrate recognition to be probed.
Figure 1.
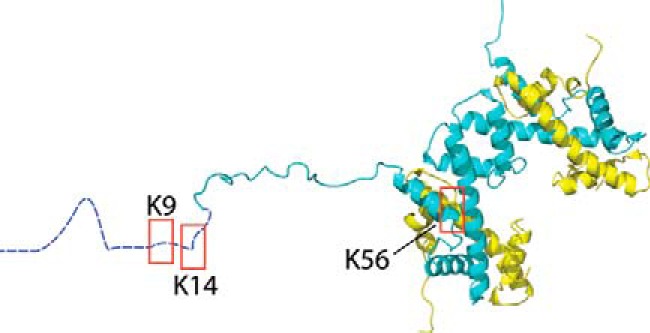
Shown is the structure of histone H3/H4 tetramer with highlighted acetylation sites. Shown is the structure of histone H3/H4 tetramer (62) with boxes around the sites that were acetylated. H3 is shown in blue and H4 in yellow. H3 residues 1–20 are shown in an extended conformation as they have no discrete fold within the crystal structure. The structure was generated from Protein Data Bank code 1AOI using VMD.
The rates of HDAC8-catalyzed deacetylation of 7-mer peptides representing the three amino acids upstream and downstream of the H3K9ac, H3K14ac, and H3K56ac acetylation sites were measured under multiple turnover (MTO) conditions, using an assay coupling acetyl-lysine deacetylation to the formation of NADH (Table 1) (55). The initial rates were linearly dependent on peptide concentration, indicating that the Km values are higher than the peptide concentrations used in this assay (>100 μm). The specificity constant (kcat/Km) is the best parameter to use for comparing the activity of HDAC8 toward multiple substrates (56–58). HDAC8 has the highest catalytic efficiency for catalyzing hydrolysis of the H3K56ac peptide (kcat/Km = 78 ± 8.0 m−1 s−1), followed by the H3K9ac (56 ± 6.0 m−1 s−1) and H3K14ac (8.0 ± 0.70 m−1 s−1) peptides (Table 2).
Table 1.
Sequences of peptides used in this study
The amino acid sequences of the peptides assayed are provided. Kac represents the acetyl-lysine residue. All peptides contain N-terminal acetyl and C-terminal carboxamide moieties.
| Peptide | 7-mer peptide sequence | 13-mer peptide sequence | 17-mer peptide sequence |
|---|---|---|---|
| H3K9ac | TARKacSTG | TKQTARKacSTGGKA | ARTKQTARKacSTGGKAPR |
| H3K14ac | TGGKacAPR | RKSTGGKacAPRKQL | |
| H3K56ac | RYQKacSTE | EIRRYQKacSTELLI |
Table 2.
Catalytic efficiencies for deacetylation of histone substrates by HDAC8
HDAC8 activity was measured, and catalytic efficiencies were determined as described under “Experimental procedures” and the legends of Figs. 2–4.
| Substrate | 7-mer peptide | 13-mer peptide | 17-mer peptide | Tetramer | Octamer | Nucleosome |
|---|---|---|---|---|---|---|
| kcat/Km (m−1 s−1) | kmax/K½ (m−1 s−1) | |||||
| H3K9ac | 56 ± 6 | 51 ± 3 | 120 ± 11 | >17,000 | 3,700 ± 100 | 28 ± 3 |
| H3K14ac | 8.0 ± 0.7 | 21 ± 4 | 2,500 ± 70 | 1,000 ± 200 | ||
| H3K56ac | 78 ± 8 | 100 ± 10 | 4,000 ± 600 | |||
To probe the importance of amino acids at further distances from the acetyl-lysine in determining substrate selectivity, longer peptides (13 and 17 amino acids) were assayed (Table 1). Increasing the length of the peptides from 7 to 13 amino acids had little to no effect on catalytic efficiency (1–3-fold change) and did not affect the substrate selectivity trend of K56ac > K9ac > K14ac. A 17-amino acid peptide representing the H3K9ac site also showed less than a 3-fold increase in kcat/Km values compared with the 7-amino acid peptide (56 ± 6.0 m−1 s−1 versus 120 ± 11 m−1 s−1; Table 2). The modest differences in activity toward the longer peptides indicate that the primary sequence surrounding the acetylated lysine residue (±3 of the acetyl-lysine) is the largest determinant of selectivity in peptide substrates, consistent with previously published data (13–16).
HDAC8 catalyzes deacetylation of H3/H4 tetramers more efficiently than corresponding peptides
To investigate the importance of long-range HDAC8–substrate interactions in substrate recognition, we compared the rates of HDAC8-catalyzed deacetylation of peptide and the corresponding full-length protein. A major challenge in identifying HDAC substrates is determining the rates of deacetylation for individual acetyl-lysine sites, because HDAC substrates, such as histones, may have multiple acetylated lysine residues. We prepared proteins with single acetyl-lysine sites using the method of recombinant, non-natural amino acid incorporation developed by Chin and co-workers (53, 54). In each case, Q-TOF LC/MS of modified histone H3 demonstrated a mass change corresponding to an added acetylated lysine (data not shown). To stabilize H3, H3 was assembled into an H3/H4 tetramer. We measured HDAC8 activity toward the singly-acetylated H3 proteins acetylated at the H3K9, H3K14, and H3K56 sites under single turnover (STO) conditions (3–15 μm HDAC8 and 0.5 μm acetylated H3/H4 tetramer) and assayed deacetylation by MS analysis. STO experiments were used to minimize the amount of singly-acetylated H3/H4 tetramer needed. An exponential decay was fit to the reaction progress curves to determine the observed rate constants, kobs (Fig. 2A). The observed rate of deacetylation of the H3K9ac/H4 tetramer was independent of the HDAC8 concentrations used in these assays indicating that the enzyme concentration is above the K½ for the reaction, even at the lowest concentration (3 μm). In contrast, the H3K14ac/H4 and H3K56ac/H4 tetramers show hyperbolic and nearly linear dependence, respectively, on the HDAC8 concentration. Assuming rapid equilibration of the HDAC8–H3/H4 complex, a hyperbolic fit to these data yields values of kmax/K½ equal to >17,000, 2,500 ± 70, and 4,000 ± 600 m −1 s−1 for the H3K9ac, H3K14ac, and H3K56ac tetramers, respectively (Fig. 2, B–D and Table 2). H3K9ac tetramer deacetylation is an order of magnitude faster than H3K56ac, followed by H3K14ac deacetylation. Each of these catalytic efficiencies is 40–300-fold faster than the corresponding peptide kcat/K½ values.
Figure 2.

Single turnover deacetylation of singly-acetylated H3/H4 tetramers. A, sample data from a deacetylation reaction (7 μm HDAC8 and 0.5 μm H3K9ac/H4 tetramer (1 μm acetyl-lysine)) measured using mass spectrometry. The time-dependent decrease in acetylated protein is best described by a single exponential. B, dependence of apparent deacetylation rate constant of H3K9ac/H4 on the concentration of HDAC8. The kobs average of 0.021 ± 0.001 s−1 shows little dependence on the [HDAC8]. Three separate hyperbolic fits are shown that bracket potential K½ values: K½ = 0.5 μm (dotted line); K½ = 1.0 μm (dashed line); and K½ = 1.5 μm (solid line). These fits demonstrate that the K½ is <1.5 μm and kmax/K½ is >17,000 m−1 s−1. The data points are from multiple measurements in a single reaction at each HDAC8 concentration. C, dependence of the deacetylation rate constant for H3K14ac/H4 on the concentration of HDAC8. The data points are from multiple measurements in a single reaction at each HDAC8 concentration. A hyperbolic fit indicates that the kmax/K½ is 2,500 ± 70 m−1 s−1 with an estimated value for kmax of 0.06 s−1. D, dependence of the deacetylation rate constant for H3K56ac/H4 on the concentration of HDAC8. The data points are from multiple measurements in a single reaction at each HDAC8 concentration. A linear fit indicates that the kmax/K½ is 4,000 ± 600 m−1 s−1.
The specificity constants indicate that substrate selectivity of HDAC8 for these H3 sites varies for the peptide (K56ac ∼ K9ac > K14ac) and protein (K9ac ≫ K56ac > K14ac) substrates. The values of kmax/K½ (measures binding through deacetylation) and kcat/Km (measures binding through dissociation) can be directly compared if product release is not rate-limiting under multiple turnover conditions. Previous data suggest that the deacetylation step is likely the rate-limiting step (see “Discussion”), also suggesting that K½ and Km reflect KD values (28, 44). To validate our assumption in comparing the STO and MTO data, we assayed the 13-mer H3K9ac peptide under both STO and MTO conditions. MALDI-MS was used to measure HDAC8-catalyzed deacetylation of the peptide, due to the high enzyme concentration and sample size constraints in the enzyme-coupled peptide deacetylation assay. Using this method, the rate constant, kmax/K½, is 153 m−1 s−1 (data not shown). This rate constant is within 3-fold of the value of the kcat/Km of 51 ± 3 m−1 s−1 measured by the fluorescence-based peptide deacetylation assay. These data suggest that the STO measurements for the peptide substrates could be increased modestly compared with the MTO data but not enough to explain the increased reactivity of the protein substrates.
Comparison of the multiple turnover data for peptides and the single turnover data measured for full-length proteins demonstrates that interactions outside of short peptide sequences are important for directing HDAC8 substrate selectivity and enhancing catalytic efficiency. Further analysis demonstrates that the H3 peptides have Km values higher than 100 μm (data not shown), whereas the H3K9ac/H4, H3K14ac/H4, and H3K56/H4 tetramers have K½ values of <1.5, 19 ± 1, and >11 μm, respectively. Therefore, one factor leading to the increase in catalytic efficiency is a decrease in the value of K½ in the STO reactions relative to Km for the peptides, suggesting enhanced binding affinity of the protein substrates. These differences suggest that long-range interactions enhance activity of HDAC8 toward full-length substrates.
The catalytic efficiency of HDAC8 toward its substrates is enhanced for all three H3Kac sites tested in the context of the tetramer compared with the corresponding peptides. However, relative HDAC8 activity for the tetramer sites compared with the peptides is different. The largest observed enhancement in catalytic efficiency is for the H3K9ac substrates (140–300-fold increase with the tetramer substrate), followed by H3K14ac (120–300-fold) and then H3K56ac (40–50-fold). In particular, the modest selectivity of HDAC8-catalyzed deacetylation of H3K56ac peptide compared with H3K9ac peptides is not maintained in the tetramer substrates, as would be expected if local sequence was the only determinant of substrate recognition.
Octamer substrates less reactive than tetramers
To further examine full-length substrate selectivity, we measured the deacetylase activity of HDAC8 toward histone octamer complexes containing single acetylation sites. We compared the local sequence of the best and worst tetramer substrates, H3K9ac and H3K14ac, in the context of the complete histone octamer. Histone octamers were reconstituted with two copies of each core histone (H2A, H2B, singly-acetylated H3, and H4). The deacetylation rate catalyzed by HDAC8 was measured under single turnover conditions and analyzed as described for the tetramer. The resulting kobs values for H3K9ac octamer are linearly dependent on the HDAC8 concentration (Fig. 3), yielding a kmax/K½ value of 3,700 ± 100 m−1 s−1. Deacetylation of the H3K14ac octamer has a hyperbolic dependence on HDAC8 concentration leading to a value of kmax/K½ of 1,000 ± 200 m−1 s−1. This catalytic efficiency is decreased 3-fold compared with the H3K14ac/H4 tetramer and is 40–120-fold faster than the deacetylation of H3K14ac peptides. The catalytic efficiency for the H3K9ac octamer site is decreased 4-fold compared with that of the H3K9ac tetramer.
Figure 3.

Single turnover deacetylation of singly-acetylated H3 octamers. A, dependence of the apparent deacetylation rate constant of H3K9ac octamer on the concentration of HDAC8. Data points are from multiple measurements in a single reaction at each HDAC8 concentration, and error bars on kobs values represent errors calculated from the exponential fits. A linear fit of the data indicates that the kmax/K½ is 3,700 ± 100 m−1 s−1. B, dependence of the apparent deacetylation rate constant of H3K14ac octamer on the concentration of HDAC8. The data points are from multiple measurements in a single reaction at each HDAC8 concentration. A hyperbolic fit indicates that the kmax/K½ is 1,000 ± 200 m−1 s−1 with a kmax value of 0.03 ± 0.02 s−1.
HDAC8-catalyzed deacetylation of acetylated nucleosome is slow
HDACs involved in transcriptional regulation are likely to encounter nucleic acid-bound substrate proteins. To test the selectivity of HDAC-catalyzed deacetylation for a substrate complex containing nucleic acids, we incorporated H3K9ac into recombinant mononucleosomes. Deacetylation of these complexes was assayed in the same manner as the tetramer and octamer substrates (Fig. 4). The addition of nucleic acids to the octamer to assemble nucleosomal substrates significantly decreased HDAC8 catalytic efficiency at the H3K9ac site, kmax/K½ = 28 ± 3 m−1 s−1. This is 2-fold slower than the kcat/Km value for the H3K9ac 7-mer peptide and 600-fold slower than deacetylation of this site in the H3/H4 tetramer. Adding the nucleosomal DNA to a Fluor-de-Lys peptide deacetylation assay resulted in an initial rate that is decreased only 25% compared with that of HDAC8 and peptide alone (data not shown); thus, the 130-fold decrease in HDAC8 activity observed between octamer and nucleosome substrates is not explained by DNA inhibition of the enzyme.
Figure 4.
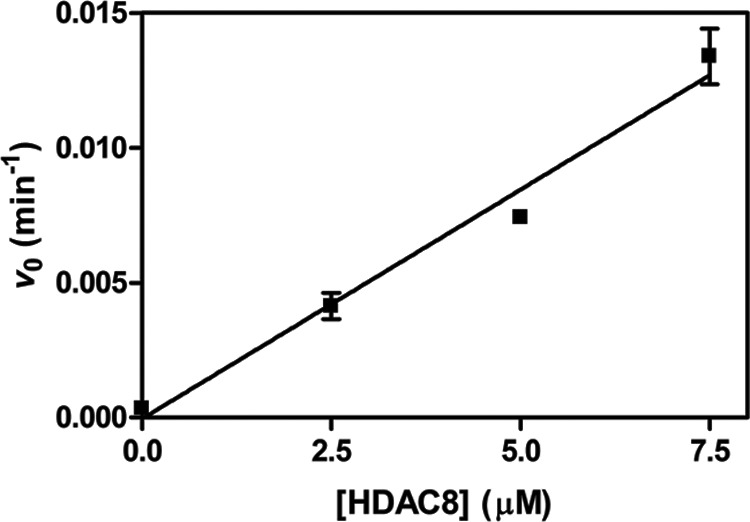
Single turnover deacetylation of singly-acetylated H3 nucleosome. The initial rate of progress curves for deacetylation of H3K9ac nucleosome catalyzed by 0–7.5 μm HDAC8 was fit linearly, and the rate constant was calculated assuming 100% deacetylated product. The data points are from multiple measurements in a single reaction at each HDAC8 concentration, and error bars represent errors calculated from the initial rate fits. A linear fit of the data indicates that kmax/K½ is 28 ± 3 m−1 s−1.
Discussion
To understand the role of HDACs in cellular regulation, it is important to determine the substrate specificity and the molecular determinants of substrate recognition for each isozyme. Until now, HDAC recognition of protein substrates has largely been tackled by studying activity toward peptide substrates, which typically interact with less than an 8 × 20-Å2 area of an ∼2,025-Å2 binding surface (22). Within this larger binding interface, there may be many more HDAC8–protein substrate contacts, including potential recognition hot spots and negative interaction sites. With a peptide, a single interaction of 0.5–2 kcal/mol can alter the catalytic efficiency by 50-fold (55). With a protein substrate, the increased number of interaction sites could overcome the several kilocalorie/mol interaction energy obtained from local contacts. Previously, distal HDAC8–substrate interactions have been observed using long peptide substrates; an upstream KRHR motif (based on histone H4) increases HDAC8-catalyzed deacetylation of an acetylated peptide (15). To elucidate the role of long-range interactions on HDAC8 substrate recognition, we measured HDAC8-catalyzed deacetylation of substrates of increasing size and complexity, from peptide to full-length protein and protein–nucleic acid complex.
To analyze HDAC8-catalyzed deacetylation of peptide and protein substrates, we compared multiple turnover reactions (kcat/Km) of peptide substrates to single turnover (kmax/K½) reactions of the protein substrates. This comparison of kcat/Km to kmax/K½ was due mainly to the challenge of preparing sufficient quantities of singly-acetylated protein substrates to measure under MTO conditions. A variety of data suggest that the kinetic mechanism for deacetylation of most peptide substrates under MTO conditions is rapid equilibrium binding followed by a slow hydrolytic step. For example, our peptide assay demonstrated comparable deacetylation rate constants (within 3-fold) under MTO and STO conditions. Additionally, HDAC8 catalyzes deacetylation of trifluoroacetate peptide substrates faster than non-fluorinated peptides (kcat), indicating that product release is not the main rate-limiting step (44). Furthermore, the kcat/Km values for peptides are significantly slower than diffusion control (102–103 m−1 s−1 versus 107–108 m−1 s−1), and the Km values are large (>100 μm), suggesting that a step other than substrate association, such as hydrolysis, is the rate-limiting step. Similarly, the values of kmax/K½ are significantly lower than diffusion-controlled values suggesting that a step after association and at or before hydrolysis is the rate-limiting step under STO conditions. Based on these data, we assume that the apparent second-order rate constants determined under MTO and STO conditions characterize the same hydrolytic step.
HDAC8 has a remarkably enhanced catalytic efficiency for protein substrates in comparison with the corresponding peptides. The varied HDAC8 catalytic efficiencies likely reflect the interactions between HDAC8 and substrate residues surrounding the acetyl-lysine, as demonstrated previously (15), differences in accessibility of the acetyl-lysine to the active site, and distal interactions between HDAC8 and protein substrates. Previous analysis of activity toward acetylated peptide substrates has shown that HDAC8 prefers substrates with aromatic amino acids on the C-terminal side of the acetyl-lysine (+1 position) (13, 14). Based on these empirical data, the mediocre catalytic efficiency of the histone H3-based peptides (10 to 102 m−1 s−1) (Table 2) was predictable due to the lack of aromatic residues. The interactions between the 7-mer peptides and HDAC8 occur within an ∼10-Å radius of the active site. HDAC8-catalyzed hydrolysis of the acetylated H3/H4 tetramers, which still lack an aromatic residue in the +1 position, is 40–400 times faster than the corresponding peptides. Thus, HDAC8–tetramer interactions that are absent with the peptide substrates enhance HDAC8 substrate recognition. The increased catalytic efficiency for deacetylation of the acetylated H3/H4 tetramer compared with peptides corresponds to a Gibbs free energy increase of 2–4 kcal/mol, indicating a lower activation energy (Equation 1) and demonstrating the importance of long-range interactions for HDAC8 substrate recognition.
| (Eq. 1) |
The Mrksich group previously demonstrated that distal HDAC8–peptide interactions can enhance deacetylation and proposed an exosite model that involves binding at the active site and at a second location elsewhere on the HDAC8 surface (15). Structures of peptides bound to HDAC8 are in an extended conformation (19, 22, 30). Consistent with this, H3K9ac and H3K14ac sites are both located on the unstructured histone tail. The H3K9ac/H4 tetramer has both the highest value of kmax/K½ and the largest increase in reactivity compared with the corresponding 9-mer peptide (300-fold), which could be attributed to one or a few strong interactions or several weak interactions. H3K14ac/H4 is the slowest of the singly-acetylated tetramers tested, but the fold difference between H3K14ac/H4 tetramer and 9-mer peptide (300-fold) is similar to that of H3K9ac/H4 tetramer. H3K9ac and H3K14ac are similarly positioned in the H3 tail, so the surprising disparity in HDAC8 activity toward these sites suggests the important role of local sequence on HDAC8 selectivity, even within the context of a full-length protein. Moreover, H3K56ac is located on an α-helix that may significantly hinder interaction with HDAC8. Consistent with this, although the H3K56ac/H4 tetramer demonstrates the second fastest reactivity, the fold difference compared with the 9-mer peptide is the smallest (50-fold). This could reflect either weak affinity with the α-helix structure or a requirement for unfolding of the helix.
The crystal structure of HDAC8 is useful in visualizing potential protein–protein interactions involved in full-length substrate recognition. In many crystal structures, HDAC8 forms a dimer at the substrate-binding interface, as part of the fundamental crystal unit. This potential protein substrate-binding interface is a flexible 45 × 45 Å2 surface containing multiple interaction sites, including 10 van der Waals interactions and 6 hydrogen bonds between the HDAC8 dimers (21, 23). The interactions observed between these two HDAC8 units provide a framework for exploring the differences in catalytic efficiency observed for peptide and full-length substrates. The 2–4 kcal/mol difference in Gibbs free energy between the peptide and tetramer deacetylation could be explained by the van der Waals interactions and/or hydrogen bonds that are observed in the dimeric crystal structures. The dimer also displays repulsive charge–charge interactions. The attractive and repulsive protein–protein interactions likely work in concert to determine HDAC8 substrate specificity. The HDAC8 substrate-binding interface is mainly composed of flexible loops. Recent crystal structures have shown conformational changes in HDAC8 loops L1 and L2 upon binding of largazole analogs, as well as different L1 and L2 loop conformations between two monomers of the same crystal structure, demonstrating the adaptability and importance of these loops in HDAC8 inhibitor and substrate binding (26).
Catalytic efficiency is not enhanced by increasing the size and complexity of the protein substrate from a tetramer to the histone octamer. The H3K14ac histone octamer was deacetylated with a similar catalytic efficiency to the corresponding tetrameric substrate, suggesting that interactions with the tetramer are sufficient to explain HDAC8 substrate interactions in that case. In contrast, the H3K9ac octamer was deacetylated at least 4-fold slower than the tetrameric substrate. This is likely due to decreased accessibility of the acetyl-lysine to the HDAC8 active site, although other effects, including protein–protein interactions and allosteric effects could be involved in the recognition of these proteins. This suggests that HDAC8 substrate recognition is highly dependent on the histone complex. A concern with assaying octamer under low-salt conditions (HDAC8 assay buffer and less than 240 mm NaCl) is that the octamer would disassemble into H3/H4 tetramer and H2A/H2B dimers. However, the sensitivity of HDAC8 toward NaCl precluded higher salt concentrations (29). HDAC8 activity is salt- and pH-sensitive, and the selected assay buffer conditions were optimal for HDAC8 activity (29). The observed kinetics for the tetramer and octamer substrates are significantly different and suggest that the octamers, once assembled, remain intact during our assays.
Addition of nucleic acid to form a nucleosome converted the most efficient protein substrate, the H3K9ac/H4 tetramer, to a substrate that is deacetylated by HDAC8 less efficiently than the corresponding peptide. The drastic decrease in kmax/K½ values for nucleosomal H3K9ac likely reflects decreased substrate accessibility to the acetyl-lysine on the H3 tail by the nucleosome. One possibility is that the positively charged histone H3 tail interacts with the negatively charged DNA in the nucleosome and is no longer accessible to HDAC8. These data are consistent with proteomic studies suggesting that histones are not physiological substrates for HDAC8 (34). However, the low reactivity observed for nucleosomal H3K9ac does not completely preclude deacetylation by HDAC8 under all conditions. The chromatin structure can be altered by transcription factors, DNA-binding proteins, chromatin-remodeling factors and other proteins, possibly complexed with HDAC, to alter the accessibility of acetylated lysines in the tail of H3.
This work presents the first report of detailed kinetics for HDAC8-catalyzed deacetylation of singly-acetylated, full-length protein substrates and adds integral information to the field of HDAC substrate specificity. The direct comparison of peptides and protein substrates reveals that additional factors alter activity of HDAC8 with protein substrates, including both increased activity due to distal protein–protein interactions and decreased activity due to decreased accessibility of the acetyl-lysine side chain. HDAC8 catalyzes deacetylation of tetrameric protein substrates with catalytic efficiencies more than 40-fold greater than corresponding peptide substrates due to enhanced protein–protein interactions. However, further increasing the protein complex size decreases catalytic efficiency, likely due to decreased side chain accessibility. These differences in catalytic efficiency represent the effects of HDAC8–protein substrate interactions that are absent in HDAC8–peptide interactions. This work provides a foundation for the study of full-length protein substrate specificity of HDACs.
Experimental procedures
Materials
Adenosine triphosphate (ATP), coenzyme A, nicotinamide adenine dinucleotide (NAD+), l-malic acid, citrate synthase, malate dehydrogenase, and propionic anhydride were purchased from Sigma. Peptides were purchased from Peptide 2.0 Inc. Zinc(II) used to reconstitute HDAC8 was purchased as an ICP standard (GFS Chemicals) or atomic spectroscopy standard (Fluka), and the acetic acid standard was purchased from Ricca Chemical Co. Chelex 100 resin was purchased from Bio-Rad. Acetyl-lysine was purchased from Chem-Impex Chemical International Inc. α-Cyano-4-hydroxycinnamic acid (CHCA) MALDI matrix was purchased from Thermo Fisher Scientific. All other materials were purchased from Fisher or Sigma and were of a purity >95% unless otherwise noted.
HDAC8 expression and purification
HDAC8 was expressed and purified using the method described previously (28, 55) with the following modifications. BL21(DE3) E. coli cells transformed with the plasmid pHD4-HDAC8-TEV-His6 were used to express HDAC8 in modified autoinduction TB medium (12 g/liter tryptone, 24 g/liter yeast extract, 8.3 g/liter Tris-HCl, 4 g/liter lactose, 1 g/liter glucose, 1% (v/v) glycerol, pH 7.4) supplemented with 100 μg/ml ampicillin and 200 μm ZnSO4. The cells were grown overnight at 30 °C and harvested 20–24 h post-inoculation (9,000 × g, 10 min, 4 °C). The cell pellet was resuspended either in low-salt DEAE buffer (50 mm HEPES, 200 μm ZnSO4, 1 mm TCEP, 50 mm NaCl, 5 mm KCl, 1 μg/ml tert-amyl methyl ether, 10 μg/ml phenylmethylsulfonyl fluoride (PMSF), pH 7.8) and lysed using a microfluidizer (Microfluidics). Nucleic acids were then precipitated by addition of 0.1% polyethyleneimine, pH 7.9, followed by centrifugation (39,000 × g, 45 min, 4 °C). HDAC8 was fractionated on a DEAE-Sepharose column with a stepwise salt elution (50 mm HEPES, 200 μm ZnSO4, 1 mm TCEP, 5–500 mm NaCl, 5 mm KCl, pH 7.8) and dialyzed against Buffer A (50 mm HEPES, 100 mm NaCl, 25 mm imidazole, pH 7.8). The eluate was dialyzed against 50 mm HEPES, 50 mm NaCl, 25 mm imidazole, pH 7.8, for 1 h at 4 °C and then incubated with Ni(II)-charged chelating Sepharose fast flow resin (GE Healthcare) for 30 min, stirring on ice. HDAC8 was eluted from the metal-affinity chromatography column by a stepwise (50–250 mm) imidazole gradient. HDAC8, together with at least 1 mg of His-tagged TEV(S219V) protease per 15 mg of protein, was dialyzed overnight against Buffer A without imidazole at 4 °C. Following the overnight TEV cleavage, HDAC8 was separated from TEV protease on a second Ni(II) column. The protein was further purified by size-exclusion chromatography using a HiPrep 16/60 Sephacryl S200 HR column (GE Healthcare) using size-exclusion buffer (30 mm HEPES, pH 8, 150 mm NaCl, 1 mm TCEP). HDAC8 was then dialyzed against metal-free chelation buffer (25 mm MOPS, pH 7.5, 1 mm EDTA, 1 mm TCEP, 5 mm KCl) overnight, followed by metal-free buffer (25 mm MOPS, pH 7.5, 1 mm TCEP, 5 mm KCl). Finally, residual EDTA was removed with a PD-10 column (GE Healthcare) eluting with storage buffer (25 mm HEPES, pH 8, 127 mm NaCl, 3 mm KCl, and 1 mm TCEP). HDAC8 was concentrated, aliquoted, and stored at −80 °C. HDAC8 activity was confirmed using the Fluor de Lys assay as described previously (28, 59, 60).
Peptide deacetylation assay
HDAC8-catalyzed deacetylation of acetylated peptides was characterized using an enzyme-coupled assay, performed as described previously (55) with a few modifications. To prepare peptide stocks, lyophilized peptides were dissolved in water, 50% acetonitrile, or 10% DMSO, depending on their solubility. Peptide solutions were chelated by incubation with Chelex resin at 4 °C for at least 3 h. Peptide concentrations were measured using the fluorescamine assay or absorbance at 280 nm, as described previously (55, 61). Peptides (0–100 μm) were incubated in HDAC8 assay buffer (50 mm HEPES, 137 mm NaCl, 3.7 mm KCl, pH 7.8) for 10 min at 30 °C before initiating reactions with the addition of 0.5 μm Zn(II)-HDAC8. Acetate formation was coupled to NADH formation measured by an increase in fluorescence (ex = 340 nm and em = 460 nm) (55). Initial rates were fit to the linear portion of the product versus time curve.
Histone expression and purification
Recombinant His6-tagged histone H3 variants containing a single acetyl-lysine were expressed and purified as described previously (54) with a few modifications. The acetyl-lysine was incorporated into expressed proteins at an amber codon site (TAG) using a tRNA–cognate tRNA synthetase pair encoded on the pAcKRS-3 plasmid (54). Amber codons were substituted for the Lys-9, Lys-14, and Lys-56 codons in the His6-tagged Xenopus histone H3 in the PCDF PyLT-1 plasmid, a generous gift from J. Chin (53, 54) using QuikChange Site-directed Mutagenesis (Agilent). BL21(DE3) cells were transformed with the mutant or wild-type PCDF PyLT-1 and pAcKRS-3 plasmids for His6-tagged H3 expression. Expression plasmids for preparation of recombinant H2A, H2B, and H4 Xenopus histones were generous gifts from Geeta Narlikar. BL21(DE3) cells were transformed with the respective H2A, H2B, and H4 plasmids and grown in LB or 2× YT supplemented with antibiotics (kanamycin and streptomycin for H3 or ampicillin for H2A, H2B, and H4) at 37 °C until reaching an A600 of 0.7. To express full-length histone H3 proteins with a single acetyl-lysine residue, 20 mm nicotinamide and 10 mm acetyl-lysine were added to the medium; 30 min later, 0.5 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to induce protein expression. For expression of the other histones, the cells were induced with 0.5 mm IPTG. The cultures were harvested 3–4 h after induction (9,000 × g, 10–15 min, 4 °C). The cell pellets were stored at −80 °C.
Histones were purified using established protocols (54, 62, 63), with H3 Ni(II) column buffers modified to include 7 m urea and 1 mm TCEP. Tetramer, octamer, and nucleosome were assembled as described previously, and nucleosomes contained a 147-bp DNA fragment containing the 601 octamer positioning sequence prepared as described (54, 62–64). The 601 plasmid was a generous gift from Yali Dou. Tetramer and octamer were purified by size-exclusion chromatography, selecting a single peak in each case. Tetramer assembly was tested using IM-MS (data not shown). Native PAGE was used to verify nucleosome consisting of DNA-bound histone protein. EDTA dialysis was used to remove any contaminating metal from histone protein complexes. Octamer and nucleosome were subsequently treated with Chelex resin for at least 1 h at 4 °C to ensure metal removal. Inductively coupled plasma mass spectrometry (ICP-MS) was used to verify less than 10% metal contamination. Nucleosome was stored in 20% glycerol, 20 mm HEPES, pH 7.5–7.8, 1 mm TCEP.
Protein deacetylation assays
Apo-HDAC8 was reconstituted with stoichiometric Zn(II) for 1 h on ice in HDAC8 assay buffer (50 mm HEPES, 137 mm NaCl, 3.7 mm KCl, pH 7.8) (28). Histone complexes were incubated in HDAC8 assay buffer for 10 min at 30 °C before initiating reactions by addition of 0–15 μm Zn(II)-HDAC8. The final concentration of NaCl in the assays with octamer was 137 or 239 mm NaCl. This salt concentration is lower than typical histone octamer storage buffer (2 m NaCl (65)), but it allows for measurement of HDAC8 activity uninhibited by salt. Reactions were quenched by addition of 25% trichloroacetic acid at each time point, incubated for 30 min on ice, and centrifuged (16,000 × g, 10 min, 4 °C), and then the pellets were washed in cold acetone twice. Acetone-washed pellets were resuspended in 2 μl of propionic anhydride and 6 μl of ammonium hydroxide (NH4OH) and incubated at 51 °C for 1 h. 30 μl of 50 mm NH4HCO3 was added to each tube, and the pH of each sample was adjusted to 7–9 using NH4OH. Then 0.2 μg of trypsin (Promega) was added for overnight digestion at 37 °C. The pH was then reduced for mass spectrometry (MS) by addition of 3.5 μl of 10% formic acid. Tryptic peptides were analyzed by MS/MS analysis in the Andrews lab at Fox Chase Cancer Center, as described previously (45, 66). Graphical analysis was done using Prism (GraphPad Software, Inc.). For each single turnover reaction, the kobs was determined by fitting a single exponential decay (Equation 2) to the fraction substrate over time determined from the MS/MS analysis. The kmax/K½ values were determined by fitting hyperbola (Equation 3) or line (Equation 4) to the dependence of kobs on HDAC8 concentration, depending on the substrate.
| (Eq. 2) |
| (Eq. 3) |
| (Eq. 4) |
MALDI-TOF-MS deacetylation assay
HDAC8-catalyzed deacetylation of the H3K9ac 13-mer peptide was measured under STO ([E] ≫ [S]) and MTO ([S] ≫ [E]) conditions using MALDI-TOF-MS. Apo-HDAC8 was reconstituted with stoichiometric zinc(II) for 1 h on ice in HDAC8 assay buffer. The H3K9ac 13-mer peptide was incubated in HDAC8 assay buffer for 10 min at 30 °C, and the deacetylation reaction was initiated with Zn(II)-HDAC8. MTO control reactions contained either 1 μm enzyme and 50 μm peptide or 150 μm enzyme and 750 μm peptide, and STO reactions contained 20 μm peptide and 0, 50, 100, and 150 μm enzyme (2.5, 5, and 7.5 ratio of [E]/substrate). At each time point, 2 μl of reaction were quenched with 2 μl of 10% HCl. Samples were stored at −80 °C prior to MS analysis. The samples were prepared by mixing the quenched reactions 1:1 with CHCA MALDI matrix followed by spotting on a Bruker MALDI-TOF-MS plate. Spectra were collected using a Bruker AutoFlex Speed MALDI-TOF mass spectrometer calibrated with a series of five peptide standards. Three random measurements from each spot were averaged, and the fraction of product formed was calculated from the area under the curve of the product and substrate peaks. The kcat/Km values for the multiple turnover reaction was determined by fitting a line to the initial rate of the reaction progress curve. The kmax/K½ values for the single turnover reaction was determined by fitting a hyperbola to the dependence of kobs on HDAC8 concentration (Equation 3). The values for kobs at each HDAC8 concentration were determined by fitting a single exponential to the time dependence of product formation (Equation 5).
| (Eq. 5) |
Author contributions
N. A. W. and C. A. F. designed the study. N. A. W. designed the assays and performed the peptide and tetramer assays. N. A. W., C. A. C., and K. R. L. purified proteins and performed octamer assays. C. A. C. and K. R. L. assembled nucleosomes and performed nucleosome assays. N. A. W., C. A. C., K. R. L., and C. A. F. analyzed the data. Y.-M. K. performed and A. J. A. designed the histone mass spectrometry analysis. N. A. W., C. A. C., K. R. L., and C. A. F. prepared the manuscript. All authors reviewed and approved the final manuscript.
Acknowledgments
We thank members of the Fierke laboratory for helpful discussions and comments on the manuscript. We thank Jeffrey E. López for collaborating on HDAC8 purification. We thank Jason Chin (Medical Research Council) for non-natural amino acid incorporation plasmids, Geeta Narlikar (University of California San Francisco) for histone expression plasmids, and Yali Dou (University of Michigan) for the nucleosome DNA plasmid. We thank John Leonard (Narlikar laboratory) and Felicia Gray (Dou laboratory) for histone purification and nucleosome assembly protocols, and we thank Ted Huston and Lubomír Dostál (University of Michigan) for performing ICP-MS to measure metal content. We thank Brandon Ruotolo and Yueyang Zhong for their determination of the histone tetramer stoichiometry.
This work was supported in part by National Science Foundation predoctoral fellowship (to N. A. W.), National Institutes of Health Grant 5-R01-GM-040602 (to C. A. F.), University of Michigan Chemistry-Biology Interface (CBI) Training Program, National Institutes of Health Grant 5T32GM008597 (to C. A. C.), Rackham Graduate School (to C. A. C.), and the Cellular Biotechnology Training Program T32-GM008353 (to N. A. W. and K. R. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HDAC
- histone deacetylase
- CHCA
- α-cyano-4-hydroxy-cinnamic acid
- H3/H4
- tetrameric histone H3/H4
- ICP-MS
- inductively coupled plasma mass spectrometry
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- KAT
- acetyltransferase
- MTO
- multiple turnover
- STO
- single turnover
- TCEP
- tris(2-carboxyethyl)phosphine hydrochloride
- TEV
- tobacco etch virus
- CHCA
- α-cyano-4-hydroxycinnamic acid.
References
- 1. Khochbin S., Verdel A., Lemercier C., and Seigneurin-Berny D. (2001) Functional significance of histone deacetylase diversity. Curr. Opin. Genet. Dev. 11, 162–166 [DOI] [PubMed] [Google Scholar]
- 2. Yang X. J., and Seto E. (2008) The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Glozak M. A., Sengupta N., Zhang X., and Seto E. (2005) Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23 [DOI] [PubMed] [Google Scholar]
- 4. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., and Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 5. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., et al. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li G., Jiang H., Chang M., Xie H., and Hu L. (2011) HDAC6 α-tubulin deacetylase: a potential therapeutic target in neurodegenerative diseases. J. Neurol. Sci. 304, 1–8 [DOI] [PubMed] [Google Scholar]
- 7. D'Mello S. R. (2009) Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 22, 513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Glozak M. A., and Seto E. (2007) Histone deacetylases and cancer. Oncogene 26, 5420–5432 [DOI] [PubMed] [Google Scholar]
- 9. Marks P., Rifkind R. A., Richon V. M., Breslow R., Miller T., and Kelly W. K. (2001) Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer 1, 194–202 [DOI] [PubMed] [Google Scholar]
- 10. Fenichel M. P. (2015) FDA approves new agent for multiple myeloma. J. Natl. Cancer Inst. 107, djv165. [DOI] [PubMed] [Google Scholar]
- 11. Poole R. M. (2014) Belinostat: first global approval. Drugs 74, 1543–1554 [DOI] [PubMed] [Google Scholar]
- 12. Mottamal M., Zheng S., Huang T. L., and Wang G. (2015) Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 20, 3898–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gurard-Levin Z. A., Kilian K. A., Kim J., Bähr K., and Mrksich M. (2010) Peptide arrays identify isoform-selective substrates for profiling endogenous lysine deacetylase activity. ACS Chem. Biol. 5, 863–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gurard-Levin Z. A., Kim J., and Mrksich M. (2009) Combining mass spectrometry and peptide arrays to profile the specificities of histone deacetylases. Chembiochem 10, 2159–2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gurard-Levin Z. A., and Mrksich M. (2008) The activity of HDAC8 depends on local and distal sequences of its peptide substrates. Biochemistry 47, 6242–6250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Riester D., Hildmann C., Grünewald S., Beckers T., and Schwienhorst A. (2007) Factors affecting the substrate specificity of histone deacetylases. Biochem. Biophys. Res. Commun. 357, 439–445 [DOI] [PubMed] [Google Scholar]
- 17. Buggy J. J., Sideris M. L., Mak P., Lorimer D. D., McIntosh B., and Clark J. M. (2000) Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem. J. 350, 199–205 [PMC free article] [PubMed] [Google Scholar]
- 18. Hu E., Chen Z., Fredrickson T., Zhu Y., Kirkpatrick R., Zhang G. F., Johanson K., Sung C. M., Liu R., and Winkler J. (2000) Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 275, 15254–15264 [DOI] [PubMed] [Google Scholar]
- 19. Dowling D. P., Gantt S. L., Gattis S. G., Fierke C. A., and Christianson D. W. (2008) Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry 47, 13554–13563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dowling D. P., Gattis S. G., Fierke C. A., and Christianson D. W. (2010) Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry 49, 5048–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vannini A., Volpari C., Filocamo G., Casavola E. C., Brunetti M., Renzoni D., Chakravarty P., Paolini C., De Francesco R., Gallinari P., Steinkühler C., and Di Marco S. (2004) Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. U.S.A. 101, 15064–15069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vannini A., Volpari C., Gallinari P., Jones P., Mattu M., Carfí A., De Francesco R., Steinkühler C., and Di Marco S. (2007) Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 8, 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Somoza J. R., Skene R. J., Katz B. A., Mol C., Ho J. D., Jennings A. J., Luong C., Arvai A., Buggy J. J., Chi E., Tang J., Sang B. C., Verner E., Wynands R., Leahy E. M., et al. (2004) Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12, 1325–1334 [DOI] [PubMed] [Google Scholar]
- 24. Marek M., Kannan S., Hauser A. T., Moraes Mourão M., Caby S., Cura V., Stolfa D. A., Schmidtkunz K., Lancelot J., Andrade L., Renaud J. P., Oliveira G., Sippl W., Jung M., Cavarelli J., et al. (2013) Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke Schistosoma mansoni. PLoS Pathog. 9, e1003645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Whitehead L., Dobler M. R., Radetich B., Zhu Y., Atadja P. W., Claiborne T., Grob J. E., McRiner A., Pancost M. R., Patnaik A., Shao W., Shultz M., Tichkule R., Tommasi R. A., Vash B., et al. (2011) Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Bioorg. Med. Chem. 19, 4626–4634 [DOI] [PubMed] [Google Scholar]
- 26. Decroos C., Clausen D. J., Haines B. E., Wiest O., Williams R. M., and Christianson D. W. (2015) Variable active site loop conformations accommodate the binding of macrocyclic largazole analogues to HDAC8. Biochemistry 54, 2126–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cole K. E., Dowling D. P., Boone M. A., Phillips A. J., and Christianson D. W. (2011) Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 133, 12474–12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gantt S. L., Gattis S. G., and Fierke C. A. (2006) Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry 45, 6170–6178 [DOI] [PubMed] [Google Scholar]
- 29. Gantt S. L., Joseph C. G., and Fierke C. A. (2010) Activation and inhibition of histone deacetylase 8 by monovalent cations. J. Biol. Chem. 285, 6036–6043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gantt S. M., Decroos C., Lee M. S., Gullett L. E., Bowman C. M., Christianson D. W., and Fierke C. A. (2016) General base–general acid catalysis in human histone deacetylase 8. Biochemistry 55, 820–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wilson B. J., Tremblay A. M., Deblois G., Sylvain-Drolet G., and Giguère V. (2010) An acetylation switch modulates the transcriptional activity of estrogen-related receptor α. Mol. Endocrinol. 24, 1349–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deardorff M. A., Bando M., Nakato R., Watrin E., Itoh T., Minamino M., Saitoh K., Komata M., Katou Y., Clark D., Cole K. E., De Baere E., Decroos C., Di Donato N., Ernst S., et al. (2012) HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489, 313–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaiser F. J., Ansari M., Braunholz D., Concepción Gil-Rodríguez M., Decroos C., Wilde J. J., Fincher C. T., Kaur M., Bando M., Amor D. J., Atwal P. S., Bahlo M., Bowman C. M., Bradley J. J., Brunner H. G., et al. (2014) Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum. Mol. Genet. 23, 2888–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Olson D. E., Udeshi N. D., Wolfson N. A., Pitcairn C. A., Sullivan E. D., Jaffe J. D., Svinkina T., Natoli T., Lu X., Paulk J., McCarren P., Wagner F. F., Barker D., Howe E., Lazzaro F., et al. (2014) An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem. Biol. 9, 2210–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Durst K. L., Lutterbach B., Kummalue T., Friedman A. D., and Hiebert S. W. (2003) The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol. Cell. Biol. 23, 607–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao J., Siddoway B., Huang Q., and Xia H. (2009) Inactivation of CREB mediated gene transcription by HDAC8 bound protein phosphatase. Biochem. Biophys. Res. Commun. 379, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Joshi P., Greco T. M., Guise A. J., Luo Y., Yu F., Nesvizhskii A. I., and Cristea I. M. (2013) The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 9, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Waltregny D., De Leval L., Glénisson W., Ly Tran S., North B. J., Bellahcène A., Weidle U., Verdin E., and Castronovo V. (2004) Expression of histone deacetylase 8, a class I histone deacetylase, is restricted to cells showing smooth muscle differentiation in normal human tissues. Am. J. Pathol. 165, 553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Waltregny D., Glénisson W., Tran S. L., North B. J., Verdin E., Colige A., and Castronovo V. (2005) Histone deacetylase HDAC8 associates with smooth muscle α-actin and is essential for smooth muscle cell contractility. FASEB J. 19, 966–968 [DOI] [PubMed] [Google Scholar]
- 40. Lee H., Sengupta N., Villagra A., Rezai-Zadeh N., and Seto E. (2006) Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (hEST1B) protein from ubiquitin-mediated degradation. Mol. Cell. Biol. 26, 5259–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van den Wyngaert I., de Vries W., Kremer A., Neefs J., Verhasselt P., Luyten W. H., and Kass S. U. (2000) Cloning and characterization of human histone deacetylase 8. FEBS Lett. 478, 77–83 [DOI] [PubMed] [Google Scholar]
- 42. Saha A., Pandian G. N., Sato S., Taniguchi J., Hashiya K., Bando T., and Sugiyama H. (2013) Synthesis and biological evaluation of a targeted DNA-binding transcriptional activator with HDAC8 inhibitory activity. Bioorg. Med. Chem. 21, 4201–4209 [DOI] [PubMed] [Google Scholar]
- 43. Lee H., Rezai-Zadeh N., and Seto E. (2004) Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol. Cell. Biol. 24, 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smith B. C., and Denu J. M. (2007) Acetyl-lysine analog peptides as mechanistic probes of protein deacetylases. J. Biol. Chem. 282, 37256–37265 [DOI] [PubMed] [Google Scholar]
- 45. Kuo Y.-M., Henry R., A., Tan S., Côté J., and Andrews A. J. (2015) Site specificity analysis of Piccolo NuA4-mediated acetylation for different histone complexes. Biochem. J. 472, 239–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wolfson N. A., Pitcairn C. A., and Fierke C. A. (2013) HDAC8 substrates: histones and beyond. Biopolymers 99, 112–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Toro T. B., and Watt T. J. (2015) KDAC8 substrate specificity quantified by a biologically relevant, label-free deacetylation assay. Protein Sci. 24, 2020–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alam N., Zimmerman L., Wolfson N. A., Joseph C. G., Fierke C. A., and Schueler-Furman O. (2016) Structure-based identification of HDAC8 non-histone substrates. Structure 24, 458–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dose A., Sindlinger J., Bierlmeier J., Bakirbas A., Schulze-Osthoff K., Einsele-Scholz S., Hartl M., Essmann F., Finkemeier I., and Schwarzer D. (2016) Interrogating substrate selectivity and composition of endogenous histone deacetylase complexes with chemical probes. Angew. Chem. Int. Ed. Engl. 55, 1192–1195 [DOI] [PubMed] [Google Scholar]
- 50. Gober I. N., and Waters M. L. (2016) Supramolecular affinity labeling of histone peptides containing trimethyllysine and its application to histone deacetylase assays. J. Am. Chem. Soc. 138, 9452–9459 [DOI] [PubMed] [Google Scholar]
- 51. Izzo A., and Schneider R. (2010) Chatting histone modifications in mammals. Brief. Funct. Genomics 9, 429–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yuan J., Pu M., Zhang Z., and Lou Z. (2009) Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle 8, 1747–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Neumann H., Peak-Chew S. Y., and Chin J. W. (2008) Genetically encoding N(ϵ)-acetyllysine in recombinant proteins. Nat. Chem. Biol. 4, 232–234 [DOI] [PubMed] [Google Scholar]
- 54. Neumann H., Hancock S. M., Buning R., Routh A., Chapman L., Somers J., Owen-Hughes T., van Noort J., Rhodes D., and Chin J. W. (2009) A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell 36, 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wolfson N. A., Pitcairn C. A., Sullivan E. D., Joseph C. G., and Fierke C. A. (2014) An enzyme-coupled assay measuring acetate production for profiling histone deacetylase specificity. Anal. Biochem. 456, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eisenthal R., Danson M. J., and Hough D. W. (2007) Catalytic efficiency and kcat/KM: a useful comparator? Trends Biotechnol. 25, 247–249 [DOI] [PubMed] [Google Scholar]
- 57. Northrop D. B. (1998) On the meaning of Km and V/K in enzyme kinetics. J. Chem. Educ. 75, 1153–1157 [Google Scholar]
- 58. Hedstrom L. (2010) Enzyme Specificity and Selectivity. John Wiley & Sons, Ltd., Chichester, UK [Google Scholar]
- 59. Wegener D., Hildmann C., Riester D., and Schwienhorst A. (2003) Improved fluorogenic histone deacetylase assay for high-throughput-screening applications. Anal. Biochem. 321, 202–208 [DOI] [PubMed] [Google Scholar]
- 60. Wegener D., Wirsching F., Riester D., and Schwienhorst A. (2003) A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem. Biol. 10, 61–68 [DOI] [PubMed] [Google Scholar]
- 61. Huang X., and Hernick M. (2011) A fluorescence-based assay for measuring N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase activity. Anal. Biochem. 414, 278–281 [DOI] [PubMed] [Google Scholar]
- 62. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., and Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 63. Luger K., Rechsteiner T. J., and Richmond T. J. (1999) Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 304, 3–19 [DOI] [PubMed] [Google Scholar]
- 64. Dyer P. N., Edayathumangalam R. S., White C. L., Bao Y., Chakravarthy S., Muthurajan U. M., and Luger K. (2004) Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44 [DOI] [PubMed] [Google Scholar]
- 65. Shahian T., and Narlikar G. J. (2012) Analysis of changes in nucleosome conformation using fluorescence resonance energy transfer. Methods Mol. Biol. 833, 337–349 [DOI] [PubMed] [Google Scholar]
- 66. Kuo Y. M., and Andrews A. J. (2013) Quantitating the specificity and selectivity of Gcn5-mediated acetylation of histone H3. PLoS ONE 8, e54896. [DOI] [PMC free article] [PubMed] [Google Scholar]