

Abstract
Congestive heart failure typically arises from cardiac myocyte necrosis/apoptosis, associated with the pathological opening of the mitochondrial permeability transition pore (mPTP). mPTP opening decreases the mitochondrial membrane potential leading to the activation of Ca2+-independent phospholipase A2γ (iPLA2γ) and the production of downstream toxic metabolites. However, the array of enzymatic mediators and the exact chemical mechanisms responsible for modulating myocardial mPTP opening remain unclear. Herein, we demonstrate that human heart failure activates specific myocardial mitochondrial phospholipases that increase Ca2+-dependent production of toxic hydroxyeicosatetraenoic acids (HETEs) and attenuate the activity of phospholipases that promote the synthesis of protective epoxyeicosatrienoic acids (EETs). Mechanistically, HETEs activated the Ca2+-induced opening of the mPTP in failing human myocardium, and the highly selective pharmacological blockade of either iPLA2γ or lipoxygenases attenuated mPTP opening in failing hearts. In contrast, pharmacological inhibition of cytochrome P450 epoxygenases opened the myocardial mPTP in human heart mitochondria. Remarkably, the major mitochondrial phospholipase responsible for Ca2+-activated release of arachidonic acid (AA) in mitochondria from non-failing hearts was calcium-dependent phospholipase A2ζ (cPLA2ζ) identified by sequential column chromatographies and activity-based protein profiling. In contrast, iPLA2γ predominated in failing human myocardium. Stable isotope kinetics revealed that in non-failing human hearts, cPLA2ζ metabolically channels arachidonic acid into EETs, whereas in failing hearts, increased iPLA2γ activity channels AA into toxic HETEs. These results mechanistically identify the sequelae of pathological remodeling of human mitochondrial phospholipases in failing myocardium. This remodeling metabolically channels AA into toxic HETEs promoting mPTP opening, which induces necrosis/apoptosis leading to further progression of heart failure.
Keywords: lipoxygenase, heart failure, phospholipase, mitochondrial permeability transition pore (mPTP), eicosanoid, cytochrome P-450
Introduction
Congestive heart failure is a public health crisis of epidemic proportions that typically results from cardiac myocyte necrosis/apoptosis leading to progressive hemodynamic compromise (1–3). In animal models, it is generally believed that cardiac myocyte cell death and heart failure are intimately associated with the pathological opening of the mitochondrial permeability transition pore (mPTP)7 (3–5). Opening of this pore dissipates the mitochondrial membrane potential and facilitates the production of toxic metabolites, the activation of membrane potential-sensitive calcium-independent phospholipase A2γ (iPLA2γ) (6), and the release/activation of proteins mediating cell death-signaling programs (e.g. cytochrome c). The resultant dropout of cardiac myocytes during heart failure by either apoptosis or necrosis leads to the progression of heart failure, further hemodynamic compromise, and ultimately death (7, 8). However, the precise chemical mechanisms, enzymatic mediators, and molecular events responsible for modulating the amount and duration of mPTP opening in both animal and human myocardium remain largely undefined.
Previous studies have demonstrated that human myocardium contains multiple different phospholipase activities encoded by distinct genes (9–11). Recently, we demonstrated that genetic knock-out of the active site of iPLA2γ (PNPLA8) results in the attenuation of mPTP opening and that iPLA2γ is largely responsible for the calcium-dependent production of oxidized fatty acid metabolites (e.g. eicosanoids, docosanoids, and oxidized linoleic acid metabolites) emanating from the mitochondrial compartment in murine hepatic and myocardial tissues (12–14). Moreover, loss of mitochondrial transmembrane potential evoked by either inhibition of mitochondrial electron transport chain complexes or prolonged calcium transients results in the further activation of iPLA2γ (6). Collectively, this sequence of events results in a feed-forward loop promoting the autoamplification of mitochondrial lipid second messenger generation. Mitochondria contain, or are associated with, multiple different types of phospholipases in humans. However, their molecular identity, their contributions to production of signaling lipids, and the effects of the resultant downstream metabolites in non-failing versus failing human myocardium are unknown.
During heart failure in animal models, increases in mitochondrial calcium concentration and changes in calcium dynamics are present resulting from alterations in sarcoplasmic reticulum (SR)-mitochondrial subcellular architecture, pathological SR calcium coupling, and reduced Ca2+ sequestration (15, 16). Because phospholipase-catalyzed arachidonic acid (AA) release is the rate-limiting step in the generation of eicosanoid-derived signaling moieties, knowledge of the types of mitochondrial phospholipases, Ca2+-mediated changes in their activities, and metabolic fates of their downstream metabolites are essential in the mechanistic understanding of the pathology resulting from the diverse array of toxic oxidized lipids produced during human heart failure.
Recent studies have demonstrated that specific eicosanoids may confer either protective or deleterious effects on myocardial function (17–19). For example, increased levels of 14,15-epoxyeicosatrienoic acid (14,15-EET) in murine myocardium produced by either overexpression of the cytochrome P450 enzyme CYP2J3 or by genetic knock-out of soluble epoxide hydrolase resulted in decreased mPTP opening probability, improved preservation of mitochondrial membrane potential (Δψmt), and attenuated damage after ischemia/reperfusion (17, 20, 21). The diverse effects of prostaglandin E2 (PGE2), a cyclooxygenase-2 product, on cardiac function have been documented, which include the reduction of cardiac ischemia/reperfusion injury and a contribution to myocardial hypertrophy via prostaglandin E2 receptor signaling (22, 23). In contrast, 12-HETE and 20-HETE induce increases in mitochondrial calcium concentration and myocardial infarct size during ischemia/reperfusion in mice (24–26).
Previously, we identified the prominent production of eicosanoids (including HETEs, EETs, and prostaglandins) in calcium-activated murine heart tissue and mitochondria that were markedly reduced by genetic ablation of iPLA2γ (13, 27). Accordingly, we hypothesized that abrupt activation of mitochondrial phospholipases by calcium uptake into the mitochondria in conjunction with the loss of membrane potential would result in the pathological activation of iPLA2γ leading to the generation of a complex array of eicosanoid metabolites and fatty acids that contribute to the opening of the mPTP and progression of human heart failure. Here, we demonstrate the dramatic increase in calcium-activated HETE production at the expense of cardioprotective EETs in failing human hearts in comparison with non-failing human myocardium. These changes collectively result in the increased sensitivity of mitochondria to calcium-induced mPTP opening and further depolarization leading to cardiac myocyte damage. Furthermore, we present primary evidence of metabolic channeling of AA into discrete eicosanoid products through the remodeling of mitochondrial phospholipase activities from cPLA2ζ in non-failing human myocardium to iPLA2γ during heart failure. Collectively, this study mechanistically identifies the pathological remodeling of cardiac phospholipases and their downstream products resulting from altered metabolic channeling as a principal mechanism increasing lipid-mediated mPTP opening leading to mitochondrial dysfunction and the progression of human heart failure.
Results
Remodeling of human mitochondrial eicosanoid production in non-failing versus failing human myocardium
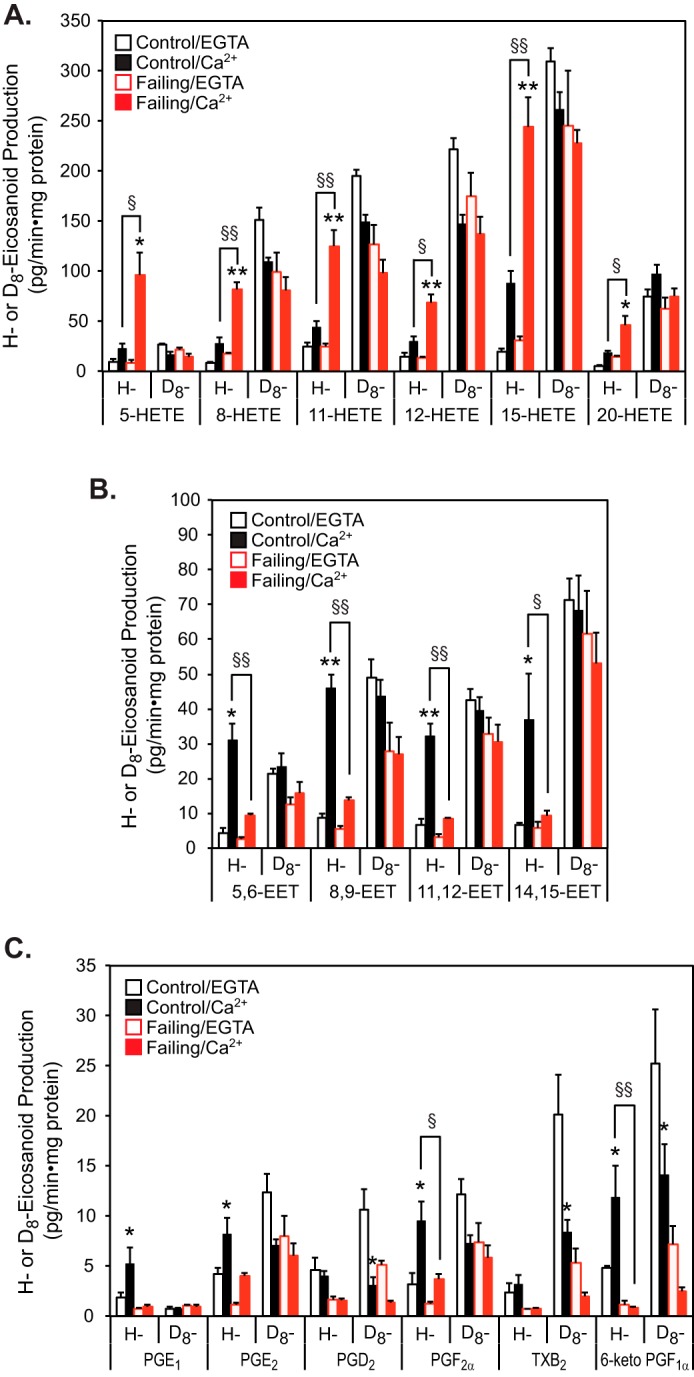
To assess whether alterations in eicosanoid molecular species are present in failing versus non-failing human heart mitochondria, we incubated mitochondria from non-failing or failing human myocardium in the absence or presence of calcium ion as described under “Experimental procedures.” Reaction products were identified and quantified using charge-switch derivatization with LC-MS/MS (14, 28) and selected reaction monitoring with HRAM (high resolution accurate mass) mass spectrometry of product ions as reported previously (14). Intriguingly, mitochondria (and their tethered/associated membranes) prepared from non-failing human myocardium exhibited a substantial increase in the initial rate of production of a wide range of eicosanoid molecular species after calcium challenge, including cytoprotective EETs (Fig. 1, A–D). In sharp contrast, similar calcium challenge of mitochondria from failing human myocardium produced robust increases in multiple lipoxygenase metabolites, including 5-, 8-, 11-, 12-, and 15-HETEs. Critically, in failing myocardium, calcium stimulation did not result in significant increases in salutary 14,15-EET production (Fig. 1, A and B), although the enzymes to catalyze the oxidation of released AA to 14,15-EET were clearly present in failing myocardium (see below). Moreover, large Ca2+-dependent increases in proinflammatory PGE2 were manifest in conjunction with decreases in cardioprotective prostacyclins (29–31) (measured as their stable end products, 6-keto-PGF1α) in mitochondria from failing hearts.
Figure 1.

Mass spectrometric analysis and quantitation of eicosanoids generated by non-failing and failing human heart mitochondria stimulated by calcium. Myocardial mitochondria were isolated from non-failing (control) and failing hearts by differential centrifugation as described under “Experimental procedures” and then sonicated in HEPES buffer (10 mm HEPES (pH 7.4) containing 10% glycerol and 1 mm DTT). Mitochondrial homogenates were incubated in the absence of Ca2+ (EGTA) or the presence of 200 μm free Ca2+ at 35 °C for 20 min. The reaction was stopped by addition of methanol (20% v/v final concentration) at 4 °C and diluted with water after addition of internal standards (250 pg each of TXB2-d4, PGE2-d4, and 12-HETE-d8). Eicosanoids were immediately isolated by solid-phase extraction, derivatized with AMPP, and analyzed by HRAM mass spectrometry. The resultant production of HETEs (A), EETs (B), and Prostaglandins (PGs) (C) is shown with the average ± S.E. (n = 6 for non-failing control hearts and n = 6 for failing hearts). *, p < 0.05, and **, p < 0.005 for control (non-failing) versus failing hearts. ‡, p < 0.005, and §, p < 0.05 for EGTA versus Ca2+. A representative LC-MS spectrum from non-failing control heart mitochondria in the absence (EGTA) or presence of Ca2+ is displayed in D.
Failing human myocardium is more susceptible to calcium-induced opening of mPTP in comparison with non-failing human myocardium
Despite the importance of the mPTP channel in mediating cardiac myocyte cell death programs, to the best of our knowledge, no previous investigations have explored alterations in mPTP opening in non-failing versus failing human hearts. Accordingly, we incubated isolated human heart mitochondria with calcium ion and measured mitochondrial swelling indicative of mPTP opening. Although high-resolution respirometry revealed that oxidative phosphorylation of mitochondria isolated from non-failing and failing hearts was not significantly different (Fig. S1), calcium challenge of mitochondria dramatically increased swelling of mitochondria from failing hearts in comparison with non-failing hearts (Fig. 2A). Inclusion of inorganic phosphate (Pi), a potent inducer of mPTP opening in animal models, only moderately increased swelling of human myocardial mitochondria without affecting the relative sensitivity of non-failing versus failing heart mitochondria to calcium-induced mPTP opening (Fig. 2B). The expression levels of known transition pore regulators (32, 33) (e.g. adenine nucleotide translocase (ANT), voltage-dependent anion channel (VDAC), and cyclophilin D (CypD)) were not different in non-failing versus failing hearts as assessed by Western blot analyses (Fig. 2C). Thus, the observed susceptibility of failing heart mitochondria to calcium-induced swelling was not due to alterations in the content of pore-regulating proteins, but rather to their maladaptive regulation by different modulators in non-failing versus failing human hearts.
Figure 2.
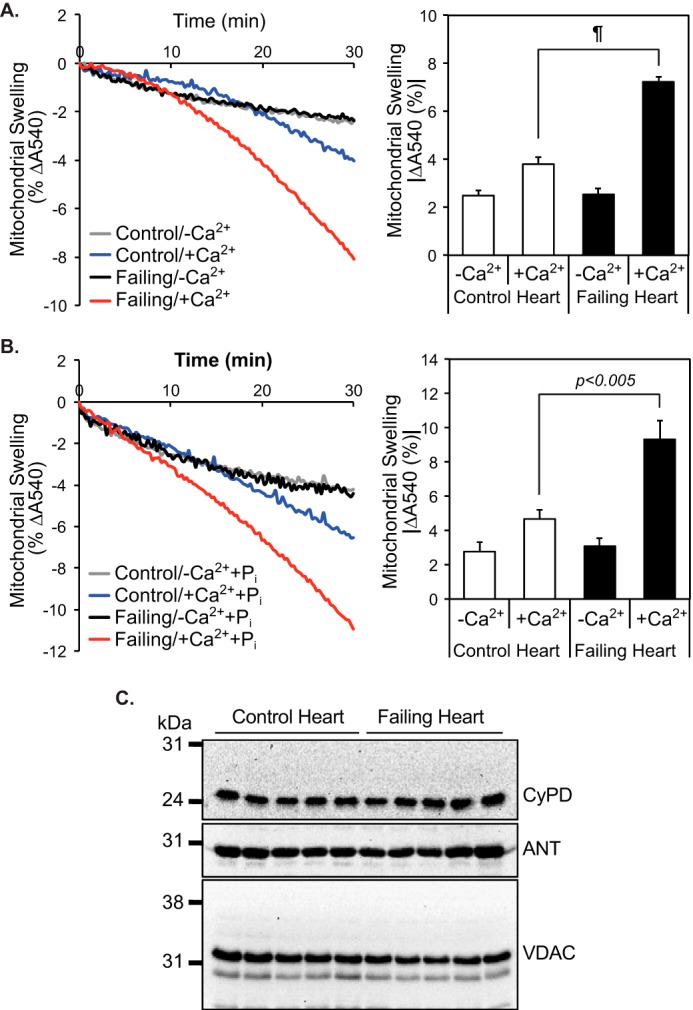
Increased sensitivity of mitochondria from failing human myocardium to calcium-induced swelling in comparison with non-failing mitochondria. Mitochondria were isolated from non-failing and failing hearts by differential centrifugation and resuspended in Mitochondrial Swelling Buffer (3 mm HEPES (pH 7.4), containing 0.23 m mannitol, 0.07 m sucrose, 5 mm succinate, and 2.5 μm rotenone) in the absence (n = 14 for non-failing controls and n = 12 for failing hearts) (A) or presence of 1 mm Pi (n = 4 for non-failing controls and n = 5 for failing hearts) (B). Mitochondrial swelling was initiated by addition of 70 μm Ca2+ and monitored by the decrease in absorbance at 540 nm (A and B, left panels). The average decrease in absorbance at 540 nm after 30 min was calculated (A and B, right panels, average ± S.E.). ¶, p < 10−6. C, protein expression levels of CypD, ANT, and VDAC in non-failing control (n = 5) and failing hearts (n = 5).
HETEs activate the calcium-induced mitochondrial permeability transition pore opening
To determine whether the dramatic increase in HETE production was mechanistically responsible for the increase in sensitivity to mitochondrial swelling, mitochondria from failing human myocardium were treated with specific inhibitors of either cyclooxygenases (COX), lipoxygenases (LOX), or cytochrome P450 monooxygenases (CYP450). Remarkably, inhibition of LOX activity with baicalein (Baic) completely abolished calcium-activated mPTP opening of failing heart mitochondria, whereas CYP450 inhibition with MSPPOH dramatically exacerbated swelling (Fig. 3A). Because MSPPOH-enhanced mitochondrial swelling appears inhibitable with cyclosporine A, inhibition of CYP450 likely facilitate cyclophilin D-dependent mPTP opening (Fig. 3B). Inhibition of COX enzymes with ibuprofen (Ibu) did not affect human failing heart mitochondrial swelling. Similarly, mPTP opening of non-failing heart mitochondria was dramatically promoted by preincubation with MSPPOH, although the induction time for mitochondrial swelling was significantly longer than that of failing heart (Fig. 3C). These distinct effects resulting from selective inhibition of discrete oxygenases demonstrate the prominent role of signaling eicosanoids in the enhancement or attenuation of mPTP opening in failing human myocardium. Mitochondrial swelling data with oxygenase-specific inhibitors are consistent with the HRAM mass spectrometric analyses of mitochondria demonstrating higher HETE levels in mitochondria from failing hearts accompanied by decreased calcium-stimulated EET production in comparison with non-failing hearts (Fig. 1). Importantly, exogenous provision of selected HETEs reversed the protection from mPTP opening conveyed by baicalein treatment (Fig. 3D). Although multiple HETEs were promoters of mitochondrial swelling (Fig. 3D), the lipoxygenase products 12-HETE and 15-HETE are likely of greater pathophysiological significance because they are prominent in human myocardium, and Ca2+ stimulation generated substantially greater amounts of these eicosanoids in failing human heart mitochondria (Fig. 1). This notion is further supported by the observation of the inability of CYP450 hydroxylase(s) to generate effective amounts of 20-HETE after calcium stimulation in either non-failing or failing human myocardial mitochondria (Fig. 1).
Figure 3.

Differential effects of eicosanoid molecular species on mPTP opening. A, isolated mitochondria from failing hearts (n = 6) were preincubated for 10 min with 10 μm baic, 20 μm Ibu, or 40 μm MSPPOH for selective inhibition of lipoxygenase, cyclooxygenase, or cytochrome P450 enzymes, respectively. After preincubation, mitochondrial swelling was initiated by addition of Ca2+. **, p < 10−5, and ¶, p < 10−6 when compared with Ca2+ alone (Ctl). B, intact mitochondria from failing hearts were preincubated with 1 μm cyclosporine A (CsA) with or without 40 μm MSPPOH for 10 min at 25 °C. Mitochondrial swelling was monitored by addition of Ca2+ (left panel). The final ΔA540 end-point measurements at 30 min are presented (right panel, n = 4–8). C, isolated mitochondria from non-failing control (n = 5) and failing hearts (n = 7) were preincubated with or without 40 μm MSPPOH for 10 min at 25 °C for inhibition of cytochrome P450(s). Mitochondrial swelling was monitored after addition of Ca2+ (left panel). The final ΔA540 end point measurements at 20 and 30 min are presented in the middle and right panels, respectively (n = 4–8). §, p < 0.005. N.S., not significant. D, isolated mitochondria from failing hearts (n = 4) were preincubated for 10 min with 10 μm baic in the absence or presence of either 5-, 12-, 15-, or 20-HETE (4 μm final concentrations). Mitochondrial swelling was initiated by addition of Ca2+. Representative tracings for mitochondrial swelling and the absorbance decreases at 540 nm after 30 min of incubation are presented. *, p < 0.05, and **, p < 0.0005.
Oxidation of exogenously added deuterated arachidonic acid versus endogenously produced arachidonic acid in mitochondria from non-failing and failing myocardium
To determine whether alterations in the enzymic activities of AA oxidases in non-failing versus failing heart mitochondria were responsible for the differences in the profiles of eicosanoid products, we compared their rate of production using deuterated isotopes of arachidonic acid examined under initial rate conditions. Mitochondrial sonicates from non-failing or failing human hearts were incubated with exogenous deuterated arachidonic acid (d8-AA) in the absence or presence of Ca2+. The results demonstrated minimal differences in deuterated oxylipin production using d8-AA in the presence of EGTA or Ca2+ treatments in either non-failing and failing myocardial mitochondria (Fig. 4). Moreover, these results demonstrate that AA availability to the downstream oxidative enzymes was rate-determining in the synthesis of the measured eicosanoids because if exogenously added d8-AA was present, no substantive differences in deuterated eicosanoid production in non-failing versus failing hearts occurred. In stark contrast, non-labeled HETE (H−) production from endogenous AA in failing heart mitochondria, even in the presence of exogenous d8-AA, was markedly higher than that in non-failing heart mitochondria (Fig. 4A). The production of virtually all non-labeled salutary EETs in failing human heart mitochondria was drastically reduced in comparison with non-failing heart mitochondria. However, substantive amounts of d8-labeled EETs (especially 14,15-EET) were readily produced when exogenous d8-AA was present. These results demonstrate that the enzymes for EET production were present in failing myocardium, but the endogenously generated AA upon calcium challenge could not be metabolically channeled to EET production (Fig. 4B). Cyclooxygenase-mediated prostaglandin E2 (PGE2) production using endogenous AA was Ca2+-dependent, and the rate of production of both thromboxane and prostacyclin was dramatically reduced in mitochondria from failing hearts (Fig. 4C). Mitochondrial preparations from failing human hearts possess less capacity for the production of prostacyclins (e.g. 6-keto-PGF1α), even after the addition of exogenous substrate, demonstrating a deficiency of prostacyclin synthase activity and/or failure of metabolic channeling of the released AA to reach its thromboxane synthase target (Fig. 4C). Collectively, these results demonstrate the importance of calcium for the production of eicosanoid products, the tight metabolic regulation of mitochondrial phospholipases in non-failing hearts, and the differential metabolic channeling of released AA in non-failing versus failing human myocardium.
Figure 4.
Deuterated (d8-) and non-labeled (H-) eicosanoid production in the presence of exogenous d8-arachidonic acid in non-failing control and failing human heart mitochondria. Isolated mitochondria (1 mg) were placed in 10 mm HEPES buffer (pH 7.4) containing 10% glycerol and 1 mm DTT, and homogenized by sonication on ice. Mitochondrial phospholipase activity was activated by addition of either 2 mm EGTA or 0.2 mm free Ca2+ in the presence of 8 nmol of d8-AA at 35 °C. After 20 min of incubation, the reaction was terminated by addition of methanol to 20% (v/v) final concentration at 4 °C. Eicosanoids were immediately extracted by solid phase extraction with internal standards, derivatized with AMPP, and analyzed by high-resolution high mass accuracy mass spectrometry. The quantities of deuterated (D8-) and non-labeled (H-) eicosanoids, including HETEs (A), EETs (B), and prostaglandins (C), were determined by calculations based on either internal standards (250 pg each of TXB2-d4 and PGE2-d4) or by calibration utilizing an external standard (250 pg of 12-HETE-d8) and are presented as the average ± S.E. (n = 4). §, p < 0.05, and §§, p < 0.01. *, p < 0.05, and **, p < 0.005 when compared with EGTA incubations.
Mitochondrial-associated phospholipases A2 in non-failing versus failing human myocardium
The remarkable differences in the rates of eicosanoid synthesis in non-failing versus failing hearts led us to further investigate the Ca2+ dependence and sensitivity of the mitochondrial phospholipase activity responsible for the release of the AA precursor for downstream oxygenation. Incubation of non-failing heart mitochondria with Ca2+ resulted in the dramatic and selective release of AA with only modest increases in other saturated and unsaturated fatty acids (Fig. 5 and Fig. S2). Concomitant with AA release, only lysophosphatidylcholines (LPCs) containing 16:0-diacyl or plasmenyl 16:0- (P16:0-) vinyl ether linkages were significantly increased upon calcium challenge. These results suggest that a highly active Ca2+-stimulated phospholipase preferentially hydrolyzes arachidonoylated phosphatidylcholine and plasmenylcholine (PC) molecular species present in the non-failing human heart (Fig. 5). In contrast, mitochondria from failing hearts demonstrated a markedly reduced capacity to release AA in response to calcium challenge with substantially less AA selectivity and attenuated LPC production (Fig. 5 and Fig. S2).
Figure 5.

Ca2+-dependent activation of phospholipase(s) in non-failing human heart mitochondria releases arachidonic acid, which is dramatically attenuated in failing human heart mitochondria. Isolated mitochondria from human non-failing control (n = 7) and failing (n = 9) hearts were placed in 10 mm HEPES buffer (pH 7.4) containing 10% glycerol and 1 mm DTT, and homogenized by sonication on ice. Activation of phospholipase(s) was measured following the addition of either 2 mm EGTA or 0.6 mm free Ca2+ at 35 °C. After 10 min of incubation, the reaction was terminated by addition of 2 ml of chloroform/methanol (1:1) in the presence of internal standards (d4-16:0-fatty acid, 17:0-LPC) followed by phase separation. Extracted fatty acid (A) and LPC (B) molecular species were identified and quantified in the negative and positive ion modes, respectively, utilizing a mass spectrometer. §, p < 0.001; **, p < 0.005, and ¶, p < 10−5 when compared with EGTA treatment. C, mitochondrial homogenates obtained from non-failing (control, n = 5) and failing (n = 5) hearts were incubated with 0 (2 mm EGTA), 5, 15, 75, 180, and 600 μm free Ca2+ for 10 min at 35 °C. AA was quantified by mass spectrometry. *, p < 0.05, and **, p < 0.005 when compared with non-failing controls.
Next, we sought to identify the major heart mitochondrial PLA2 enzyme(s) responsible for the release of AA from human mitochondria from non-failing and failing human myocardium. The results demonstrated that ∼80% of Ca2+-activated AA release and LPC production containing either 16:0 fatty acyl or plasmenyl residue in non-failing heart mitochondria were inhibited by a non-selective serine hydrolase inhibitor, methyl arachidonyl fluorophosphonate (MAFP) but not by either (S)-BEL ((S)-bromoenol lactone, a selective calcium-independent phospholipase A2β inhibitor), (R)-BEL (a selective iPLA2γ inhibitor), Pyr (pyrrolidine derivative, a cPLA2α-specific inhibitor), or AACOCF3 (arachidonyl trifluoromethyl ketone, a generic phospholipase inhibitor) (Fig. 6, A and B). In stark contrast, 50% of Ca2+-dependent AA release in failing heart mitochondria was inhibited by (R)-BEL (Fig. 6C) with lesser inhibition with (S)-BEL, indicating both the presence and increased activity of iPLA2γ in the failing heart. Subsequent highly sensitive Western analyses demonstrated that the calcium-independent phospholipase A2β (iPLA2β), calcium-dependent phospholipase A2α (cPLA2α), and cPLA2β were not detectable in non-failing heart mitochondria (data not shown), whereas the level of a 74-kDa iPLA2γ isoform associated with mitochondria was moderately increased in failing hearts (Fig. S3). Consistent with the results of PLA2 activity assay, mPTP opening in failing myocardial mitochondria was substantially inhibited by the iPLA2γ inhibitor (R)-BEL, whereas (S)-BEL was less effective. Furthermore, a cPLA2α-specific inhibitor did not affect the calcium-induced opening of the mPTP (Fig. 6D). Collectively, these data indicate that a loss of the majority of mitochondrial calcium-dependent PLA2 activity occurs in failing human heart resulting in a decline of protective downstream eicosanoids (e.g. CYP450 products and prostacyclin), whereas iPLA2γ emerges as the predominant mitochondrial phospholipase activity in failing myocardium leading to the production of toxic fatty acids (e.g. palmitic acid by its sn-1 acyl chain specificity (11)) and HETEs predisposing to mPTP opening during heart failure.
Figure 6.
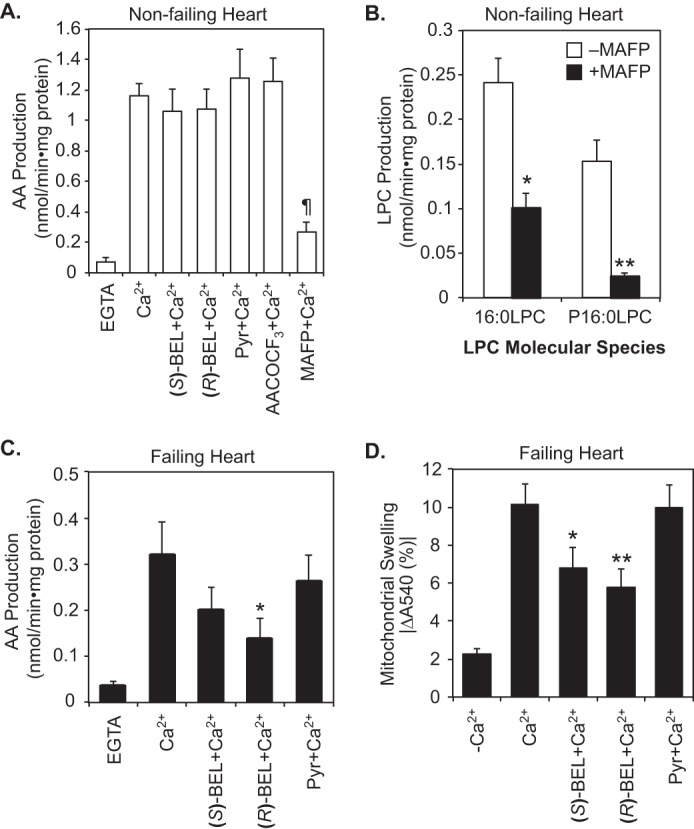
Pharmacological inhibition of PLA2 activities differentiates non-failing myocardial mitochondrial PLA2s from those present in failing human heart. Isolated mitochondria from human non-failing control (n = 5) (A and B) and failing hearts (n = 8) (C) were placed in 10 mm HEPES buffer (pH 7.4) containing 10% glycerol and 1 mm DTT and homogenized by sonication on ice. Mitochondrial homogenates were preincubated with 10 μm (S)-BEL, 10 μm (R)-BEL, 2 μm Pyr, or 10 μm MAFP for 10 min or with 25 μm AACOCF3 for 30 min at 25 °C. Mitochondrial phospholipase reactions were initiated by addition of 2 mm EGTA or 0.6 mm Ca2+ at 35 °C. After 10 min of incubation, the reactions were terminated by addition of chloroform/methanol (1:1, v/v). The lipid products including AA, 16:0-LPC, and 16:0 plasmenyl LPC (P16:0 LPC) were extracted by a modified Bligh-Dyer method in the presence of lipid internal standards and quantified by mass spectrometry. ¶, p < 10−5 in A when compared with Ca2+ treatment. *, p < 0.01, and **, p < 0.001 in B. *, p < 0.05 in C when compared with Ca2+-activated AA release. D, intact mitochondria isolated from human failing hearts (n = 9) were placed in swelling buffer and preincubated with vehicle alone, 10 μm (S)-BEL, 10 μm (R)-BEL, or 2 μm Pyr for 10 min at 25 °C. Mitochondrial swelling was initiated by addition of 10 μm EGTA (−Ca2+) or 70 μm Ca2+. Final decreases in absorbance at 540 nm indicative of mPTP opening after 30 min of incubation are shown as the mean ± S.E. *, p < 0.05, and **, p < 0.01 when compared with Ca2+-induced mitochondrial swelling.
Identification of the predominant calcium-activated phospholipase activity in non-failing human mitochondria as cPLA2ζ by sequential column chromatographies followed by activity-based protein profiling
In aggregate, these results suggested the presence of a previously unidentified phospholipase activity in human myocardial mitochondria that is calcium-sensitive and AA-selective and is present in either greater abundance or possesses a higher specific activity in non-failing relative to failing myocardium. These intriguing observations led us on a campaign to identify this unknown mitochondrial PLA2 in non-failing human hearts. Our strategy employed protein extraction at high pH for solubilization and sequential column chromatographies, including DEAE, chromatofocusing (Mono P), and Mono Q FPLC (Fig. 7A, Fig. S4, and Table 1) followed by activity-based protein profiling using a serine hydrolase inhibitor as probe. The highly purified active fractions collected from the final Mono Q column were loaded onto an SDS-polyacrylamide gel, silver-stained, and either trypsinized in situ or utilized for activity-based protein profiling with desthiobiotin-fluorophosphonate (Fig. 7B). Trypsinolysis of the active protein from the Mono Q column and analysis of the resultant peptides by ESI-nano LC-MS/MS identified multiple signature fragmentation peptides of cPLA2ζ (i.e. 526YGAYVPTELFGSELFMGR543 and 553ICYLQGMWGSAFATSLDEIFLKpTAGpSGLSFLEWpYR587) (Fig. 8). Furthermore, the peptides from trypsinolysis of SDS-polyacrylamide gel (red dotted box in Fig. 7) were also identified as cPLA2ζ fragmentation (i.e. 191GDGpTAPREEYGSRQLQLAVPGAYEK215 and 1MLWALWPRWLADK13) (Fig. S5). These results clearly indicated that cPLA2ζ was the unknown mitochondrial phospholipase in non-failing myocardium responsible for the production of the majority of arachidonate release (Fig. 8). Although the protein that co-chromatographed with the PLA2 activity was not readily detectable by silver staining due to its high specific activity and low abundance, the power of activity-based protein profiling with desthiobiotin labeling followed by biotin immunoblotting confirmed the identity of cPLA2ζ as the major PLA2 in non-failing human myocardial mitochondria.
Figure 7.

Purification of the predominant mitochondrial Ca2+-dependent PLA2 in non-failing human myocardial mitochondria through activity-based protein profiling following multiple chromatographic purification steps. To detect the putative serine hydrolase(s) responsible for the observed PLA2 activity, mitochondria isolated from non-failing human myocardium were resuspended in highly basic buffer (20 mm Na2CO3 (pH 11.4)), and the soluble fraction was collected by ultracentrifugation at 100,000 × g. Next, the mitochondrial extract was applied to sequential DEAE and chromatofocusing Mono P columns as described under “Experimental procedures” (Fig. S4). Fractions with phospholipase A2 activity (identified using [14C]PAPC as substrate) were collected at each chromatographic step for high-resolution separation utilizing a Mono Q FPLC column. A, elution profile of the identified PLA2 activity following Mono Q chromatography as monitored by UV absorbance (280 nm) and release of [14C]arachidonic acid from [14C]PAPC. Active fractions from the Mono Q FPLC column were resolved by SDS-PAGE and visualized by silver staining (B) or were incubated with desthiobiotin-fluorophosphonate for labeling of serine hydrolases prior to separation by SDS-PAGE and subsequent Western blot analysis to visualize resultant biotinylated proteins (C). A protein band was identified in which the intensity of labeling by desthiobiotin-fluorophosphonate correlated well with the observed PLA2 catalytic activity in each fraction (fractions 25–27 in the red dotted box in B). Hsp70, 70-kDa heat shock protein; MCAD, medium-chain acyl-CoA dehydrogenase; SCAD, short-chain acyl-CoA dehydrogenase.
Table 1.
Purification table for the predominant human mitochondrial phospholipase A2
Myocardial mitochondria (200 mg) were isolated from human non-failing human heart, resuspended in 20 mm Na2CO3 buffer (pH 11.4, containing 10% glycerol, 2 mm EGTA, and 2 mm EDTA), and homogenized by brief sonication. Phospholipase A2 activity in the supernatant after ultracentrifugation was purified by multiple chromatographic steps, including DEAE, Mono P chromatofocusing, and Mono Q FPLC through following the release of [14C]AA from [14C]PAPC substrate. It should be noted that the initial yield of crude homogenates assigned as 100% is certainly underestimated due to substantial isotope dilution of the radioactive substrate by endogenous lipids present in the mitochondrial homogenate. The amount of protein in the active fractions from each chromatographic step was determined by a Bradford protein assay with BSA as standard. The details for each protein purification step are described under “Experimental procedures.”
| Mitochondria (200 mg) | Total protein | Total activity | Specific activity | Purification | Yield |
|---|---|---|---|---|---|
| mg | nmol/min | nmol/min·mg | -fold | % | |
| Crude homogenates | 200 | 0.94 | 0.0047 | 1 | 100 |
| pH 11.4 extraction | 62 | 0.53 | 0.0085 | 1.8 | 53 |
| DEAE | 6.8 | 2.9 | 0.43 | 91 | 306 |
| Mono P | 0.5 | 1.3 | 2.7 | 510 | 138 |
| Mono Q | 0.018 | 1.6 | 87 | 18,596 | 170 |
Figure 8.

Identification of the purified human myocardial mitochondrial PLA2 as cPLA2ζ by mass spectrometric proteomic analysis. Active fractions from the Mono Q FPLC column (Fig. 7) were trypsinized, and the resultant peptides were analyzed by mass spectrometry (NanoLC-MS/MS) utilizing a protein sequence database using the SEQUEST algorithm. An excellent correlation between observed and predicted b and y ions for the tryptic peptides corresponding to residues 526–543 (A) and 553–587 (B) of human cPLA2ζ was obtained. Interestingly, multiple phosphorylated residues were identified in the 553–587 peptide. p, phosphorylation; #, oxidation.
Discussion
Myocardial mitochondria undergo marked changes in size, lipid content, lipid second messenger generation, and bioenergetics in animal models after prolonged opening of the mPTP by various pathological stimuli (e.g. ischemia/reperfusion). These changes result from maladaptive calcium dynamics, the dissipation of membrane potential (Δψmt), and the increase utilization of ketone bodies that feed into the tricarboxylic acid cycle at succinate likely increasing the generation of reactive oxygen species (7, 34–36). However, previous reports as well as results from this study did not demonstrate alterations in mitochondrial oxygen consumption, respiratory coupling, or different responses to various respiratory agonists and antagonists in mitochondria from failing human hearts (37, 38). Accordingly, the conclusions from those studies suggested that alterations in mitochondrial function were not responsible for the progression of human heart failure. However, no previous work has investigated alterations in human mitochondrial lipid content, lipid second messenger generation, or the identity and changes in the enzymic mediators responsible for maladaptive lipid signaling upon pathological stimuli (e.g. calcium overload and decreased membrane potential) manifest in heart failure. The present results demonstrate the following: 1) the profound remodeling of Ca2+-dependent mitochondrial phospholipase activities during heart failure from cPLA2ζ to iPLA2γ; 2) the dramatic alterations in lipid second messenger production in failing hearts resulting in the generation of toxic HETEs and decreases in salutary EETs; 3) the differential metabolic channeling of AA released by cPLA2ζ in comparison with iPLA2γ in non-failing versus failing myocardial mitochondria; and 4) the ability of HETEs to promote Ca2+-induced mPTP opening further activating iPLA2γ and its production of toxic HETEs in an autoamplification process (Fig. 9). Collectively, these results are the first to identify human mitochondrial Ca2+-activated phospholipases as mediators of alterations in the metabolic channeling of AA to discrete oxidized metabolites during heart failure and the bidirectional roles of oxidized arachidonic acid in either the protection or damage to human mitochondria challenged by calcium.
Figure 9.

Schematic comparison of phospholipases responsible for Ca2+-activated arachidonic acid release and its differential downstream eicosanoid metabolites in non-failing versus failing human myocardium mitochondria. The predominant PLA2 activity in non-failing human heart mitochondria is from cPLA2ζ, which releases AA that is channeled to cardioprotective EET production. In stark contrast, iPLA2γ in failing heart mitochondria is a major PLA2 activity responsible for the dramatic increase in toxic HETEs upon calcium challenge facilitating mPTP opening. Deuterated eicosanoid metabolite profiles generated by the oxidation of exogenous d8-AA did not differ significantly in non-failing versus failing myocardial mitochondria suggesting the presence of differential metabolic channeling of endogenously released AA mediated by pathological remodeling of mitochondrial PLA2 isoforms. P450, cytochrome P450.
Alterations in the amplitude and duration of calcium transients in failing myocardium/myocytes have been found to be elevated during diastole, decreased during systole, and prolonged during diastolic relaxation in animal models (39, 40). The delay in the relaxation phase has been correlated with deficient calcium uptake due to decreases in sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) expression and activity as well as decreased phosphorylation of phospholamban during heart failure (39, 40). The resultant alterations in calcium uptake and delayed sequestration during heart failure likely predispose heart mitochondria to mPTP opening through Ca2+ activation of mitochondrial phospholipase(s) and subsequent production of cardiotoxic lipid second messengers. Consistent with this notion, our recent findings have demonstrated that murine myocardial mitochondrial iPLA2γ is activated by calcium and that iPLA2γ loss of function in cardiac myocytes markedly attenuates Ca2+-induced mPTP opening, generation of proinflammatory oxidized lipid metabolites (e.g. eicosanoids, docosanoids, and oxidized linoleic acid metabolites), and membrane potential dissipation resulting in reduction of infarct size during cardiac I/R in mice (13, 27).
The cardioprotective effects of CYP450 products and the toxic effects of certain HETEs produced by both CYP450 and LOX enzymes have been studied in animal models of heart failure. For example, EETs and a soluble epoxide hydrolase inhibitor have been demonstrated to reduce infract size during I/R and improve cardiac function by preventing cardiac fibrosis (20, 41, 42). In contrast, cardiac overexpression of 12/15-LOX was reported to result in systolic dysfunction, inflammation, cardiac fibrosis, and macrophage infiltration (43, 44).
The results of this study identify dramatic increases in both the rate of production and steady-state amounts of toxic HETEs in mitochondria from failing human myocardium. As far as we are aware, this study is the first to demonstrate the prominent roles of HETEs in regulating mPTP opening in either experimental animals or in human myocardium after calcium challenge. Our results obtained by utilizing selective pharmacological inhibitors suggest the intriguing possibility that the sensitivity of failing myocardium to stress and its predisposition to necrosis/apoptosis is increased by the up-regulation of Ca2+-activated phospholipase-initiated HETE production at the expense of salutary EETs.
A completely unanticipated finding in this study is the remodeling of mitochondrial phospholipases during heart failure. It was originally thought that AA precursor release for subsequent eicosanoid production in human hearts would be detrimental to cardiac function on multiple levels, including lethal arrhythmias, inflammation, and hemodynamic performance. However, the results of initial rate analyses using stable isotopes in conjunction with HRAM mass spectrometry indicate that the generation of endogenous AA by mitochondrial phospholipase(s) and the resultant downstream eicosanoid metabolites in non-failing hearts are remarkably different from those in human failing hearts. Accordingly, it became important to identify the salutary mitochondrial phospholipase(s) in non-failing hearts. Through sequential chromatographies in conjunction with activity-based protein profiling and high mass accuracy proteomics (45, 46), we quite unexpectedly identified the predominant phospholipase activity associated with non-failing human heart mitochondria as cPLA2ζ. In stark contrast, the predominant mitochondrial phospholipase activity in failing hearts was catalyzed by iPLA2γ generating toxic HETEs which, in combination with a prominent decrease in protective EETs, likely results in alterations in surface charge and potential in the mitochondrial membrane (27, 47). Moreover, previous studies by our group reported the regiospecific PLA1 activity of iPLA2γ with phospholipids containing polyunsaturated fatty acids at the sn-2 position. This results in palmitate release in the plane of the inner membrane. Palmitate has previously been shown to be a potent activator of mPTP opening that, when generated in the plane of the inner membrane, could rapidly diffuse laterally to amplify mPTP opening (11, 27, 48). Thus, a single enzyme, iPLA2γ, has dual regulating pathways for opening of the mPTP, each of which are dependent upon the generation of lipid-signaling molecules (e.g. palmitate and oxidized PUFAs). Furthermore, these results identify inhibition of iPLA2γ as a high value pharmacological target to prevent the progression of heart failure by attenuating cardiac myocyte dropout from necrosis and apoptosis.
A second unexpected outcome of this study is the demonstration that AA generated in human myocardial mitochondria is not due to cPLA2α, which is considered to be the canonical pathway for AA generation as reported in numerous other studies. Instead, AA release in mitochondria from non-failing human myocardium is largely the result of cPLA2ζ that is under tight control to release AA that is channeled into the production of cardiac protective metabolites. These findings are particularly relevant considering that mitochondria represent 30% of myocardial volume, and human mitochondrial phospholipids contain large amounts of esterified AA, thereby emphasizing the important roles of mitochondrial phospholipases in human heart function during health and disease. Although cPLA2ζ from mouse has been cloned (49, 50), its primary structure is substantially different (only 73% homology to the human isoform) and there have not been any studies published to the best of our knowledge on human cPLA2ζ. Critically, failing myocardium has a deficiency in generating or directing the cPLA2ζ-mediated release of arachidonic acid to the synthesis of beneficial eicosanoid metabolites. Furthermore, these results show that the necessary oxygenase enzymes for conversion of AA to salutary eicosanoids products are present in failing human myocardium, but that they cannot be accessed by AA release from phospholipases active in failing human hearts. We note that cPLA2ζ has not been previously reported in human myocardium nor has its utility in the production of salutary eicosanoids been recognized.
Collectively, this study demonstrates the profound remodeling of mitochondrial phospholipase activities and metabolic channeling of arachidonic acid in non-failing versus failing human myocardium resulting in dramatic alterations in eicosanoid second messenger generation and metabolite profiles in failing hearts. Accordingly, myocardial phospholipases are prime targets for pharmacological intervention because they likely initiate a series of cellular events that collectively conspire to accelerate the progression of human heart failure. Considering the activation of iPLA2γ mediated by calcium or membrane potential and its resultant production of toxic HETEs, which open the mPTP, future studies should target the prevention of the activation of human myocardial iPLA2γ and/or the generation of its detrimental downstream toxic products.
Experimental procedures
Reagents
Ibuprofen, baicalein, MSPPOH, AACOCF3, BEL, MAFP, 5-HETE, 12-HETE, 15-HETE, 20-HETE, arachidonic acid, d8-arachidonic acid, TXB2-d4, PGE2-d4, and 12-HETE-d8 were purchased from Cayman Chemical. N-((2S,4R)-4-(Biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl)-3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl]acrylamide (cPLA2α inhibitor, a pyrrolidine derivative) was obtained from EMD Millipore. Desthiobiotin-fluorophosphonate was purchased from Thermo Fisher Scientific. Lipid internal standards, including 17:0-LPC and d4-16:0-fatty acid, were purchased from Avanti Polar Lipids, Inc., and Cambridge Isotope Laboratories, Inc. A rabbit polyclonal antibody directed against human iPLA2γ was generated in our laboratory as described previously (12). Antibodies against biotin, VDAC, ANT, and CypD were obtained from Santa Cruz Biotechnology. Unless otherwise indicated, all reagents for chromatographic protein purification, proteomic analysis, and mitochondrial studies were obtained from Sigma or Thermo Fisher Scientific.
Collection of human non-failing and failing heart tissue
All human tissue was collected with the written informed consent received from participants as a part of an Institutional Review Board- (IRB)approved protocol at Washington University School of Medicine. Clinical variables from a variety of fields were collected on each heart failure patient by the clinical heart failure and heart transplant service as a part of a separate IRB-approved protocol. Human heart tissue was obtained from Mid-America Transplant (St. Louis, MO) and the Washington University Translational Cardiovascular Tissue Core. Tissue from the left ventricular apex of failing human hearts was collected at the time of ventricular assist device (VAD) implantation. Tissue from the left ventricle and left ventricular apex of non-failing human hearts was obtained from organ donors having a heart deemed unsuitable for transplantation due to donor age, epicardial (non-occlusive) coronary vascular disease, or high risk behavioral profile. All tissue was trimmed of excess fat and washed in cold phosphate-buffered saline and then placed in ice-cold mitochondrial isolation buffer or freeze-clamped at the temperature of liquid nitrogen for later mass spectral and metabolic analyses.
Isolation of human heart mitochondria
Human heart tissue from non-failing and VAD (failing) donors was collected in Mitochondrial Isolation Buffer (MIB: 0.21 m mannitol, 70 mm sucrose, 0.1 mm potassium-EDTA, 1 mm EGTA, 10 mm Tris-HCl, 0.5% fatty acid-free BSA (pH 7.4)) at 4 °C. Fat and fibrous tissues were removed prior to extensive washing of the myocardial sample with isolation buffer to remove residual blood. After cutting into small pieces and chopping briefly with a razor blade on ice (4 °C ambient temperature) in mitochondrial isolation buffer, the heart tissue was homogenized using 12–15 passes with a Teflon homogenizer at a rotational speed of 120 rpm. Next, the homogenate was centrifuged for 5 min at 850 × g, and the supernatant was collected and centrifuged at 7200 × g for 10 min. The mitochondrial pellet was resuspended in MIB without BSA and EGTA and then centrifuged again for 7200 × g for 10 min. The resultant pellet was resuspended in either mitochondrial respiration buffer, mitochondrial swelling buffer, or HEPES buffer (for the determination of PLA2 activity).
Mitochondrial swelling assay
Isolated human myocardial mitochondria were placed in Mitochondrial Swelling Buffer (3 mm HEPES (pH 7.4), containing 0.23 m mannitol, 0.07 m sucrose, 5 mm succinate, and 2.5 μm rotenone in the presence or absence of 1 mm KH2PO4) at 4 °C. For experiments examining the effects of PLA2 inhibition or arachidonic acid oxygenase inhibition, mitochondria were pretreated with the following inhibitors: 10 μm (R)- or (S)-BEL ((E)-6-(bromomethylene)-3-(1-naphthalenyl)-2H-tetrahydropyran-2-one), 2 μm pyrrolidine cPLA2α inhibitor, 10 μm baicalein (5,6,7-trihydroxyflavone), 40 μm MSPPOH (N-(methylsulfonyl)-2-(2-propynyloxy)- benzenehexanamide), 20 μm ibuprofen (2-(4-isobutylphenyl)propanoic acid), or dimethyl sulfoxide (DMSO) vehicle alone (0.5% v/v) for 10 min at 23 °C. Mitochondrial swelling was initiated by addition of 70 μm CaCl2 (final concentration) or 10 μm EGTA as control. Decreases in the absorbance at 540 nm indicative of mitochondrial swelling were measured at 15-s intervals using a SpectraMax M5e microplate reader (Molecular Devices, Sunnyvale, CA).
High-resolution mitochondrial respirometry
Human heart tissue collected from control (non-failing) and VAD (failing) donors was placed in ice-cold Mitochondrial Isolation Buffer (MIB: 0.21 m mannitol, 0.070 m sucrose, 0.1 mm K-EDTA, 10 mm Tris-HCl, 1 mm EGTA, 0.5% BSA (pH 7.4)) in a Petri dish on ice. Heart tissue was immediately diced into small millimeter-sized pieces with a razor blade, transferred to a 15-ml Potter-Elvehjem Teflon-pestle glass-mortar tissue grinder with 7 ml of MIB. The tissue was homogenized using a motor-driven pestle operated at 120 rpm. The homogenate was then centrifuged in 10 ml of MIB for 7 min at 850 × g. The supernatant was carefully collected and centrifuged at 10,000 × g for 10 min. The final pellet was resuspended in MIB with no BSA. High-resolution respirometry was performed using a 2-ml chamber OROBOROS® Oxygraph 2K (Innsbruck, Austria). Respiratory measurements were performed at 30 °C utilizing 50 μg of mitochondria in MiRO5 buffer. The rate of oxygen consumption was calculated as a time derivative of oxygen concentration using the DatLab Analysis software (OROBOROS®, Innsbruck, Austria). Respiration was started by the addition of either pyruvate (5 mm)/malate (5 mm), palmitoylcarnitine (20 μm)/malate, glutamate (10 mm)/malate, or pyruvate (5 mm)/glutamate (10 mm)/malate (State 2) followed by sequential addition of ADP (1.25 mm) (State 3), succinate (5 mm) (State 3 Max), rotenone (0.5 μm), oligomycin (2.5 μm) (State 4), N,N,N′,N′-tetramethyl-p-phenylenediamine (5 μm) with ascorbate (0.5 mm), and antimycin A (1 μm). All values reflect the subtraction of residual oxygen consumption after addition of antimycin A. The quantity of mitochondrial protein was determined using a PierceTM BCA protein assay kit according to the manufacturer's instructions.
Determination of phospholipase activity in mitochondrial homogenates
Isolated human myocardial mitochondria placed in HEPES buffer (10 mm HEPES (pH 7.4) containing 1 mm DTT and 10% glycerol) were sonicated for 20 s with a Branson sonicator using 1-s pulses at 30% power. The protein contents of the sonicated mitochondria were then determined using a Bradford protein assay (Bio-Rad) with bovine serum albumin as standard. For experiments examining the effects of PLA2 inhibition, mitochondria were preincubated with the inhibitors (10 μm (R)-BEL, 10 μm (S)-BEL, 25 μm arachidonyl trifluoromethyl ketone (AACOCF3), 10 μm methyl arachidonoyl fluorophosphonate (MAFP), or 2 μm pyrrolidine) or DMSO vehicle alone (1% v/v, final) for 15 min at 23 °C. Mitochondrial phospholipase activity was initiated by adding either 2 mm EGTA or CaCl2 at the indicated concentrations at 35 °C for 10 min, and then reactions were terminated by sequential addition of 2 ml of chloroform/methanol (1:1, v/v), internal standards (d4-16:0 free fatty acid and 17:-0-LPC), and 700 μl of 10 mm LiCl. After vigorous vortexing, samples were centrifuged to promote phase separation, and the chloroform layer was collected and dried under nitrogen stream. The remaining lipids were then re-extracted with 3 ml of chloroform/methanol/water (1:1:1, v/v/v), and those lipids present in the chloroform layer were dried under nitrogen stream. Identification and quantitation of extracted free fatty acid and LPC molecular species were performed in the negative and positive ion modes, respectively, utilizing a TSQ Quantum Ultra mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with an automated nanospray apparatus (Advion Bioscience, Ithaca, NY) as described previously (11). In some cases, fatty acids were also analyzed in positive ion mode after chemical derivatization with AMPP under the same reaction conditions as eicosanoid AMPP derivatization (see below).
Identification and quantitation of eicosanoids by LC-MS/MS
Mitochondrial homogenates in HEPES buffer at the indicated concentrations were incubated in the presence of either 2 mm EGTA or 0.2 mm free Ca2+ for 20 min at 35 °C. The reaction was stopped by addition of methanol (20% v/v final concentration) at 4 °C and diluted with water after addition of internal standards (250 pg each of TXB2-d4, PGE2-d4, and 12-HETE-d8). The solution was immediately applied to a Strata-X solid-phase extraction cartridge preconditioned with 1 ml of methanol followed by 1 ml of 10% methanol. The cartridge was then washed with 1 ml of 5% methanol twice, and the remaining solvent was flushed out with N2 at a pressure of 5 p.s.i. Bound eicosanoids were eluted with 1 ml of methanol containing 0.1% glacial acetic acid. All cartridge steps were carried out using a vacuum manifold attached to a house vacuum line. After evaporation of the organic solvent with a SpeedVac, isolated eicosanoids were derivatized with N-(4-aminomethylphenyl)pyridinium (AMPP), as described previously (28). Briefly, eicosanoids were dissolved in 12.5 μl of ice-cold acetonitrile/N,N-dimethylformamide (4:1, v/v) followed by addition of 12.5 μl of ice-cold 640 mm (3-(dimethylamino)propyl)ethyl carbodiimide hydrochloride in HPLC grade water. Eicosanoid derivatization was initiated by adding 25 μl of 5 mm N-hydroxybenzotriazole and 15 mm AMPP and heating at 60 °C for 30 min. The derivatized eicosanoids were separated by RP-HPLC utilizing a C18 reverse-phase column (Ascentis Express, 2.7-μm particles, 150 × 2.1 mm) at 23 °C using a linear mobile phase gradient with solvent A (0.1% glacial acetic acid in water) and solvent B (0.1% glacial acetic acid in acetonitrile) at a flow rate of 0.2 ml/min. The solvent gradient was programmed as follows: 0.0–5.0 min, 25% B; 5.0–7.0 min, 25–35% B; 7.0–20.0 min, 35–60% B; 20.0–20.1 min, 60–100% B; 20.1–24.0 min, 100% B; 24.0–25.0 min, 100% B to 25% B and 10-min isocratic hold at 25% B (14). The separated derivatized eicosanoids were analyzed after infusion into a hybrid tandem mass spectrometer (LTQ-Orbitrap, Thermo Fisher Scientific) via selected reaction monitoring in the positive ion mode with sheath, auxiliary, and sweep gas flows (arbitrary units) of 30, 5, and 1, respectively. The capillary temperature was set to 275 °C, and the electrospray voltage was 4.1 kV. Capillary voltage and the tube lens voltage were set to 2 and 100 V, respectively. Instrument control and data acquisition were performed using Thermo Xcalibur Version 2.1 software. For stable isotope labeling experiments utilizing d8-arachidonic acid (d8-AA), it is critical to first remove residual oxidized d8-AA from the original stock by reverse-phase HPLC. In addition, mitochondrial phospholipase activity assay described above was performed in the presence of 8 nmol of d8-AA, which was purified by HPLC to remove residual oxidized AA, during the 20-min reaction time. Quantitation of eicosanoids was performed by using either internal standards, including 250 pg each of TXB2-d4, PGE2-d4, or calibration of an external standard such as 250 pg of d8-12-HETE.
Purification of human heart mitochondrial phospholipase A2
Mitochondria isolated from non-failing human hearts were placed in 20 mm Na2CO3 buffer ((pH 11.4) containing 10% glycerol, 2 mm EGTA, and 2 mm EDTA) and sonicated for 20 s with 1-s pulses at 30% power. Insoluble membrane material was removed by ultracentrifugation at 100,000 × g for 1 h, and the soluble fraction was diluted with 20 mm Tris-Cl (pH 7.4), loaded to a 1-ml HiTrap-DEAE (GE Healthcare) column equilibrated with the same buffer and eluted with a stepwise NaCl gradient (0–0.5 m NaCl). Active fractions were identified by a PLA2 assay utilizing l-α-1-palmitoyl-2-arachidonoyl-[arachidonoyl-1-14C]-sn-glycero-3-phosphocholine ([14C]PAPC) (PerkinElmer Life Sciences)/1-(1Z-octadecenyl)-2-oleoyl-sn-glycero-3-phosphoethanolamine (7:5) as substrate. The released [14C]AA was extracted into chloroform using the Bligh-Dyer method and isolated by TLC, and its quantity was determined by liquid scintillation counting. After desalting active fractions using a gel filtration column (Bio-Rad) with Mono P equilibration buffer (20 mm bis-Tris-Cl (pH 6.3) containing 2% glycerol), the active PLA2 was loaded onto a Mono P chromatofocusing column (GE Healthcare) and resolved with a linear gradient from pH 6.0 to 4.0 by utilizing Polybuffer 74 (GE Healthcare). Mono P active fractions determined by radioactive PLA2 assay were diluted with Mono Q equilibration buffer (20 mm Tris-Cl (pH 7.4) containing 10% glycerol), loaded onto Mono Q column (GE Healthcare), and resolved with a linear NaCl gradient (0-0.4 m). Active Mono Q fractions were determined using [14C]PAPC as substrate. The amount of protein in active fractions from each of the chromatographic steps was determined using a Bradford Protein Assay (Bio-Rad) with BSA as standard. For activity-based protein profiling, the fractions from the Mono Q column were incubated with 0.5 μm desthiobiotin-fluorophosphonate for 30 min in the presence of 1 mm Ca2+. Reactions were terminated by addition of SDS-PAGE sample loading buffer. Following electrophoresis, immunoblot analysis was performed to identify biotinylated target proteins.
In-gel digestion of proteins for identification by mass spectrometric analyses
Protein bands visualized with silver staining were excised from the gel, cut into small pieces (∼1 mm3), and destained twice by incubating with a freshly prepared 1:1 (v/v) solution (300 μl) of 30 mm potassium ferricyanide and 100 mm sodium thiosulfate for 8 min. The solution was discarded, and the gel pieces were washed four times with 1 ml of water. After incubation with 100% acetonitrile for 15 min at 23 °C, the solution was discarded. The gel pieces were dried utilizing a SpeedVac, rehydrated in 25 mm ammonium bicarbonate (pH 7.8) containing 12.5 μg/ml trypsin, and then incubated on ice for 45 min. Excess liquid was removed, and 25 mm ammonium bicarbonate (pH 7.8) was added to cover the gel pieces. Following overnight incubation at 37 °C, the supernatant was removed and transferred to a siliconized microcentrifuge tube on ice. Next, 30 μl of 25 mm ammonium bicarbonate (pH 7.8) was added and incubated for 20 min at 23 °C. The supernatant was removed and combined with the supernatant from the previous step. Finally, 30 μl of 50% acetonitrile containing 5% formic acid was added to the gel pieces, which were then incubated for 20 min at 23 °C. The resultant supernatant was combined with the supernatants from the previous steps. This extraction step was repeated once, and the peptide extracts were combined and evaporated to near dryness using a SpeedVac. The peptide extract was reconstituted in 5% acetonitrile containing 0.1% formic acid for analysis by ESI-NanoLC-MS/MS.
ESI-NanoLC-MS/MS analysis
Trypsinized peptide samples were loaded onto a PepMap100 C18 precolumn (300 μm × 1 cm, Dionex, Sunnyvale, CA) by a Surveyor autosampler (Thermo Fisher Scientific, San Jose, CA). The precolumn was then washed with 0.1% formic acid for 5 min. Trypsinized peptides from the precolumn were eluted with a gradient from 95% of mobile phase A (0.1% formic acid in water) and 5% of mobile phase B (0.1% formic acid in acetonitrile) to 10% of A and 90% of B onto a reverse-phase C18 analytical PepMap100 Nano-LC column (75 μm × 15 cm, Dionex, Sunnyvale, CA) at a flow rate of 200 nl/min for 150 min. The eluted peptides were injected into an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) by a TriVersaTM Nanomate system (Advion, Ithaca, NY) at a constant spray voltage of 1.7 kV. The mass spectrometer was operated in a data-dependent acquisition mode. Each full mass scan (from m/z 300 to 1600) in the Orbitrap was followed by tandem mass product ion analysis of the five most intense peaks from the full mass spectrum in the ion trap. The mass spectrometer was calibrated using the manufacturer's recommended positive mode calibration solution containing l-methionyl-arginyl-phenylalanyl-alanine acetate, Ultramark 1621, and caffeine. The mass accuracy was within 5 ppm at mass values from m/z 130 to 2000. Resolving powers (at m/z 400 Th) of 30,000 in full scan mode and 15,000 in MS2 were used. For MS2 product ion analyses, the normalized collision energy of 30% was applied; the activation time was set at 30 ms with an activation parameter q = 0.25, and precursor ions were isolated within a mass window range of 2 Th. The acquired data were searched against a customized protein sequence database using the SEQUEST algorithm. All positive identifications were individually confirmed.
Statistical analyses
A two-tailed Student t test was routinely performed to determine the significance of differences between two groups. A p value of <0.05 was considered significant. All data were reported as average ± S.E.
Author contributions
S. H. M., A. M. C., C. M. J., and R. W. G designed the research. S. H. M., M. A. K., and J. K. routinely collected fresh human heart tissues from the transplantation facility, isolated mitochondria, and managed tissue samples. A. M. C. and S. M. J. analyzed clinical data. S. H. M. performed mitochondrial swelling assay, immunoblotting analysis, phospholipase activity assay using radioactive substrate, and stable isotope experiments. S. H. M. and K. Y. conducted phospholipase activity assay by using mass spectrometry. X. L. analyzed regular and deuterated eicosanoids from human heart tissue and mitochondria by using HRAM mass spectrometry. S. H. M. purified human myocardial mitochondrial phospholipase by utilizing sequential chromatographies followed by activity-based protein profiling, and X. L. performed ESI-NanoLC-MS/MS analyses to obtain proteomic data and identify the purified protein. M. A. K. conducted high-resolution respirometry experiments. S. H. M., C. M. J., and R. W. G contributed to interpretation of experimental data and manuscript preparation. All authors reviewed and approved the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grants RO1HL133178 and RO1HL118639. R. W. G. has financial relationships with LipoSpectrum and Platomics. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S5.
- mPTP
- mitochondrial permeability transition pore
- AA
- arachidonic acid
- HETE
- hydroxyeicosatetraenoic acid
- EET
- epoxyeicosatrienoic acid
- PGE2
- prostaglandin E2
- TXB2
- thromboxane B2
- MAFP
- methyl arachidonyl fluorophosphonate
- iPLA2γ
- calcium-independent phospholipase A2γ
- cPLA2ζ
- calcium-dependent phospholipase A2ζ
- cPLA2α
- calcium-dependent phospholipase A2α
- LOX
- lipoxygenase
- CYP450
- cytochrome P450
- HRAM
- high resolution accurate mass
- ESI
- electrospray ionization
- BEL
- bromoenol lactone
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- AACOCF3
- arachidonyl trifluoromethyl ketone
- Ibu
- ibuprofen
- Baic
- baicalein
- MSPPOH
- N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide
- LPC
- lysophosphatidylcholine
- CypD
- cyclophilin D
- [14C]PAPC
- l-α-1-palmitoyl-2-arachidonoyl-[arachidonoyl-1-14C]-sn-glycero-3-phosphocholine
- SR
- sarcoplasmic reticulum
- ANT
- adenine nucleotide translocase
- VDAC
- voltage-dependent anion channel
- AMPP
- N-(4-aminomethylphenyl)pyridinium
- Pyr
- pyrrolidine
- VAD
- ventricular assist device.
References
- 1. Whelan R. S., Kaplinskiy V., and Kitsis R. N. (2010) Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 72, 19–44 10.1146/annurev.physiol.010908.163111 [DOI] [PubMed] [Google Scholar]
- 2. Kung G., Konstantinidis K., and Kitsis R. N. (2011) Programmed necrosis, not apoptosis, in the heart. Circ. Res. 108, 1017–1036 10.1161/CIRCRESAHA.110.225730 [DOI] [PubMed] [Google Scholar]
- 3. Nakayama H., Chen X., Baines C. P., Klevitsky R., Zhang X., Zhang H., Jaleel N., Chua B. H., Hewett T. E., Robbins J., Houser S. R., and Molkentin J. D. (2007) Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Invest. 117, 2431–2444 10.1172/JCI31060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karamanlidis G., Lee C. F., Garcia-Menendez L., Kolwicz S. C. Jr., Suthammarak W., Gong G., Sedensky M. M., Morgan P. G., Wang W., and Tian R. (2013) Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 18, 239–250 10.1016/j.cmet.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lim S. Y., Hausenloy D. J., Arjun S., Price A. N., Davidson S. M., Lythgoe M. F., and Yellon D. M. (2011) Mitochondrial cyclophilin-D as a potential therapeutic target for post-myocardial infarction heart failure. J. Cell. Mol. Med. 15, 2443–2451 10.1111/j.1582-4934.2010.01235.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rauckhorst A. J., Pfeiffer D. R., and Broekemeier K. M. (2015) The iPLA2γ is identified as the membrane potential sensitive phospholipase in liver mitochondria. FEBS Lett. 589, 2367–2371 10.1016/j.febslet.2015.07.016 [DOI] [PubMed] [Google Scholar]
- 7. Kwong J. Q., and Molkentin J. D. (2015) Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 21, 206–214 10.1016/j.cmet.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tait S. W., and Green D. R. (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632 10.1038/nrm2952 [DOI] [PubMed] [Google Scholar]
- 9. Hazen S. L., Hall C. R., Ford D. A., and Gross R. W. (1993) Isolation of a human myocardial cytosolic phospholipase A2 isoform. Fast atom bombardment mass spectroscopic and reverse-phase high pressure liquid chromatography identification of choline and ethanolamine glycerophospholipid substrates. J. Clin. Invest. 91, 2513–2522 10.1172/JCI116487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hazen S. L., and Gross R. W. (1992) Identification and characterization of human myocardial phospholipase A2 from transplant recipients suffering from end-stage ischemic heart disease. Circ. Res. 70, 486–495 10.1161/01.RES.70.3.486 [DOI] [PubMed] [Google Scholar]
- 11. Yan W., Jenkins C. M., Han X., Mancuso D. J., Sims H. F., Yang K., and Gross R. W. (2005) The highly selective production of 2-arachidonoyl lysophosphatidylcholine catalyzed by purified calcium-independent phospholipase A2γ: identification of a novel enzymatic mediator for the generation of a key branch point intermediate in eicosanoid signaling. J. Biol. Chem. 280, 26669–26679 10.1074/jbc.M502358200 [DOI] [PubMed] [Google Scholar]
- 12. Moon S. H., Jenkins C. M., Kiebish M. A., Sims H. F., Mancuso D. J., and Gross R. W. (2012) Genetic ablation of calcium-independent phospholipase A2γ(iPLA2γ) attenuates calcium-induced opening of the mitochondrial permeability transition pore and resultant cytochrome c release. J. Biol. Chem. 287, 29837–29850 10.1074/jbc.M112.373654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moon S. H., Jenkins C. M., Liu X., Guan S., Mancuso D. J., and Gross R. W. (2012) Activation of mitochondrial calcium-independent phospholipase A2γ (iPLA2γ) by divalent cations mediating arachidonate release and production of downstream eicosanoids. J. Biol. Chem. 287, 14880–14895 10.1074/jbc.M111.336776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu X., Moon S. H., Mancuso D. J., Jenkins C. M., Guan S., Sims H. F., and Gross R. W. (2013) Oxidized fatty acid analysis by charge-switch derivatization, selected reaction monitoring, and accurate mass quantitation. Anal. Biochem. 442, 40–50 10.1016/j.ab.2013.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lopez-Crisosto C., Pennanen C., Vasquez-Trincado C., Morales P. E., Bravo-Sagua R., Quest A. F. G., Chiong M., and Lavandero S. (2017) Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 14, 342–360 10.1038/nrcardio.2017.23 [DOI] [PubMed] [Google Scholar]
- 16. Hasenfuss G., and Pieske B. (2002) Calcium cycling in congestive heart failure. J. Mol. Cell. Cardiol. 34, 951–969 10.1006/jmcc.2002.2037 [DOI] [PubMed] [Google Scholar]
- 17. Imig J. D. (2012) Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol. Rev. 92, 101–130 10.1152/physrev.00021.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lundqvist A., Sandstedt M., Sandstedt J., Wickelgren R., Hansson G. I., Jeppsson A., and Hultén L. M. (2016) The arachidonate 15-lipoxygenase enzyme product 15-HETE is present in heart tissue from patients with ischemic heart disease and enhances clot formation. PLoS ONE 11, e0161629 10.1371/journal.pone.0161629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suzuki H., Kayama Y., Sakamoto M., Iuchi H., Shimizu I., Yoshino T., Katoh D., Nagoshi T., Tojo K., Minamino T., Yoshimura M., and Utsunomiya K. (2015) Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes 64, 618–630 10.2337/db13-1896 [DOI] [PubMed] [Google Scholar]
- 20. Motoki A., Merkel M. J., Packwood W. H., Cao Z., Liu L., Iliff J., Alkayed N. J., and Van Winkle D. M. (2008) Soluble epoxide hydrolase inhibition and gene deletion are protective against myocardial ischemia-reperfusion injury in vivo. Am. J. Physiol. Heart Circ. Physiol. 295, H2128–H2134 10.1152/ajpheart.00428.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gross G. J., Hsu A., Pfeiffer A. W., and Nithipatikom K. (2013) Roles of endothelial nitric oxide synthase (eNOS) and mitochondrial permeability transition pore (MPTP) in epoxyeicosatrienoic acid (EET)-induced cardioprotection against infarction in intact rat hearts. J. Mol. Cell. Cardiol. 59, 20–29 10.1016/j.yjmcc.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hishikari K., Suzuki J., Ogawa M., Isobe K., Takahashi T., Onishi M., Takayama K., and Isobe M. (2009) Pharmacological activation of the prostaglandin E2 receptor EP4 improves cardiac function after myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 81, 123–132 10.1093/cvr/cvn254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frias M. A., Rebsamen M. C., Gerber-Wicht C., and Lang U. (2007) Prostaglandin E2 activates Stat3 in neonatal rat ventricular cardiomyocytes: A role in cardiac hypertrophy. Cardiovasc. Res. 73, 57–65 10.1016/j.cardiores.2006.09.016 [DOI] [PubMed] [Google Scholar]
- 24. Nithipatikom K., Gross E. R., Endsley M. P., Moore J. M., Isbell M. A., Falck J. R., Campbell W. B., and Gross G. J. (2004) Inhibition of cytochrome P450 ω-hydroxylase: a novel endogenous cardioprotective pathway. Circ. Res. 95, e65–e71 10.1161/01.RES.0000146277.62128.6f [DOI] [PubMed] [Google Scholar]
- 25. Nazarewicz R. R., Zenebe W. J., Parihar A., Parihar M. S., Vaccaro M., Rink C., Sen C. K., and Ghafourifar P. (2007) 12(S)-Hydroperoxyeicosatetraenoic acid (12-HETE) increases mitochondrial nitric oxide by increasing intramitochondrial calcium. Arch. Biochem. Biophys. 468, 114–120 10.1016/j.abb.2007.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lv X., Wan J., Yang J., Cheng H., Li Y., Ao Y., and Peng R. (2008) Cytochrome P450 ω-hydroxylase inhibition reduces cardiomyocyte apoptosis via activation of ERK1/2 signaling in rat myocardial ischemia-reperfusion. Eur. J. Pharmacol. 596, 118–126 10.1016/j.ejphar.2008.08.008 [DOI] [PubMed] [Google Scholar]
- 27. Moon S. H., Mancuso D. J., Sims H. F., Liu X., Nguyen A. L., Yang K., Guan S., Dilthey B. G., Jenkins C. M., Weinheimer C. J., Kovacs A., Abendschein D., and Gross R. W. (2016) Cardiac myocyte-specific knock-out of calcium-independent phospholipase A2γ (iPLA2γ) decreases oxidized fatty acids during ischemia/reperfusion and reduces infarct size. J. Biol. Chem. 291, 19687–19700 10.1074/jbc.M116.740597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bollinger J. G., Thompson W., Lai Y., Oslund R. C., Hallstrand T. S., Sadilek M., Turecek F., and Gelb M. H. (2010) Improved sensitivity mass spectrometric detection of eicosanoids by charge reversal derivatization. Anal. Chem. 82, 6790–6796 10.1021/ac100720p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Egan K. M., Lawson J. A., Fries S., Koller B., Rader D. J., Smyth E. M., and Fitzgerald G. A. (2004) COX-2-derived prostacyclin confers atheroprotection on female mice. Science 306, 1954–1957 10.1126/science.1103333 [DOI] [PubMed] [Google Scholar]
- 30. Shinmura K., Tamaki K., Sato T., Ishida H., and Bolli R. (2005) Prostacyclin attenuates oxidative damage of myocytes by opening mitochondrial ATP-sensitive K+ channels via the EP3 receptor. Am. J. Physiol. Heart Circ. Physiol. 288, H2093–H2101 10.1152/ajpheart.01003.2004 [DOI] [PubMed] [Google Scholar]
- 31. Thiemermann C., and Zacharowski K. (2000) Selective activation of E-type prostanoid(3)-receptors reduces myocardial infarct size. A novel insight into the cardioprotective effects of prostaglandins. Pharmacol. Ther. 87, 61–67 10.1016/S0163-7258(00)00069-3 [DOI] [PubMed] [Google Scholar]
- 32. Baines C. P., Kaiser R. A., Purcell N. H., Blair N. S., Osinska H., Hambleton M. A., Brunskill E. W., Sayen M. R., Gottlieb R. A., Dorn G. W., Robbins J., and Molkentin J. D. (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 10.1038/nature03434 [DOI] [PubMed] [Google Scholar]
- 33. Rodriguez-Enriquez S., He L., and Lemasters J. J. (2004) Role of mitochondrial permeability transition pores in mitochondrial autophagy. Int. J. Biochem. Cell Biol. 36, 2463–2472 10.1016/j.biocel.2004.04.009 [DOI] [PubMed] [Google Scholar]
- 34. Lesnefsky E. J., Chen Q., Tandler B., and Hoppel C. L. (2017) Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annu. Rev. Pharmacol. Toxicol. 57, 535–565 10.1146/annurev-pharmtox-010715-103335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hausenloy D. J., and Yellon D. M. (2003) The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion. J. Mol. Cell. Cardiol. 35, 339–341 10.1016/S0022-2828(03)00043-9 [DOI] [PubMed] [Google Scholar]
- 36. Aubert G., Martin O. J., Horton J. L., Lai L., Vega R. B., Leone T. C., Koves T., Gardell S. J., Krüger M., Hoppel C. L., Lewandowski E. D., Crawford P. A., Muoio D. M., and Kelly D. P. (2016) The failing heart relies on ketone bodies as a fuel. Circulation 133, 698–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Holzem K. M., Vinnakota K. C., Ravikumar V. K., Madden E. J., Ewald G. A., Dikranian K., Beard D. A., and Efimov I. R. (2016) Mitochondrial structure and function are not different between nonfailing donor and end-stage failing human hearts. FASEB J. 30, 2698–2707 10.1096/fj.201500118R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cordero-Reyes A. M., Gupte A. A., Youker K. A., Loebe M., Hsueh W. A., Torre-Amione G., Taegtmeyer H., and Hamilton D. J. (2014) Freshly isolated mitochondria from failing human hearts exhibit preserved respiratory function. J. Mol. Cell. Cardiol. 68, 98–105 10.1016/j.yjmcc.2013.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meyer M., Schillinger W., Pieske B., Holubarsch C., Heilmann C., Posival H., Kuwajima G., Mikoshiba K., Just H., and Hasenfuss G. (1995) Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 92, 778–784 10.1161/01.CIR.92.4.778 [DOI] [PubMed] [Google Scholar]
- 40. Schwinger R. H., Böhm M., Schmidt U., Karczewski P., Bavendiek U., Flesch M., Krause E. G., and Erdmann E. (1995) Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca2+-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation 92, 3220–3228 10.1161/01.CIR.92.11.3220 [DOI] [PubMed] [Google Scholar]
- 41. Merabet N., Bellien J., Glevarec E., Nicol L., Lucas D., Remy-Jouet I., Bounoure F., Dreano Y., Wecker D., Thuillez C., and Mulder P. (2012) Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J. Mol. Cell. Cardiol. 52, 660–666 10.1016/j.yjmcc.2011.11.015 [DOI] [PubMed] [Google Scholar]
- 42. Sirish P., Li N., Liu J. Y., Lee K. S., Hwang S. H., Qiu H., Zhao C., Ma S. M., López J. E., Hammock B. D., and Chiamvimonvat N. (2013) Unique mechanistic insights into the beneficial effects of soluble epoxide hydrolase inhibitors in the prevention of cardiac fibrosis. Proc. Natl. Acad. Sci. U.S.A. 110, 5618–5623 10.1073/pnas.1221972110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kayama Y., Minamino T., Toko H., Sakamoto M., Shimizu I., Takahashi H., Okada S., Tateno K., Moriya J., Yokoyama M., Nojima A., Yoshimura M., Egashira K., Aburatani H., and Komuro I. (2009) Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J. Exp. Med. 206, 1565–1574 10.1084/jem.20082596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wen Y., Gu J., Peng X., Zhang G., and Nadler J. (2003) Overexpression of 12-lipoxygenase and cardiac fibroblast hypertrophy. Trends Cardiovasc. Med. 13, 129–136 10.1016/S1050-1738(03)00027-6 [DOI] [PubMed] [Google Scholar]
- 45. Cravatt B. F., Wright A. T., and Kozarich J. W. (2008) Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 77, 383–414 10.1146/annurev.biochem.75.101304.124125 [DOI] [PubMed] [Google Scholar]
- 46. Liu Y., Patricelli M. P., and Cravatt B. F. (1999) Activity-based protein profiling: the serine hydrolases. Proc. Natl. Acad. Sci. U.S.A. 96, 14694–14699 10.1073/pnas.96.26.14694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Broekemeier K. M., and Pfeiffer D. R. (1995) Inhibition of the mitochondrial permeability transition by cyclosporin A during long time frame experiments: relationship between pore opening and the activity of mitochondrial phospholipases. Biochemistry 34, 16440–16449 10.1021/bi00050a027 [DOI] [PubMed] [Google Scholar]
- 48. Sparagna G. C., Hickson-Bick D. L., Buja L. M., and McMillin J. B. (2000) A metabolic role for mitochondria in palmitate-induced cardiac myocyte apoptosis. Am. J. Physiol. Heart Circ. Physiol. 279, H2124–H2132 [DOI] [PubMed] [Google Scholar]
- 49. Ohto T., Uozumi N., Hirabayashi T., and Shimizu T. (2005) Identification of novel cytosolic phospholipase A2s, murine cPLA(2)δ, ϵ, and ζ, which form a gene cluster with cPLA2β. J. Biol. Chem. 280, 24576–24583 10.1074/jbc.M413711200 [DOI] [PubMed] [Google Scholar]
- 50. Ghosh M., Loper R., Ghomashchi F., Tucker D. E., Bonventre J. V., Gelb M. H., and Leslie C. C. (2007) Function, activity, and membrane targeting of cytosolic phospholipase A2ζ in mouse lung fibroblasts. J. Biol. Chem. 282, 11676–11686 10.1074/jbc.M608458200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.