

Publisher's Note: There is an Inside Blood Commentary on this article in this issue.
Key Points
Regionally localized PD-L1+ macrophages form a specialized microenvironmental niche for Hodgkin Reed-Sternberg cells in cHL.
Abstract
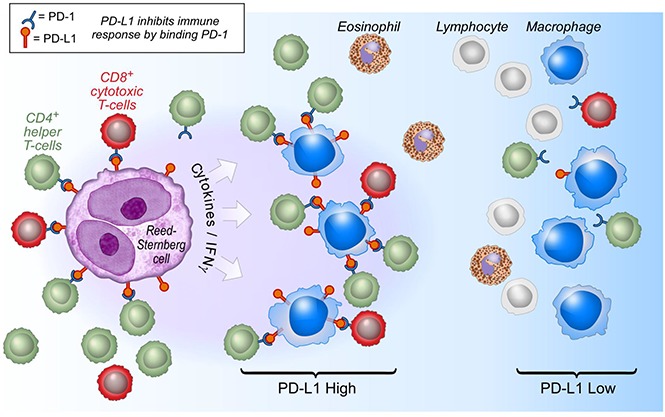
Signaling between programmed cell death protein 1 (PD-1) and the PD-1 ligands (PD-L1, PD-L2) is essential for malignant Hodgkin Reed-Sternberg (HRS) cells to evade antitumor immunity in classical Hodgkin lymphoma (cHL). Copy number alterations of 9p24.1/CD274(PD-L1)/PDCD1LG2(PD-L2) contribute to robust PD-L1 and PD-L2 expression by HRS cells. PD-L1 is also expressed by nonmalignant tumor-associated macrophages (TAMs), but the relationships among PD-L1+ HRS cells, PD-L1+ TAMs, and PD-1+ T cells remain undefined. We used multiplex immunofluorescence and digital image analysis to examine the topography of PD-L1+ and PD-1+ cells in the tumor microenvironment (TME) of cHL. We find that the majority of PD-L1 in the TME is expressed by the abundant PD-L1+ TAMs, which physically colocalize with PD-L1+ HRS cells in a microenvironmental niche. PD-L1+ TAMs are enriched for contacts with T cells, and PD-L1+ HRS cells are enriched for contacts with CD4+ T cells, a subset of which are PD-1+. Our data define a unique topology of cHL in which PD-L1+ TAMs surround HRS cells and implicate CD4+ T cells as a target of PD-1 blockade.
Visual Abstract
Introduction
Classical Hodgkin lymphoma (cHL) is a unique subtype of lymphoma in which the malignant Hodgkin Reed-Sternberg (HRS) cells represent only a small proportion of the overall tumor cellularity (1%-5%).1 The tumor microenvironment (TME) is predominantly composed of inflammatory cells, including macrophages, CD4+ and CD8+ T cells, plasma cells, eosinophils, and other immune cells, yet antitumor immunity fails to effectively recognize and eliminate the malignant cells. HRS cells achieve immune evasion by multiple mechanisms including enhanced expression of programmed cell death-1 ligands (PD-L1 and PD-L2) that bind PD-1 (CD279) on the surface of antigen-experienced T cells to suppress T-cell activation, and diminished or absent expression of major histocompatibility complex (MHC) class I to prevent recognition by the adaptive immune response.2-5
The critical role for PD-1:PD-1 ligand interactions in cHL was established with recent trials of monoclonal antibodies directed against PD-1.6-8 Treatment with nivolumab, a fully human immunoglobulin (Ig) G4 antibody, in a phase 1 study resulted in an overall response rate of 87% and complete remission rate of 17% in a series of patients with multiply relapsed/refractory cHL.6 In an expanded phase 2 trial comprising patients with relapsed/refractory disease after brentuximab vedotin and stem cell transplant, an objective response was seen in 66.3% of patients, with a progression-free survival at 6 months of 76.9%, including patients with durable remissions. Importantly, patients with the highest PD-L1 expression among HRS cells had the best clinical response.7 Similar clinical response rates were found in trials of pembrolizumab, a distinct antibody that also targets PD-1.8,9
PD-1 ligand expression by HRS cells is attributable, in large part, to characteristic copy gains of chromosome 9p24.1, which includes the PD-L1, PD-L2, and JAK2 loci and results in a direct increase in PD-L1 and PD-L2 transcripts and proteins, as well as an indirect increase resulting from augmented JAK-STAT signaling.2,3 Critically, high-level PD-L1/PD-L2 copy gains (amplification) in HRS cells is associated with advanced stage disease and an inferior outcome after standard induction therapy.3 However, not all PD-L1 protein within the cHL TME is associated with HRS cells. We have shown that PD-L1 is also expressed by tumor-associated macrophages (TAMs).10 This observation is of interest, as increased TAMs and a macrophage-related gene expression signature predict poor clinical response to combination chemotherapy in patients with advanced stage disease.11 The number of TAMs that express PD-L1, the relative contribution of TAMs and HRS cells to the overall pool of PD-L1 in the TME, and the geographic distribution of PD-L1-expressing cells within the TME are undefined. Similarly, the numbers and types of T cells that express PD-1 and their geographic distribution are unknown, despite the striking clinical effectiveness of PD-1 blockade.
Methods
Tissue samples
Formalin-fixed, paraffin-embedded whole tissues from tumors were derived from the archives of Brigham & Women’s Hospital, Boston, MA, with institutional review board approval (2014P001721). Hematoxylin & eosin-stained tissue sections and the original diagnostic reports were reviewed by an expert hematopathologist (S.J.R.). Twenty cases were selected for the study, based on the availability of high-quality, whole lymph node excision biopsy tissue (12 Epstein-Barr virus negative; 8 Epstein-Barr virus positive), including nodular sclerosing HL (n = 11), mixed cellularity HL (n = 6), lymphocyte-rich HL (n = 1), and cHL, not otherwise specified (n = 2) subtypes (supplemental Table 1, available on the Blood Web site).
Multiplexed immunofluorescence
Multiplexed immunofluorescence (IF) was performed by staining 4-µm-thick formalin-fixed, paraffin-embedded whole tissue sections with standard, primary antibodies sequentially and paired with a unique fluorochrome followed by staining with nuclear counterstain/4′,6-diamidino-2-phenylindole per published protocols.12-14 For example, deparaffinized slides were incubated with anti-PD-L1 antibody (clone 9A11; Cell Signaling Technology, Danvers, MA) for 40 minutes and then treated with anti-mouse horseradish peroxidase-conjugated (HRP) secondary antibody (EnVision plus, Dako; Agilent Technologies, Carpinteria, CA) for 30 minutes. IF labeling was developed for a strictly observed 5 minutes, using Opal-520 amplification reagent (PerkinElmer, Hopkinton, MA) per manufacturer’s direction. Slides were washed in Tris buffer (5 minutes) and then transferred to preheated citrate solution (90°C) before being heat-treated using a microwave set at 20% of maximum power for 15 minutes. Slides were cooled in the same solution to room temperature. Between all steps, the slides were washed with Tris buffer. The same process was repeated for the following antibodies/fluorescent dyes, in order: anti-CD30 (clone BerH2, Dako)/Opal-540, anti-CD68 (clone PGM1, Dako)/Opal-650, anti-CD163 (clone 10D6, Thermo Fisher)/Opal-690. Each slide was then treated with 2 drops NucBlue Fixed cell ReadyProbes reagent (#P36965; Life Technologies, Carlsbad, CA), washed in distilled water, and manually coverslipped. Slides were air dried, mounted with Prolong Diamond Anti-fade mounting medium (#P36965; Life Technologies), and stored in a lightproof box at 4°C before imaging. The target antigens, antibody clones, and dilutions for markers included in this report and details of controls are listed in supplemental Table 2 and the supplemental Methods.
Image acquisition
Test regions for multiplex IF analysis were identified in matched tissue sections stained for CD30 by chromogenic immunohistochemistry. Two geographically distinct regions were selected for each tumor to best represent the overall tissue and to include CD30+ HRS tumor cells, and these regions were imaged using the Vectra multispectral imaging platform (Vectra 3, PerkinElmer, Hopkinton, MA) at 4 times resolution. Areas with nontumor or residual normal tissue (ie, residual lymph node) were excluded from the analysis. For each region, 4 tiled fields of view were acquired at 20× resolution as multispectral images. Further details are provided in the supplemental Methods.
Cell identification
After image capture, the fields of view were spectrally unmixed and analyzed, using supervised machine learning algorithms within Inform 2.1 (PerkinElmer), which assigns phenotypes to all cells in the image, according to a combination of IF characteristics associated with segmented nuclei (4′,6-diamidino-2-phenylindole signal). Each cell-phenotype-specific algorithm is based on an iterative training/test process, whereby a small number of cells (training phase, typically 15-20 cells) are manually selected as being most representative of each phenotype of interest and the algorithm then predicts the phenotype for all remaining cells (testing phase).12 The decisions made by the software can be overruled to improve accuracy until phenotyping is optimized. Unique phenotyping was performed for each tumor and then applied to both tiled study images to account for intersample variability of signal intensities. Thresholds for “positive” staining and the accuracy of phenotypic algorithms were confirmed by the pathologist (S.J.R.) for each case. Inform (PerkinElmer) automatically derives maps of cell membranes and Cartesian coordinates for each phenotyped cell within the image.
Quantification of the microenvironment
The shortest Euclidian distance from each cell of one phenotype (“A”) toward the nearest cell of a second phenotype (“B”) was calculated using the Cartesian coordinates. These minimum distances from each cell of type A were then averaged to calculate the average nearest neighbor distance between cell types A and B (NNAB).
Physical interactions between two cells were determined on the basis of the membrane maps that are provided by Inform 2.1. We examined each HRS cell, determined the numbers of each of the defined cell types interacting with it (ie, CD4+ T cells, CD8+ T cells), and then took the average result for all HRS cells. In a similar manner, we also calculated percentages of populations that were within the immediate vicinity of a given cell type (eg, the percentage of cell types that were not physically interacting but were within a defined distance from any HRS cell). Additional details are provided in the supplemental Methods.
Results
Cell-specific expression and localization of PD-L1+ TAMs in relation to PD-L1+ HRS cells
Sequential IF staining of 20 cases of cHL revealed the expected patterns of cellular staining, with anti-CD30 delineating cells morphologically consistent with HRS cells, anti-CD68 delineating cells morphologically consistent with TAMs, and anti-PD-L1 highlighting subsets of cells that coexpress either CD30 or CD68 (Figure 1A). The number, distribution, and morphology of CD30+ HRS cells and CD68+ TAMs observed with multiplex IF were indistinguishable from those observed with chromogenic immunohistochemistry (supplemental Figure 1 and not shown). A machine-learning algorithm trained on the morphological and staining characteristics of selected cells identified HRS cells and TAMs accurately, as judged by visual review, and confirmed the presence of PD-L1+ HRS cells and TAMs in every case tested (Figure 1B). We quantified the relative contribution of HRS cells and TAMs to total PD-L1 staining (calculated as percentage of total fluorescence units) and found that, for every case, TAMs contributed the majority of PD-L1 in the tumor microenvironment (TME) (Figure 1B). A mean of 78.5% of the total PD-L1 expression within the TME was contributed by TAMs across the series (range, 50.4%-98.5%; standard deviation, 14.8).
Figure 1.

Expression of PD-L1 by HRS cells and TAMs. (A) Multiplex IF staining (40× resolution, case P6) for CD30 (top left, orange) to highlight HRS cells, CD68 (bottom left, magenta) to highlight TAMs, and PD-L1 (green) to show colocalization of PD-L1 and CD30 (top right, colocalization, yellow) and PD-L1 and CD68 (bottom right). Each image includes a nuclear counterstain/4′,6-diamidino-2-phenylindole (blue). (B) The relative amount of total PD-L1 per tumor (calculated as percentage of total fluorescence units), contributed by HRS cells (red) and TAMs (blue). The cases are ordered by the percentage of PD-L1 attributed to HRS cells, from highest to lowest. Cell lineage assignments (HRS cell; TAM) are based on pathologist-trained algorithms and include data from all fluorescent-channels (“Methods”). DAPI, 4′,6-diamidino-2-phenylindole.
Visual inspection of stained tissue sections suggested possible enrichment of PD-L1+ TAMs in the vicinity of PD-L1+ HRS cells (exemplified by case P6; Figure 2A; supplemental Figure 2). To quantify this observation, we used the Cartesian coordinates for each of the thousands of cells of interest within each tissue section (Figure 2B) and calculated the respective distances from each PD-L1+ TAM to the nearest PD-L1+ HRS cell (Figure 2C) and the distances from each PD-L1− TAM to the nearest PD-L1+ HRS cell (Figure 2D). In all 20 cases, the mean distance from PD-L1+ TAMs to the nearest PD-L1+ HRS cell was significantly less than the mean distance from PD-L1− TAMs to the nearest PD-L1+ HRS cell (Figure 2E). This difference was highly significant across the case series (P < .0001, paired Student t test). Conversely, the mean distance from PD-L1+ HRS cells to PD-L1+ TAMs was shorter than the mean distance from PD-L1+ HRS cells to PD-L1− TAMs in 18 of 20 cases (supplemental Figure 2G). This difference was also highly significant across the case series (P = .0002, paired Student t test).
Figure 2.

Distances from PD-L1+TAMs and PD-L1−TAMs to the nearest PD-L1+HRS cells. (A) Representative multiplex IF image (20× resolution; case P6) showing staining for CD30 (orange), CD68 (magenta), and PD-L1 (green). (B) Cellular phenotype map of the image shown in A depicting locations of PD-L1+ HRS cells (orange dots), PD-L1+ TAMs (purple dots), and PD-L1− TAMs (pink dots). (C) Ray plot depicting the distance from each PD-L1+ TAM to the nearest PD-L1+ HRS cell. (D) Ray plot depicting the distance from each PD-L1− TAM to the nearest PD-L1+ HRS cell. (E) The mean distances (microns) and standard errors for all 20 study tumors, divided into mean distance from PD-L1− TAMs to the nearest PD-L1+ HRS cells (red) and mean distance from PD-L1+ TAMs to the nearest PD-L1+ HRS cells (blue). The tumors are ordered by the distances from PD-L1− TAMs to PD-L1+ HRS cells, from highest to lowest. P value (<.0001) was calculated by paired Student t test. NN, nearest neighbor.
To ensure the specificity of the analysis, we also optimized IF staining for CD163, a macrophage marker with a more restricted expression pattern than CD68 (supplemental Figure 3). We found that the majority of cells with positive staining for CD68 were also positive for CD163, as expected (supplemental Figure 4A-D). By quantitative analysis, we found that the mean distance from PD-L1+ CD68+ CD163+ TAMs to the nearest PD-L1+ HRS cell was significantly less than the mean distance from PD-L1− CD68+ CD163+ TAMs to the nearest PD-L1+ HRS cell in all cases (P < .0001, paired Student t test; supplemental Figure 4E), consistent with coordinate localization of PD-L1+ TAMs with PD-L1+ HRS cells.
T-cell-specific expression and localization of PD-1
Immunostaining for PD-1 revealed positive staining of a subset of lymphocytes in cHL (supplemental Figure 5). Quantitative analysis confirmed that PD-1+ cells accounted for a minority of the T-cell population across the series (median, 9% of CD4+ T cells and 18% of CD8+ T cells). In addition, we observed that levels of cellular PD-1 expression were lower among positive-staining lymphocytes in the vicinity of HRS cells compared with positive-staining lymphocytes within the residual germinal centers that were found in a subset of cases (supplemental Figure 5). Quantitative analysis confirmed that the mean PD-1 expression by positive-staining CD3+ T cells in the vicinity of HRS cells was lower than among the PD-1high follicular helper T cells within reactive germinal centers; consistent with the notion that PD-1+ T cells in the cHL TME express PD-1 at low to intermediate levels (supplemental Figure 5E).15,16
PD-1+ T cells in relation to PD-L1+ TAMs
Visual inspection and cell phenotyping revealed that a subset of PD-1+ cells within the cHL TME are CD4+ and that these cells appeared enriched in the vicinity of PD-L1+ TAMs (exemplified by case N10; Figure 3A-B). Quantitative analysis revealed that the mean distance from PD-1+ CD4+ T cells to the nearest PD-L1+ TAM was less than the mean distance from PD-1+ CD4+ T cells to the nearest PD-L1− TAM in 16 of 20 cases (Figure 3C). This difference in distances was highly significant across the case series (P = .004; Figure 3C). Similarly, we observed that the mean distance from PD-1+ CD8+ T cells to the nearest PD-L1+ TAM (exemplified by case P6; Figure 3D-E) was less than the mean distance from PD-1+ CD8+ T cells to the nearest PD-L1− TAM in 15 of 20 cases (Figure 3F). This difference was highly significant for the series (P = .005) and, overall, consistent with coordinate regional localization of PD-1+ T cells and PD-L1+ TAMs. The mean distance from PD-1+ T cells to PD-L1+ TAMs was also weakly, but positively, correlated with the mean distance from PD-L1+ TAMs to PD-L1+ HRS cells for the series (r = 0.269 for CD4+ T cells and 0.283 for CD8+ T cells, respectively).
Figure 3.

Distances from PD-1+CD4+and PD-1+CD8+T cells to the nearest PD-L1+TAMs. (A) Representative multiplex IF image (20× resolution; case N10) showing staining for CD4 (cyan), PD-1 (yellow), CD68 (magenta), and PD-L1 (green). (B) Cellular phenotype map of image shown in A depicting the locations of PD-1+ CD4+ T cells (green dots), PD-L1+ TAMs (purple dots), PD-L1− TAMs (pink dots), and undefined cells (gray dots). (C) The mean distances (microns) and standard errors for all 20 study tumors, divided into mean distance from PD-1+ CD4+ T cells to the nearest PD-L1+ TAMs (blue) and mean distance from PD-1+ CD4+ T cells to the nearest PD-L1− TAMs (red). Tumors are ordered by the distance between PD-1+ CD4+ T cells and PD-L1− TAMs, from highest to lowest. P value (.004) was calculated by paired Student t test. (D) Representative multiplex IF image (20× resolution; case P6) showing staining for CD8 (red), PD-1 (yellow), CD68 (magenta), and PD-L1 (green). (E) Cellular phenotype map of image shown in D depicting the locations of PD-1+ CD8+ T cells (red dots), PD-L1+ TAMs (purple dots), PD-L1− TAMs (pink dots), and undefined cells (gray dots). (F) The mean distances (microns) and standard error for all 20 study tumors, divided into mean distance from PD-1+ CD8+ T cells to the nearest PD-L1+ TAMs (blue) and mean distance from PD-1+ CD8+ T cells to the nearest PD-L1− TAMs (red). Tumors are ordered by the distance from PD-1+ CD8+ T cells to the nearest PD-L1− TAMs, from highest to lowest. P value (.005) was calculated by paired Student t test.
PD-1+ T cells in relation to PD-L1+ HRS cells
We further wished to determine whether PD-1+ T cells were preferentially oriented in proximity to PD-L1+ HRS cells. Quantitative analysis revealed that the mean distance from PD-1+ CD4+ T cells to the nearest PD-L1+ HRS cell was less than the mean distance from PD-1+ CD4+ T cells to the nearest PD-L1− HRS cell in 15 of 20 cases (supplemental Figure 6A-C). This difference was significant across the case series (P = .03). The mean distance from PD-1+ CD8+ T cells to the nearest PD-L1+ HRS cell (exemplified by case P6, supplemental Figure 6D-E) was less than the mean distance from PD-1+ CD8+ T cells to the nearest PD-L1− HRS cell in 13 of 20 cases (supplemental Figure 6F). The difference did not reach significance for the series (P = .1).
T cells in direct contact with TAMs
Close visual inspection of stained tissue sections indicated PD-1+ expression on a subset of CD4+ T cells and a subset of CD8+ T cells in direct contact with TAMs (Figure 4A-D). CD4+ T cells were more likely than CD8+ T cells to be in direct contact with TAMs across the series (28% [95% confidence interval (CI), 22%-34%] vs 7% [95% CI, 6%-9%], respectively; Figure 4E). Moreover, CD4+ T cells and CD8+ T cells, as proportions of the cellularity, were both significantly higher at the points of contact with TAMs than at points without contact (>75 µm distant; P = .01 and <.01, respectively; Figure 4E).
Figure 4.

T-cell subsets in contact with TAMs. (A) Representative image (40× resolution; case N13) showing CD4+ T cells (left panel, green) with coexpression of PD-1 (right panel, yellow) touching CD68+ TAMs (left and right panels, magenta). (B) Membrane map depicting CD4+ T cells (PD-1+ dark green; PD-1− light green), and PD-L1+ TAMs (purple). Cells are generally only outlined, with the exceptions of PD-1+ CD4+ T cells and PD-L1+ TAMs that are in contact, which are filled to highlight the interaction. (C) Representative image (40× resolution; case N13) showing CD8+ T cells (left panel, red) with coexpression of PD-1 (right panel, yellow) touching CD68+ TAMs (left and right panels, magenta). (D) Membrane map depicting CD8+ T cells (PD-1+ dark red; PD-1− light red), and PD-L1+ TAMs (purple). Cells are generally only outlined, with the exceptions of PD-1+ CD8+ T cells and PD-L1+ TAMs that are in contact, which are filled. (E) Mean and standard error of the proportion of cells that are CD4+ T cells, CD8+ T cells, or undefined that are in contact with TAMs (red bars), within 75 µm of TAMs (blue bars), or more than 75 µm from TAMs (green bars), respectively. P values calculated by the Wilcoxon test. (F) Mean and standard error of the proportion of cells that are PD-1+ CD4+ T cells, PD-1− CD4+ T cells, PD-1+ CD8+ T cells, or PD-1− CD8+ T cells and that are in contact with PD-L1+ TAMs (red bars), within 75 µm of PD-L1+ TAMs (blue bars), or more than 75 µm from PD-L1+ TAMs (green bars), respectively. P values calculated by the Wilcoxon test.
PD-1+ CD4+ T cells also exceeded PD-1+ CD8+ T cells in contact with PD-L1+ TAMs across the series (3.9% [95% CI, 1.6%-6.3%] vs 1.8% [95% CI, 1.0%-2.7%], respectively). Similar to T cells in general, PD-1+ CD4+ T cells and PD-1+ CD8+ T cells, as proportions of the cellularity, were significantly higher at the points of contact with PD-L1+ TAMs than at points without contact (>75 µm distant; P = .04 and <.01, respectively; Figure 4F), consistent with the notion that PD-1+ CD4+ T cells and PD-1+ CD8+ T cells in contact with PD-L1+ TAMs are a locally enriched population.
T cells in direct contact with HRS cells
We also observed PD-1 on subsets of CD4+ T cells and CD8+ T cells in direct contact with HRS cells (Figure 5A-D). CD4+ T cells exceeded CD8+ T cells in contact with HRS cells across the series (35% of all contacts [95% CI, 28%-42%] vs 4% of all contacts [95% CI, 3%-6%], respectively; Figure 5E). CD4+ T cells, as proportions of the cellularity, were significantly higher at the points of contact with HRS cells than at points without contact (P < .01; Figure 5E). In contrast, CD8+ T cells, as proportions of the cellularity, were not significantly different at the points of contact with HRS cells and at points without contact (P = .78; Figure 5E).
Figure 5.

T-cell subsets in contact with HRS cells. (A) Representative image (40× resolution; case N12) showing CD4+ T cells (left panel, green) with coexpression of PD-1 (right panel, yellow) touching a CD30+ HRS cell (left and right panels, orange). (B) Membrane map depicting CD4+ T cells (PD-1+ dark green; PD-1− light green), and PD-L1+ HRS cells (orange). Cells are generally only outlined, with the exceptions of PD-1+ CD4+ T cells and PD-L1+ HRS cells that are in contact, which are filled to highlight the interaction. (C) Representative image (40× resolution; case P6) showing CD8+ T cells (left panel, red) with coexpression of PD-1 (right panel, yellow) touching CD30+ HRS cells (left and right panels, orange). (D) Membrane map depicting CD8+ T cells (PD-1+ dark red; PD-1− light red), and PD-L1+ HRS cells (orange). Cells are generally only outlined, with the exceptions of PD-1+ CD8+ T cells and PD-L1+ HRS cells that are in contact, which are filled. (E) Mean and standard error of the proportion of cells that are CD4+ T cells, CD8+ T cells, or undefined and that are in contact with HRS cells (red bars), within 75 µm of HRS cells (blue bars), or more than 75 µm from the HRS cells (green bars), respectively. P values calculated by the Wilcoxon test. (F) Mean and standard error of the proportion of cells that are PD-1+ CD4+ T cells, PD-1− CD4+ T cells, PD-1+ CD8+ T cells, or PD-1− CD8+ T cells in contact with PD-L1+ HRS cells (red bars), within 75 µm of PD-L1+ HRS cells (blue bars), or more than 75 µm from PD-L1+ HRS cells (green bars), respectively. P values calculated by the Wilcoxon test.
When the PD-1 status of the T cells and PD-L1 status of the HRS cells were considered, we found that PD-1+ CD4+ T cells exceeded PD-1+ CD8+ T cells in contact with PD-L1+ HRS cells (4.7% of all contacts [95% CI, 2.3%-7.1%] vs 1.8% [95% CI, 0.8%-2.8%], respectively; Figure 5F). Similar to CD4+ T cells in general, PD-1+ CD4+ T cells were a higher proportion of the cellularity at the points of contact with PD-L1+ HRS than at points without contact (P < .01; Figure 5F). In contrast, the proportion of PD-1+ CD8+ T cells was not significantly different between the points of contact with PD-L1+ HRS cells and at points without contact (P = .37; Figure 5F), consistent with the notion that PD-1+ CD4+ T cells, but not PD-1+ CD8+ T cells, are enriched in immediate proximity to PD-L1+ HRS cells.
Discussion
PD-1 blockade is especially effective in cHL, where 65% to 85% of patients with relapsed/refractory disease demonstrate clinical response.6-9 The sensitivity of cHL to PD-1 blockade is determined, in part, by genetic gains of PD-L1 and PD-L2 within the malignant HRS cells that result in robust expression of the PD-1 ligands that, in turn, engage PD-1 on infiltrating immune cells.2,3,10,17 Here, we defined the expression and topographic distribution of PD-L1+ and PD-1+ nonmalignant cells in the cHL microenvironment. We characterized the complex cellular TME in cHL using formalin-fixed, paraffin-embedded diagnostic biopsies, simultaneously identifying tens of thousands of cells per sample across large regions of interest, including malignant HRS cells, TAMs, and T cells. In addition, we developed and employed the analytic means to quantify the relative proportion and location of cells expressing PD-L1 and PD-1 and the spatial relationships between specific cell populations.
By these methods, we detected PD-L1 expression by at least a subset of HRS cells and TAMs in all cHLs, as in our previous studies using chromogenic immunohistochemistry.10 In all tumors, the majority of tissue PD-L1 was expressed by TAMs. This result is consistent with the observation that TAMs are, in general, far more common than HRS cells. Moreover, we find that TAMs are not randomly distributed; instead, PD-L1+ TAMs lie in greater proximity to PD-L1+ HRS cells than PD-L1− TAMs. The biological importance of this microenvironmental niche is supported by the preferential localization of PD-1+ T cells in proximity to and for contact with PD-L1+ TAMs. Taken together, our results suggest a model in which the inflammatory microenvironment of cHL is highly organized with PD-L1+ TAMs immediately surrounding HRS cells to engage PD-1+ T cells and augment immunosuppression (Figure 6).
Figure 6.

Model of PD-1:PD-L1 Interactions in cHL. HRS cells (purple) express PD-L1 (and PD-L2) in part as a result of copy gain of chromosome 9p24.1 which includes PD-L1/PD-L2/JAK2. Tumor-associated macrophages (blue) that are in proximity to HRS cells express high levels of PD-L1, likely in response to local cytokine production, and thereby significantly increase the total amount of PD-L1 in the vicinity of the malignant cells. Both TAMs’ and HRS cells’ PD-L1 is available to bind PD-1 on CD4+ T cells (green) and CD8+ T cells (red). CD4+ T cells and PD-1+ CD4+ T cells are in greater numbers and are specifically enriched in the vicinity of PD-L1+ HRS cells compared with CD8+ T cells and PD-1+ CD8+ T cells and may indicate a preferential role for CD4+ T cells during PD-1 blockade. IFNγ, interferon-γ.
Whether PD-L1 expression by TAMs is directly dependent on the presence of HRS cells is unknown, but the PD-L1+ TAMs are likely programmed as a consequence of the local cytokine milieu. Macrophages demonstrate marked phenotypic plasticity in response to their environment,18 and the induction of PD-L1 can be mediated by a variety of cytokines, including interferon γ and granulocyte-macrophage colony-stimulating factor.17,19,20 These and other pro-inflammatory cytokines are produced by HRS cells, but also the T cells, natural killer cells, and myeloid cells within the TME.19,21 In this respect, the inflammatory TME of cHL resembles that of certain solid tumors, in which PD-L1 expression by nonmalignant cells, including macrophages, is prominent.22 It will be of interest to microdissect and interrogate regions rich in PD-L1+ TAMs to characterize the spectrum of cytokines and chemokines that define this specialized niche in greater detail. It will also be of interest to specifically isolate PD-L1+ TAMs to determine whether they express additional phenotypic markers of immunosuppression, such as indoleamine 2,3-dioxygenase 1, which might be targetable and thus synergize with PD-1 blockade.18,23
To be effective, PD-L1 must engage PD-1 to inhibit antitumor immunity. Despite the marked clinical efficacy of checkpoint blockade, the critical cell populations that express PD-1 and effect antitumor immunity in cHL have remained undefined. We examined the expression of PD-1 on T cells and found that those within the TME of cHL express PD-1 at intermediate levels. Prior studies have established that T cells with intermediate or low levels of PD-1 expression are antigen-experienced, “exhausted” T cells that are primed for reactivation, whereas those with the highest levels of PD-1 include follicular helper T cells cells in germinal centers and T cells with an irreversibly “exhausted” phenotype in the periphery.15,16,24 Thus, our data suggest that the majority of PD-1+ T cells within the TME of cHL have a PD-1 phenotype primed for reactivation.
CD4+ and CD8+ T cells, including a subset that are PD-1+, are enriched in the vicinity of, and in contact with, PD-L1+ TAMs. This observation is consistent with the role of TAMs as professional antigen-presenting cells that process and present exogenous antigens, including those potentially from HRS cells, to CD4+ T cells by the MHC class II pathway and, through cross-presentation, to CD8+ T cells by the MHC class I pathway.18 As a consequence, the PD-L1+ TAMs may both promote antitumor immunity through antigen presentation to T cells and to immunosuppression through the engagement of PD-1. Validation of these proposed activities will require functional studies.
We find that CD4+ T cells are more often in contact with HRS cells than CD8+ T cells, consistent with previous studies.25 Moreover, CD4+ T cells in contact with HRS cells represent a locally enriched population, whereas CD8+ T cells do not. Regions with HRS cells can be locally dense in inflammatory cells, a characteristic that can affect spatial analysis. This is particularly true in the nodular sclerosis subtype compared with the mixed cellularity subtype of cHL. Regardless of subtype, however, we find that PD-L1+ macrophages and CD4+ T cells are enriched relative to PD-L1− macrophages and CD8+ T cells in the vicinity of and for contact with HRS cells.
Similarly, we find that PD-1+ CD4+ T cells, but not PD-1+ CD8+ T cells, in contact with PD-L1+ HRS cells represent a locally enriched population. These observations are of particular interest, given that HRS cells more generally express MHC class II than MHC class I.4,26 Inactivating somatic mutations in β2-microglobulin (β2M) is a frequent genetic lesion among HRS cells, and the reduction and loss of expression of the β2M/MHC class I complex might be expected to compromise the ability of HRS cells to engage CD8+ T cells.4,5,26 The high percentage of cHLs with reduction or complete loss of the β2M/ MHC class I protein complex (79%) also indicates that CD8+ T cells are unlikely to be the only effector cells associated with the efficiency of PD-1 blockade (65%-85%) in cHL.4
Indeed, our data suggest that PD-1+ CD4+ T cells may play a more important role in the antitumor response than previously anticipated. Recent studies indicate that CD4+ T cells may themselves be able to directly kill tumor cells (even those lacking MHC-II), using mechanisms that are more traditionally associated with CD8+ CTLs.27,28 In subsequent studies, it will be important to employ additional phenotypic markers to further define the PD-1+ CD4+ T-cell population, including those that identify CD4+ cytotoxic T cells, Th1-type and Th2-type T cells, and T-regulatory cells.29 It will be useful to determine whether these cells express additional immunoregulatory proteins, such as LAG-3, which are also targetable with novel therapies.30 These data also suggest the importance of determining the distribution and extended functional phenotypes of additional cell lineages, such as natural killer cells, natural killer/T cells, and γδ T cells, which can have prominent roles in executing antitumor immunity in the absence of MHC class I.31,32 The methods described in this study can also be applied to B cells, plasma cells, and other components of humoral immunity.
Finally, the systematic analysis of a large cohort of diagnostic biopsy specimens, preferably in the context of a clinical trial, will be necessary to determine whether the topological arrangements we observe are associated with response to therapy. In addition, it will be essential to analyze biopsy samples taken from patients while receiving PD-1 inhibitor therapy to positively identify cell populations that are primarily responsible for HRS cell killing.
In summary, we have quantified PD-L1:PD-1 interactions in a series of cHL and find a common architectural framework in which the majority of PD-L1 in the microenvironment is derived from TAMs that, like HRS cells, are in extensive contact with PD-1+ T cells. We propose that HRS tumor cells survive within a specialized cellular niche, an even more localized microenvironment within the broader tumor mass. This expands the overall pool of available PD-L1 surrounding HRS cells and increases the potential for functional downregulation of PD-1+ T cells before or at the time that they interact with HRS cells. Given that PD-L1+ and PD-1+ cells are in immediate proximity and in contact with HRS cells, it appears that the immune-suppressive and immune-stimulatory mechanisms governing antitumor immunity exist in a delicate and dynamic equilibrium. Further defining this immunologically privileged niche may uncover additional therapeutic targets.
Acknowledgments
This work was supported in part by Leukemia & Lymphoma Society SCOR 7009-12 (S.J.R.), National Institutes of Health, National Cancer Institute grant R01 CA161026 (M.A.S.), International Immune Oncology Network of Bristol-Myers Squibb (F.S.H., M.A.S., S.J.R.), the Center for Immuno-Oncology (F.S.H., S.J.R.), the Medical Research Council/Engineering and Physical Sciences Research Council Newcastle Molecular Pathology Node, Bloodwise, Bright Red and North East Promenaders against Cancer (C.D.C.), and National Institutes of Health, National Cancer Institute grant P50CA101942 (G.J.F.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.D.C., D.G., F.S.H., M.A.S., and S.J.R. designed research; C.D.C., D.G., M.L., M.G.M.R., E.C.S., and E.G. performed research; G.J.F. and F.S.H. contributed vital reagents and tools; C.D.C., D.G., M.L., M.G.M.R., E.C.S., E.G., X.H., R.R., D.N., X.S.L., M.A.S., and S.J.R. collected, analyzed, and interpreted data; X.H., R.R., D.N., and X.S.L. performed statistical analysis; and C.D.C. and S.J.R. wrote the manuscript.
Conflict-of-interest disclosure: F.S.H., M.A.S., S.J.R. receive research funding from Bristol-Myers Squibb and G.J.F. has patents/pending royalties on the PD-1 pathway from Bristol-Myers-Squibb, Roche, Merck, EMD-Serono, Boehringer-Ingelheim, AstraZeneca, and Novartis. The remaining authors declare no competing financial interests.
Correspondence: Scott J. Rodig, Department of Pathology, Brigham & Women's Hospital, Boston, MA 02215; e-mail: srodig@partners.org.
References
- 1.Swerdlow SH, Campo E, Harris NL, et al. . WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. [Google Scholar]
- 2.Green MR, Monti S, Rodig SJ, et al. . Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116(17):3268-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roemer MGM, Advani RH, Ligon AH, et al. . PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol. 2016;34(23):2690-2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roemer MGM, Advani RH, Redd RA, et al. . Classical Hodgkin lymphoma with reduced β2M/MHC class I expression is associated with inferior outcome independent of 9p24.1 status. Cancer Immunol Res. 2016;4(11):910-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reichel J, Chadburn A, Rubinstein PG, et al. . Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood. 2015;125(7):1061-1072. [DOI] [PubMed] [Google Scholar]
- 6.Ansell SM, Lesokhin AM, Borrello I, et al. . PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Younes A, Santoro A, Shipp M, et al. . Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: a multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol. 2016;17(9):1283-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armand P, Shipp MA, Ribrag V, et al. . Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. J Clin Oncol. 2016;34(31):3733-3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen R, Zinzani PL, Fanale MA, et al. ; KEYNOTE-087. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol. 2017;35(19):2125-2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen BJ, Chapuy B, Ouyang J, et al. . PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. 2013;19(13):3462-3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steidl C, Lee T, Shah SP, et al. . Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362(10):875-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng Z, Puri S, Moudgil T, et al. . Multispectral imaging of formalin-fixed tissue predicts ability to generate tumor-infiltrating lymphocytes from melanoma. J Immunother Cancer. 2015;3:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tóth ZE, Mezey E. Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. J Histochem Cytochem. 2007;55(6):545-554. [DOI] [PubMed] [Google Scholar]
- 14.Bogusz AM, Baxter RHG, Currie T, et al. . Quantitative immunofluorescence reveals the signature of active B-cell receptor signaling in diffuse large B-cell lymphoma. Clin Cancer Res. 2012;18(22):6122-6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorfman DM, Brown JA, Shahsafaei A, Freeman GJ. Programmed death-1 (PD-1) is a marker of germinal center-associated T cells and angioimmunoblastic T-cell lymphoma. Am J Surg Pathol. 2006;30(7):802-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26(1):677-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5(1):3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skinnider BF, Mak TW. The role of cytokines in classical Hodgkin lymphoma. Blood. 2002;99(12):4283-4297. [DOI] [PubMed] [Google Scholar]
- 20.Thorn M, Guha P, Cunetta M, et al. . Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther. 2016;23(6):188-198. [DOI] [PubMed] [Google Scholar]
- 21.Farrar MA, Schreiber RD. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol. 1993;11(1):571-611. [DOI] [PubMed] [Google Scholar]
- 22.Taube JM, Anders RA, Young GD, et al. . Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chevolet I, Speeckaert R, Schreuer M, et al. . Characterization of the in vivo immune network of IDO, tryptophan metabolism, PD-L1, and CTLA-4 in circulating immune cells in melanoma. OncoImmunology. 2015;4(3):e982382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14(12):1212-1218. [DOI] [PubMed] [Google Scholar]
- 25.Morris CS, Stuart AE. Reed-Sternberg/lymphocyte rosette: lymphocyte subpopulations as defined by monoclonal antibodies. J Clin Pathol. 1984;37(7):767-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diepstra A, Niens M, Vellenga E, et al. . Association with HLA class I in Epstein-Barr-virus-positive and with HLA class III in Epstein-Barr-virus-negative Hodgkin’s lymphoma. Lancet. 2005;365(9478):2216-2224. [DOI] [PubMed] [Google Scholar]
- 27.Kitano S, Tsuji T, Liu C, et al. . Enhancement of tumor-reactive cytotoxic CD4+ T cell responses after ipilimumab treatment in four advanced melanoma patients. Cancer Immunol Res. 2013;1(4):235-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haabeth OAW, Tveita AA, Fauskanger M, et al. . How do CD4(+) T cells detect and eliminate tumor cells that either lack or express MHC class II molecules? Front Immunol. 2014;5(APR):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28(1):445-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3--potential mechanisms of action. Nat Rev Immunol. 2015;15(1):45-56. [DOI] [PubMed] [Google Scholar]
- 31.Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27(45):5932-5943. [DOI] [PubMed] [Google Scholar]
- 32.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235-271. [DOI] [PubMed] [Google Scholar]