

Abstract
The cross-talk between dynamic microtubules and the cell cortex plays important roles in cell division, polarity, and migration. A critical adaptor that links the plus ends of microtubules with the cell cortex is the KANK N-terminal motif and ankyrin repeat domains 1 (KANK1)/kinesin family member 21A (KIF21A) complex. Genetic defects in these two proteins are associated with various cancers and developmental diseases, such as congenital fibrosis of the extraocular muscles type 1. However, the molecular mechanism governing the KANK1/KIF21A interaction and the role of the conserved ankyrin (ANK) repeats in this interaction are still unclear. In this study, we present the crystal structure of the KANK1·KIF21A complex at 2.1 Å resolution. The structure, together with biochemical studies, revealed that a five-helix-bundle–capping domain immediately preceding the ANK repeats of KANK1 forms a structural and functional supramodule with its ANK repeats in binding to an evolutionarily conserved peptide located in the middle of KIF21A. We also show that several missense mutations present in cancer patients are located at the interface of the KANK1·KIF21A complex and destabilize its formation. In conclusion, our study elucidates the molecular basis underlying the KANK1/KIF21A interaction and also provides possible mechanistic explanations for the diseases caused by mutations in KANK1 and KIF21A.
Keywords: cell adhesion, kinesin, microtubule, scaffold protein, structural biology, ankyrin repeat, KANK1/KIF21A complex, microtubule-cell adhesion cross-talk
Introduction
Cell migration plays a crucial role in diverse cellular processes, including embryonic development, immune response, tissue remodeling, and angiogenesis (1–3). Directional cell migration requires tightly coordinated cellular morphological changes and dynamically regulated assembly/disassembly of integrin-based focal adhesions (FAs)4 (3–5). Malfunction of cell migration can lead to inflammatory diseases, psychiatric disorders, and cancers (1, 2, 6).
Increasing evidence supports the importance of the microtubule (MT) networks in spatial and temporal control of turnover of FAs by serving as tracks for the exocytosis/endocytosis of specific vesicles/proteins required for adhesion functions (7–10). The search-and-capture model has been proposed, in which the MTs dynamically probe the intracellular space of cells and are captured and stabilized by specific interactions between the plus ends of MTs and cell cortices at the cell-cell adhesions and FAs (8, 11, 12). A variety of plus-end–tracking proteins (+TIPs), including the cytoplasmic linker proteins (CLIPs), the CLIP-associating proteins (CLASPs), adenomatous polyposis coli, etc., have been implicated in linking MTs to cell cortices (13–18). Specifically, CLASPs anchor MTs to the cell cortex by simultaneously binding to the MT plus-end–tracking protein EB1 and the cortical phosphoinositide-binding protein LL5β (19). Notably, the CLASP·LL5β complex, together with liprin-α/β and ELKS, forms the core plasma membrane–associated platforms for cortical MTs attachment (20, 21).
Recently, the KANK1·KIF21A complex has emerged as another key adaptor mediating the cross-talk between MTs and cell cortices (20–22). KANK1, a member of the KANK family of proteins (KANK1/2/3/4), was originally identified as a tumor suppressor for renal cell carcinoma (23–25). KANK1 has been shown to be an important regulator of cell polarity and migration via modulating the Rho GTPase signaling pathway (26–29). Mutations of the KANK family of proteins have been associated with congenital cerebral palsy, nephrotic syndrome, and various cancers (23, 28, 30). KANK1 contains a KANK N-terminal (KN) motif, the central coiled-coil domains, and the C-terminal ankyrin (ANK) repeats. KANK1 can bind to liprin-α/β via the central coiled-coil domains, which links KANK1 to the core cortical MT stabilization complexes (20, 31). KANK1 further recruits the kinesin-4 member KIF21A to the cell cortex through its ANK repeats (20, 32). KIF21A is an inhibitor of MT polymerization and prevents MT growth at the cell margin (20). Interestingly, KIF21A adopts a closed conformation with the motor activity blocked by the central coiled-coil regions (20). The congenital fibrosis of the extraocular muscles type 1 (CFEOM1)-associated mutations of KIF21A (e.g. R954W) relieve its autoinhibited structure, thus not only making KIF21A constitutively active at the plus ends of MTs but also leading to the abnormal accumulation of KANK1·KIF21A complex at the cell cortex, which is expected to interfere with the normal MT–cell cortex cross-talk (20, 32). Accordingly, destruction of the dynamic MT organization and proper MT–cell cortex connection may contribute to CFEOM1 pathogenesis. Most recently, the KANK1·KIF21A complex has been associated with the regulation of the cross-talk between MTs and FAs via the binding of the KN motif of KANK1/2 to the key FA protein talin (33, 34). Therefore, the KANK1·KIF21A complex mediates diverse signaling pathways linking MTs with cell adhesions.
Given the critical roles of the KANK1·KIF21A complex both in assembly of the MT–cell cortex macromolecular complexes and etiologies of developmental diseases and cancers, it is important to elucidate the mechanistic basis governing the interaction between KANK1 and KIF21A. In this work, we characterize the KANK1/KIF21A interaction in detail. The high-resolution crystal structure of the KANK1·KIF21A complex solved here reveals that the highly conserved fragment immediately preceding the ANK repeats of KANK1 adopts a folded domain structure and directly interacts with its ANK repeats, forming a structural and functional supramodule capable of specifically binding to an evolutionarily conserved peptide fragment located in the middle of KIF21A. We further show that mutations in KANK1 and KIF21A present in cancer patients destabilize or even eliminate the complex formation of the two proteins, thus providing possible mechanistic explanations for the diseases caused by the mutations of the two proteins.
Results
The interaction between KANK1 and KIF21A
KANK1 recruits KIF21A to cell cortex by binding to a 38-amino acid (aa) fragment in the middle of KIF21A (aa 1142–1180; human KIF21A: NP_001166935) through its ANK repeats (20) (Fig. 1A). We first confirmed the KANK1/KIF21A interaction by showing that a KANK1 fragment (aa 1081–1360, NP_852069; referred to as the KANK1 ANK domain), composed of several ANK repeats and an extended fragment N-terminal to the ANK repeats, binds to a fragment of mouse KIF21A (aa 1127–1169, NP_001102512), which covers the previously discovered 38-residue sequence of human KIF21A with a Kd value of ∼0.22 μm (Fig. 1B). Using a truncation-based approach, a 19-residue fragment of KIF21A (aa 1138–1156; referred to as the KANK1-binding domain (KBD)) was sufficient to bind to the KANK1 ANK domain (Kd ∼0.58 μm) (Fig. 1, B and C). Further deletion of the KIF21A KBD at its C terminus (aa 1138–1148) showed no detectable binding to the KANK1 ANK domain (Fig. 1B). We further demonstrated that the KANK1 ANK domain and the KIF21A KBD form a stable 1:1 stoichiometric complex in solution (Fig. 1D).
Figure 1.

Characterization of the interaction between KANK1 and KIF21A. A, schematic diagram showing the domain organizations of KANK1 and KIF21A. The interaction between KANK1 and KIF21A is indicated by a two-way arrow. Talin, liprins, and IRSp53 bind to the KN domain and the coiled-coil domain of KANK1, respectively. The middle coiled-coil domain forms an intramolecular interaction with the motor domain of KIF21A. CFEOM1-causing mutants (e.g. R954W, indicated by a red asterisk) would release the autoinhibited conformation of KIF21A. B, mapping the KANK1/KIF21A interaction via ITC-based measurements. n.d., not detectable. C, ITC assay showing that the KBD of KIF21A bound with the ANK domain of KANK1 with a Kd value of ∼0.58 μm. D, analytical gel filtration analysis showing that KANK1 ANK and KIF21A KBD form a 1:1 stoichiometric complex. SDS-PAGE analysis of the protein components from the eluted peaks (12–16 ml) is also shown. mau, milliabsorbance units.
Crystal structure of the KANK1·KIF21A complex
To understand the mechanistic basis of the KANK1/KIF21A interaction, we solved the crystal structure of the KANK1 ANK domain in complex with a synthetic KIF21A KBD peptide at 2.1 Å resolution using the molecular replacement method (Table 1). Each asymmetric unit contains one complex with 1:1 stoichiometry that is consistent with our biochemical analysis (Figs. 1D and 2A).
Table 1.
Data collection and refinement statistics
Numbers in parentheses represent the value for the highest resolution shell. RMSD, root mean square deviation.
| Data collection and processing | |
| Source | SSRF-BL19U1 |
| Wavelength (Å) | 0.97538 |
| Space group | P212121 |
| Unit cell | |
| a, b, c (Å) | 35.513, 51.958, 136.984 |
| α, β, γ (degrees) | 90, 90, 90 |
| Resolution range (Å) | 50.00–2.10 (2.14–2.10) |
| No. of unique reflections | 14,123 (484) |
| Redundancy | 4.3 (2.5) |
| I/σ (I) | 11.3 (2.0) |
| Completeness (%) | 92.2 (65.2) |
| Rmerge (%)a | 10.4 (42.2) |
| Wilson B | 27.1 |
| Structure refinement | |
| Resolution (Å) | 41.39–2.11 (2.19–2.11) |
| Rworkb/Rfreec (%) | 18.04 (20.93)/24.56 (26.20) |
| RMSD bonds (Å)/angles (degrees) | 0.013/1.21 |
| No. of reflections | |
| Working set | 12,160 (553) |
| Test set | 601 (25) |
| No. of protein atoms | 2048 |
| No. of solvent atoms | 98 |
| Average B factor protein/peptide (Å2) | 32.64/33.30 |
| Ramachandran plot (%) | |
| Most favored regions | 97.4 |
| Additionally allowed | 2.6 |
| Generously allowed | 0 |
a Rmerge = ∑|Ii − Im|/∑Ii, where Ii is the intensity of the measured reflection, and Im is the mean intensity of all symmetry-related reflections.
b Rcryst = Σ‖Fobs| − |Fcalc‖/Σ|Fobs|, where Fobs and Fcalc are observed and calculated structure factors.
c Rfree = ΣT‖Fobs| − |Fcalc‖/ΣT|Fo|, where T is a test data set of about 5% of the total reflections randomly chosen and set aside before refinement.
Figure 2.

Overall structure of the KANK1·KIF21A complex. A and B, ribbon diagram (A) and cylinder (B) representations of the KANK1·KIF21A complex structure. In the drawing, the capping domain and ANK repeats of KANK1 are shown in light blue and orange, respectively. The KBD peptide of KIF21A is shown in green. C, superposition of the structures of the KANK2-ANK (PDB entry 4HBD, light blue) and the KANK1·KIF21A complex (this work). D, omit map of the KIF21A KBD peptide bound to KANK1 ANK. The map is countered at the level of 1.0 σ in PyMOL.
In the complex structure, the KANK1 ANK domain contains five ANK repeats with the typical helix-turn-helix conformation (Fig. 2, A and B). Interestingly, a capping domain immediately preceding the ANK repeats adopts a compact helix bundle structure composed of five α-helices (Fig. 2 (A and B) and Figs. S1 and S2). To the best of our knowledge, this is the first case where a folded domain functions as a capping structural element for ANK repeats. We performed a homologous structure search using the DALI server (35). No homologous structure was found during the search. Further amino acid sequence alignment analysis of the capping domain from the KANK family of proteins reveals that both the secondary structure and the residues forming the hydrophobic core of the capping domain are highly conserved (Figs. S1 and S2), implying that all members of the KANK family of proteins contain a similar capping domain before their ANK repeats. The crystal structure of the KANK2 ANK domain has been deposited as part of a Structural Genomics Consortium project (PDB entry 4HBD), with no associated publication. We superposed the ANK domains of KANK1 and KANK2 and found that the ANK domain of KANK2 has an architecture similar to that of KANK1 (Fig. 2C).
The KIF21A KBD peptide adopts two short α-helices connected by a short loop and occupies the elongated concave groove formed by the inner αA helices and finger-like hairpin loops (finger loops) connecting two consecutive ANK repeats of KANK1 (Fig. 2, A and B). It should be noted that the first finger loop connects the capping domain and the first ANK repeat of KANK1 (Fig. 2, A and B). The electron densities of the residues of the KIF21A peptide are clearly defined in the complex (Fig. 2D).
The interface of KANK1·KIF21A complex
The interaction interface between KANK1 ANK and KIF21A KBD can be arbitrarily divided into two regions based on the interaction property: 1) the polar interaction site and 2) the hydrophobic interaction site (Fig. 3, A–C). The binding in site I is mainly mediated by the hydrogen-bonding interactions and the electrostatic interactions (Fig. 3A). For example, the side chain of Arg-1143KIF21A forms salt bridges with the side chains of Glu-1284KANK1 and Asp-1306KANK1 and forms a hydrogen bond with Ser-1276KANK1. The side chain of Glu-1284KANK1, in turn, interacts with the side chain of Arg-1144KIF21A, which is further stabilized by the intramolecular hydrogen-bonding interaction between Arg-1144KIF21A and Asn-1140KIF21A (Fig. 3, A and C). In addition, the side chain of Asp-1308KANK1 interacts with the side chain of Lys-1141KIF21A; the side chain of His-1285KANK1 forms a hydrogen bond with the side chain of Thr-1146KIF21A. Consistent with the interactions observed in the complex structure, mutations that disrupt these polar interactions weakened or even abolished the KANK1/KIF21A interaction (Fig. 3, E and F).
Figure 3.

Detailed interface of the KANK1·KIF21A complex. A and B, the KANK1/KIF21A interface is divided into two sites corresponding to site I (the polar interaction site) (A) and site II (the hydrophobic interaction site) (B). The interaction details between KANK1 and KIF21A in two sites are shown in stereo view. C, the combined surface and ribbon representations of the KANK1·KIF21A complex. The hydrophobic residues, positively charged residues, and negatively charged residues of KANK1 are colored yellow, blue, and red, respectively. D, structure-based sequence alignment of the KBD peptide of KIF21A from different species. In this alignment, the absolutely conserved and conserved residues are colored red and green, respectively. Residues involved in binding to KANK1 are annotated below as blue dots. E and F, summary of ITC-based measurements of binding affinities between wild type or mutants of KANK1 ANK and KIF21A KBD. n.d., not detectable.
Several key residues (i.e. Thr-1147, Leu-1152, Leu-1153, and Tyr-1154) at the C-terminal half of the KIF21A KBD peptide contact with the hydrophobic patch formed by Tyr-1176, Met-1209, Leu-1210, Leu-1213, and Leu-1248 from KANK1 (Fig. 3, B and C). Substitution of Tyr-1176KANK1 or Leu-1248KANK1 with Asp totally disrupted the interaction (Fig. 3E). Reciprocally, either the L1152D or the L1153A mutant of the KIF21A KBD peptide eliminated its binding to the KANK1 ANK domain (Fig. 3F).
Importantly, all of the residues that contribute to the interaction are absolutely conserved in KANK1 and KIF21A from different species (Fig. 3D and Fig. S1), implicating the indispensable functions of the KANK1/KIF21A interaction throughout the evolution. To confirm that binding of KIF21A is a common feature among all of the KANK family proteins, we performed careful sequence analysis of KANK1–4 ANKs. We noticed that the ANK domain of KANK2 has essentially the same KIF21A KBD–binding residues as that of KANK1 (Fig. S1), indicating that the KANK2 ANK domain may also bind to KIF21A. Satisfyingly, the KIF21A KBD peptide bound to KANK2 ANK with an affinity comparable with that for its binding to KANK1 ANK (Fig. 4A). However, the residues corresponding to those involved in the KIF21A binding of KANK1/2 ANKs include some variations in KANK3/4 ANKs (Fig. S1). In line with this observation, the ANK domains of KANK3 and KANK4 displayed no detectable binding to the KIF21A KBD peptide (Fig. 4, B and C). Collectively, these data confirm the prediction from the structural analysis that only KANK1 and KANK2 are capable of binding to KIF21A.
Figure 4.

The binding specificities of KANK family proteins to KIF21A KBD. A–C, ITC-based measurements of binding affinities between the KIF21A KBD and the ANK domains from the KANK family proteins, KANK2 (A), KANK3 (B), and KANK4 (C). n.d., not detectable.
The capping domain is essential for the KANK1/KIF21A interaction
An interesting structural feature revealed by the complex structure is that an α-helical capping domain couples tightly with the ANK repeats of KANK1 (Fig. 5A). The first finger loop connects the α3-α5 of the capping domain and the α1A-α1B of the first ANK repeat (ANK1) of KANK1 (Figs. 2A and 5A). The coupling interface is mainly mediated by the hydrophobic interactions (e.g. Trp-1130, Val-1144, and Ile-1148 from α3-α4 of the capping domain; Ile-1185, Leu-1188, and Leu-1189 from α1B of the ANK1) (Fig. 5, A and B). In addition to the hydrophobic packing interface, several polar interactions also contribute to the capping domain/ANK repeats (CAP/ANK) interaction. For example, the side chain of Asn-1182 forms a hydrogen bond with the main chain of Gln-1136; the main chain of Ser-1135 forms a hydrogen bond with the side chain of His-1180 (Fig. 5B). Intriguingly, the residues involved in the domain coupling are conserved in the KANK family of proteins (Fig. S1), suggesting that the stable folding of the CAP-ANK tandem is a common feature for all of the KANKs.
Figure 5.

The capping domain is essential for the intact KANK1/KIF21A binding. A, the combined surface and ribbon representations of the coupling interface between the capping domain and the ANK repeats of KANK1. B, stereo views of the molecular details of the capping domain/ANK repeats interface. C, ITC assay showing that the ANK repeats alone bound to the KIF21A KBD peptide with a Kd value of ∼64.9 μm.
Given that the KANK1/KIF21A interface is remote from the CAP/ANK coupling site, one would expect that the capping domain is not directly required for KANK1 to bind to KIF21A. However, the ANK repeats of KANK1 alone showed a ∼110-fold lower binding affinity to the KIF21A KBD peptide (Kd ∼64.9 μm) compared with that of the intact ANK domain of KANK1 (Figs. 1C and 5C). Moreover, the CAP domain of KANK1 was insoluble when expressed alone (data not shown), suggesting that the CAP domain may structurally couple with the ANK repeats of KANK1 and provide overall stability of KANK1 ANK domain. Taken together, these data implied that the CAP domain and the ANK repeats of KANK1 form into a structural and functional supramodule in binding to KIF21A.
Functional implications of the disease-causing mutations in the KANK1/KIF21A interface
A collection of evidence shows that loss of function of the KANK family of proteins has been implicated in various carcinomas and other diseases, including obsessive-compulsive disorder, cerebral palsy, and nephrotic syndrome (23, 28, 36). Likewise, several missense mutations or deletions in KIF21A have been reported in the patients with CFEOM1, a severe neuronal developmental disease (37–39). Moreover, point mutations of KIF21A are also implicated in the pathogenesis of various cancers (40–42). The complex structure solved here provides insights into the mechanistic basis of the diseases caused by these mutations.
Notably, all of the CFEOM1 mutations cluster within the motor domain or the predicted coiled-coil domains preceding the KBD domain of KIF21A. Thus, these mutations are unlikely to affect the KANK1/KIF21A interaction. A total of five missense mutations involving four residues are located at the interface of the KANK1·KIF21A complex. For example, the R1143Q and R1143P mutations of KIF21A were found in the patients with colorectal adenocarcinoma and head and neck squamous cell carcinoma, respectively (40–43). In the complex structure, the side chain of Arg-1143KIF21A forms hydrogen-bonding and charge-charge interactions with the side chains of Ser-1276KANK1, Glu-1284KANK1, and Asp-1306KANK1 (Figs. 3A and 6A). The R1143Q and R1143P mutations would disrupt the polar interaction network observed in the complex structure. As expected, either the R1143Q or the R1143P mutant of the KIF21A KBD peptide abolished its binding to the KANK1 ANK domain (Fig. 6B). Moreover, the Y1176C mutation of KANK1 was found to associate with skin cutaneous melanoma (43). Tyr-1176KANK1 is involved in the hydrophobic interactions between KANK1 ANK and KIF21A KBD (Figs. 3B and 6A). Substitution of Tyr-1176KANK1 with Cys would weaken these hydrophobic interactions, thus disturbing the formation of the KANK1·KIF21A complex. Indeed, the Y1176C mutant of KANK1 ANK showed impaired binding to the KBD peptide of KIF21A (Fig. 6B). To our surprise, the R1254W mutation of KANK1 found in the patients with stomach adenocarcinoma showed a decreased binding affinity toward KIF21A compared with the wild-type protein (Fig. 6B). The Arg-1254 from α3B of KANK1 is not localized at the interface of the KANK1·KIF21A complex. However, the side chain of Arg-1254KANK1 forms an intramolecular hydrogen bond with the main chain of Ala-1214 from α2A of KANK1 (Fig. 6A). Substitution of Arg-1254KANK1 with Trp may induce the structural instability of the ANK domain of KANK1, thus weakening the formation of the KANK1·KIF21A complex. Given the high similarities of sequence and KIF21A-binding property between KANK1 and KANK2, our structure also provides the possible mechanistic basis of the mutation of KANK2 (S676F) in patients with nephrotic syndrome (28). The main chain instead of the side chain of Ser-1179KANK1 (equivalent to Ser-676 in KANK2) forms a hydrogen bond with the side chain of Gln-1149KIF21A (Fig. 6A). Therefore, the substitution of Ser-676 with Phe is not expected to affect the KANK2/KIF21A interaction significantly. Indeed, the S676F variant of KANK2 showed a similar binding affinity toward KIF21A compared with the wild-type KANK2 (Figs. 4A and 6B). Thus, our structural and biochemical analyses suggest that the nephrotic syndrome-causing mutation in KANK2 is not likely to disturb the KANK2/KIF21A interaction, but instead interferes with the binding(s) of KANK2 to other target(s).
Figure 6.
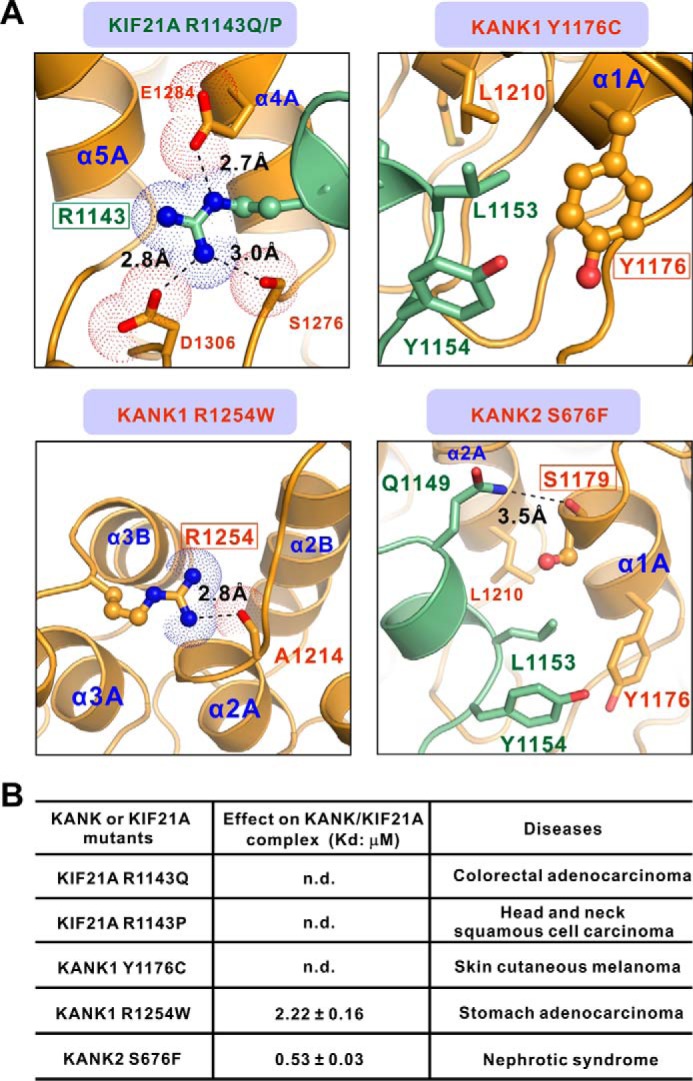
The effects of disease-associated mutations on the formation of the KANK1·KIF21A complex. A, the combined ribbon and stick-dot representations showing the detailed roles of disease-causing mutations in KANK1 ANK and KIF21A KBD. In this drawing, the side chains of key residues associated with diseases are shown in the stick mode, and the related hydrogen bonds and salt bridges are indicated by black dashed lines. B, effects of various disease-associated mutations on KANK1·KIF21A complex formation based on ITC assays. n.d., not detectable.
Potential regulation of the KANK1/KIF21A interaction by phosphorylation
How is the KANK1·KIF21A-mediated MT–cell cortex cross-talk regulated? We noted with extreme interest that the KIF21A KBD bears several conserved typical phosphorylation sites for the protein kinases, such as PKA, Akt, and CaMKII (44) (Fig. 7A). Based on the complex structure, all of these potential phosphorylation sites (i.e. Thr-1046, Thr-1047, and Thr-1048) are exposed to the solvent and available for possible phosphorylations. Satisfyingly, all of the phosphomimicking mutants of KIF21A (i.e. T1046E, T1047E, and T1048E) showed largely decreased bindings to KANK1 (Fig. 7, B–D), implying that phosphorylation of each of these three sites would impair the KANK1/KIF21A interaction. Notably, KANK1 was shown to be phosphorylated by Akt and involved in the negative regulation of cell migration through inhibiting Rho signaling pathway (26). It is tempting to speculate that the phosphorylation-dependent regulation of the KANK1/KIF21A interaction may provide a switch to the dynamic MT–cell cortex cross-talk. Future studies are required to validate whether such a switch is indeed employed in the physiological environments and, if so, which kinase(s) is involved in the phosphorylation of KIF21A.
Figure 7.

Phosphomimicking mutants of KIF21A impair their bindings to KANK1 ANK. A, consensus phosphorylation sites (PKA and Akt) located in the KBD fragment of KIF21A. B–D, ITC-based measurements of binding affinities between the ANK domain of KANK1 and different phosphomimicking mutants of KIF21A KBD.
Discussion
A variety of scaffold proteins play crucial roles in the cross-talk between dynamic MTs and cell adhesions, a process that is critical for cell division, polarity, and migration. In the symmetric/asymmetric cell divisions, the evolutionarily conserved LGN·NuMA complex functions as a bridge linking the cortical polarity cue(s) (e.g. the Par complex, Discs large, cell adhesion molecules, lipids, etc.) with the astral MTs, thus facilitating the proper spindle orientation of cell divisions (45–47). The most remarkable adaptors between MTs and the cortical membrane are the +TIPs that are tracked at the plus ends of MTs mainly via their bindings to the end binding (EB) family of proteins. The +TIPs, in turn, bind to the core cortical attachment LL5β·ELKS·liprin-α/β complex. A recent study demonstrated that the tumor suppressor KANK1 co-clusters with the liprin-α/β and LL5β-containing cortical MT attachment complexes at the cell periphery (20). The MT growth inhibitor KIF21A is further recruited to the cell cortex by KANK1, forming the MTs-KIF21A·KANK1-cell cortex axis. Most recently, KANK1/2 was shown to associate with the integrin-activating protein talin at the FAs, leading to the connection between MTs and FAs (33, 34). It should be noted that depletion of KIF21A had no impact on localization of KANK1, whereas the cortical localization of KIF21A absolutely depended on KANK1 (20). In line with these observations, KANK3 and KANK4, which did not bind to KIF21A, still localized at the FA belts (34) (Fig. 4).
An interesting discovery of this work is that, unlike the common capping sequence of ANK repeats, which forms into a single α-helix, the capping sequence of KANK1 ANK repeats adopts a compact five-helix bundle structure. We further demonstrated that the capping domain is tightly coupled with the ANK repeats of KANK1 and essential for the intact KANK1/KIF21A interaction (Fig. 5). It is possible that the structural and functional supramodule assembly between the ANK repeats and a folded domain (e.g. the CAP domain in KANK1) may also exist in other ANK repeat-containing proteins. Therefore, the diverse capping sequences N- or C-terminal to the canonical ANK repeats would provide further binding specificity toward distinct targets.
Several missense mutations of KANK1 and KIF21A were reported in patients with various cancers and other diseases (Fig. 6). Here, we demonstrated that these cancer-related variants weakened or even disrupted the KANK1/KIF21A interaction in vitro (Fig. 6). Future work is required to investigate whether these disease-causing mutations interfere with the MT–cell cortex cross-talk and then affect the regulations of cell polarity and migration in vivo. Interestingly, all of the CFEOM1-causing mutations locate at the motor domain or the coiled-coil domains rather than the KBD domain of KIF21A. Several CFEOM1-associated substitutions (e.g. R954W) would relieve the autoinhibition of KIF21A and lead to enhanced motor activity, suggesting that CFEOM1 pathology is due to a gain of function of KIF21A (20, 37). Therefore, the small KBD peptide of KIF21A derived from this work may provide a valuable tool to evaluate the function of the CFEOM1-causing enhanced KANK1/KIF21A interaction in vivo. Taken together, our biochemical and structural studies provide not only the mechanistic basis governing the specific KANK1/KIF21A interaction but also the functional implications of the disease-causing mutations in the KANK1/KIF21A interface.
Experimental procedures
Protein expression and purification
The coding sequences of the ANK domain of KANK1 and the KBD of KIF21A were PCR-amplified from the mouse brain cDNA library. All of the point mutations of KANK1 ANK and KIF21A KBD were created based on the standard PCR-based mutagenesis method using the Phanta Max superfidelity DNA polymerase (Vazyme Biotech Co., Ltd., catalogue no. P505) and confirmed by DNA sequencing. All of the wild-type and mutants were cloned into a modified version of pET32a vector containing an N-terminal Trx tag and His6 tag and expressed in BL21 (DE3) Escherichia coli cells for 18 h at 16 °C. The recombinant proteins were purified by Ni2+-nitrilotriacetic acid-agarose affinity chromatography (GE Healthcare), followed by size-exclusion chromatography (Superdex-200 26/60, GE Healthcare) in buffer containing 50 mm Tris, pH 8.0, 100 mm NaCl, 1 mm EDTA, and 1 mm DTT. The N-terminal tag of each recombinant protein was further cleaved by the human rhinovirus 3C protease and removed by the size-exclusion chromatography.
Isothermal titration calorimetry assay
Isothermal titration calorimetry (ITC) measurements were carried out on a MicroCal iTC200 system (Malvern) at 25 °C. All of the proteins were dissolved in a buffer containing 50 mm Tris, pH 8.0, 100 mm NaCl, 1 mm EDTA, and 1 mm DTT. The KIF21A KBD and its mutants with high concentrations (500–600 μm) were loaded into the syringe, whereas the KANK1 ANK and its mutants with low concentrations (30–40 μm) were placed in the cell. Each titration was completed by injecting a 1-μl aliquot of syringe protein into the protein in the cell at a time interval of 120 s to ensure that the titration peak returned to the baseline. The titration data were analyzed using the program Origin version 7.0 from MicroCal with the one-site binding model.
Analytical gel filtration chromatography
Analytical gel filtration chromatography was carried out on an AKTA pure L system (GE Healthcare). Protein samples were loaded onto a Superose 12 10/300 GL column (GE Healthcare) equilibrated with a buffer containing 50 mm Tris, 100 mm NaCl, 1 mm EDTA, and 1 mm DTT at pH 8.0.
Crystallography
Freshly purified KANK1 ANK domain (∼0.3 mm) was mixed with a saturating amount of the commercially synthesized KIF21A KBD peptide (China Peptides Co., Ltd.). Crystals of the KANK1·KIF21A complex (∼10 mg/ml) were obtained by the sitting drop vapor diffusion method at 16 °C. The complex crystals were grown in 0.1 m sodium formate, pH 8.0, 14% PEG 3350. Ethylene glycol (25%) was added as the cryoprotectant. Molecular replacement was used to solve the structure using the ANK domain of KANK2 (PDB entry 4HBD) as the search model using the software suites of Phaser (48) in CCP4 (49). Refinements were performed using phenix.refinement (50). The KIF12A KBD peptide was manually built. Model buildings and adjustments were carried out using Coot (51). The structural diagrams were prepared by PyMOL.
Author contributions
J. Z. and R. Z. designed the experiments. Z. W. and J. Z. performed the experiments. Y. S. and D. Y. contributed to X-ray data collection and structure determination. R. Z. and J. Z. analyzed the data. J. Z. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank the Shanghai Synchrotron Radiation Facility (SSRF) BL18U1 and BL19U1 for X-ray beam time, the Protein Expression and Purification System at the National Centre for Protein Science Shanghai for instrument support and technical assistance, and Prof. Mingjie Zhang (Hong Kong University of Science and Technology) for critical reading of the manuscript.
This work was supported by National Natural Science Foundation of China Grants 31470733 and U1532121, Shanghai Municipal Science and Technology Commission, China (“Yang-Fan program”) Grant 14YF1406700 (to J. Z.), and Strategic Priority Research Program of the Chinese Academy of Sciences Grant XDB08030104 (to R. Z.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2.
The atomic coordinates and structure factors (code 5YBE) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- FA
- focal adhesion
- MT
- microtubule
- +TIP
- plus-end–tracking protein
- CLIP
- cytoplasmic linker protein
- CLASP
- CLIP-associating protein
- KN
- KANK N-terminal
- ANK
- ankyrin
- CFEOM1
- congenital fibrosis of the extraocular muscles type 1
- aa
- amino acid(s)
- KBD
- KANK1-binding domain
- ITC
- isothermal titration calorimetry
- PDB
- Protein Data Bank.
References
- 1. Friedl P., and Gilmour D. (2009) Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 10, 445–457 10.1038/nrm2720 [DOI] [PubMed] [Google Scholar]
- 2. Mayor R., and Etienne-Manneville S. (2016) The front and rear of collective cell migration. Nat. Rev. Mol. Cell Biol. 17, 97–109 10.1038/nrm.2015.14 [DOI] [PubMed] [Google Scholar]
- 3. Petrie R. J., Doyle A. D., and Yamada K. M. (2009) Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 10, 538–549 10.1038/nrm2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boettiger D. (2012) Mechanical control of integrin-mediated adhesion and signaling. Curr Opin Cell Biol. 24, 592–599 10.1016/j.ceb.2012.07.002 [DOI] [PubMed] [Google Scholar]
- 5. Gardel M. L., Schneider I. C., Aratyn-Schaus Y., and Waterman C. M. (2010) Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26, 315–333 10.1146/annurev.cellbio.011209.122036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamaguchi H., Wyckoff J., and Condeelis J. (2005) Cell migration in tumors. Curr. Opin. Cell Biol. 17, 559–564 10.1016/j.ceb.2005.08.002 [DOI] [PubMed] [Google Scholar]
- 7. Stehbens S. J., Paszek M., Pemble H., Ettinger A., Gierke S., and Wittmann T. (2014) CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat. Cell Biol. 16, 561–573 10.1038/ncb2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stehbens S., and Wittmann T. (2012) Targeting and transport: how microtubules control focal adhesion dynamics. J. Cell Biol. 198, 481–489 10.1083/jcb.201206050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Theisen U., Straube E., and Straube A. (2012) Directional persistence of migrating cells requires Kif1C-mediated stabilization of trailing adhesions. Dev. Cell 23, 1153–1166 10.1016/j.devcel.2012.11.005 [DOI] [PubMed] [Google Scholar]
- 10. de Curtis I., and Meldolesi J. (2012) Cell surface dynamics: how Rho GTPases orchestrate the interplay between the plasma membrane and the cortical cytoskeleton. J. Cell Sci. 125, 4435–4444 10.1242/jcs.108266 [DOI] [PubMed] [Google Scholar]
- 11. Kirschner M., and Mitchison T. (1986) Beyond self-assembly: from microtubules to morphogenesis. Cell 45, 329–342 10.1016/0092-8674(86)90318-1 [DOI] [PubMed] [Google Scholar]
- 12. Akhmanova A., Stehbens S. J., and Yap A. S. (2009) Touch, grasp, deliver and control: functional cross-talk between microtubules and cell adhesions. Traffic 10, 268–274 10.1111/j.1600-0854.2008.00869.x [DOI] [PubMed] [Google Scholar]
- 13. Drabek K., van Ham M., Stepanova T., Draegestein K., van Horssen R., Sayas C. L., Akhmanova A., Ten Hagen T., Smits R., Fodde R., Grosveld F., and Galjart N. (2006) Role of CLASP2 in microtubule stabilization and the regulation of persistent motility. Curr. Biol. 16, 2259–2264 10.1016/j.cub.2006.09.065 [DOI] [PubMed] [Google Scholar]
- 14. Kodama A., Karakesisoglou I., Wong E., Vaezi A., and Fuchs E. (2003) ACF7: an essential integrator of microtubule dynamics. Cell 115, 343–354 10.1016/S0092-8674(03)00813-4 [DOI] [PubMed] [Google Scholar]
- 15. Perez F., Diamantopoulos G. S., Stalder R., and Kreis T. E. (1999) CLIP-170 highlights growing microtubule ends in vivo. Cell 96, 517–527 10.1016/S0092-8674(00)80656-X [DOI] [PubMed] [Google Scholar]
- 16. Näthke I. S. (2004) The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu. Rev. Cell Dev. Biol. 20, 337–366 10.1146/annurev.cellbio.20.012103.094541 [DOI] [PubMed] [Google Scholar]
- 17. Akhmanova A., and Steinmetz M. O. (2008) Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 9, 309–322 10.1038/nrm2369 [DOI] [PubMed] [Google Scholar]
- 18. Kumar P., and Wittmann T. (2012) +TIPs: SxIPping along microtubule ends. Trends Cell Biol. 22, 418–428 10.1016/j.tcb.2012.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lansbergen G., Grigoriev I., Mimori-Kiyosue Y., Ohtsuka T., Higa S., Kitajima I., Demmers J., Galjart N., Houtsmuller A. B., Grosveld F., and Akhmanova A. (2006) CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5β. Dev. Cell 11, 21–32 10.1016/j.devcel.2006.05.012 [DOI] [PubMed] [Google Scholar]
- 20. van der Vaart B., van Riel W. E., Doodhi H., Kevenaar J. T., Katrukha E. A., Gumy L., Bouchet B. P., Grigoriev I., Spangler S. A., Yu K. L., Wulf P. S., Wu J., Lansbergen G., van Battum E. Y., Pasterkamp R. J., et al. (2013) CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev. Cell 27, 145–160 10.1016/j.devcel.2013.09.010 [DOI] [PubMed] [Google Scholar]
- 21. Astro V., and de Curtis I. (2015) Plasma membrane-associated platforms: dynamic scaffolds that organize membrane-associated events. Sci. Signal. 8, re1 10.1126/scisignal.aaa3312 [DOI] [PubMed] [Google Scholar]
- 22. Akhmanova A., and Steinmetz M. O. (2015) Control of microtubule organization and dynamics: two ends in the limelight. Nat. Rev. Mol. Cell Biol. 16, 711–726 10.1038/nrm4084 [DOI] [PubMed] [Google Scholar]
- 23. Kakinuma N., Zhu Y., Wang Y., Roy B. C., and Kiyama R. (2009) Kank proteins: structure, functions and diseases. Cell Mol. Life Sci. 66, 2651–2659 10.1007/s00018-009-0038-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu Y., Kakinuma N., Wang Y., and Kiyama R. (2008) Kank proteins: a new family of ankyrin-repeat domain-containing proteins. Biochim. Biophys. Acta 1780, 128–133 10.1016/j.bbagen.2007.09.017 [DOI] [PubMed] [Google Scholar]
- 25. Sarkar S., Roy B. C., Hatano N., Aoyagi T., Gohji K., and Kiyama R. (2002) A novel ankyrin repeat-containing gene (Kank) located at 9p24 is a growth suppressor of renal cell carcinoma. J. Biol. Chem. 277, 36585–36591 10.1074/jbc.M204244200 [DOI] [PubMed] [Google Scholar]
- 26. Kakinuma N., Roy B. C., Zhu Y., Wang Y., and Kiyama R. (2008) Kank regulates RhoA-dependent formation of actin stress fibers and cell migration via 14-3-3 in PI3K-Akt signaling. J. Cell Biol. 181, 537–549 10.1083/jcb.200707022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roy B. C., Kakinuma N., and Kiyama R. (2009) Kank attenuates actin remodeling by preventing interaction between IRSp53 and Rac1. J. Cell Biol. 184, 253–267 10.1083/jcb.200805147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gee H. Y., Zhang F., Ashraf S., Kohl S., Sadowski C. E., Vega-Warner V., Zhou W., Lovric S., Fang H., Nettleton M., Zhu J. Y., Hoefele J., Weber L. T., Podracka L., Boor A., et al. (2015) KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J. Clin. Invest. 125, 2375–2384 10.1172/JCI79504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li C. C., Kuo J. C., Waterman C. M., Kiyama R., Moss J., and Vaughan M. (2011) Effects of brefeldin A-inhibited guanine nucleotide-exchange (BIG) 1 and KANK1 proteins on cell polarity and directed migration during wound healing. Proc. Natl. Acad. Sci. U.S.A. 108, 19228–19233 10.1073/pnas.1117011108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lerer I., Sagi M., Meiner V., Cohen T., Zlotogora J., and Abeliovich D. (2005) Deletion of the ANKRD15 gene at 9p24.3 causes parent-of-origin-dependent inheritance of familial cerebral palsy. Hum. Mol. Genet. 14, 3911–3920 10.1093/hmg/ddi415 [DOI] [PubMed] [Google Scholar]
- 31. Luo M., Mengos A. E., Mandarino L. J., and Sekulic A. (2016) Association of liprin beta-1 with kank proteins in melanoma. Exp. Dermatol. 25, 321–323 10.1111/exd.12933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kakinuma N., and Kiyama R. (2009) A major mutation of KIF21A associated with congenital fibrosis of the extraocular muscles type 1 (CFEOM1) enhances translocation of Kank1 to the membrane. Biochem. Biophys. Res. Commun. 386, 639–644 10.1016/j.bbrc.2009.06.109 [DOI] [PubMed] [Google Scholar]
- 33. Bouchet B. P., Gough R. E., Ammon Y. C., van de Willige D., Post H., Jacquemet G., Altelaar A. M., Heck A. J., Goult B. T., and Akhmanova A. (2016) Talin-KANK1 interaction controls the recruitment of cortical microtubule stabilizing complexes to focal adhesions. Elife 5, e18124 10.7554/eLife.18124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun Z., Tseng H. Y., Tan S., Senger F., Kurzawa L., Dedden D., Mizuno N., Wasik A. A., Thery M., Dunn A. R., and Fässler R. (2016) Kank2 activates talin, reduces force transduction across integrins and induces central adhesion formation. Nat. Cell Biol. 18, 941–953 10.1038/ncb3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holm L., and Sander C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123–138 10.1006/jmbi.1993.1489 [DOI] [PubMed] [Google Scholar]
- 36. Willour V. L., Yao Shugart Y., Samuels J., Grados M., Cullen B., Bienvenu O. J. 3rd, Wang Y., Liang K. Y., Valle D., Hoehn-Saric R., Riddle M., and Nestadt G. (2004) Replication study supports evidence for linkage to 9p24 in obsessive-compulsive disorder. Am. J. Hum. Genet. 75, 508–513 10.1086/423899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng L., Desai J., Miranda C. J., Duncan J. S., Qiu W., Nugent A. A., Kolpak A. L., Wu C. C., Drokhlyansky E., Delisle M. M., Chan W. M., Wei Y., Propst F., Reck-Peterson S. L., Fritzsch B., and Engle E. C. (2014) Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron 82, 334–349 10.1016/j.neuron.2014.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heidary G., Engle E. C., and Hunter D. G. (2008) Congenital fibrosis of the extraocular muscles. Semin. Ophthalmol. 23, 3–8 10.1080/08820530701745181 [DOI] [PubMed] [Google Scholar]
- 39. Yamada K., Andrews C., Chan W. M., McKeown C. A., Magli A., de Berardinis T., Loewenstein A., Lazar M., O'Keefe M., Letson R., London A., Ruttum M., Matsumoto N., Saito N., Morris L., et al. (2003) Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat. Genet. 35, 318–321 10.1038/ng1261 [DOI] [PubMed] [Google Scholar]
- 40. Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sveen A., Johannessen B., Tengs T., Danielsen S. A., Eilertsen I. A., Lind G. E., Berg K. C. G., Leithe E., Meza-Zepeda L. A., Domingo E., Myklebost O., Kerr D., Tomlinson I., Nesbakken A., Skotheim R. I., and Lothe R. A. (2017) Multilevel genomics of colorectal cancers with microsatellite instability-clinical impact of JAK1 mutations and consensus molecular subtype 1. Genome Med. 9, 46 10.1186/s13073-017-0434-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Giannakis M., Mu X. J., Shukla S. A., Qian Z. R., Cohen O., Nishihara R., Bahl S., Cao Y., Amin-Mansour A., Yamauchi M., Sukawa Y., Stewart C., Rosenberg M., Mima K., Inamura K., et al. (2016) Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 15, 857–865 10.1016/j.celrep.2016.03.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ubersax J. A., and Ferrell J. E. Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8, 530–541 10.1038/nrm2203 [DOI] [PubMed] [Google Scholar]
- 45. Tuncay H., and Ebnet K. (2016) Cell adhesion molecule control of planar spindle orientation. Cell Mol. Life Sci. 73, 1195–1207 10.1007/s00018-015-2116-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. di Pietro F., Echard A., and Morin X. (2016) Regulation of mitotic spindle orientation: an integrated view. EMBO Rep. 17, 1106–1130 10.15252/embr.201642292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu J., Wen W., Zheng Z., Shang Y., Wei Z., Xiao Z., Pan Z., Du Q., Wang W., and Zhang M. (2011) LGN/mInsc and LGN/NuMA complex structures suggest distinct functions in asymmetric cell division for the Par3/mInsc/LGN and Gαi/LGN/NuMA pathways. Mol. Cell 43, 418–431 10.1016/j.molcel.2011.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., and Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.