

Abstract
RcnR, a transcriptional regulator in Escherichia coli, derepresses the expression of the export proteins RcnAB upon binding Ni(II) or Co(II). Lack of structural information has precluded elucidation of the allosteric basis for the decreased DNA affinity in RcnR's metal-bound states. Here, using hydrogen–deuterium exchange coupled with MS (HDX-MS), we probed the RcnR structure in the presence of DNA, the cognate metal ions Ni(II) and Co(II), or the noncognate metal ion Zn(II). We found that cognate metal binding altered flexibility from the N terminus through helix 1 and modulated the RcnR–DNA interaction. Apo–RcnR and RcnR–DNA complexes and the Zn(II)–RcnR complex exhibited similar 2H uptake kinetics, with fast-exchanging segments located in the N terminus, in helix 1 (residues 14–24), and at the C terminus. The largest difference in 2H incorporation between apo- and Ni(II)- and Co(II)-bound RcnR was observed in helix 1, which contains the N terminus and His-3, and has been associated with cognate metal binding. 2H uptake in helix 1 was suppressed in the Ni(II)- and Co(II)-bound RcnR complexes, in particular in the peptide corresponding to residues 14–24, containing Arg-14 and Lys-17. Substitution of these two residues drastically affected DNA-binding affinity, resulting in rcnA expression in the absence of metal. Our results suggest that cognate metal binding to RcnR orders its N terminus, decreases helix 1 flexibility, and induces conformational changes that restrict DNA interactions with the positively charged residues Arg-14 and Lys-17. These metal-induced alterations decrease RcnR–DNA binding affinity, leading to rcnAB expression.
Keywords: metal homeostasis, metal ion-protein interaction, metalloprotein, transcription regulation, hydrogen exchange mass spectrometry
Introduction
Transition metals are essential to all organisms for the proper function of enzymes, yet are toxic, and therefore must be regulated to maintain an optimum level in the cell (1). Tight regulation of transition metals often involves a metal-trafficking system composed of proteins that generate biological responses to the specific binding of one or more metal ions (cognate metals) and not to other metal ions (non-cognate metals) (1–4). The nickel-trafficking pathway in Escherichia coli supplies Ni(II) for the maturation of hydrogenases under anaerobic conditions (5, 6) and includes an importer, NikABCDE (7, 8), metallochaperones and accessory proteins for metal incorporation (3), HypA (9, 10), HypB (11, 12), and SlyD (13, 14), and the exporter RcnAB (15, 16). The combined activity of two transcriptional regulators, NikR (nickel-responsive regulator) (17), which controls expression of the importer, and RcnR (resistance to cobalt and nickel repressor) (18, 19), which regulates the expression of the exporter, maintain nickel homeostasis in a manner functionally similar to transcriptional regulators involved in the regulation of other metal ions, such as Zn(II) (Zur/ZntR) (20, 21).
E. coli RcnR is a 40-kDa α-helical tetrameric transcriptional repressor that is a founding member of the RcnR/CsoR family (DUF156 family) of DNA-binding proteins (22). Apo-RcnR binds to DNA, repressing the expression of the export proteins, RcnA and RcnB (23). Many metal ions bind to RcnR in vitro, but only binding Ni(II) or Co(II) (cognate metals) decreases the affinity of RcnR for DNA and induces the transcription of the exporter (18). Other family members sense different metal ions or respond to non-metal signals, including Mycobacterium tuberculosis CsoR (Cu(I)), which has 40% sequence similarity to RcnR (∼20% identity) (24) or E. coli FrmR (formaldehyde), which is 65% homologous to RcnR (40% identity) (25). Family members are tetramers composed of α-helical monomers and have no structural homology to well-studied DNA-binding motifs, such as the winged helix, helix-turn-helix, and ribbon-helix-helix motifs, indicating a unique DNA-binding mode (22).
Tetrameric RcnR recognizes a pair of TACT-G6-N-AGTA motifs located in the rcnA-rcnR intergenic region (22). RcnR makes contacts with the minor groove of the TACT inverted repeats, whereas the G-tracts might provide a structural component facilitating the interaction (22). The junction of A-form DNA, characteristic of the G-tracts, and B-form DNA, characteristic of the TACT motif, likely introduces a structural kink that might facilitate DNA recognition and wrapping (22, 26, 27). Models of the frmRAB promoter region with the apo-EcFrmR crystal structure suggested that Lys-10, Arg-14, Arg-16, and Arg-17 (Lys-10, Arg-14, Ser-16, and Lys-17 in RcnR sequence) interact with the major grooves of the DNA, and Lys-91 (Lys-90 in RcnR) is packed into the minor groove, and that these interactions are disrupted when Pro-2 is cross-linked to Cys-35 by formaldehyde in the N terminus (25).
In contrast to FrmR, there is a lack of detailed information regarding how EcRcnR interacts with DNA and the nature of the allostery induced by cognate metal binding that affects the protein–DNA interaction. Herein, we report the use of hydrogen–deuterium exchange coupled with mass spectrometry (HDX-MS)2 to probe the RcnR structure in the presence of DNA, cognate metal ions (Ni(II)/Co(II)), and the non-cognate metal ion, Zn(II). The results provide information regarding the effects on protein dynamics of the binding of cognate versus non-cognate metal ions and, coupled with a mutagenic study, lead to a proposed mechanism for how binding of cognate metals decrease DNA affinity in RcnR. This proposed mechanism is supported by the structure of FrmR and studies in CsoR and reveal how the allosteric response may be adapted from formaldehyde cross-linking to metal binding to evolve transcriptional regulators that respond to distinct chemical signals.
Results
RcnR-DNA titration experiments using native mass spectrometry
The interaction of RcnR with DNA was investigated using a titration employing site 1 DNA, a 24-mer DNA molecule with a single TACT-G6-N-AGTA recognition site, coupled with native ESI-MS (28). Native ESI-MS measurements allow for the unambiguous assignment of DNA:RcnR stoichiometry over a range of ligand concentrations, although differences in ionization efficiencies for the different complexes and potential gas phase artifacts make determination of binding constants difficult. As shown in Fig. 1, the data clearly reveal the formation of 1:1 and 2:1 DNA–RcnR tetramer complexes, with the latter favored by increasing DNA concentrations. The 2:1 complex is detectable at a molar ratio of 1:1, indicating a significant affinity for a second site 1 DNA molecule at the protein and DNA concentrations used in this experiment. This can be understood in terms of a homology model (24) that reveals a band of positive charge circumscribing the RcnR tetramer, and thus provides two surfaces for specific interactions with two site 1 DNA molecules. This result is also consistent with nonspecific binding of a second site 1 DNA, as would occur in DNA wrapping by RcnR (22).
Figure 1.

Titration of RcnR with site 1 DNA monitored by ESI-MS. The concentrations and resulting RcnR:DNA ratios used in each experiment are indicated. Circles represent RcnR monomers and rectangles represent site 1 DNA molecules. The y-axes of the spectra are normalized intensity. The figure is colored to reflect the species involved (black = RcnR without DNA bound, blue = 1:1 DNA–RcnR complex, red = 2:1 DNA–RcnR complex). Inset: an expansion of the m/z = 3000–5500 region containing information regarding 1:1 and 1:2 DNA complexes.
Global protein HDX analysis
A comparison of RcnR complexes with site 1 DNA versus site 1 + 2 DNA, a 44-mer DNA molecule with the full core RcnR-binding site (22), using HDX-MS showed that the two complexes have remarkably similar global exchange kinetics (Fig. 2). RcnR has 88 backbone amide hydrogen atoms, 20% of which are rapidly exchanged (within 30 s) in apo-RcnR. The exchange increases to 47% after 30 min of D2O exposure. Remarkably, the protection of protein amide protons afforded by DNA binding is minimal, suggesting that the DNA complexes are quite dynamic. The site 1 DNA–RcnR complex has a slightly slower rate of deuterium incorporation than apo-RcnR, suggesting that site 1 DNA provides some protection to the backbone amide protons, but the fastest exchanging group is still accessible. Similarly, for the site 1 + 2 DNA–RcnR complex, the overall exchange rate decreases only marginally from that of the site 1 DNA–RcnR complex, indicating a little more protection in the larger complex, but the rapidly exchanging protons are not affected. Among these faster exchanging complexes, all have the same initial amount of deuterium uptake at 30 s, but the uptake slows at longer exposure time compared with apo-RcnR.
Figure 2.
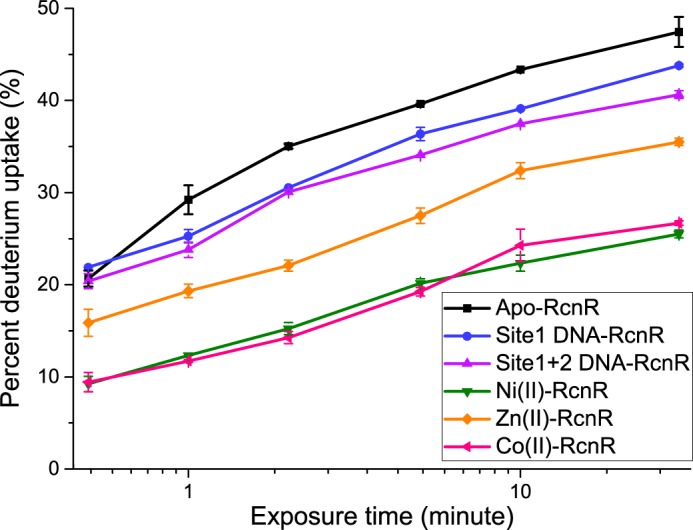
The rate of global protein 2H uptake for apo-RcnR, and site 1 DNA-, site 1 + 2 DNA-, Ni(II)-, Co(II)-, and Zn(II)–RcnR complexes. Studies were performed on 7.5 μm DNA–RcnR tetramer complexes and 5 μm tetramer apo- and metal–RcnR complexes. The molar ratios shown are 1 DNA molecule:1 RcnR tetramer, and 1 metal ion:1 RcnR monomer. The error bars are the mean ± S.D. calculated from three individual measurements.
In contrast, cognate metal binding dramatically increases the protection of backbone amide protons (Fig. 2). The initial exchange of only ∼10% protons represents a ∼50% decrease relative to apo-RcnR or the DNA complexes, indicating that some of the protons in the fast-exchanging group are protected by cognate metal binding. Only 26% of protons were exchanged in 30 min, and the exchange rate is essentially identical for Ni(II) and Co(II). Given the small effect associated with DNA binding, the change in exchange rate indicates altered protein dynamics that reflect a more rigid structure in the cognate metal complexes.
The global deuterium exchange kinetics for the Zn(II)–RcnR complex are similar to those of the apoprotein and DNA complexes, but feature a smaller number (16%) of rapidly exchanged protons and a smaller number of protons exchanged over 30 min. Thus, the non-cognate metal complex affords more protection and is consistent with a less dynamic RcnR structure than in the DNA complexes, but not nearly so much as observed in the cognate metal complexes.
Local protein HDX analysis of RcnR allosteric changes
To identify specific regions of the protein that are protected in the DNA and metal complexes, localized HDX-MS was performed. Pepsin digestion following HDX was done online for 4.5 min and 44 peptides that provided 98.9% coverage of RcnR were selected. The proteolytic peptide map is shown in Fig. 3. The HDX kinetics for six peptides are shown in Fig. 4, with the remaining peptides shown in Fig. S1.
Figure 3.

The proteolytic peptide coverage map for local protein HDX experiments. This coverage map was generated using ProteinLynx Global Server 3.0.1. 44 peptides (blue bars indicate specific peptides) were found with 98.9% coverage and 6.99% redundancy. The amino acid sequence of RcnR is labeled on the top. White vertical lines between blue bars indicate pepsin cleavage sites.
Figure 4.

Deuterium uptake kinetics for selected peptides. Error bars are the mean ± S.D. from triplicate measurements. The location of each peptide in the tetramer is indicated in red on the homology model of RcnR from reference (24). Missing data (e.g. for Zn (orange) in panel c and site 1 + 2 DNA (purple) in panels c and e) indicates that this particular peptide was not found in the specific experiment or that the data quality was not good. However, redundancies in the coverage means that the same information can be found from other peptides (e.g. for aa 26–37 and 25–36) in the Fig. S1. Color scheme for the different samples is indicated in panel d, and is the same as described in the legend to Fig. 2.
In apo-RcnR, the largest change in deuterium incorporation is associated with the N terminus (residues 2–13), helix 1 (residues 14–24), and the C terminus (residues 80–87) (Fig. 5). The N-terminal region and helix 1 have distinct deuterium uptake kinetics (Fig. 4, a and b). The N terminus reaches the maximum deuterium uptake level, ∼35%, within 30 s and thus constitutes one fast-exchange group. The helix 1 peptide reaches 17% of its maximum exchange in 30 s, and continues to incorporate deuterium, reaching 40% after 1.5 h of D2O exposure, indicating that the N terminus is more dynamic.
Figure 5.

a, deuterium uptake heat map shown for RcnR monomers of apo-RcnR and site 1 DNA-, site 1 + 2 DNA-, Ni(II)–, Co(II)–, and Zn(II)–RcnR complexes. The heat map data were generated by DynamX2.0 (Waters) using the peptides shown in Fig. 4 and Fig. S1. Blue indicates a region of less 2H uptake associated with a less flexible structure or less solvent accessible backbone amide protons, whereas red indicates a region with more 2H incorporation or high structural flexibility. b, the secondary structure for apo-RcnR based on the homology model in Ref. 24.
The deuterium uptake kinetics for the site 1 DNA–RcnR complex is very similar to those of apo-RcnR and features the same rapid deuterium uptake at N terminus (Fig. 4a). However, a small decrease in the deuterium incorporation rates in the peptides corresponding to residues 2–24, 37–52, and 80–87 is observed in the complex (Fig. 4 and Fig. S1). The site 1 + 2 DNA–RcnR complex shows more protection in these three regions, suggesting a potential DNA-binding site that aligns well with the location of positively charged residues (Arg-14, Arg-46, and Lys-17) identified by mutagenesis (see below).
The data regarding the metal complexes reveal that cognate metal binding induces minor protection of amide protons for most of the peptides, and a significant level of protection in peptides corresponding to residues in the N terminus and helix 1 (residues 14–24) that is virtually identical for Ni(II) and Co(II) and not observed in the Zn(II)–RcnR complex (Fig. 4, a and b). There is <10% deuterium incorporation at the N terminus after 90 min of D2O exposure, indicating that the N terminus is much less flexible in either cognate metal complex. This result is supported by the overall deuterium incorporation heat map (Fig. 5). This analysis highlights a segment corresponding to residues 2–13 that shows low levels of protection in apo-RcnR, the DNA– and the Zn(II)–RcnR complexes, but is protected in the cognate metal complexes. The results are consistent with the use of the N-terminal amine as a ligand in the cognate metal-binding site (29) (see below), and with the observation of N-terminal folding in CsoR upon binding Cu(I) (30) and in FrmR in the presence of formaldehyde (25).
Other peptides, such as those containing residues 26–39, 53–72, and 53–78 also show some protection upon cognate metal binding, but the differences are relatively small compared with the N-terminal region, and mutations, including R74A, have little effect on DNA affinity (Figs. 4 and 6 and Fig. S1) (18). However, it is worth noting that these peptides span loop regions in a homology model of RcnR and contain some of the other known metal ligands, including Cys-35 and His-64 (18, 31). This is consistent with the metal-binding site being largely pre-formed in apo-RcnR and where the binding of the flexible N terminus acts as an allosteric “switch” (32).
Figure 6.
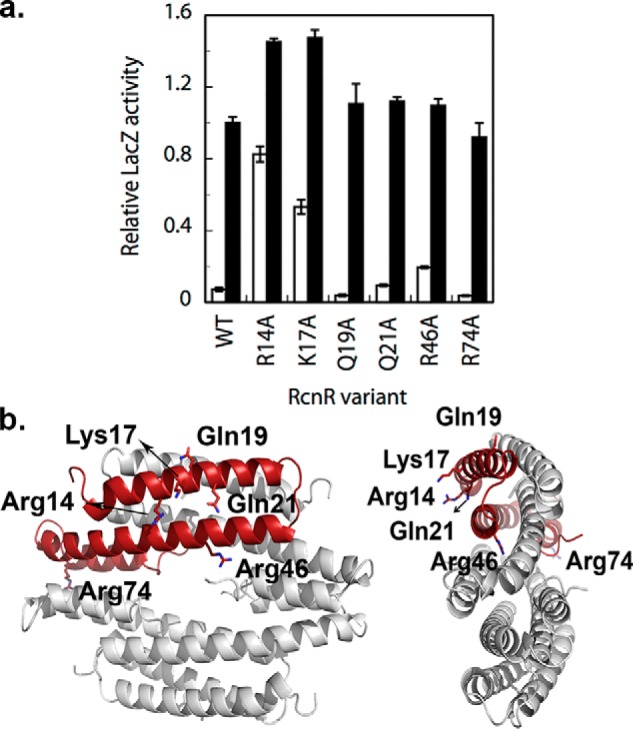
a, the results of LacZ reporter assays of RcnR variants. Proteins with high activity in the absence of added nickel (white bars) are indicative of impaired DNA-binding. All variants tested here retained nickel responsiveness (black bars). b, the location of the point mutations in the RcnR tetramer. The figure was made using the homology model from Ref. 24.
Although most regions of RcnR have low deuterium exchange rates in the presence of the cognate metals, part of helix 2, particularly in peptides 45–53 and 40–55 (Fig. 4 and Fig. S1), show a slightly higher exchange rate compared with apo-RcnR. This is consistent with prior studies of CsoR and FrmR that show helix repacking upon binding of the signaling moiety (Cu(I) for CsoR and formaldehyde for FrmR) (25, 30).
LacZ transcription reporter assays of important DNA-binding residues
The HDX-MS results for the peptides corresponding to residues 14–24 and 2–24 show that about half of the backbone amide protons are protected in the cognate metal complexes relative to apo-RcnR (Fig. 4b and Fig. S1). This region contains two highly conserved residues, Arg-14 and Lys-17. Mutagenesis was used to assess the roles of specific Arg and Lys residues in binding DNA by preparing several RcnR variants that were subsequently assayed for repressor activity in vivo using a LacZ transcription reporter assay (Fig. 6) (18). The R14A and K17A variants showed high levels of transcription in the absence of any added nickel (≥0.5 fractional activity of WT + nickel), suggesting DNA binding was strongly impaired. Other potential DNA-binding residues did not show a strong effect (Fig. 6). The R46A variant also showed some loss of repression in the absence of added metal (∼0.2 fractional activity of WT + nickel). This low level of de-repression is similar to what was observed previously for two variants in the C terminus, D77A and Y88F, in a study of RcnR variants that focused on nickel and cobalt responsiveness (18), although mutation of these residues might also affect DNA affinity. Arg-14, Gln-21, and Arg-46 are conserved between E. coli RcnR and FrmR, whereas a conservative replacement of Lys-17 to Arg is found in FrmR (Fig. 7).
Figure 7.

Multiple sequence alignment of RcnR, FrmR, CsoR, and InrS from various organisms using an online Clustal Omega program. Highly conserved residues are highlighted in blue, and conserved residues between RcnR and FrmR are highlighted in red.
Discussion
This work is one of the few examples utilizing HDX to study protein–DNA interactions (33, 34), and the only one that we are aware of to examine the effects of metal and DNA binding on protein dynamics. According to previous studies employing X-ray absorption spectroscopy as a structural probe and LacZ assays to assess function (18, 29, 32), Co(II) and Ni(II) bind to RcnR with slightly different ligand sets, employing His-3 for Co(II) but not for Ni(II) (29). Ni(II) is ligated by the N terminus, Glu-34, Cys-35, His-64, Glu-63, and one other protein ligand, whereas Co(II) binds N terminus, Cys-35, His-3, Glu-63, His-64, and Glu-34 (18, 29, 32). The complex defined in this way for Co(II) resembles a tris-bidentate chelate in that pairs of neighboring residues are employed as ligands (32). The cognate metal-binding sites were found to be distinct from the non-cognate sites in that the latter do not employ the N-terminal amine or His-3 as ligands (29).
The HDX-MS studies reported here show that formation of the cognate metal complexes orders the N-terminal motif, resulting in a much less dynamic N terminus that is reflected in the decreased rate of HDX. Formation of the Zn(II) complex does not induce the same ordering of the N terminus, consistent with the fact that the N terminus does not provide ligands to the Zn(II) site (29). Other peptides that contain metal-binding residues, such as peptides containing residues 26–39, 53–72, and 53–78, show some protection upon cognate metal binding, but the differences are relatively small compared with the N-terminal region (Figs. 4 and Fig. S1). This is consistent with the metal-binding site being largely pre-formed in apo-RcnR and where the binding of the flexible N terminus is involved in the allosteric switch that results in lower DNA affinity (32). Reduced flexibility of the N-terminal region upon cognate metal binding has also been observed upon Cu(I) binding in the copper sensor, CsoR (30). In this manner, the cognate metal-binding site in EcRcnR serves the same function as the Pro2–Cys35 formaldehyde cross-link in FrmR: to order the N terminus and bring the N-terminal amine in close proximity to Cys-35 (25).
The HDX-MS data also reveal a change in the dynamics of helix 1 in the cognate metal complexes that leads to less flexibility, and is not observed to the same extent in either the Zn(II) complex or the DNA complexes (Figs. 4 and Fig. S1). Helix 1 contains a large number of positively charged residues including, Arg-6, Lys-8, Lys-10, Lys-12, Arg-14, and Lys-17. Arg-14 and Lys-17 are highly conserved across the CsoR/RcnR family (Fig. 7), and were shown to be crucial for DNA binding to EcRcnR and CsoR (35) by mutagenesis. It seems likely that these two residues affect DNA affinity through nonspecific contacts that are altered by cognate metal binding, although in the absence of detailed structural information regarding the protein–DNA complex, there is nothing known about specific protein–DNA interactions that would lead to DNA sequence recognition. This too is similar to the effects of formaldehyde cross-linking seen in the structure of EcFrmR (25). In the structure of EcFrmR, the distance separating a pair of Arg-14 residues on one face of the FrmR tetramer shifts from 35 to 45 Å upon formaldehyde cross-linking, changing the distribution of positive charge on the protein surface and inhibiting the interaction with DNA (25). Thus, the available data suggest a possible general mechanism wherein changes in the ability of DNA to interact with Arg-14 and Lys-17 are associated with a chemical signal, which in the case of EcRcnR is cognate metal binding.
The change in the positions of positively charged residues is likely associated with helix 1 and helix 2 repacking, as illustrated by the EcFrmR structure (25) and by NMR studies of GtCsoR that revealed the formation of a kink in response to Cu(I) binding in the helix 2 region between Ala-76 and Ile-84, including the key Cu(I)-binding residue, Cys-79 (30). The deuterium uptake curve of RcnR peptides containing residues 40–53, 40–55, 45–53, 42–52, 44–53, and 45–55 (helix 2 region, Fig. 4 and Fig. S1) show faster deuterium uptake kinetics in Co(II) and Ni(II)–RcnR complexes relative to apo-RcnR, but not in Zn(II)–RcnR and DNA–RcnR complexes, consistent with more solvent exposure and a similar repacking of helices 1 and 2 in RcnR upon cognate metal binding.
The studies presented reveal details of the mechanism by which cognate metals (Co(II) and Ni(II)) induce structural changes in RcnR that affect DNA binding affinity and lead to de-repression of rcnAB. Binding of cognate metals to the N terminus lead to an ordering of the N-terminal metal-binding domain that brings the N-terminal amine in close proximity to Cys-35. In this way, the metal-binding site serves the same function as formaldehyde cross-linking of Pro-2 and Cys-35 in EcRcnR, and reflects an adaptation that allows for detection of a larger variety of chemical signals. The ordering of the N terminus decreases the flexibility of helix 1 and repacks helices 1 and 2, changing the distribution of surface positive charge on the protein and decreasing the affinity for DNA.
Experimental procedures
Materials
All chemicals besides those specified were purchased from Fisher Chemical. TEAA buffer was prepared with acetic acid and triethylamine (TEA).
RcnR overexpression and purification
RcnR was overexpressed and purified according to published procedures (32).
RcnR mutagenesis and β-galactosidase reporter experiments
RcnR variants R14A, K17A, Q19A, Q21A, R46A, and R74A were constructed in plasmid pRcnR as previously described (18), using the primers listed in Table 1 with the codon change shown in bold (only coding sequence shown from 5′ to 3′; obtained from either Operon Biotechnologies, Huntsville, AL, or Invitrogen). All pRcnR variants were sequenced (Seqwright, Houston, TX) to verify that only the desired mutation was present.
Table 1.
Primers used in making RcnR variants
| Mutants | |
|---|---|
| R14A | cag aaa ctg aaa gcg gct gcc agt aag att cag ggc |
| K17A | ctg aaa gcg cgt gcc agt gcg att cag ggc cag gtg gtg |
| Q19A | gcg cgt gcc agt aag att gcg ggc cag gtc gtg gcg |
| Q21A | gcc agt aag att cag ggc gcg gtc gtg gcg ctc aag |
| R46A | cag att gct gct atc gct ggc gcg gta aac gg |
| R74A | ggg gat gag cta aaa gct gaa gaa gat ctg gat g |
β-Galactosidase reporter experiments were conducted using two plasmids (pJI115 and pRcnR) as described previously for RcnR metal-sensing variants (29, 31). To assay reporter activity, transformed cells were grown anaerobically (37 °C, 14–16 h) in LB medium containing ampicillin (100 mg/ml) and chloramphenicol (34 mg/ml) with or without nickel added to 500 μm final concentration.
dsDNA purification
The forward and reversed single strands of DNA containing a single RcnR-binding site (site 1 DNA, forward strand sequence: aatctactggggggtagtatcagg) (22) and DNA containing the dyad-symmetric binding (site 1 + 2 DNA, forward strand sequence: aatctactggggggtagtatcaggtactgggggggagtagaatc) (22) were purchased from Eurofins Genomics. Equal amounts of forward and corresponding reversed strands of site 1 DNA and site 1 + 2 DNA were annealed by heating to 95 °C for 10 and 20 min, respectively, then slowly cooling down to room temperature and incubated on ice. Ethanol precipitation was performed and the dried dsDNA pellet was resuspended in a buffer with 0.1 m TEAA, pH 7.0, and 5% acetonitrile. The sample was then purified on an Agilent Zorbax 300SB-C18 column using an Agilent 1100 HPLC system. An 5–15% acetonitrile gradient with 0.1 m TEAA, pH 7.0, was run and the dsDNA was eluted at around 12% acetonitrile concentration. Following lyophilization, the DNA was resuspended in a buffer containing 300 mm ammonium acetate, pH 7.0, or a buffer with 20 mm HEPES (Gold Biotechnology), 300 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Soltec Ventures), 10% glycerol, pH 7.0, depending on the following experiments.
Protein-DNA titration mass spectrometry
Wild-type RcnR was desalted into 300 mm ammonium acetate buffer, pH 7.0, using a Zeba desalting column (Thermo Fisher) and exchanged several times using an Amicon Ultra Centrifugal Filter (Millipore) to remove NaCl. 10 μm RcnR tetramer with 0, 5, 10, 20, and 40 μm ds-site 1 DNA molecule were loaded into the nanospray ESI capillaries, and the masses of the complexes were determined using a QSTAR-XL (SCIEX) Q-TOF mass spectrometer. Instrument source conditions, primarily the declustering and focusing potentials, were adjusted to lower values to preserve the RcnR tetramer in the gas phase.
Global protein HDX-MS
Wild-type RcnR was buffer exchanged into 20 mm HEPES, 300 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, 10% glycerol, pH 7.0 using a Zeba desalting column (Thermo Fisher). For DNA–RcnR complexes, 1 eq (based on RcnR tetramer concentration) of ds-site 1 DNA or ds-site 1 + 2 DNA molecules were added to the RcnR solution, with the final DNA–RcnR complex concentration at 75 μm. For metal–RcnR complexes, 1.2 equivalents (based on RcnR monomer concentration) of NiCl2, CoCl2, or ZnCl2 were added, with the final metal–RcnR complex concentration at 200 μm monomer complex (or 50 μm tetramer complex), and the samples were incubated at room temperature for 30 min.
HDX reactions were initiated by diluting 2 μl of RcnR–DNA or RcnR–metal complexes, which were pre-equilibrated at room temperature for 2 min, with 18 μl of deuterium buffer containing 20 mm HEPES, pD 7.4, 300 mm NaCl at room temperature. The sample concentration at exchanging conditions were 7.5 μm for DNA–RcnR tetramer complexes and 20 μm for apo- and metal–RcnR monomer complexes (5 μm tetramer complex).
The deuterium buffer preparation procedure is as follows: 20 mm HEPES and 300 mm NaCl were dissolved in 99.5% D2O (Cambridge Isotope), and the pHobs was measured by an electrode that was pre-equilibrated with 99.5% D2O for 20 min. pD = pHobs + 0.4 was used to calculate the desired pHobs. 0.5 m NaOH was dissolved in 99.5% D2O and was used to adjust the pH to the desired pHobs.
At selected exposure times (0, 0.5, 1, 2, 5, 10, and 30 min), the HDX reaction was then quenched and RcnR was denatured by injected into a switching valve attached in a cooling box to maintain the temperature at 0 °C to reduce the rate of back-exchange. The masses of RcnR samples at different deuterium exposure times were measured using an ABI-SCIEX QSTAR-XL mass spectrometer equipped with an Agilent 1100 HPLC system and a 2.1 × 10-mm MassPREP online desalting Cartridge (Waters). For apo-RcnR, site 1 DNA–RcnR and metal–RcnR complex samples, the desalting Cartridge was equilibrated with 5% acetonitrile, and the sample was desalted and eluted using 5–95% acetonitrile gradient with 0.1% formic acid within 1 min. For the site 1 + 2 DNA–RcnR complex, the desalting Cartridge was equilibrated with 20% acetonitrile, and 20–95% acetonitrile gradient with 0.1% formic acid within 1 min was used to elute the DNA molecule prior to RcnR elution.
Local protein HDX-MS
RcnR–DNA and RcnR–metal complexes were prepared as described above for global protein HDX, but with only 1% glycerol in the buffer system. The HDX reaction was initiated by diluting 3.8 μl of 7.5 μm DNA–RcnR tetramer complex and 30 μm metal–RcnR monomer complexes (or 7.5 μm metal–RcnR tetramer complex) with 52.2 μl of deuterium buffer. Because the KD of DNA–RcnR and metal–RcnR complexes are at the nanomolar range (17, 32), the concentration used here is sufficient to keep the percentage of the complex in the solution more than 90%. To double check that the dilution is not causing the dissociation of the complexes, manual HDX initiation processes using Zeba desalting column (Thermo Fisher), which was pre-equilibrated with D2O buffer, at 15 and 60 min time points were performed. The deuterated samples were then injected into a Waters nanoACQUITY UPLC equipped with HDX technology, including a 2.1 × 30-mm pepsin immobilized Enymate column (Waters), trapping column (Waters Acquity Vanguard BEH C18 2.1 × 5 mm), and an analytical HSS T3 column (Waters). By comparing the result from manual injection (data not shown) and the automatic system, the uptake curves are very similar, especially for those peptides that have important information. Therefore, for the rest of the exposure time points (0, 0.5, 1, 2, 15, 30, 60, and 90 min), the experiment was done by the HDX automation system (LEAP technology) and the data were collected using a Synapt G2Si mass spectrometer. The resulting peptides were identified in undeuterated control samples using the Waters MSE and Waters ProteinLynx Global Server 3.0.1 (PLGS). Peptic peptide maps were generated by DynamX2.0 software (Waters). The percent of deuterium exchange was calculated by dividing the mass differences between deuterated and non-deuterated samples by the number of backbone amides. For peptides, the number of backbone amides was calculated after subtracting one at the N terminus and one at each proline residue. The data were not corrected for the back-exchange.
Author contributions
H.-T. H., C. E. B., and J. S. I. formal analysis; H.-T. H. and J. S. I. investigation; H.-T. H. visualization; H.-T. H., C. E. B., J. S. I., and I. A. K. methodology; H.-T. H. writing-original draft; C. E. B., J. S. I., P. T. C., I. A. K., and M. J. M. writing-review and editing; M. J. M. conceptualization; M. J. M. supervision; M. J. M. funding acquisition; M. J. M. project administration.
Supplementary Material
Acknowledgment
We gratefully acknowledge Dr. Steve Eyles for technical assistance in obtaining mass spectra.
This work was supported by National Institutes of Health Grant R01-GM069696 (to M. J. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- HDX-MS
- hydrogen-deuterium exchange–mass spectrometry
- ESI
- electrospray ionization
- RcnR
- resistance to cobalt and nickel repressor
- TEA
- trimethylamine
- TEAA
- trimethylamine with acetic acid.
References
- 1. Waldron K. J., Rutherford J. C., Ford D., and Robinson N. J. (2009) Metalloproteins and metal sensing. Nature 460, 823–830 10.1038/nature08300 [DOI] [PubMed] [Google Scholar]
- 2. Foster A. W., Osman D., and Robinson N. J. (2014) Metal preferences and metallation. J. Biol. Chem. 289, 28095–28103 10.1074/jbc.R114.588145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Higgins K. A., Carr C. E., and Maroney M. J. (2012) Specific metal recognition in nickel trafficking. Biochemistry 51, 7816–7832 10.1021/bi300981m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reyes-Caballero H., Campanello G. C., and Giedroc D. P. (2011) Metalloregulatory proteins: metal selectivity and allosteric switching. Biophys. Chem. 156, 103–114 10.1016/j.bpc.2011.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Böck A., King P. W., Blokesch M., and Posewitz M. C. (2006) Maturation of hydrogenases. Adv. Microb. Physiol. 51, 1–71 10.1016/S0065-2911(06)51001-X [DOI] [PubMed] [Google Scholar]
- 6. Forzi L., and Sawers R. G. (2007) Maturation of [NiFe]-hydrogenases in Escherichia coli. BioMetals 20, 565–578 10.1007/s10534-006-9048-5 [DOI] [PubMed] [Google Scholar]
- 7. de Pina K., Navarro C., McWalter L., Boxer D. H., Price N. C., Kelly S. M., Mandrand-Berthelot M. A., and Wu L. F. (1995) Purification and characterization of the periplasmic nickel-binding protein NikA of Escherichia coli K12. Eur. J. Biochem. 227, 857–865 10.1111/j.1432-1033.1995.tb20211.x [DOI] [PubMed] [Google Scholar]
- 8. Heddle J., Scott D. J., Unzai S., Park S. Y., and Tame J. R. (2003) Crystal structures of the liganded and unliganded nickel-binding protein NikA from Escherichia coli. J. Biol. Chem. 278, 50322–50329 10.1074/jbc.M307941200 [DOI] [PubMed] [Google Scholar]
- 9. Chan Chung K. C., and Zamble D. B. (2011) Protein interactions and localization of the Escherichia coli accessory protein HypA during nickel insertion to [NiFe] hydrogenase. J. Biol. Chem. 286, 43081–43090 10.1074/jbc.M111.290726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hube M., Blokesch M., and Böck A. (2002) Network of hydrogenase maturation in Escherichia coli: role of accessory proteins HypA and HybF. J. Bacteriol. 184, 3879–3885 10.1128/JB.184.14.3879-3885.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maier T., Lottspeich F., and Böck A. (1995) GTP hydrolysis by HypB is essential for nickel insertion into hydrogenases of Escherichia coli. Eur. J. Biochem. 230, 133–138 10.1111/j.1432-1033.1995.0133i.x [DOI] [PubMed] [Google Scholar]
- 12. Leach M. R., Sandal S., Sun H., and Zamble D. B. (2005) Metal binding activity of the Escherichia coli hydrogenase maturation factor HypB. Biochemistry 44, 12229–12238 10.1021/bi050993j [DOI] [PubMed] [Google Scholar]
- 13. Kaluarachchi H., Zhang J. W., and Zamble D. B. (2011) Escherichia coli SlyD, more than a Ni (II) reservoir. Biochemistry 50, 10761–10763 10.1021/bi201590d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J. W., Butland G., Greenblatt J. F., Emili A., and Zamble D. B. (2005) A role for SlyD in the Escherichia coli hydrogenase biosynthetic pathway. J. Biol. Chem. 280, 4360–4366 10.1074/jbc.M411799200 [DOI] [PubMed] [Google Scholar]
- 15. Rodrigue A., Effantin G., and Mandrand-Berthelot M. A. (2005) Identification of rcnA (yohM), a nickel and cobalt resistance gene in Escherichia coli. J. Bacteriol. 187, 2912–2916 10.1128/JB.187.8.2912-2916.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blériot C., Effantin G., Lagarde F., and Mandrand-Berthelot M. A. (2011) RcnB is a periplasmic protein essential for maintaining intracellular Ni and Co concentrations in Escherichia coli. J. Bacteriol. 193, 3785–3793 10.1128/JB.05032-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leitch S., Bradley M. J., Rowe J. L., Chivers P. T., and Maroney M. J. (2007) Nickel-specific response in the transcriptional regulator, Escherichia coli NikR. J. Am. Chem. Soc. 129, 5085–5095 10.1021/ja068505y [DOI] [PubMed] [Google Scholar]
- 18. Iwig J. S., Leitch S., Herbst R. W., Maroney M. J., and Chivers P. T. (2008) Ni(II) and Co(II) sensing by Escherichia coli RcnR. J. Am. Chem. Soc. 130, 7592–7606 10.1021/ja710067d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blaha D., Arous S., Blériot C., and Dorel C. (2011) The Escherichia coli metallo-regulator RcnR represses rcnA and rcnR transcription through binding on a shared operator site: insights into regulatory specificity towards nickel and cobalt. Biochimie 93, 434–439 10.1016/j.biochi.2010.10.016 [DOI] [PubMed] [Google Scholar]
- 20. Choi S., and Bird A. J. (2014) Zinc'ing sensibly: controlling zinc homeostasis at the transcriptional level. Metallomics 6, 1198–1215 10.1039/C4MT00064A [DOI] [PubMed] [Google Scholar]
- 21. Outten C. E., and O'Halloran T. V. (2001) Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science 292, 2488–2492 10.1126/science.1060331 [DOI] [PubMed] [Google Scholar]
- 22. Iwig J. S., and Chivers P. T. (2009) DNA recognition and wrapping by Escherichia coli RcnR. J. Mol. Biol. 393, 514–526 10.1016/j.jmb.2009.08.038 [DOI] [PubMed] [Google Scholar]
- 23. Iwig J. S., Rowe J. L., and Chivers P. T. (2006) Nickel homeostasis in Escherichia coli: the rcnR-rcnA efflux pathway and its linkage to NikR function. Mol. Microbiol. 62, 252–262 10.1111/j.1365-2958.2006.05369.x [DOI] [PubMed] [Google Scholar]
- 24. Musiani F., Zambelli B., Bazzani M., Mazzei L., and Ciurli S. (2015) Nickel-responsive transcriptional regulators. Metallomics 7, 1305–1318 10.1039/C5MT00072F [DOI] [PubMed] [Google Scholar]
- 25. Denby K. J., Iwig J., Bisson C., Westwood J., Rolfe M. D., Sedelnikova S. E., Higgins K., Maroney M. J., Baker P. J., Chivers P. T., and Green J. (2016) The mechanism of a formaldehyde-sensing transcriptional regulator. Sci. Rep. 6, 38879 10.1038/srep38879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu X.-J., Shakked Z., and Olson W. K. (2000) A-form conformational motifs in ligand-bound DNA structures. J. Mol. Biol. 300, 819–840 10.1006/jmbi.2000.3690 [DOI] [PubMed] [Google Scholar]
- 27. Hud N. V., and Plavec J. (2003) A unified model for the origin of DNA sequence-directed curvature. Biopolymers 69, 144–158 10.1002/bip.10364 [DOI] [PubMed] [Google Scholar]
- 28. Kaltashov I. A., Bobst C. E., and Abzalimov R. R. (2013) Mass spectrometry-based methods to study protein architecture and dynamics. Protein Sci. 22, 530–544 10.1002/pro.2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Higgins K. A., Chivers P. T., and Maroney M. J. (2012) Role of the N-terminus in determining metal-specific responses in the E. coli Ni- and Co-responsive metalloregulator, RcnR. J. Am. Chem. Soc. 134, 7081–7093 10.1021/ja300834b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chang F.-M., Coyne J. H., Cubillas C., Vinuesa P., Fang X., Ma Z., Ma D., Helmann J. D., los Santos A., Wang Y.-X., Dann C. E., and Giedroc D. P. (2014) Cu(I)-mediated allosteric switching in a copper-sensing operon repressor (CsoR). J. Biol. Chem. 289, 19204–19217 10.1074/jbc.M114.556704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Higgins K. A., Hu H. Q., Chivers P. T., and Maroney M. J. (2013) Effects of select histidine to cysteine mutations on transcriptional regulation by Escherichia coli RcnR. Biochemistry 52, 84–97 10.1021/bi300886q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carr C. E., Musiani F., Huang H.-T., Chivers P. T., Ciurli S., and Maroney M. J. (2017) Glutamate ligation in the Ni(II)- and Co(II)-responsive Escherichia coli transcriptional regulator, RcnR. Inorg. Chem. 56, 6459–6476 10.1021/acs.inorgchem.7b00527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang J., Chalmers M. J., Stayrook K. R., Burris L. L., Wang Y., Busby S. A., Pascal B. D., Garcia-Ordonez R. D., Bruning J. B., Istrate M. A., Kojetin D. J., Dodge J. A., Burris T. P., and Griffin P. R. (2011) DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat. Struct. Mol. Biol. 18, 556–563 10.1038/nsmb.2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nevin P., Lu X., Zhang K., Engen J. R., and Beuning P. J. (2015) Noncognate DNA damage prevents the formation of the active conformation of the Y-family DNA polymerases DinB and DNA polymerase κ. FEBS J. 282, 2646–2660 10.1111/febs.13304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chang F.-M., Martin J. E., and Giedroc D. P. (2015) Electrostatic occlusion and quaternary structural ion pairing are key determinants of Cu(I)-mediated allostery in the copper-sensing operon repressor (CsoR). Biochemistry 54, 2463–2472 10.1021/acs.biochem.5b00154 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.