

Abstract
Cardiac development and function require actin–myosin interactions in the sarcomere, a highly organized contractile structure. Sarcomere assembly mediated by formin homology 2 domain-containing 3 (Fhod3), a member of formins that directs formation of straight actin filaments, is essential for embryonic cardiogenesis. However, the role of Fhod3 in the neonatal and adult stages has remained unknown. Here, we generated floxed Fhod3 mice to bypass the embryonic lethality of an Fhod3 knockout (KO). Perinatal KO of Fhod3 in the heart caused juvenile lethality at around day 10 after birth with enlarged hearts composed of severely impaired myofibrils, indicating that Fhod3 is crucial for postnatal heart development. Tamoxifen-induced conditional KO of Fhod3 in the adult heart neither led to lethal effects nor did it affect sarcomere structure and localization of sarcomere components. However, adult Fhod3-deleted mice exhibited a slight cardiomegaly and mild impairment of cardiac function, conditions that were sustained over 1 year without compensation during aging. In addition to these age-related changes, systemic stimulation with the α1-adrenergic receptor agonist phenylephrine, which induces sustained hypertension and hypertrophy development, induced expression of fetal cardiac genes that was more pronounced in adult Fhod3-deleted mice than in the control mice, suggesting that Fhod3 modulates hypertrophic changes in the adult heart. We conclude that Fhod3 plays a crucial role in both postnatal cardiac development and functional maintenance of the adult heart.
Keywords: actin, cardiac development, cardiac hypertrophy, formin, heart, myofibril, sarcomere
Introduction
Normal cardiac function requires the accurately regulated actin–myosin interaction, which can be achieved by the precisely assembled and maintained sarcomere, the highly organized arrays of thin actin filaments and thick myosin filaments (1). During sarcomere assembly in the embryonic heart, actin cytoskeleton undergoes dynamic rearrangement by action of various actin-binding proteins (2), including tropomodulin, troponin T, α-tropomyosin, and formins; genetic deletion of these proteins causes failure of heart development and thereby results in embryonic lethality (3–7).
The structural maintenance of mature sarcomeres is also required for normal cardiac function. Although it was thought that actin filaments in mature muscle cells are relatively stable and do not easily turn over, recent studies have revealed dynamic exchange of cardiac actin thin filaments, i.e. a free actin is incorporated into actin filaments in mature sarcomeres (8, 9). It remains uncertain, however, how actin is exchanged in and out of thin actin filaments in the heart under continuously beating conditions. Because actin filaments in the sarcomere are repetitively pulled into the array of myosin filaments by the periodic actin–myosin interaction, unregulated actin turnover would be directly linked to cardiac dysfunction. Under beating conditions, cardiomyocytes can increase their cell volume by expanding the contractile unit sarcomere in response to physiological demands and pathological changes such as hypertension (10). Although the two actin dynamics regulators Wdr1 (11) and Lmod2 (12) are known to participate in postnatal cardiac development and maintenance of adult hearts, the regulatory mechanism for actin dynamics during development and maintenance of the heart has largely remained to be elucidated (2).
The formin family proteins are structurally characterized by the presence of the formin homology (FH)3 domains 1 and 2 and constitute a group of actin nucleation factors that play pivotal roles in controlling actin polymerization (13–15). The FH2 domain directly binds to two actin molecules to facilitate actin filament nucleation and remains associated with the barbed end of the filament to promote polymerization, which is accelerated by FH1-mediated recruitment of profilin complexed with an actin monomer (16). Through cooperation of the FH1 and FH2 domains, formins direct formation of straight actin filaments, thereby modulating the actin dynamics in various actin structures, such as lamellipodia, filopodia, and contractile fibers (17). Recent studies have revealed that formins, including mDia1, Daam1, and Fmn2, are critical for morphogenesis and organogenesis during development (18–20).
Fhod3 (previously known as Fhos2), a formin that is abundantly expressed in the heart (21), plays an essential role in the regulation of actin assembly in cardiac myofibrils (22–24). We have recently shown that genetic deletion of Fhod3 in mice confers embryonic lethality with defects in cardiogenesis (6). In Fhod3-null embryos, premyofibrils are formed once but fail to mature, suggesting that Fhod3 is essential for myofibrillogenesis, particularly for myofibril maturation. Indeed, Fhod3 accumulates at the central region of the sarcomere before myofibrillogenesis is completed, and its actin-assembling activity is required for myofibril maturation (25). However, the involvement of Fhod3 in postnatal cardiac development is unknown. Similarly, the role of Fhod3 in sarcomere maintenance in the fully-developed heart remains to be elucidated, although its significance can be expected from recent reports showing that some genetic variants of the Fhod3 gene are associated with human adult-onset cardiomyopathy (26, 27).
Here we have generated a floxed allele of Fhod3 (Fhod3flox) for conditional knock-out of Fhod3. By crossing Fhod3flox mice with muscle creatine kinase (MCK)-Cre mice and α-myosin heavy chain (αMHC)-MerCreMer mice, we examined the effect of Fhod3 depletion at perinatal and adult stages, respectively. In addition, by continuous infusion of phenylephrine, an α1-adrenergic receptor agonist, we evaluated the role of Fhod3 in modulation of hypertrophic response.
Results
Cardiac-specific deletion of Fhod3 in perinatal mice
We have previously reported that Fhod3 is indispensable for cardiogenesis, especially for myofibrillogenesis (6). In contrast, the role of Fhod3 in the fully-developed heart has remained elusive, although the Fhod3 mRNA is abundant in the adult heart as well as in the fetal heart (Fig. 1A) (21, 24). To address the question, we first tested the protein level of Fhod3 in various developmental stages of mice. Immunoblot analysis revealed that the Fhod3 protein was expressed in the heart at comparable levels throughout the lifetime from the embryo to the senior adult (Fig. 1B). The expression of Fhod3 is mostly restricted to the heart (Fig. 1, C and D). To investigate how Fhod3 functions in the developed heart, we generated the Fhod3flox allele by introduction of loxP sites into the introns surrounding the exon 18 (Fig. 2), which encodes the entire FH1 and the first part of FH2 domains (the core domains for actin polymerization). Cre-mediated recombination leads to deletion of the exon 18 and introduces a frameshift mutation, thereby resulting in a premature stop codon 7 amino acids downstream from the deletion.
Figure 1.
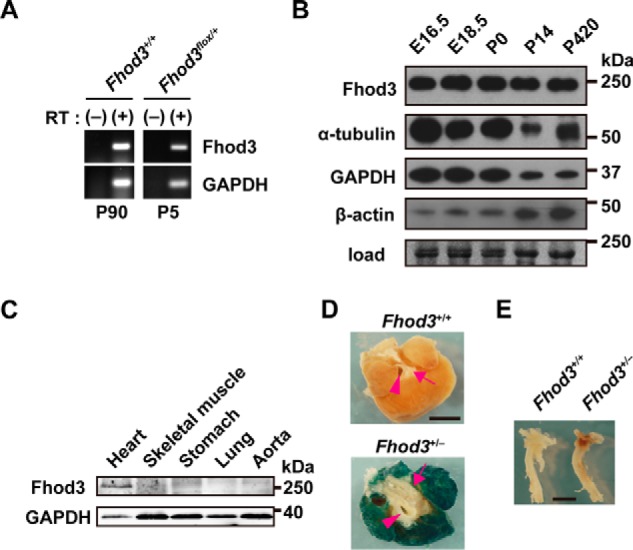
Fhod3 expression in mouse heart. A, total RNAs prepared from mouse heart at the indicated postnatal days were analyzed by reverse transcription-PCR with specific primers for Fhod3 and GAPDH. RT, reverse transcriptase. B, proteins prepared from the heart of wild-type mice at the indicated age were analyzed by immunoblot with anti-Fhod3-C20, anti-α-tubulin, anti-GAPDH, and anti-β-actin antibodies. The loading amount was verified by myosin heavy chain bands stained with fast green. Positions for marker proteins are indicated in kDa. C, proteins prepared from the indicated tissues of wild-type mice at 16 weeks of age were analyzed by immunoblot with anti-Fhod3-C20 and anti-GAPDH antibodies. Positions for marker proteins are indicated in kDa. D and E, lacZ staining of the heart (D, top view) and aorta (E, lateral view) of Fhod3+/+ and Fhod3+/− (Fhod3+/lacZ) mice at 30 weeks. Roots of aortic and pulmonary arteries were indicated by arrowheads and arrows, respectively. Scale bars, 2 mm.
Figure 2.

Generation of Fhod3 floxed mice. A, schematic representation of the targeting strategy for conditional knock-out of Fhod3. Exon 18 of the Fhod3 gene is represented as a yellow box. Green and blue bars indicate probes used for Southern blot analysis, and expected sized of fragments obtained after SpeI or BstEII digestion are indicated in base pairs. Small black arrows indicate primers for PCR genotyping. B, PCR analyses of tail DNAs from wild-type and heterozygous mice for the targeted allele. The PCR product from the wild-type allele was 329 bp in length, and the PCR products from the targeted allele were 387 and 645 bp in length. C, Southern blot analyses of tail DNAs from the wild-type and heterozygous mice for the targeted allele. SpeI-digested DNAs and BstEII-digested DNAs were probed with 5′-probe (green in A) and 3′-probe (blue in A), respectively. The wild-type allele was detected as 12.2- and 12.1-kb fragments by 5′- and 3′-probes, respectively. The targeted allele was detected as 8.4- and 14.1-kb fragments by 5′- and 3′-probes, respectively.
To delete Fhod3 from the neonatal heart, we crossed mice carrying the Fhod3flox allele to MCK-Cre mice, which express Cre recombinase in the heart, skeletal muscle, and some types of the smooth muscle from the late-embryonic stage to adulthood (28, 29). Because Fhod3 is hardly expressed in the skeletal muscle and vascular smooth muscle (Fig. 1, C–E) (21, 24), MCK-Cre-mediated recombination is expected to induce a cardiac-specific depletion of the Fhod3 protein in the perinatal period. Perinatal Fhod3 cKO mice (Fhod3flox/−;MCK-Cre+), in which the Cre-mediated recombination occurred around birth (Fig. 3A), were born in the expected Mendelian ratio (25%) and were indistinguishable in appearance from control littermates at birth. However these mice died by around postnatal day (P) 15 (Fig. 3B). As demonstrated by immunoblot analysis of heart extracts (Fig. 3C), the Fhod3 protein in Fhod3 cKO mice (Fhod3flox/−;MCK-Cre+) began to decrease from around birth and became nearly undetectable at P8. Macroscopically, Fhod3 cKO neonates began to exhibit growth retardation at around P6, and the difference in the body size between Fhod3 cKO and control neonates became apparent with time (Fig. 3, D and E). In contrast, the heart of Fhod3 cKO neonates was markedly enlarged in comparison with that of control ones (Fig. 3D), suggesting that lethality is associated with cardiac defects.
Figure 3.

Perinatal deletion of Fhod3 results in juvenile lethality. A, PCR analyses of genomic DNAs from hearts of Fhod3flox/+;MCK-Cre+ and Fhod3flox/+;MCK-Cre− mice. The PCR product from the wild-type allele (Fhod3+) or the null allele (Fhod3−) was 329 bp in length (#3). The PCR product from the floxed allele (Fhod3flox) was 387 bp in length (#2). The 549-bp fragment was produced by Cre-mediated recombination (#1). Primers used are shown in Fig. 2A. B, survival curves of Fhod3 cKO (Fhod3flox/−;MCK-Cre+, n = 205) and control littermate (Fhod3flox/+;MCK-Cre+, n = 227; Fhod3flox/−;MCK-Cre−, n = 226; and Fhod3flox/+;MCK-Cre−, n = 245) mice. C, detection of the Fhod3 protein by immunoblot analysis. Proteins prepared from the heart of Fhod3 cKO (Fhod3flox/−;MCK-Cre+) and control littermate (Fhod3flox/+;MCK-Cre−) mice at the indicated embryonic or postnatal days were analyzed by immunoblot with the anti-Fhod3-C20. The loading amounts were verified by immunoblot with anti-α-tubulin or by fast green staining. D, representative images of Fhod3 cKO (Fhod3flox/−;MCK-Cre+) and control littermate (Fhod3flox/+;MCK-Cre−) mice at E17.5, P2, P6, and P10. The excised hearts are shown in right panels. Scale bars: left, 1 cm; right, 2 mm. E, body weight of Fhod3 cKO (Fhod3flox/−;MCK-Cre+) (n = 40) and control littermate (Fhod3flox/+;MCK-Cre−) (n = 34) mice from P0 to P15.
Depletion of Fhod3 in the neonatal heart induces disruption of the sarcomere structure
When myocardial sections prepared from Fhod3 cKO mice were stained with hematoxylin and eosin (Fig. 4A), they showed signs of myofibrillar degeneration such as wavy appearance of myofibrils and myocyte swelling with an increase in vacuoles. To further investigate myocardial changes associated with depletion of Fhod3, we performed immunofluorescence staining for Fhod3 and sarcomeric α-actinin, a marker for myofibril assembly, and we found that Fhod3 was not detected at the protein level (Fig. 4B). Regularly-striated alignment of sarcomeric α-actinin was largely maintained in Fhod3-deleted myofibrils, although some myofibrils had abnormal α-actinin signals, which are aggregated or continuous along the sarcolemma. F-actin staining exhibited more pronounced myofibrillar changes with continuous aggregates of F-actin with α-actinin, a protein necessary for F-actin attachment to the Z-lines in cardiac sarcomeres (Fig. 4C). Transmission electron microscopic analysis also showed breakdown of sarcomere structure in the neonatal heart of Fhod3 cKO mice (Fig. 4, D and E). In contrast, cardiac sarcomeres in Fhod3 cKO mice were normally developed before birth (Fig. 4F). Thus, neonatal depletion of Fhod3 likely disrupts the sarcomere which has been once properly formed before birth, indicating that Fhod3 participates in postnatal development and maintenance of the cardiac sarcomere.
Figure 4.

Perinatal deletion of Fhod3 induces disruption of cardiac sarcomeres. A, histological analyses of Fhod3 cKO (Fhod3flox/−;MCK-Cre+) and control littermate (Fhod3flox/+;MCK-Cre−) mice at P7. Paraffin-embedded sections of neonatal hearts were stained with hematoxylin and eosin. Scale bars, 30 μm. Yellow arrowheads indicate vacuoles. B and C, confocal fluorescence micrographs of hearts of Fhod3 cKO (Fhod3flox/−;MCK-Cre+) and control littermate (Fhod3flox/+;MCK-Cre−) mice at P6. Cryosections of neonatal hearts were stained with the anti-α-actinin antibody (green) and the anti-Fhod3-(650–802) antibody (magenta) (B) or with the anti-α-actinin antibody (magenta) phalloidin (green) (C). Scale bars, 5 μm. Abnormal α-actinin signals aggregated (yellow arrowheads) or continuous along the sarcolemma (blue arrowheads) are indicated. Magenta arrowheads indicate continuous aggregates of F-actin. D, quantitative analysis of myofibrillar changes was performed by counting the number of continuous F-actin aggregates in randomly selected fields (Fhod3flox/−;MCK-Cre+, n = 28; and Fhod3flox/+;MCK-Cre−, n = 29). ‡, p < 0.001. E, electron micrographs of thin sections of hearts of Fhod3 cKO and control littermate mice at P6. Scale bars, 1 μm. These experiments have been repeated four times on three different pairs of cKO and control mice with similar results. Magenta arrowheads indicate Z lines or Z line-like structures. F, myofibrils in the embryonic heart. Confocal fluorescence micrographs of hearts of Fhod3 cKO (Fhod3flox/−;MCK-Cre+) and control littermate (Fhod3flox/+; MCK-Cre−) embryos at E17.5. Sections of embryonic hearts were stained with the anti-MyBPC antibody (magenta) and the anti-α-actinin antibody (green) (upper panels), the anti-tropomodulin1 (Tmod) antibody (magenta) and the anti-α-actinin antibody (green) (middle panels), or the anti-Fhod3-(650–802) antibody (magenta) and the anti-α-actinin antibody (green) (lower panels). Scale bars, 10 μm. G, confocal fluorescence micrographs of quadriceps muscles of Fhod3 cKO (Fhod3flox/−;MCK-Cre+) and control littermate (Fhod3flox/+; MCK-Cre−) mice at P6. Sections of quadriceps muscles were stained with the anti-α-actinin antibody (green) and phalloidin (magenta). Lower panels show the magnified views of the boxed areas in upper panels. Scale bars, 20 μm. H, in vivo phosphorylation of Fhod3. Proteins of lysates prepared from mouse heart at the indicated postnatal days were immunoprecipitated (IP) with anti-Fhod3 or anti-cMyBP-C antibodies, and the precipitants were subjected to SDS-PAGE followed by immunoblot with anti-phosphoserine antibodies or staining with Coomassie Brilliant Blue (CBB).
However, the sarcomeric structure of the skeletal muscle showed no obvious differences between Fhod3 cKO and control neonates (Fig. 4G), supporting the idea that lethality is primarily associated with defects in the cardiac sarcomere. We further investigated the phosphorylation status of Fhod3 in the neonatal heart, because it has been reported that the activity of Fhod3 is regulated by phosphorylation (30). As shown in Fig. 4H, only weak bands of phosphorylated Fhod3 were detected both in the neonatal and adult heart under conditions where cMyBP-C, a sarcomeric phosphoprotein (31), was substantially phosphorylated.
Cardiac-specific deletion of Fhod3 in adult mice
Fhod3 plays a crucial role in not only the embryonic heart (6) but also the neonatal heart (Figs. 3 and 4). However, it was difficult to assess whether Fhod3 functions in the fully-developed adult heart by using MCK-Cre-mediated cKO mice, because Fhod3 cKO mice did not fully survive until adulthood (Fig. 3). To delineate the role of Fhod3 in the adult heart, we crossed mice carrying the Fhod3flox allele with transgenic mice expressing MerCreMer under the control of the αMHC promoter (32), thereby generating mice with the Fhod3 alleles that can be deleted specifically in the adult heart by administration of TAM. Fhod3flox/−;αMHC-MerCreMer+ mice were born at the expected Mendelian frequency and were indistinguishable in appearance from control littermates.
To delete Fhod3 specifically in the adult heart, 4–6-week-old mice were orally administered TAM for 2 weeks. The occurrence of Cre-mediated, heart-specific recombination was confirmed by PCR analysis using genomic DNAs extracted from the heart and kidney in adult mice treated with TAM; the recombinant allele was detected only in the heart of Cre-positive mice but not in the kidney of Cre-positive mice or the heart of Cre-negative mice (Fig. 5A).
Figure 5.

Tamoxifen-induced deletion of Fhod3 in the adult heart does not lead to lethality. A, PCR analyses of genomic DNAs from hearts and kidneys of TAM-treated Fhod3 iKO (Fhod3flox/−;MerCreMer+) and control littermate (Fhod3flox/−;MerCreMer−) mice. The PCR product from the wild-type allele (Fhod3+) or the null allele (Fhod3−) was 329 bp in length. The PCR product from the floxed allele (Fhod3flox) was 387 bp in length. The 549-bp fragment was produced by Cre-mediated recombination. Primers used are shown in Fig. 2A. B, detection of the Fhod3 protein by immunoblot analysis. Indicated amount of proteins prepared from the heart of TAM-treated Fhod3 iKO (Fhod3flox/−;MerCreMer+) and control littermate (Fhod3flox/−;MerCreMer−) mice were analyzed by immunoblot with anti-Fhod3. The loading amount was verified by myosin heavy chain bands stained with fast green. C, detection of the Fhod3 protein by immunoblot analysis. Proteins prepared from the heart of TAM-treated Fhod3 iKO (Fhod3flox/−;MerCreMer+) and TAM-treated control littermate (Fhod3flox/−;MerCreMer−) mice were analyzed by immunoblot with the three different anti-Fhod3 antibodies (anti-Fhod3-(650–802), anti-Fhod3-(873–974), and anti-Fhod3-C20) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody. D, survival curves of Fhod3 iKO (Fhod3flox/−;MerCreMer+, n = 72) and control littermate (Fhod3flox/−;MerCreMer−, n = 51; Fhod3flox/+;MerCreMer+, n = 42; Fhod3flox/+;MerCreMer−, n = 33) mice treated with TAM for the first 2 weeks. E, representative images of hearts of TAM-treated Fhod3 iKO (Fhod3flox/−;MerCreMer+) and TAM-treated control littermate (Fhod3flox/−;MerCreMer−) mice. These experiments have been repeated three times with similar results. Scale bars, 4 mm. F, histological analyses of hearts of TAM-treated Fhod3 iKO and TAM-treated control littermate mice. Longitudinal sections of hearts were stained with hematoxylin and eosin. Scale bars, 3 mm. G, heart-to-body weight and kidney-to-body weight ratios after TAM administration were determined. *, p < 0.05; N.S., not significant. H, quantitative real-time PCR analysis of Fhod1 gene expression in Fhod3 iKO and control littermate mice.
The protein expression level of Fhod3 in Fhod3 iKO mice was reduced to ∼5% or less of that of wild-type mice, as demonstrated by immunoblot analysis of lysates of isolated left ventricular tissue (Fig. 5, B and C). Thus, TAM administration effectively induces Cre-mediated, cardiac-specific recombination of the Fhod3 allele to significantly reduce the protein expression level of Fhod3 in the adult heart.
Depletion of Fhod3 in the adult heart does not lead to lethality
Despite the reduction of Fhod3 in the heart from TAM-treated iKO mice, no significant difference in the long-term survival up to 60 weeks was observed among all the genotype groups (Fig. 5C). Some mice died during the 2 weeks of TAM administration, probably due to acute cardiotoxicity (33). Adult Fhod3-deleted mice showed a slightly higher mortality rate than other genotype mice but with no statistical significance. We next evaluated the effect of Fhod3 depletion in the adult heart at 8–10 weeks after TAM administration; the period was selected because the transient impairment of cardiac function is known to be fully recovered until 4 weeks after the cessation of TAM administration (34).
The size of the adult heart in TAM-treated Fhod3 iKO mice was slightly larger than that in control mice (Fig. 5E), which was supported by histological analysis (Fig. 5F). Left ventricular weight-to-body weight ratio of adult Fhod3-deleted mice was slightly but significantly higher than that of control mice, whereas there was no difference in kidney weight-to-body weight ratio between adult Fhod3-deleted and control mice (Fig. 5G). On the other hand, we were not able to detect any compensatory increase in the expression of Fhod1, the only other member of the Fhod subfamily (Fig. 5H). Thus deletion of Fhod3 in the adult heart confers a slight cardiomegaly.
Depletion of Fhod3 in the adult heart does not cause any detectable changes in the sarcomere structure
We next investigated the effect of Fhod3 depletion in the adult heart on the sarcomeric structure. In contrast to the finding that the neonatal heart of Fhod3 cKO mice shows disrupted structures of sarcomeres (Fig. 4, B–E), the adult heart of TAM-treated Fhod3 iKO mice showed apparently normal sarcomeric structures (Fig. 6). There were no significant differences in sarcomeric structures, including the Z-line marked with the antibody to α-actinin (Fig. 6, A and B), thin actin filaments stained with phalloidin (Fig. 6, B and C), and thick myosin filaments visualized with the antibody to myosin-binding protein C (MyBPC) (Fig. 6D), albeit specific signals for Fhod3 were absent in the Fhod3-deleted adult heart (Fig. 6, A, C, and D). Immunohistochemical staining of tropomodulin, a protein that caps actin filament at the pointed end, showed no detectable differences between adult Fhod3-deleted and control hearts (Fig. 6E), suggesting that the length of thin actin filaments and the capping state of pointed ends are unaffected. Intact sarcomeres in the Fhod3-deleted adult heart were also observed by transmission electron microscopy (Fig. 6F). Furthermore, intercalated disc structure, which is disrupted in several mouse models of dilated cardiomyopathy (35), was not altered in the Fhod3-deleted adult heart, as estimated by immunohistochemical staining of vinculin, a component of intercalated discs, and by transmission electron microscopy (Fig. 6, G and H). Thus, adult Fhod3 deletion hardly affects the structure of sarcomeres and intercalated discs.
Figure 6.
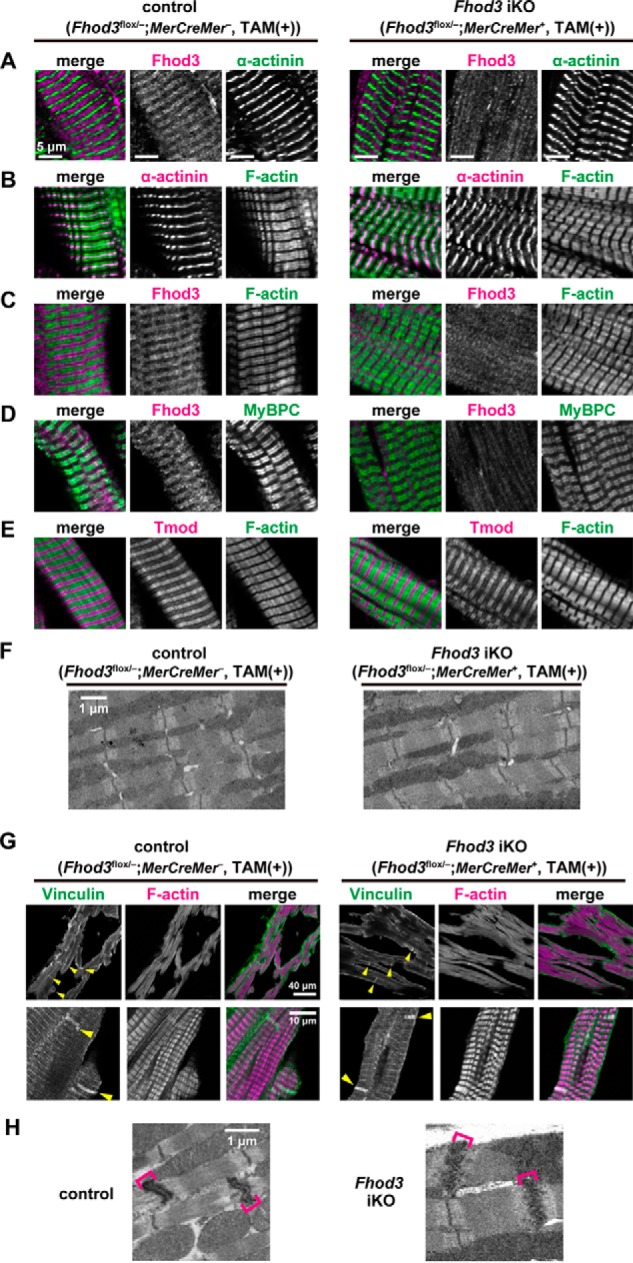
Tamoxifen-induced deletion of Fhod3 in the adult heart does not affect sarcomeric structures. A–E, confocal fluorescence micrographs of hearts of TAM-treated Fhod3 iKO and TAM-treated control littermate mice. Sections of adult hearts were stained with the following anti-Fhod3-(650–802) antibody (red) and the anti-α-actinin antibody (green) (A); anti-α-actinin antibody (red) and phalloidin (green) (B); anti-Fhod3-(650–802) antibody (red) and phalloidin (green) (C); anti-Fhod3-(650–802) antibody (magenta) and the anti-MyBPC antibody (green) (D); and anti-tropomodulin1 (Tmod) antibodies (red) and phalloidin (green) (E). Scale bars, 5 μm. F, electron micrographs of thin sections of TAM-treated Fhod3 iKO and TAM-treated control littermate mice. Bar, 1 μm. Over 10 images from each genotype were analyzed. G, intercalated discs in the heart. Sections of adult hearts were stained with the anti-vinculin antibody (green) and phalloidin (magenta). Yellow arrowheads indicate intercalated discs. Scale bars, 40 μm (upper panels) and 10 μm (lower panels). H, electron micrographs of thin sections of hearts of TAM-treated Fhod3 iKO (Fhod3flox/−;MerCreMer+) and TAM-treated control littermate (Fhod3flox/+;MCK-Cre−) mice. Brackets indicate intercalated discs. Scale bar, 1 μm.
Mice lacking cardiac Fhod3 exhibit prolonged morphological and functional changes at advanced ages
Morphological and functional characteristics of Fhod3 cKO mice were followed until 1 year of age for evaluation of late effects of Fhod3 deletion in the adult heart. As shown in Fig. 7A, the left ventricular chamber of aged Fhod3-deleted mice was slightly enlarged, suggesting that structural changes induced by Fhod3 deletion was sustained but not corrected over a follow-up period of 1 year. Consistent with these findings, ventricular dilation with thinned walls was observed in the heart of aged Fhod3-deleted mice by echocardiographic measurement of left ventricular dimensions and posterior wall thickness (Fig. 7B). The measurement also revealed significant reduction of left ventricular ejection fraction in aged Fhod3-deleted mice (Fig. 7B). These macroscopic anatomical changes suggest that cardiac remodeling is induced by Fhod3 depletion in the adult heart. To test the possibility, we performed quantitative real-time PCR analysis to examine activation of cardiac remodeling-associated fetal genes that encode atrial natriuretic factor (ANF), B-type natriuretic peptide (BNP), and β-myosin heavy chain (βMHC). As shown in Fig. 7C, mRNA expression of the three genes in aged Fhod3-deleted mice was elevated when compared with those in age-matched control mice, although the increase in ANF mRNA was not statistically significant. Picrosirius red staining revealed that fibrotic changes such as accumulation of collagen I or collagen III also occurred in the heart of aged Fhod3-deleted mice (Fig. 7D), which is consistent with cardiac increase in mRNA for the genes encoding the α1 chain of collagen I (Col1a1) as demonstrated by real-time PCR analysis (Fig. 7E). Thus, morphological changes and functional impairment by depletion of Fhod3 are only slightly induced but not compensated during aging, which leads to ventricular dilation with wall thinning and fibrosis at advanced age.
Figure 7.

Fhod3 deletion induces cardiac dilatation and dysfunction at advanced age. A, histological analyses of Fhod3 iKO (Fhod3flox/−;MerCreMer+) and TAM-treated control littermate (Fhod3flox/−;MerCreMer−) mice at 50 weeks after TAM administration. Longitudinal sections of hearts were stained with hematoxylin and eosin. Scale bars, 3 mm. B, echocardiography analysis of Fhod3 iKO and control littermate mice at 50 weeks after TAM administration. LVDd, left ventricular dimension in diastole; LVDs, left ventricular dimension in systole; PW, end diastolic posterior wall thickness; LVEF, left ventricular ejection fraction. C, quantitative real-time PCR analysis of hypertrophy-related gene expression in Fhod3 iKO and control littermate mice at 50 weeks after TAM administration. Nppa, encoding ANF; Gapdh, encoding GAPDH; Nppb, encoding BNP; Myh7, encoding βMHC. D, picrosirius red staining of left ventricle septum of Fhod3 iKO and control littermate mice at 50 weeks after TAM administration. Scale bars, 100 μm. Blue arrowheads indicate interstitial accumulation of collagen. The degrees of fibrosis were evaluated using polarization microscopy (Fhod3 iKO, n = 30 images from three mice; control, n = 30 images form three mice). E, quantitative real-time PCR analysis of fibrosis-related gene expression in Fhod3 iKO and control littermate mice at 50 weeks after TAM administration. Col1a1, encoding collagen type I α1. *, p < 0.05; †, p < 0.01; ‡, p < 0.001.
Mice lacking cardiac Fhod3 show a hypertrophic response to phenylephrine infusion but exhibit no substantial changes in sarcomeric structures
A modest but significant cardiac dysfunction in adult Fhod3-deleted mice (Fig. 7) may implicate that Fhod3 regulates cardiac function under pathological conditions. To test the possibility, we treated adult Fhod3-deleted and control mice with phenylephrine, an α1-adrenergic agonist that induces sustained hypertension and development of hypertrophy (36). Infusion of phenylephrine using osmotic mini-pumps for 2 weeks led to the death of a minor portion of mice probably due to intolerance of acute hemodynamic changes, whereas mice without the infusion survived uneventfully (Fig. 8A). The mortality of phenylephrine-infused Fhod3 cKO mice was not significantly different from that of phenylephrine-infused control mice. At the time point after continuous phenylephrine infusion for 2 weeks, the heart of treated mice markedly enlarged (Fig. 8, B and C). Consistent with this, the ratios of left ventricular to body weight in the phenylephrine-infused mice were significantly higher when compared with those in mice without phenylephrine infusion (Fig. 8D). There were no significant differences in the ratios between Fhod3-deleted and control mice after phenylephrine infusion. However, the heart in phenylephrine-treated Fhod3-deleted mice showed a tendency to be more enlarged than that of phenylephrine-treated control mice, raising the possibility that Fhod3 is involved in modulation of hypertrophic processes.
Figure 8.

Continuous infusion of phenylephrine induces hypertrophic changes in the Fhod3-deleted heart. A, survival curves after continuous infusion of phenylephrine (PE) for 2 weeks. TAM-treated Fhod3 iKO and TAM-treated control littermate mice were infused with PE (100 mg/kg/day) or saline for 2 weeks. B, representative images of hearts of TAM-treated Fhod3 iKO and TAM-treated control littermate mice after PE infusion. These experiments have been repeated three times with similar results. C, histological analyses of hearts of TAM-treated Fhod3 iKO (Fhod3flox/−;MerCreMer+, TAM(+)) and TAM-treated control littermate (Fhod3flox/−;MerCreMer−, TAM(+)) mice after PE infusion. Longitudinal sections of hearts were stained with hematoxylin and eosin. Scale bars, 3 mm. D, heart-to-body weight and kidney-to-body weight ratios after PE infusion were determined. †, p < 0.01; ‡, p < 0.001.
To test this, we investigated whether Fhod3 expression is altered under hypertrophic conditions. As shown by immunoblot analysis (Fig. 9A), the amount of Fhod3 in the heart was not up-regulated in phenylephrine-treated mice, whereas phenylephrine treatment markedly increased βMHC expression in both Fhod3-deleted and control mice. Immunofluorescence staining confirmed the phenylephrine-induced accumulation of βMHC in the Fhod3-deleted heart (Fig. 9B), indicating that phenylephrine treatment indeed induces hypertrophic changes in Fhod3-deleted mice as well as control mice. Phenylephrine infusion did not induce significant changes in sarcomeric structures in both Fhod3-deleted and control mice, as demonstrated by immunofluorescence staining (Fig. 9C) and by transmission electron microscopy (Fig. 9D). Thus, the adult Fhod3-deleted heart is sensitive to phenylephrine infusion, although Fhod3 deletion does not induce detectable changes in sarcomeric structures even after phenylephrine treatment.
Figure 9.

Phenylephrine infusion does not induce sarcomeric changes in the Fhod3-deleted heart. A, detection of the Fhod3 protein by immunoblot analysis. TAM-treated Fhod3 iKO and TAM-treated control mice were continuously infused with PE for 2 weeks. Proteins prepared from the heart of the PE-infused mice were analyzed by immunoblot with the anti-Fhod3-C20, anti-βMHC, and anti-GAPDH antibodies. B, confocal fluorescence micrographs of hearts of TAM-treated Fhod3 iKO and TAM-treated control littermate mice after PE infusion. Sections of hearts were stained with the anti-βMHC antibody (green) and phalloidin (magenta). Scale bars, 100 μm. C, confocal fluorescence micrographs of hearts of TAM-treated Fhod3 iKO and age-matched control mice after PE infusion. Sections of embryonic hearts were stained with the anti-α-actinin antibody (green) and the anti-Fhod3-(650–802) antibody (magenta). Scale bars, 5 μm. These experiments have been repeated three times on two different pairs of iKO and control mice with similar results. D, electron micrographs of thin sections of hearts of TAM-treated Fhod3 iKO and TAM-treated control littermate mice after PE infusion. Bar, 1 μm. Over 10 images from each genotype and treatment were analyzed.
Deletion of Fhod3 in the adult heart negatively affects cardiac function and promotes hypertrophic responses
We finally examined the effect of Fhod3 depletion on cardiac function under hypertrophic conditions by echocardiography. Before phenylephrine treatment, the left ventricular chamber in adult Fhod3-deleted mice was enlarged compared with that in control mice (Fig. 10A), and the left ventricular function was significantly impaired in adult Fhod3-deleted mice (Fig. 10B). These observations are consistent with the present macroscopic and histological analyses showing that Fhod3 deletion in the adult heart induces slight but significant cardiomegaly (Fig. 5, E–G). After 2 weeks of phenylephrine infusion, left ventricular chamber enlargement and functional impairment were further developed in both Fhod3-deleted and control mice (Fig. 10, A and B), although some of the parameter differences were not statistically significant. At the same time, wall thickening and increments in left ventricular mass were also observed in both Fhod3-deleted and control mice (Fig. 10C), indicating that Fhod3-deleted mice have an ability to induce hypertrophy. Consistent with this, the cell width of individual cardiomyocytes was increased in response to phenylephrine treatment in the heart of Fhod3-deleted mice (Fig. 10D), although the extent in Fhod3-deleted mice tended to be smaller than that in control mice. Next, we tested the effect of phenylephrine on expression of cardiac remodeling-associated fetal genes relative to that of Gapdh. It should be noted that Gapdh expression is not affected by phenylephrine treatment (Fig. 9A) (37). As shown in Fig. 10E, phenylephrine treatment enhanced the expression of mRNAs for ANF, BNP, and βMHC in the heart of Fhod3-deleted mice more profoundly than that of control mice. Thus, Fhod3 is likely able to modulate cardiac hypertrophic responses.
Figure 10.

Fhod3-deleted mice shows more pronounced hypertrophic response to phenylephrine infusion. A–C, echocardiography analysis of hearts of TAM-treated Fhod3 iKO and TAM-treated control littermate mice before and after PE infusion. LVDd, left ventricular dimension in diastole; LVDs, left ventricular dimension in systole; PW, end diastolic posterior wall thickness; LVEF, left ventricular ejection fraction. D, width of cardiomyocyte at the nuclei level was estimated by wheat germ agglutinin staining of left ventricle septum from TAM-treated Fhod3 iKO mice with PE infusion (n = 71 from two mice) or without PE infusion (n = 68 from one mouse) and TAM-treated control littermate mice with PE infusion (n = 67 from two mice) or without PE infusion (n = 59 from one mouse). E, quantitative real-time PCR analysis of hypertrophy-related gene expression in hearts of TAM-treated Fhod3 iKO and TAM-treated control littermate mice with and without PE infusion. Nppa, encoding ANF; Gapdh, encoding GAPDH; Nppb, encoding BNP; Myh7, encoding βMHC. *, p < 0.05; †, p < 0.01; ‡, p < 0.001; N.S., not significant.
Discussion
In this study, we show that Fhod3 plays a crucial role in postnatal development of the heart; depletion of Fhod3 in the neonatal heart results in juvenile lethality associated with cardiac defects (Figs. 3 and 4). In embryos, Fhod3 is dispensable for initiation of spontaneous contraction of the developing heart, composed of premyofibrils, but it is absolutely required for subsequent myofibril maturation in cardiogenesis (6), indicating that Fhod3 is responsible for expanding the basic contractile unit called sarcomere during embryonic heart development (6, 25). During the early postnatal period, greater circulatory demands of the growing organism induce a 2-fold or more increase in left ventricular mass, which mainly results from an increase in the size of cardiomyocytes with expansion of their contractile apparatus (38, 39). It is therefore likely that Fhod3, an actin-assembling factor (22), functions via expanding the sarcomere during heart development in neonates as well as embryos. In addition to sarcomere expansion, Fhod3 may also be involved in maintenance of sarcomere structure in the neonatal stage, because myofibrils once formed during embryonic cardiogenesis become degenerated after birth in perinatally Fhod3-deleted mice (Fig. 4). Thus Fhod3-mediated actin assembly appears to contribute to postnatal cardiac development via expansion and maintenance of the contractile apparatus sarcomere.
In contrast to the life-threatening effect of embryonic and perinatal deletion of Fhod3, its deletion in the adult stage does not affect mortality (Fig. 5). This is consistent with the present finding that sarcomeric structures are largely maintained in the adult heart of Fhod3-deleted mice (Fig. 6). A weak but significant cardiac remodeling, however, occurs with aging in adult Fhod3-deleted mice, as indicated by macroscopic and functional changes, signs of fibrosis, and expression of the cardiac remodeling-associated fetal genes (Fig. 7). Furthermore, systemic infusion of the α1-stimulant phenylephrine induces more severe hypertrophic processes in the heart of adult Fhod3-deleted mice than in the Fhod3-expressing heart (Figs. 8–10), indicating that Fhod3 is involved in hypertrophic responses to cardiac stress in the adult heart. Although juvenile lethality is induced by perinatal deletion of Fhod3 (Fig. 3), Fhod3 knock-out in adult mice does not result in a significant increase in morbidity after phenylephrine treatment (Fig. 8). Thus, Fhod3 plays a major role in neonatal cardiac development but not in stress-induced cardiac hypertrophy during adulthood. The difference is likely due to the fact that cardiomyocytes increase their size more than 2-fold under physiological conditions in the neonatal stage (40), but the cell size grows to a limited extent in response to pathological stimuli in the adult (39). The greater increase in cardiomyocyte size during neonatal cardiac development appears to explain the reason for the greater dependence on Fhod3, because this actin-assembling factor is crucial for cardiac sarcomere expansion, which leads to an increase in cardiomyocyte size, as discussed above.
Although this study supports the idea that Fhod3 regulates actin assembly for sarcomere integrity in the heart (6, 21, 24), the molecular mechanism underlying the regulation remains largely unknown. The precise site where Fhod3 functions in the cardiac sarcomere is a controversial issue. We have provided several lines of evidence showing that Fhod3 localizes to the center of sarcomeres, specifically the zone of the thin actin filaments that overlap the thick myosin filaments; localization of endogenous Fhod3 to this specific zone in the embryonic and adult heart (24, 25) as well as in cultured cardiomyocytes (22) is demonstrated by immunochemical analysis using three anti-Fhod3 antibodies, each recognizing distinct regions of Fhod3 (21); and the same localization of HA-tagged Fhod3 is observed using an anti-HA antibody (22). In contrast, other groups have reported that Fhod3 localizes to the Z-line, where the barbed ends of actin filaments are anchored, in the adult heart (23, 30, 41). Although both patterns of Fhod3 localization are possible, this study strengthens our conclusion by confirming the authenticity of the three distinct antibodies to Fhod3; their signals disappear from the zone containing both thin and thick filaments in neonatal and adult Fhod3-deleted hearts (Figs. 4B, 6D, and 9C). Further studies are required to address the question how Fhod3 functions at the zone of the actin filaments that is superimposed by the myosin filaments.
According to a recent assessment (41), the Fhod3 mRNA is expressed abundantly in the embryonic stage but not after birth. In contrast, the Fhod3 mRNA is similarly detected in the postnatal and adult heart in this study (Fig. 1A). Although the reason for the discrepancy is presently unknown, the protein level of Fhod3 in the heart does not change significantly throughout the lifetime from the embryo to the senior adult (Fig. 1B), consistent with the critical role of Fhod3 after birth. Besides Fhod3, other formin family proteins also seem to participate in regulation of actin dynamics in cardiomyocytes (41). Actin polymerization by formins is generally accelerated via FH1 domain-mediated recruitment of the profilin–actin complexes (16), which is consistent with the recent finding that profilin-1 participates in actin dynamics in cardiomyocytes, including sarcomeric organization and cell size control (42), probably via cooperation with formins. Daam1, another major cardiac formin (41), has been reported to be required for myocardial maturation and sarcomere assembly (7, 43). In contrast to the localization of Fhod3 at the central region of sarcomeres, mouse Daam1 in the cardiomyocytes mainly localizes to the cell membrane not in a sarcomeric pattern (7), suggesting that Daam1 and Fhod3 regulate cardiac actin dynamics in a different manner.
In contrast to the failure of the α1-adrenergic agonist phenylephrine to induce expression of Fhod3 in the heart (Fig. 9A), Zhou et al. (44) have very recently reported that treatment with angiotensin II increases Fhod3 expression, and ROCK2-mediated phosphorylation of Fhod3 participates in the angiotensin II-induced hypertrophy. Although neonatal cardiac development requires Fhod3 but does not seem to involve its phosphorylation, because Fhod3 is only marginally phosphorylated in the developing neonatal hearts (Fig. 4H). Future studies are needed to evaluate the contribution of Fhod3 and its phosphorylation to the development of cardiac hypertrophy under various pathological conditions.
In conclusion, we report here that Fhod3 plays an important role in neonatal cardiac development and in functional maintenance and stress-induced hypertrophic response of the developed heart. Taken together with the recent findings that Fhod3 variants are associated with hypertrophic and dilated cardiomyopathy (26, 27), Fhod3 might represent a potential therapeutic target for cardiac disease associated with failure in myofibril maintenance.
Experimental procedures
Generation of Fhod3 conditional knock-out mice
Mice carrying the Fhod3 floxed allele (accession no. CDB0927K) were generated according to the protocols of RIKEN Center for Life Science Technologies. In brief, the 5′-homologous arm (3.5 kb), the 3′-homologous arm (1.3 kb), and the flox body containing exon 18 (1.4 kb) were subcloned into pENTR conditional FW for Gateway cassette to generate the targeting vector (Fig. 2A). The linearized targeting vector was electroporated into HK3i embryonic stem cells (45), and G418-resistant clones were screened by PCR analysis and confirmed by Southern blot analysis to identify ones with correct homologous recombination. Chimeric mice were generated with the recombinant embryonic stem cell clones and mated with the C57BL/6 strain to generate heterozygous animals (Fhod3flox-Neo/+), which were subsequently crossed with mice expressing flippase (46) to obtain animals heterozygous for the floxed Fhod3 allele (Fhod3flox/+) (Fig. 2A). Genotyping was performed using the following primers shown in Fig. 2A: a, 5′-GGGTTGCAGAGAATCACCCAG-3′; b, 5′-ACAGCTGAGCAGTCAGTCCAG-3′; c, 5′-CCAGACTCGAGCACCTGTTTG-3′; d, 5′-TGGTCCCTCACTTGAGGTGAC-3′; and e, 5′-CTGACCGCTTCCTCGTGCTTTACG-3′. Two mutant strains generated from two independent recombinant embryonic stem cell clones were analyzed. No phenotypic differences between the two strains were observed.
Mice heterozygous for the constitutive null Fhod3 allele (Fhod3+/−) were generated by replacement of exon 1 with LacZ as described previously (6). Transgenic mice expressing Cre recombinase under the control of muscle myosin kinase (MCK) promoter (MCK-Cre mice, B6.Cg-Tg(Ckmm-cre)5Khn/J; 006475) and transgenic mice expressing Cre recombinase fused to two mutated estrogen receptors under the control of α-myosin heavy chain (αMHC) promoter (αMHC-MerCreMer mice, B6.Cg-Tg(Myh6-cre/Esr1*)1Jmk/J; 005657) were purchased from The Jackson Laboratory and were maintained on a C57BL/6J genetic background.
For perinatal deletion of Fhod3 in the heart, homozygous Fhod3flox/flox mice were crossed with Fhod3+/−;MCK-Cre+ mice to obtain Fhod3 conditional knock-out (cKO) mice (Fhod3flox/−;MCK-Cre+). For inducible deletion of Fhod3 in the heart, Fhod3flox/flox mice were crossed with Fhod3+/− MerCreMer+ mice to obtain TAM-iKO mice (Fhod3flox/−;MerCreMer+). Fhod3flox/− mice negative for Cre transgene (Fhod3flox/−;Cre−) or age-matched wild-type C57BL/6J mice were used as controls. The control Fhod3flox/− mice were phenotypically normal and showed no phenotypic differences from mice heterozygous for the constitutive Fhod3-null allele (Fhod3+/−), which were phenotypically normal even after pressure overload by surgical transverse aortic constriction (6).
All experimental protocols were approved by the Animal Care and Use Committee of Kyushu University (Permit No. A26-102) and the Animal Care and Use Committee of Miyazaki University (Permit No. 2014-526-3). All mice were housed and maintained in a specific pathogen-free animal facility at Kyushu University, and all efforts were made to minimize the number of animals used and their suffering. All experiments were performed in strict accordance with the guidelines for Proper Conduct of Animal Experiments (Science Council of Japan) and the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
LacZ staining
LacZ staining of heterozygous Fhod3+/− mice was performed as described previously (6). Briefly, organs of mice were fixed at 4 °C by immersion in phosphate-buffered saline (PBS) (137 mm NaCl, 2.68 mm KCl, 8.1 mm Na2HPO4, and 1.47 mm KH2PO4, pH 7.4) containing 1% formaldehyde, 0.2% glutaraldehyde, 0.02% Nonidet P-40, and 1 mm MgCl2. Fixed organs were incubated at 37 °C in PBS containing 1 mg/ml X-Gal, 5 mm K3Fe(CN)6, 5 mm K4Fe(CN)6, and 2 mm MgCl2.
Tamoxifen administration
TAM pellets containing 400 mg of TAM citrate per kg diet (daily administration of TAM of ∼40 mg per kg body weight) was obtained from Oriental Yeast Co., Ltd. After TAM administration of 2 weeks, the mice were fed with standard diet over 4 weeks to avoid interference with transient Cre expression and were used on all experiments.
Antibodies
Rabbit anti-Fhod3 polyclonal antibodies were raised against three different regions of Fhod3, namely anti-Fhod3-(650–802), anti-Fhod3-(873–974), and anti-Fhod3-C20, followed by affinity purification, as described previously (21). Monoclonal antibodies were purchased from commercial sources as follows: clone EA-53 against α-actinin from Sigma; clone G-7 against myosin-binding protein C (MyBPC) from Santa Cruz Biotechnology; NOQ7.5.4D against β-myosin heavy chain (βMHC) from Abcam; clone hVIN-1 against vinculin from Sigma; and clone 6C5 against GAPDH from Chemicon. Rabbit polyclonal antibodies against tropomodulin1 were purchased from ProteinTech Group; and rabbit polyclonal anti-phosphoserine antibodies were from Chemicon. Alexa Fluor 488-conjugated F(ab′)2 fragment of anti-mouse IgG and Alexa Fluor 555-conjugated F(ab′)2 fragment of anti-rabbit IgG were purchased from Cell Signaling Technology.
Immunoblot analysis
Immunoblot analysis was performed as described previously (25). Briefly, the hearts of neonates were homogenized and sonicated at 4 °C in a lysis buffer composed of 10% glycerol, 135 mm NaCl, 5 mm EDTA, and 20 mm Hepes, pH 7.4, containing protease inhibitor mixture (Sigma). In the case of adult mice, the ventricular tissue samples of mice were snap-frozen, crushed using SK-Mill (SK-100, FUNAKOSHI), and dissolved in a buffer composed of 9 m urea, 2% SDS, 2% Triton X-100, 1% dithiothreitol, and 10 mm Tris-HCl, pH 6.8, containing Protease Inhibitor Mixture (Sigma). The lysates were applied to SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Millipore). Protein transfer was immediately confirmed by fast green staining (loading). The membrane was probed with the antibody, followed by development using ECL-plus (GE Healthcare) for visualization of the antibodies.
In vivo phosphorylation assay
Immunoprecipitation was performed, as described previously (47), with minor modifications. Briefly, the heart tissues were snap-frozen, crushed using SK-Mill (SK-100, FUNAKOSHI), and dissolved in a lysis buffer (10% glycerol, 135 mm NaCl, 5 mm EDTA, and 20 mm Hepes, pH 7.4) containing 0.1% Triton X-100, Protease Inhibitor Mixture (Sigma), and PhosSTOP phosphatase inhibitor mixture (Sigma). The lysates were precipitated with the anti-Fhod3-C20 or anti-cMyBP-C antibodies in the presence of protein G-magnetic beads. After washing three times with the lysis buffer, the precipitants were applied to SDS-PAGE and transferred to a polyvinylidene difluoride membrane. The membrane was probed with the anti-phosphoserine, anti-Fhod3-C20, or anti-cMyBP-C antibodies, followed by development using ECL-prime (GE Healthcare) for visualization of the antibodies.
Histological analysis
Histological analysis was performed as described previously (6). Briefly, mice were sacrificed via cervical dislocation, and the whole heart was harvested. The harvested tissues were fixed by immersion in a solution containing 3.7% formaldehyde in PBS. The fixed tissues were dehydrated in ethanol, embedded in paraffin, sectioned, and stained with hematoxylin and eosin or picrosirius red. Images were taken with BZ-9000 microscope (Keyence). For estimation of fibrosis level, picrosirius red-stained sections were examined with a microscope (BX-51; Olympus) coupled to an image processing system (NIS-Element-D3.2; Nikon) and semi-quantified using an image analysis software (WinRoof; Mitani).
Immunofluorescence staining
Immunofluorescence staining was performed according to the previously described method (6) with minor modifications. Briefly, mice were deeply anesthetized with an intraperitoneal injection of pentobarbital (50 mg/kg/body weight) and sevoflurane inhalation. After exposure of the heart, PEM buffer (1 mm EGTA, 1 mm MgCl2, and 100 mm PIPES, pH 6.9) containing 100 mm 2,3-butanedione monoxime (BDM) was administered from the left ventricular apex, followed by the administration of 3.7% formaldehyde in the PEM buffer. In the case of preparation of heart tissues, for facilitation of selective coronary perfusion, the ascending aorta was clamped with a hemostat, and the right atrium was clipped with surgical scissors before the perfusion. The perfused tissues were removed from the deceased mice, cut into small pieces, and immersed for 90 min at 4 °C in the same fixative. The fixed tissues were washed in PBS, subjected to osmotic dehydration overnight at 4 °C in 30% sucrose, and embedded in OCT compound (Sakura Finetek). The blocks were frozen and cut into 5-μm sections using a cryostat (HM550; Thermo Fisher Scientific). Sections were then washed with PBS containing 0.1% Triton X-100 and blocked with a blocking buffer (Blocking One Histo; Nacalai Tesque) for 5 min at room temperature. Sections were labeled overnight at 4 °C with primary antibodies diluted in a dilution buffer (PBS containing 3% bovine serum albumin, 2% goat serum, and 0.1% Triton X-100) and then labeled for 2 h at room temperature with a fluorescein-conjugated secondary antibody mixture in the same buffer. For Fhod3 immunofluorescence staining, anti-Fhod3-(650–802) antibody was used. Actin filaments were stained with Alexa Fluor 488 phalloidin (Invitrogen). Nuclei were stained with Hoechst 33342 (Invitrogen). Cell membranes were stained with FITC-labeled WGA (J-OIL MILLS). Images were taken with LSM700 or LSM780 confocal scanning laser microscope (Carl Zeiss MicroImaging).
Phenylephrine administration
Continuous subcutaneous infusion of phenylephrine (100 mg/kg/day) or saline was conducted with Alzet osmotic mini-pumps (model 2002, DURECT Corp.). Mice were anesthetized with an intraperitoneal injection of pentobarbital (50 mg/kg/body weight) and sevoflurane inhalation, and then the pump was subcutaneously implanted at the back. After continuous administration for 2 weeks, the mice were analyzed.
Transmission electron microscopic analysis
Transmission electron microscopy of thin sections was performed according to the previously described method (6) with minor modifications. Briefly, mice were deeply anesthetized with an intraperitoneal injection of pentobarbital and sevoflurane inhalation. After exposure of the heart, the PEM buffer containing 100 mm BDM was administered from the left ventricular apex, followed by the administration of a fix buffer (2.5% glutaraldehyde, 0.1 m sucrose, 3 mm CaCl2, and 0.1 m sodium cacodylate, pH 7.4) for 1 h, followed by rinse in PBS overnight at 4 °C. Then the heart was postfixed for 1 h with 1% OsO4, dehydrated in ethanol and propylene oxide, and embedded in the Epon 812 resin. Thin sections containing the heart were stained for 5 min with 2% uranyl acetate and for 10 min with Sato's lead mixture and then examined with a JEM-2000EX (JOEL).
Detection of mRNA by RT-PCR and quantitation of mRNA levels by real-time RT-PCR
Total RNAs were extracted from the left ventricular tissue using TRIzol reagent (Invitrogen). Complementary DNAs were synthesized using TaqMan reverse transcription reagent (Applied Biosystems). Expression of Fhod3 in the postnatal heart was determined by PCR using specific primers: 5′-CTGTGGTCAAAACTGGAACCC-3′ (forward primer) and 5′-TGCACTGCCCAACGGCACTTC-3′ (reverse primer) for Fhod3; 5′-GGAAGCCCATCACCATCTTCCA-3′ (forward primer) and 5′-CCTTCTCCATGGTGGTGAAGAC-3′ (reverse primer) for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Quantitative real-time PCR were performed using SYBR Premix Ex Ta II (TaKaRa Bio) on the Roter-Gene 6200 system (Corbett life Science) with the following primer pairs for ANF, BNP, βMHC, collagen type 1a, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal standard: ANF, forward 5′-TTCCTCGTCTTGGCCTTTTG-3′ and reverse 5′-CCTCATCTTCTACCGGCATC-3′; BNP, forward 5′-GTCAGTCGTTTGGGCTGTAAC-3′ and reverse 5′-AGACCCAGGCAGAGTCAGAA-3′; βMHC, forward 5′-CGCATCAAGGAGCTCACC-3′ and 5′-CTGCAGCCGCAGTAGGTT-3′; collagen I (α1 chain), forward 5′-GACTGGCAACCTCAAGAAGG-3′ and reverse 5′-GACTGTCTTGCCCCAAGTTC-3′; and GAPDH, forward 5′-GGAAGCCCATCACCATCTTCCA-3′ and reverse 5′-CCTTCTCCATGGTGGTGAAGAC-3′.
Echocardiography
Serial echocardiographic examinations were performed non-invasively using a Vevo 2100 (Visual Sonics). Under anesthesia with 1–2% isoflurane inhalation, two-dimensional targeted M-mode images were obtained from the short-axis view at the level of papillary muscles. Echocardiographic images were analyzed by the analysis software of Vevo 2100. Left ventricular diastolic and systolic diameters (LVDd and LVDs) and end diastolic posterior wall thickness were measured following the guidelines of American Society of Echocardiography. The left ventricular ejection fraction (LVEF) was calculated according to the Teichholz method, and the left ventricular mass was calculated by the Devereux formula.
Statistical analysis
All data were expressed as mean ± S.E. Two groups were compared by paired or unpaired Student's t test. Multiple groups were compared by analysis of variance followed by post hoc Tukey test. A p value of <0.05 was considered to be statistically significant. GraphPad Prism 5.0 (GraphPad Software Inc., San Diego) was used for all statistical analysis.
Author contributions
T. U., R. T., and H. S. designed and coordinated the study; T. U., N. F., S. M., M. K., H. K., G. S., Y. K., and R. T. performed experiments; S. Y. provided reagents and animals; T. U., R. T., and H. S. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Acknowledgments
We thank Masato Tanaka (Kyushu University) for manipulation of mouse embryos; Kanako Motomura (Kyushu University) for histological analysis; Ryo Ugawa (Kyushu University) for transmission electron microscopic analysis; Drs. Atsushi Yamashita and Yujiro Asada (University of Miyazaki) for polarization microscopic analysis; Shoko Miura (Kyushu University) and Ami Matsuda (Miyazaki University) for technical assistance. We also appreciate the technical support from the Research Support Center, Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences, and the Laboratory for Technical Support, Medical Institute of Bioregulation, Kyushu University.
This work was supported in part by Grant-in-aid for Scientific Research on Innovative Areas “Oxygen Biology: a new criterion for integrated understanding of life” Grant 26111009 (to H. S.), Japan Society for the Promotion of Science (JSPS) KAKENHI Grant 26460371 (to R. T.), SHINGAKUJUTSU Grant 25117515 (to R. T.), grants from the Takeda Science Foundation (to R. T.) and the Institute of Seizon and Life Sciences (to R. T.), and the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University (to R. T.). The authors declare that they have no conflicts of interest with the contents of this article.
- FH
- formin homology
- ANF
- atrial natriuretic factor
- BNP
- B-type natriuretic peptide
- cKO
- conditional knockout
- MCK
- muscle creatine kinase
- αMHC
- α-myosin heavy chain
- βMHC
- β-myosin heavy chain
- TAM
- tamoxifen
- BDM
- 2,3-butanedione monoxim
- LVDd
- left ventricular dimension in diastole
- LVDs
- left ventricular dimension in systole
- LVEF
- left ventricular ejection fraction
- P
- postnatal day
- PE
- phenylephrine
- iKO
- inducible knock-out.
References
- 1. Moss R. L., Razumova M., and Fitzsimons D. P. (2004) Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circ. Res. 94, 1290–1300 10.1161/01.RES.0000127125.61647.4F [DOI] [PubMed] [Google Scholar]
- 2. Ono S. (2010) Dynamic regulation of sarcomeric actin filaments in striated muscle. Cytoskeleton 67, 677–692 10.1002/cm.20476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fritz-Six K. L., Cox P. R., Fischer R. S., Xu B., Gregorio C. C., Zoghbi H. Y., and Fowler V. M. (2003) Aberrant myofibril assembly in tropomodulin1 null mice leads to aborted heart development and embryonic lethality. J. Cell Biol. 163, 1033–1044 10.1083/jcb.200308164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nishii K., Morimoto S., Minakami R., Miyano Y., Hashizume K., Ohta M., Zhan D.-Y., Lu Q.-W., and Shibata Y. (2008) Targeted disruption of the cardiac troponin T gene causes sarcomere disassembly and defects in heartbeat within the early mouse embryo. Dev. Biol. 322, 65–73 10.1016/j.ydbio.2008.07.007 [DOI] [PubMed] [Google Scholar]
- 5. McKeown C. R., Nowak R. B., Gokhin D. S., and Fowler V. M. (2014) Tropomyosin is required for cardiac morphogenesis, myofibril assembly, and formation of adherens junctions in the developing mouse embryo. Dev. Dyn. 243, 800–817 10.1002/dvdy.24115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kan-O M, Takeya R., Abe T., Kitajima N., Nishida M., Tominaga R., Kurose H., and Sumimoto H. (2012) Mammalian formin Fhod3 plays an essential role in cardiogenesis by organizing myofibrillogenesis. Biol. Open 1, 889–896 10.1242/bio.20121370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li D., Hallett M. A., Zhu W., Rubart M., Liu Y., Yang Z., Chen H., Haneline L. S., Chan R. J., Schwartz R. J., Field L. J., Atkinson S. J., and Shou W. (2011) Dishevelled-associated activator of morphogenesis 1 (Daam1) is required for heart morphogenesis. Development 138, 303–315 10.1242/dev.055566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Littlefield R. S., and Fowler V. M. (2008) Thin filament length regulation in striated muscle sarcomeres: pointed-end dynamics go beyond a nebulin ruler. Semin. Cell Dev. Biol. 19, 511–519 10.1016/j.semcdb.2008.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Skwarek-Maruszewska A., Hotulainen P., Mattila P. K., and Lappalainen P. (2009) Contractility-dependent actin dynamics in cardiomyocyte sarcomeres. J. Cell Sci. 122, 2119–2126 10.1242/jcs.046805 [DOI] [PubMed] [Google Scholar]
- 10. Hill J. A., and Olson E. N. (2008) Cardiac plasticity. N. Engl. J. Med. 358, 1370–1380 10.1056/NEJMra072139 [DOI] [PubMed] [Google Scholar]
- 11. Yuan B., Wan P., Chu D., Nie J., Cao Y., Luo W., Lu S., Chen J., and Yang Z. (2014) A cardiomyocyte-specific Wdr1 knockout demonstrates essential functional roles for actin disassembly during myocardial growth and maintenance in mice. Am. J. Pathol. 184, 1967–1980 10.1016/j.ajpath.2014.04.007 [DOI] [PubMed] [Google Scholar]
- 12. Pappas C. T., Mayfield R. M., Henderson C., Jamilpour N., Cover C., Hernandez Z., Hutchinson K. R., Chu M., Nam K.-H., Valdez J. M., Wong P. K., Granzier H. L., and Gregorio C. C. (2015) Knockout of Lmod2 results in shorter thin filaments followed by dilated cardiomyopathy and juvenile lethality. Proc. Natl. Acad. Sci. U.S.A. 112, 13573–13578 10.1073/pnas.1508273112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chesarone M. A., DuPage A. G., and Goode B. L. (2010) Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 11, 62–74 10.1038/nrm2816 [DOI] [PubMed] [Google Scholar]
- 14. Campellone K. G., and Welch M. D. (2010) A nucleator arms race: cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 11, 237–251 10.1038/nrm2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Skau C. T., and Waterman C. M. (2015) Specification of architecture and function of actin structures by actin nucleation factors. Annu. Rev. Biophys. 44, 285–310 10.1146/annurev-biophys-060414-034308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paul A. S., and Pollard T. D. (2009) Review of the mechanism of processive actin filament elongation by formins. Cell Motil. Cytoskeleton 66, 606–617 10.1002/cm.20379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blanchoin L., Boujemaa-Paterski R., Sykes C., and Plastino J. (2014) Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 94, 235–263 10.1152/physrev.00018.2013 [DOI] [PubMed] [Google Scholar]
- 18. Liu R., Linardopoulou E. V., Osborn G. E., and Parkhurst S. M. (2010) Formins in development: orchestrating body plan origami. Biochim. Biophys. Acta 1803, 207–225 10.1016/j.bbamcr.2008.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Randall T. S., and Ehler E. (2014) A formin-g role during development and disease. Eur. J. Cell Biol. 93, 205–211 10.1016/j.ejcb.2013.11.004 [DOI] [PubMed] [Google Scholar]
- 20. Thumkeo D., Watanabe S., and Narumiya S. (2013) Physiological roles of Rho and Rho effectors in mammals. Eur. J. Cell Biol. 92, 303–315 10.1016/j.ejcb.2013.09.002 [DOI] [PubMed] [Google Scholar]
- 21. Kanaya H., Takeya R., Takeuchi K., Watanabe N., Jing N., and Sumimoto H. (2005) Fhos2, a novel formin-related actin-organizing protein, probably associates with the nestin intermediate filament. Genes Cells 10, 665–678 10.1111/j.1365-2443.2005.00867.x [DOI] [PubMed] [Google Scholar]
- 22. Taniguchi K., Takeya R., Suetsugu S., Kan-O M., Narusawa M., Shiose A., Tominaga R., and Sumimoto H. (2009) Mammalian formin Fhod3 regulates actin assembly and sarcomere organization in striated muscles. J. Biol. Chem. 284, 29873–29881 10.1074/jbc.M109.059303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iskratsch T., Lange S., Dwyer J., Kho A. L., dos Remedios C., and Ehler E. (2010) Formin follows function: a muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. J. Cell Biol. 191, 1159–1172 10.1083/jcb.201005060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kan-O M, Takeya R., Taniguchi K., Tanoue Y., Tominaga R., and Sumimoto H. (2012) Expression and subcellular localization of mammalian formin Fhod3 in the embryonic and adult heart. PLoS ONE 7, e34765 10.1371/journal.pone.0034765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujimoto N., Kan-O M., Ushijima T., Kage Y., Tominaga R., Sumimoto H., and Takeya R. (2016) Transgenic expression of the formin protein Fhod3 selectively in the embryonic heart: role of actin-binding activity of Fhod3 and its sarcomeric localization during myofibrillogenesis. PLoS ONE 11, e0148472 10.1371/journal.pone.0148472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wooten E. C., Hebl V. B., Wolf M. J., Greytak S. R., Orr N. M., Draper I., Calvino J. E., Kapur N. K., Maron M. S., Kullo I. J., Ommen S. R., Bos J. M., Ackerman M. J., and Huggins G. S. (2013) Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 6, 10–18 10.1161/CIRCGENETICS.112.965277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arimura T., Takeya R., Ishikawa T., Yamano T., Matsuo A., Tatsumi T., Nomura T., Sumimoto H., and Kimura A. (2013) Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ. J. 77, 2990–2996 10.1253/circj.CJ-13-0255 [DOI] [PubMed] [Google Scholar]
- 28. Brüning J. C., Michael M. D., Winnay J. N., Hayashi T., Hörsch D., Accili D., Goodyear L. J., and Kahn C. R. (1998) A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2, 559–569 10.1016/S1097-2765(00)80155-0 [DOI] [PubMed] [Google Scholar]
- 29. Gotthardt M., Hammer R. E., Hübner N., Monti J., Witt C. C., McNabb M., Richardson J. A., Granzier H., Labeit S., and Herz J. (2003) Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. J. Biol. Chem. 278, 6059–6065 10.1074/jbc.M211723200 [DOI] [PubMed] [Google Scholar]
- 30. Iskratsch T., Reijntjes S., Dwyer J., Toselli P., Dégano I. R., Dominguez I., and Ehler E. (2013) Two distinct phosphorylation events govern the function of muscle FHOD3. Cell. Mol. Life Sci. 70, 893–908 10.1007/s00018-012-1154-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gautel M., Zuffardi O., Freiburg A., and Labeit S. (1995) Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 14, 1952–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sohal D. S., Nghiem M., Crackower M. A., Witt S. A., Kimball T. R., Tymitz K. M., Penninger J. M., and Molkentin J. D. (2001) Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 89, 20–25 10.1161/hh1301.092687 [DOI] [PubMed] [Google Scholar]
- 33. Davis J., Maillet M., Miano J. M., and Molkentin J. D. (2012) Lost in transgenesis: a user's guide for genetically manipulating the mouse in cardiac research. Circ. Res. 111, 761–777 10.1161/CIRCRESAHA.111.262717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koitabashi N., Bedja D., Zaiman A. L., Pinto Y. M., Zhang M., Gabrielson K. L., Takimoto E., and Kass D. A. (2009) Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ. Res. 105, 12–15 10.1161/CIRCRESAHA.109.198416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perriard J.-C., Hirschy A., and Ehler E. (2003) Dilated cardiomyopathy: a disease of the intercalated disc? Trends Cardiovasc. Med. 13, 30–38 10.1016/S1050-1738(02)00209-8 [DOI] [PubMed] [Google Scholar]
- 36. Mori J., Basu R., McLean B. A., Das S. K., Zhang L., Patel V. B., Wagg C. S., Kassiri Z., Lopaschuk G. D., and Oudit G. Y. (2012) Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: a metabolic contribution to heart failure with normal ejection fraction. Circ. Heart Fail. 5, 493–503 10.1161/CIRCHEARTFAILURE.112.966705 [DOI] [PubMed] [Google Scholar]
- 37. Farivar R. S., Crawford D. C., Chobanian A. V., and Brecher P. (1995) Effect of angiotensin II blockade on the fibroproliferative response to phenylephrine in the rat heart. Hypertension 25, 809–813 10.1161/01.HYP.25.4.809 [DOI] [PubMed] [Google Scholar]
- 38. Hew K. W., and Keller K. A. (2003) Postnatal anatomical and functional development of the heart: a species comparison. Birth Defects Res. B. Dev. Reprod. Toxicol. 68, 309–320 10.1002/bdrb.10034 [DOI] [PubMed] [Google Scholar]
- 39. Maillet M., van Berlo J. H., and Molkentin J. D. (2013) Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 14, 38–48 10.1038/nrm3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li F., Wang X., Capasso J. M., and Gerdes A. M. (1996) Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 28, 1737–1746 10.1006/jmcc.1996.0163 [DOI] [PubMed] [Google Scholar]
- 41. Rosado M., Barber C. F., Berciu C., Feldman S., Birren S. J., Nicastro D., and Goode B. L. (2014) Critical roles for multiple formins during cardiac myofibril development and repair. Mol. Biol. Cell 25, 811–827 10.1091/mbc.E13-08-0443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kooij V., Viswanathan M. C., Lee D. I., Rainer P. P., Schmidt W., Kronert W. A., Harding S. E., Kass D. A., Bernstein S. I., Van Eyk J. E., and Cammarato A. (2016) Profilin modulates sarcomeric organization and mediates cardiomyocyte hypertrophy. Cardiovasc. Res. 110, 238–248 10.1093/cvr/cvw050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ajima R., Bisson J. A., Helt J.-C., Nakaya M.-A., Habas R., Tessarollo L., He X., Morrisey E. E., Yamaguchi T. P., and Cohen E. D. (2015) DAAM1 and DAAM2 are co-required for myocardial maturation and sarcomere assembly. Dev. Biol. 408, 126–139 10.1016/j.ydbio.2015.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou Q., Wei S.-S., Wang H., Wang Q., Li W., Li G., Hou J.-W., Chen X.-M., Chen J., Xu W.-P., Li Y.-G., and Wang Y.-P. (2017) Crucial role of ROCK2-mediated phosphorylation and up-regulation of FHOD3 in the pathogenesis of angiotensin II-induced cardiac HypertrophyNovelty and significance. Hypertension 69, 1070–1083 10.1161/HYPERTENSIONAHA.116.08662 [DOI] [PubMed] [Google Scholar]
- 45. Kiyonari H., Kaneko M., Abe S., and Aizawa S. (2010) Three inhibitors of FGF receptor, ERK, and GSK3 establishes germline-competent embryonic stem cells of C57BL/6N mouse strain with high efficiency and stability. Genesis 48, 317–327 [DOI] [PubMed] [Google Scholar]
- 46. Kanki H., Suzuki H., and Itohara S. (2006) High-efficiency CAG-FLPe deleter mice in C57BL/6J background. Exp. Anim. 55, 137–141 10.1538/expanim.55.137 [DOI] [PubMed] [Google Scholar]
- 47. Takeya R., Taniguchi K., Narumiya S., and Sumimoto H. (2008) The mammalian formin FHOD1 is activated through phosphorylation by ROCK and mediates thrombin-induced stress fibre formation in endothelial cells. EMBO J. 27, 618–628 10.1038/emboj.2008.7 [DOI] [PMC free article] [PubMed] [Google Scholar]