

Abstract
Patients recovering from sepsis have higher rates of CNS morbidities associated with long-lasting impairment of cognitive functions, including neurodegenerative diseases. However, the molecular etiology of these sepsis-induced impairments is unclear. Here, we investigated the role of the receptor for advanced glycation end products (RAGE) in neuroinflammation, neurodegeneration-associated changes, and cognitive dysfunction arising after sepsis recovery. Adult Wistar rats underwent cecal ligation and perforation (CLP), and serum and brain (hippocampus and prefrontal cortex) samples were obtained at days 1, 15, and 30 after the CLP. We examined these samples for systemic and brain inflammation; amyloid-β peptide (Aβ) and Ser-202–phosphorylated Tau (p-TauSer-202) levels; and RAGE, RAGE ligands, and RAGE intracellular signaling. Serum markers associated with the acute proinflammatory phase of sepsis (TNFα, IL-1β, and IL-6) rapidly increased and then progressively decreased during the 30-day period post-CLP, concomitant with a progressive increase in RAGE ligands (S100B, Nϵ-[carboxymethyl]lysine, HSP70, and HMGB1). In the brain, levels of RAGE and Toll-like receptor 4, glial fibrillary acidic protein and neuronal nitric-oxide synthase, and Aβ and p-TauSer-202 also increased during that time. Of note, intracerebral injection of RAGE antibody into the hippocampus at days 15, 17, and 19 post-CLP reduced Aβ and p-TauSer-202 accumulation, Akt/mechanistic target of rapamycin signaling, levels of ionized calcium-binding adapter molecule 1 and glial fibrillary acidic protein, and behavioral deficits associated with cognitive decline. These results indicate that brain RAGE is an essential factor in the pathogenesis of neurological disorders following acute systemic inflammation.
Keywords: amyloid-β (Aβ), neurodegeneration, neuroinflammation, receptor for advanced glycation end products (RAGE), sepsis, Tau protein (Tau)
Introduction
Sepsis is defined as a life-threatening organ dysfunction caused by dysregulated host responses to an infection (1). At admission, up to 71% of septic patients develop potentially irreversible acute cerebral dysfunction (2, 3), a condition caused by systemic inflammation without brain infection and clinically characterized by slowing of mental processes, impaired attention, disorientation, delirium, or coma (4). Recent studies have shown that long-lasting consequences following sepsis recovery often include brain disorders (5). Even after full recovery, animals subjected to sepsis induced by cecal ligation and perforation (CLP)2 demonstrated significant difficulties in performing behavioral tasks, indicating cognitive deficits (6). In recovered patients, persistent deficits in functional abilities and general quality of life are observed (7), which may be associated with long-lasting impairment in cognitive capacities associated with memory and executive function (8).
This long-term impairment in brain function was suggested to result from neurodegenerative or ischemic mechanisms triggered by systemic inflammation (9). Peripherally produced cytokines can enter the central nervous system (CNS), as sepsis is able to induce transient disruption of the blood-brain barrier (BBB) (10, 11). This in turn leads to microglia/astrocyte activation and local production of pro-inflammatory mediators and reactive species (12, 13). However, details of the molecular cascades linking systemic inflammation to neuroinflammation and brain dysfunction still need to be better understood. Comprehension of these mechanisms in detail may reveal valuable information that can be used as a basis to develop new strategies to treat sepsis co-morbidities. In addition, unveiling new details on the molecular events linking systemic inflammation to brain dysfunction may also uncover new insights to the understanding of the onset of neurodegeneration itself (14).
Neurodegenerative processes may evolve over the course of many years, and the diagnosis is generally performed only in advanced or late stages, when brain function is impaired due to significant neuronal loss. Neurodegeneration is characterized by progressive neuronal death associated with the accumulation of misfolded, aberrant forms of cellular proteins or peptides with neurotoxic activity. In Alzheimer's disease (AD), neuronal death occurs concomitant to progressive formation of neurofibrillary tangles and amyloid (senile) plaques. Neurofibrillary tangles are formed due to aberrant hyperphosphorylation of the microtubule-stabilizing protein Tau, whereas amyloid plaques originate by hydrophobic aggregates of misfolded amyloid-β peptide (Aβ). Hyperphosphorylation of Tau occurs in at least 22 other brain conditions, such as amyotrophic lateral sclerosis, Down's syndrome, and prion diseases (15), and because disruption of Tau homeostasis is associated with cognitive deficits (16), modulation of Tau phosphorylation by pathogenic processes may also be associated with the onset of neurodegenerative processes. Similarly, cleavage of the amyloid precursor protein (AβPP) by different secretases may generate small peptides, including a variety of Aβ peptides (17). Necrotic and apoptotic neuronal death is induced by extracellular Aβ both through activation of death receptors in neurons and induction of microglial pro-inflammatory activation and astrocytosis (18). Pro-inflammatory activation, in turn, further induces Aβ aggregation due to amino acid oxidation and peptide destabilization (19).
The receptor for advanced glycation end products (RAGE) is a multiligand pattern-recognition receptor, belonging to the immunoglobulin superfamily of proteins (20). Although it was first described to be activated by advanced glycation end products (AGEs), RAGE can interact with a number of different classes of agonists, including S100 family proteins (e.g. S100B), high-mobility group box protein 1 (HMGB1), 70-kDa heat-shock protein (HSP70), and Nϵ-(carboxymethyl)lysine (CML) (21), among others. RAGE is expressed only by lungs and endothelial cells in a constitutive fashion and is generally repressed in other tissues; however, an increase in the concentration of circulating RAGE ligands induces the expression of this receptor in most cell types (20). RAGE activation triggers different signaling cascades for immune responses, including the ERK1/2-dependent activation of NF-κB and consequent transcriptional activation of pro-inflammatory genes (22), whereas RAGE inhibition was demonstrated to have a protective role against sepsis and LPS-induced endotoxemia (23–25). In the CNS, RAGE expression has been linked to neuroinflammation, Aβ influx through the BBB, and neurodegeneration associated with AD, among other conditions (26, 27). It was previously demonstrated that HMGB1, a RAGE ligand, mediates cognitive dysfunction in sepsis survivors (28) and that RAGE is increased in the brain of rats 30 days after sepsis induction by CLP (29, 30). Following the observation that Aβ binds and activates RAGE (31), RAGE was identified as an important link in the intertwined signaling of inflammatory, amyloidogenic, and pro-apoptotic cascades during the progression of AD, as it maintains a chronic pro-inflammatory state via Aβ-dependent stimulation of microglia (32). Nonetheless, a possible role of RAGE in other CNS disorders, including those associated with pro-inflammatory states, has been relatively neglected.
This study was performed to evaluate whether RAGE is associated with the changes in CNS homeostasis that arise after the recovery of the acute pro-inflammatory phase of sepsis. Therefore, the levels of pro-inflammatory markers, Aβ, phosphorylated Tau, and RAGE-associated molecules (including RAGE, several RAGE ligands, and intracellular downstream targets) were monitored in serum and brain from 24 h to 30 days after CLP surgery. The serum was evaluated for peripheral inflammatory markers and RAGE ligands, and the hippocampus and prefrontal cortex were evaluated because these structures are directly associated with cognitive dysfunctions observed in this rat model (6, 33). RAGE signaling was blocked by immune neutralization in the hippocampus between 15 and 19 days after CLP, and the cognitive function (inhibitory avoidance and object recognition tasks), neuroinflammatory (RAGE, Iba-1, and GFAP), and neurodegenerative markers (Aβ and phosphorylated Tau) were evaluated 30–31 days after CLP. The results presented here indicate the following: (i) RAGE signaling increases as acute pro-inflammatory markers decrease over the 30 days following CLP, in serum, and CNS; and (ii) blocking of RAGE in the hippocampus inhibits neuroinflammatory and neurodegenerative markers in this brain region, as well as cognitive deficits that are observed 30 days after CLP. Overall these data suggest that RAGE signaling in the CNS exerts an important role in the progressive impairment of brain function that arises after the recovery from the acute phase of sepsis, and it may be involved in the long-term development of brain dysfunction and neurodegeneration triggered by episodes of acute systemic inflammation, including polymicrobial sepsis.
Results
Neuroinflammation and neurodegeneration markers increase after the acute phase of sepsis
Increased levels of IL-1β, TNF-α, and IL-6 were observed in serum 24 h after sepsis induction (Fig. 1A). Fifteen days after CLP, IL-1β and TNF-α were still increased, but no differences were detected between sham and CLP animals 30 days after surgery. These results confirmed that CLP induced an acute increase in systemic pro-inflammatory mediators and that the differences in cytokine levels between sham-operated and animals undergoing CLP decreased with time. As animals subjected to CLP often present with brain dysfunction after full recovery from inflammation, biochemical markers of neuroinflammation in hippocampus and prefrontal cortex were evaluated at early and late periods after sepsis induction. One day after CLP, IL-1β, IL-6, and TNF-α levels were increased in the hippocampus (Fig. 1B) and the prefrontal cortex (Fig. 1C). Fifteen days after CLP, the levels of all cytokines were still increased, but 30 days after CLP, TNF-α had returned to control values in the hippocampus, whereas IL-1β and IL-6 levels returned to values similar to sham-operated animals in the prefrontal cortex (Fig. 1, B and C). Toll-like receptor 4 (TLR4), a membrane receptor important in the modulation of pro-inflammatory responses, increased in both brain structures only at 30 days after CLP (Fig. 2, A and D). A similar profile was observed with the content of GFAP, a marker of astrocyte activation (Fig. 2, B and E). However, the neuronal nitric-oxide synthase (nNOS) content did not change in the hippocampus (Fig. 2C), although it increased in the prefrontal cortex 15 and 30 days after CLP (Fig. 2F).
Figure 1.

Content of pro-inflammatory cytokines in serum, hippocampus, and prefrontal cortex at 1, 15, and 30 days after CLP. The content of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α was assessed by ELISA in serum (A), hippocampus (B), and prefrontal cortex (C). Values represent relative quantification considering control (sham group) as 100%. Scattered individual data points (n = 6) and standard deviations are represented. Differences between sham and CLP groups on each day were considered significant when p < 0.05 according Student's t test (two-tailed) analysis (*, p < 0.05, and **, p < 0.001).
Figure 2.

Content of pro-inflammatory markers in hippocampus and prefrontal cortex at 1, 15, and 30 days after CLP. TLR4, GFAP, and nNOS protein levels in hippocampus (A–C, respectively) and prefrontal cortex (D–F, respectively) were evaluated by Western blotting. Scattered individual data points (n = 6) and standard deviation are represented. Representative Western blots are demonstrated with graphs. Differences between sham and CLP groups in each day were considered significant when p < 0.05 according to Student's t test (two-tailed) analysis. Individual p values are depicted when differences were detected.
Increased formation of Aβ from AβPP cleavage and aberrant phosphorylation of the microtubule-stabilizing protein Tau are key events leading to the formation of amyloid plaques and neurofibrillary tangles, respectively. In the hippocampus, increased Aβ immunodetection and enhanced Tau phosphorylation were observed only at 30 days after CLP (Fig. 3, A and B, respectively). In the prefrontal cortex, Aβ immunocontent increased 15 and 30 days after CLP (Fig. 3C), and Tau phosphorylation at Ser-202 transiently increased 15 days after CLP and then returned to basal levels at 30 days (Fig. 3D). Confirming this observation, immunofluorescence visualization of these markers 30 days after CLP showed increased immunostaining of Aβ and phospho-Tau in the hippocampus (Fig. 3E) and enhanced immunostaining of Aβ as well as remnant presence of phospho-Tau in the prefrontal cortex (Fig. 3F).
Figure 3.

Content of Aβ and phosphorylated Tau in hippocampus and prefrontal cortex of animals at 1, 15, and 30 days after CLP. The levels of Aβ and Tau phosphorylated at Ser-202 (p-TauSer-202) in hippocampus (A and B, respectively) and prefrontal cortex (C and D, respectively) were evaluated by Western blotting. Scattered individual data points (n = 6) and standard deviation are represented. Representative Western blots are demonstrated. Differences between sham and CLP groups in each day were considered significant when p < 0.05 according to Student's t test (two-tailed) analysis; individual p values are depicted. Immunofluorescence-based visualization of Aβ and p-TauSer-202 was performed in hippocampus (E) and prefrontal cortex (F) samples of animals 30 days after CLP. DAPI was used for nuclear staining. Magnification bar length is 100 μm. Insets show staining details.
Circulating RAGE ligands and brain RAGE increase as animals recover from CLP
To evaluate a possible relationship between RAGE signaling and brain function impairment in sepsis, the content of several biochemical markers associated with RAGE was assessed. The content of various RAGE ligands (CML, HMGB1, HSP70, and S100B) was determined in the serum (Fig. 4A). CML levels increased in all periods after CLP; HMGB1 increased 15 and 30 days after CLP, whereas HSP70 levels were increased only at 30 days. No differences in S100B levels were detected. Levels of the soluble form of RAGE (sRAGE) in the serum were analyzed, as RAGE shedding and release off the cell membrane is an important regulatory mechanism of signaling termination. No differences in serum sRAGE levels were detected at any period after CLP (Fig. 4A). The content of RAGE ligands and RAGE was also evaluated in the brain. In the hippocampus, CML levels were unaltered, whereas HMGB1 and HSP70 levels decreased 15 days after CLP (Fig. 4B). In the prefrontal cortex, CML levels were increased only 1 day after CLP (Fig. 4C); HMGB1 and HSP70 levels were decreased 15 days after CLP, but no differences were observed at other periods (Fig. 4C). The content of RAGE in both structures increased at 15 and 30 days after CLP (Fig. 4, B and C).
Figure 4.

RAGE ligands, sRAGE, and RAGE in serum and brain. The circulating content of the RAGE ligands S100B, CML, HSP70, and HMGB1 and of the sRAGE isoform in serum was assessed by ELISA (A). Values represent relative quantification considering control (sham group) as 100%. The immunocontent of CML, HMGB1, HSP70, and RAGE in hippocampus (B) and prefrontal cortex C) was assessed by Western blotting. Representative Western blots are demonstrated. Scattered individual data points (n = 6) and standard deviation are represented for all data. Differences between sham and CLP groups were considered significant when p < 0.05 according to Student's t test (two-tailed) analysis (*, p < 0.05, and **, p < 0.001).
Hippocampal RAGE antibody injection inhibits neuroinflammation and neurodegeneration markers
The levels of circulating RAGE ligands and brain RAGE are more prominent after the acute phase of sepsis, when most pro-inflammatory markers are already decreased or declining to levels similar to sham-operated animals. In this context, the role of RAGE in changes observed in the brain 30 days after CLP was investigated by selective blocking of RAGE in the hippocampus with anti-RAGE antibody (RAGEab, 100 μg/kg). RAGEab was administered via cannula consecutively at days 15, 17, and 19 after CLP. At 30 days after CLP, the endogenous content of RAGE in the hippocampus decreased in CLP-subjected animals receiving RAGEab as visualized by immunofluorescence microscopy (Fig. 5A). Besides, immunolocalization of Iba-1 (Fig. 5B) and GFAP (Fig. 5C) indicated that RAGEab administration inhibited CLP-induced microglial and astrocyte activation, respectively. The increases in hippocampal Aβ immunostaining (Fig. 5D) and Tau phosphorylation (Fig. 5E) observed 30 days after CLP were also inhibited by RAGEab administration to hippocampus. Quantification of fluorescence intensity of RAGE, Iba-1, GFAP, Aβ, and p-Tau immunostaining and statistical analysis confirmed these observations (Table 1).
Figure 5.

Effects of hippocampal RAGEab injection over RAGE and markers of neuroinflammation and neurodegeneration in hippocampus of animals submitted to CLP. RAGEab was administered bilaterally into the hippocampus at 100 μg/kg at days 15, 17, and 19 after CLP. Control animals received 100 μg/kg of isotype IgG. At day 30 after CLP, the hippocampus was prepared for immunofluorescence detection of RAGE (A), Iba-1 (B), GFAP (C), Aβ (D), and phospho-Tau (E). DAPI was used for nuclear staining. Magnification bar length is 400 μm in the bigger panel and 100 μm in all other panels.
Table 1.
Values of fluorescence immunostaining intensity (fold-induction)
Mean ± S.D. values of fluorescence intensity units from eight animals per group. ****, p < 0.0001, ***, p < 0.001, and **, p < 0.01 were compared with sham group. ####, p < 0.0001, ###, p < 0.001, and ##, p < 0.01 were compared with CLP group. One-way ANOVA was done with Tukey's post hoc test.
| Hippocampus |
Prefrontal cortex |
|||||
|---|---|---|---|---|---|---|
| Sham | CLP | CLP + RAGEab | Sham | CLP | CLP + RAGEab | |
| RAGE | 1 ± 0.44 | 28.5 ± 5.29**** | 2.54 ± 1.37#### | 1 ± 1.02 | 35.39 ± 11.7**** | 4.25 ± 2.51#### |
| Iba-1 | 1 ± 0.51 | 28.2 ± 2.02**** | 7.64 ± 1.82#### | 1 ± 0.97 | 4.97 ± 2.39** | 1.39 ± 0.67## |
| GFAP | 1 ± 0.28 | 8.49 ± 2.95**** | 1.58 ± 0.14#### | 1 ± 0.48 | 17.9 ± 6.34**** | 2.15 ± 0.68#### |
| Aβ | 1 ± 0.30 | 53.08 ± 10.91** | 17.63 ± 5.18#### | 1 ± 0.98 | 28.17 ± 13.83*** | 10.12 ± 5.01## |
| p-Tau | 1 ± 0.14 | 6.08 ± 1.45** | 2.97 ± 0.85### | 1 ± 0.57 | 0.99 ± 0.64 | 0.65 ± 0.38 |
| NeuN | 1 ± 0.10 | 0.53 ± 0.11**** | 1.01 ± 0.15#### | 1 ± 0.31 | 0.55 ± 0.19** | 1.18 ± 0.20### |
Interestingly, hippocampal RAGEab administration also had effects in the prefrontal cortex. Mean values and statistical analysis of fluorescence quantification in hippocampus are shown in Table 1. The increase in RAGE induced by CLP was inhibited in prefrontal cortex of animals treated with RAGEab, as observed by fluorescent immunolocalization (Fig. 6A). The number of cells with positive staining for both Iba-1 (Fig. 6B) and GFAP (Fig. 6C) was also decreased by hippocampal RAGE inhibition, and a similar effect was observed with Aβ immunostaining (Fig. 6D). No significant effect was observed on Tau phosphorylation, as this parameter was not significantly altered in the prefrontal cortex 30 days after CLP (Fig. 6E). To further confirm this observation, a double staining was performed with the neuronal marker NeuN. Hippocampal Tau phosphorylation is possible to observe at the subcellular level and at different magnifications along with NeuN-positive neurons. Distribution of p-Tau and NeuN staining within cells varies according to the irradiation pattern of neuritis/axons from the perinuclear area, which is the site of NeuN expression (Fig. 7A). In the prefrontal cortex, however, staining for phosphorylated Tau is not observed, although NeuN staining suggests a decrease in the number of neurons at both structures in the CLP group, which is rescued by RAGEab (Fig. 7B and Table 1).
Figure 6.

Effects of hippocampal RAGEab injection in prefrontal cortex RAGE and markers of neuroinflammation and neurodegeneration in animals submitted to CLP. RAGEab was administered bilaterally into the hippocampus at 100 μg/kg at days 15, 17, and 19 after CLP. Control animals received 100 μg/kg of isotype IgG. At day 30 after CLP, the prefrontal cortex was prepared for immunofluorescence detection of RAGE (A), Iba-1 (B), GFAP (C), Aβ (D), and phospho-Tau (E). DAPI was used for nuclear staining. Magnification bar length is 100 μm.
Figure 7.

Effects of hippocampal RAGEab injection over phospho-Tau and NeuN staining in hippocampus and prefrontal cortex of animals submitted to CLP. RAGEab was administered bilaterally into the hippocampus at 100 μg/kg at days 15, 17, and 19 after CLP. Control animals received 100 μg/kg isotype IgG. At day 30 after CLP, the hippocampus (A) and the prefrontal cortex (B) were prepared for immunofluorescence detection of phospho-Tau (red) and the neuron nuclear marker NeuN (green). DAPI was used for nuclear staining. Magnification bar length in hippocampus: left panels, 1000 μm; central panels, 200 μm; and right panels, 100 μm. In prefrontal cortex panels, the magnification bar length is 100 μm.
Aβ and phosphorylated Tau immunofluorescence was observed in isolated cells to identify morphological staining patterns that could be associated with extracellular deposition or intracellular accumulation. Staining of Aβ antibodies in isolated neurons of hippocampus and prefrontal cortex from CLP and CLP + RAGEab groups resembles neuronal bodies and neurite projections (Fig. 8, A and B). Plaque-like structures were not observed. A similar examination was performed with phosphorylated Tau with double staining for NeuN. The morphological pattern displayed at a cellular level in hippocampus does not resemble extracellular deposition of structures such as neurofibrillary tangles, but they are suggestive of intracellular fibrillary constitution (Fig. 9A). Neurons of the prefrontal cortex, in contrast, do not present p-Tau reactivity, as observed above (Fig. 9B).
Figure 8.

Morphological observation of Aβ immunofluorescence staining in hippocampus and prefrontal cortex 30 days after CLP. Sections of tissues from CLP and CLP + RAGEab groups are shown at left panels (magnification bar length is 100 μm). White arrows indicate isolated cells detailed in augmented visualization. DAPI-merged and isolated Aβ stainings are compared. Morphology is indicative of intraneuronal localization of Aβ. Letters and numbers in upper left of images identify panels that originated the detailed images of isolated cells.
Figure 9.

Morphological observation of phospho-Tau and NeuN immunofluorescence double staining in hippocampus and prefrontal cortex 30 days after CLP. Sections of tissues from CLP group are shown at upper left panels (magnification bar length is 100 μm). Costaining with phospho-Tau (red) and the neuronal nuclear marker NeuN (green) are used to confirm neuronal localization of phospho-Tau. Details of highlighted areas are compared for each type of staining, including DAPI and merged images. Morphology is indicative of intraneuronal localization of filamentous phospho-Tau deposition in hippocampus. Prefrontal cortex did not show phospho-Tau staining, confirming Western blotting and immunofluorescence data for 30 days post-CLP animals. Letters and numbers in upper left of images identify panels that originated the detailed images of isolated cells.
Long-term effects of RAGE in brain involves modulation of Akt and mTOR
RAGE and TLR4 modulate inflammatory and neurodegenerative responses through activation of specific protein kinase cascades. The phosphorylation of ERK1/2 and Akt, which participate in different signal cascades evoked by RAGE and TLR4, was verified. In the hippocampus, ERK1/2 phosphorylation was increased 24 h and 15 days after CLP but returned to levels similar to sham animals 30 days after CLP (Fig. 10A). In the prefrontal cortex, ERK1/2 phosphorylation was increased 24 h after CLP but returned to basal levels at later periods (Fig. 10D). In contrast, Akt had an opposite profile, presenting increased phosphorylation only 15 and 30 days after CLP in both structures (Fig. 10, B and E). Considering this profile of transient activation of ERK1/2 and late sustained activation of Akt, the phosphorylation state of Akt downstream target mTOR was analyzed. Phosphorylation of mTOR in the hippocampus was significantly decreased compared with sham animals at 30 days after CLP, with no alterations at earlier times (Fig. 10C). In the prefrontal cortex, mTOR phosphorylation was increased 1 day after CLP but displayed an opposite profile at 15 and 30 days following CLP, presenting decreased phosphorylation compared with sham animals (Fig. 10F). These results suggest that RAGE signaling is associated with Akt phosphorylation and mTOR dephosphorylation, as these effects occurred concomitantly after the acute inflammatory phase of sepsis.
Figure 10.

ERK1/2, Akt, and mTOR phosphorylation in hippocampus and prefrontal cortex of animals at 1, 15, and 30 days after CLP. The phosphorylated levels of hippocampal ERK1/2 (A), Akt (B), and mTOR (C), and the phosphorylated levels of prefrontal cortex ERK1/2 (D), Akt (E), and mTOR (F) relative to their total respective protein levels were assessed by Western blotting. Scattered individual data points (n = 6) and standard deviation are represented for all data. Representative Western blots are demonstrated. Differences between sham and CLP groups in each day were considered significant when p < 0.05 according student's t test (two-tailed) analysis; individual p values are depicted.
To confirm this hypothesis, the effect of hippocampal RAGE inhibition over Akt and mTOR phosphorylation states 30 days after CLP in both structures was analyzed. RAGEab treatment to the hippocampus significantly inhibited the increase in Akt phosphorylation (Fig. 11A) and reversed the inhibition of mTOR phosphorylation (Fig. 11B) observed 30 days after CLP. Furthermore, quantification of RAGE levels in these same samples confirmed that RAGEab injection to the hippocampus down-regulated RAGE protein levels in this structure (Fig. 11C), suggesting a link for RAGE, Akt, and mTOR. The immunocontent of Aβ and phosphorylated Tau was also assessed in these samples (Fig. 11, D and E, respectively). Hippocampal RAGEab administration also affected RAGE signaling in the prefrontal cortex. Changes in prefrontal cortex Akt and mTOR observed in the CLP group were significantly reversed by RAGEab injection into the hippocampus (Fig. 11, F and G, respectively). Similarly, levels of RAGE in these samples were significantly inhibited by RAGEab administration (Fig. 11H). Finally, the effect of hippocampal RAGEab injection on Aβ immunodetection (Fig. 11I) and phosphorylated Tau (Fig. 11J) levels also repeated the pattern seen in the prefrontal cortex immunolocalization.
Figure 11.

Akt and mTOR phosphorylation are associated with RAGE and markers of neurodegeneration in brain. RAGEab was administered bilaterally into the hippocampus at 100 μg/kg at days 15, 17, and 19 after CLP. Hippocampus was isolated, and the levels of phosphorylated Akt (A) and mTOR (B), RAGE (C), Aβ (D), and phosphorylated Tau (E) 30 days after CLP were analyzed by Western blotting. Similarly, in isolated prefrontal cortex, phosphorylated Akt (F) and mTOR (G), RAGE (H), Aβ (I), and phosphorylated Tau (J) were assessed. Scattered individual data points (n = 6) and standard deviation are represented for all data. Representative Western blottings are demonstrated. Differences between groups were considered significant when p < 0.05 according to one-way ANOVA with Tukey's post hoc test; individual p values are depicted.
Hippocampal RAGEab injection restores impaired cognitive performance
Finally, the effect of hippocampal RAGE immune neutralization in cognitive performance was evaluated 30 days after CLP. Sepsis induces a significant impairment in memory retention tasks, and animals treated with RAGEab administration had performance similar to control animals (Fig. 12, A and B). These results, altogether, suggest that administration of RAGEab into hippocampus is able to restore some of the alterations in brain function that are observed after recovery from the acute phase of sepsis.
Figure 12.
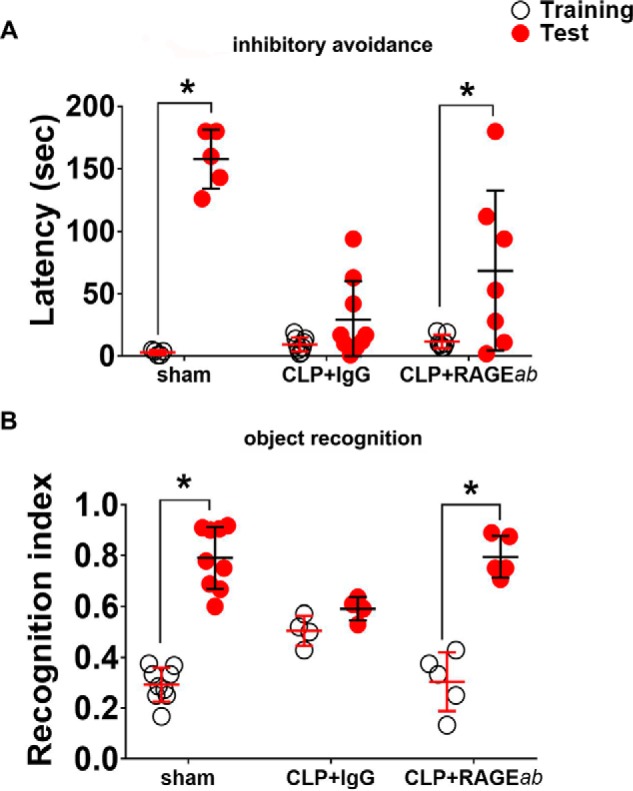
Effect of RAGEab injection into hippocampus over cognitive tests in animals subjected to CLP. RAGEab was administered bilaterally into the hippocampus at 100 μg/kg at days 15, 17, and 19 after CLP. For inhibitory avoidance tasks (A), training sessions were performed at day 30 after surgery. Test sessions were carried out 24 h after training, and the step-down latency was used as a measure of retention. For object recognition task (B), training at day 30 after CLP was conducted by placing rats in the field with two identical objects (objects A1 and A2). Twenty four hours later, animals were allowed to explore the field in the presence of the familiar object A and a novel object C. A recognition index was calculated as the ratio TB/(TA + TB), with TA = time spent exploring the familiar object A; TB = time spent exploring the novel object B. Scattered individual data points with mean and standard deviation from animals from two different experiments are represented; comparisons among groups were performed using the Mann-Whitney U test. For behavioral analyses, individual groups were compared by the Wilcoxon tests. Animals that presented unusual locomotor activity or any other sign of altered behavior during training sessions were excluded from tests. Differences were considered significant when p < 0.05 (*).
Discussion
In this study, animals surviving sepsis presented brain alterations commonly associated with the onset of neurodegenerative processes, and RAGE was demonstrated to play a key role in the progression of these changes. Sepsis enhances transcription of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6 (4), which affect the arrangement of tight junctions in BBB endothelial cells (34–36). This increases BBB permeability, contributing to neuroinflammation and cell death (11). The present model of polymicrobial sepsis induced by CLP requires the use of antibiotics to maintain survival around 40%. However, the antibiotic therapy, as observed here and in previous works, does not prevent the progression of sepsis, although it significantly enhances survival (37, 38). The variation in pro-inflammatory cytokines at the systemic level presented a typical pattern here. Pro-inflammatory cytokines may accumulate in brain due to saturable influx transport, retrograde axonal transport systems, or simple diffusion in areas where BBB is impaired. A similar pattern was observed here, as brain pro-inflammatory markers increased, systemic cytokines decreased following acute inflammation recovery. All cytokines were elevated in the brain 24 h after CLP and decreased with time, except TNF-α in the prefrontal cortex (which peaked at 15 days and then decreased). In contrast, alterations in TLR4, GFAP, and nNOS, which are markers associated with inflammatory activation in specific cells, were not observed earlier than 15 days after CLP. Indeed, TLR4 and GFAP levels were altered only 30 days after CLP. These data unveil a bimodal profile of inflammation in the CNS following sepsis, with a more prominent role of pro-inflammatory cytokines at earlier stages, probably resulting from transient BBB disruption during the acute phase of sepsis, followed by resident activation of local cells as BBB is restored and animals recover from acute inflammation. As mentioned above, BBB is disrupted during the acute phase of sepsis, but it restores its integrity and selective permeability as animals recover from acute inflammation (11, 39).
Previous work has demonstrated that CLP induces microglial activation, and this is associated with long-term cognitive dysfunction caused by sepsis (13). A similar profile was observed here for astrocytes, as GFAP levels increased late after CLP. Altogether, these observations suggest that the glia exert an important role in the chronic neuroinflammation and long-term impairments in brain function observed after sepsis recovery. In this context, the late increase in TLR4 levels (30 days after CLP) may be associated with late astrocyte/microglial activation instead of polymicrobial-dependent up-regulation. TLR4 triggers production of cytokines, nitric oxide (NO), and reactive oxygen species in microglia and astrocytes (40). Abnormal Tau phosphorylation is associated with several neurodegenerative conditions, including AD and other tauopathies where formation of highly dense hydrophobic aggregates of misfolded proteins causes neuronal death (41). Tau presents at least 85 potential sites of phosphorylation, which are regulated by several different protein kinases and phosphatases. The specific phosphorylation of some of these sites has been associated with particular physiological processes, whereas the overstimulation of some sites has been suggested to be more critical for the induction of abnormal aggregation (42–44). Abnormal stimulation of Tau Ser-202 phosphorylation correlates with AD in adult humans and is associated with neurofibrillary tangle formation (43). The involvement of aberrant Tau phosphorylation in brain function impairment following sepsis is consistent with previous observations, including progressive decline in cognitive functions and oxidative damage to CNS (45, 46). RAGE expression in the adult CNS is more often associated with microglia and astrocytes, although it may be induced in neurons as well. The mechanistic relationship between RAGE and Tau phosphorylation in the brain may involve RAGE up-regulation as a consequence of microglia and astrocyte activation, release of pro-inflammatory mediators to the extracellular milieu, and consequent activation of neurotoxic pathways in neurons, including GSK-3β- and CDK5-regulated cascades that participate in Tau phosphorylation (47). This hypothesis is supported by our finding that in the hippocampus RAGE up-regulation precedes Tau phosphorylation at 15 days.
The immunostaining profile of phosphorylated Tau in the hippocampus is indicative of intracellular sublocalization, whereas no evident formation of tangles was detected. Extracellular accumulation of neurofibrillary tangles is characteristically observed in post-mortem analyses of AD brains, being normally associated with late stages of neurodegeneration. However, Tau abnormal phosphorylation, aggregation, and proteolysis constitute earlier steps in the formation of neurofibrillary tangles, being crucial events in the pathogenesis of AD and other sporadic tauopathies (48, 49). It is postulated that the process of Tau abnormal phosphorylation and aggregation in tauopathies follows a progression through three stages: pre-tangle formation, when soluble oligomers and small aggregates are observed as intracellular inclusions; intracellular neurofibrillary tangle stage, when it is possible to observe phosphorylated Tau in the form of intracellular filaments; and extraneuronal neurofibrillary tangles stage, when neurons that originated the processes are no longer viable (48). Our observations are suggestive that the first two stages are taking place in hippocampus. In prefrontal cortex, however, Tau phosphorylation had already returned to basal levels 30 days after CLP, although NeuN staining suggests a decreased number of neurons.
The progressive accumulation of Aβ is also consistent with this scenario. AβPP metabolism is normally accelerated in AD and related diseases and may lead to Aβ generation (50). However, the amyloid plaques observed in pathological analysis of AD brains are believed to take decades to form (51). Accumulation of Aβ in amyloid plaques is affected by transcriptional regulation of AβPP, modifications in expression and/or activity of secretases involved in AβPP cleavage, and oxidative damage resulting from glial activation (19). Interestingly, secretases responsible for AβPP cleavage also act in RAGE shedding (52, 53), indicating that RAGE and Aβ share important regulatory steps, and their homeostasis may be disrupted by common mechanisms. RAGE is also a major regulator of systemic Aβ translocation into CNS through BBB (54), and Aβ-RAGE interaction enhances Tau phosphorylation (27). Brain accumulation of Aβ following CLP recovery has been previously associated with long-term cognitive impairment (30). Here, a progressive increase in Aβ immunodetection was observed with time after CLP and RAGE immune neutralization in hippocampus was able to inhibit this effect in both structures at 30 days, suggesting a link between RAGE and Aβ induction. This is in agreement with previous observations showing that RAGE inhibition prevented Aβ production, inflammation, oxidative stress, and cognitive deficits in an AD mouse model and in rats receiving intrahippocampal AGEs (31, 55).
Immunofluorescence images display a staining pattern not indicative of plaque-like structures but an intraneuronal location varying between cell bodies and neurites and, apparently, axons in some cells. Although deposition of Aβ in AD senile plaques is postulated to occur in the course of years, or perhaps decades, the formation, aggregation, and deposition of Aβ in traumatic brain injury (TBI) can be highly accelerated, taking place in a span of hours from an acute episode of trauma (56). The pattern of Aβ staining in hippocampus and prefrontal cortex observed here does not indicate an advanced stage of neurodegeneration commonly associated with the presence of amyloid plaques but intraneuronal accumulation of Aβ or, more likely, its full-length precursor protein, considering the morphological pattern of staining. Early intraneuronal accumulation of Aβ in neurons that are particularly vulnerable in AD were observed in AD brains, as well as Down's syndrome and numerous transgenic mouse models of AD (57). This “pre-plaque” accumulation takes place in intraneuronal endosomal vesicles, reportedly located in cell soma and, at higher extent, distal neurites and synapses (58, 59). Recently, the hypothesis that Aβ extracellular plaque deposits are the remnants of these degenerating neurites and synapses, as well as neuron cell soma (neurites and synapses also exist at neuron cell bodies), has been increasingly supported by detailed studies on subcellular localization of Aβ formation, oligomerization, and subsequent deposition in the progression of neurodegeneration (57). Our results are in accordance with this hypothesis, as Aβ staining in our model seems to follow a pattern of intracellular localization, not associated with plaques and not as extensive as would be expected following an acute TBI episode. Notwithstanding, in non-transgenic Wistar rats, intraneuronal Aβ accumulation in response to neuroinflammation is not usually as high as observed here, especially considering the intensity of the effect and the age of the animals used in this study (3-month-old young adults). In this context, a possible cross-reactivity with full-length AβPP in our immunofluorescence images should not be ruled out. This is a plausible hypothesis to explain why CLP induces this extent of Aβ labeling in young animals at such short times after surgery. Importantly, this possibility does not invalidate the current interpretation of our observations, considering that Aβ has originated from AβPP cleavage and thus a marked increase in AβPP could constitute an earlier step of this process. Although detailed molecular steps of these events remain to be elucidated, the occurrence of augmented Aβ and Tau phosphorylation in the hippocampus and prefrontal cortex, together with previous evidence mentioned above, are highly suggestive of neurotoxic processes that potentially represent early molecular steps of neurodegenerative cascades in the course of activation. Besides, the observation that RAGE immunoneutralization inhibited Aβ immunodetection and Tau phosphorylation represents valuable information concerning the sequence of events by which sepsis and systemic inflammation trigger long-term neurodegenerative processes.
Circulating RAGE ligands were increased in serum, and RAGE was up-regulated in brain structures. These data could indicate that the influx of peripheral RAGE ligands to the CNS is occurring, as sepsis-impaired BBB allows unspecific transport of pro-inflammatory mediators to the brain (10, 11, 60). Although the determination of RAGE ligands in total tissue does not reflect the extracellular content of these molecules in the CNS, they presented little or no variations, corroborating the hypothesis that RAGE in CNS is activated and up-regulated by peripherally-produced ligands and cytokines. As a consequence, intracellular effects triggered downstream RAGE activation that may take place in a similar time frame to RAGE protein up-regulation. Importantly, a correlation between Akt/mTOR and RAGE signaling is observed. Akt phosphorylation in brain is increased concomitantly with the increase in RAGE markers in serum and brain, and this relationship was further reinforced by the observation that RAGEab inhibited Akt phosphorylation. ERK1/2 and Akt are important components in neurodegenerative and inflammatory pathways and cross-talk to counteract, enhance, or suppress signaling by each other (61). In neurodegenerative diseases (especially AD), neuronal ERK1/2 activation is generally associated with GSK3-β cascade regulating Tau phosphorylation, but in glia these kinases are associated with inflammation (26, 62). A ubiquitous consequence of RAGE binding is NF-κB activation through ERK1/2, which in turn activates inflammatory cytokines and RAGE transcription. Alternatively, Akt activation is associated with survival responses, including inhibition of pro-inflammatory activation and autophagic clearance of protein aggregates (63, 64). Nonetheless, both exhibit differential states of basal activity and response to stimuli in different brain areas. Recently, it was observed that ERK1/2- and Akt-mediated pathways share little common regulation in different brain structures of AβPP/PS1 transgenic mice, despite simultaneous dysfunctional processes (65). ERK1/2 phosphorylation was increased in earlier periods and returned to basal levels later; in contrast, Akt phosphorylation exhibited a pattern of progressive increase. These data point to a shift from ERK1/2-mediated pathways, in earlier periods, to Akt signaling in later periods. Also, these results are in accordance with the decrease in acute pro-inflammatory markers observed during CLP recovery. Thus, a scenario where pro-inflammatory cytokines increase in the CNS causing ERK1/2 phosphorylation during the acute phase of sepsis followed by a second phase where RAGE up-regulation evokes Akt-dependent responses as pro-inflammatory cytokines decrease is consistent with our data. In neurodegenerative conditions, mTOR is generally associated with control of autophagy and apoptosis in response to protein misfolding and aggregation. Akt and ERK1/2 affect mTOR by modulating the activation of the mTOR-inhibitory TSC1–TSC2 complex (66). Akt also controls mTOR by phosphorylation (67, 68). Here, mTOR phosphorylation decreased concomitant to stimulation of Akt. It is possible that the late effect of CLP on mTOR phosphorylation in brain is related to inhibitory feedback mechanisms of RAGE involving Akt late phosphorylation. The observation that RAGEab reversed the effects of CLP on mTOR and Akt phosphorylation states after 30 days is consistent with this interpretation. Besides, it is also important to note that HSP70 levels in both structures were decreased 15 days after CLP. HSP70 is a molecular chaperone important for the control of autophagy as its function is directly related to protein misfolding and turnover. Decreased levels of HSP70 could be related to a deficit in autophagic processing, and this, in turn, could contribute to enhance the accumulation of damaged or misfolded proteins.
The time course of events observed here indicates a trend pattern. The decrease in acute systemic inflammation characteristic of sepsis occurs with progressive increase in RAGE-associated markers in serum and brain over time, suggesting a shift from acute inflammation toward chronic neuroinflammation. In different chronic diseases related to organ function degeneration, RAGE expression is elevated and associated with the support of a chronic inflammatory state (20, 69). The results presented here suggest that RAGE signaling increases as the acute pro-inflammatory signaling decreases as animals recover from sepsis. The spatial and temporal differences in the expression of markers associated with acute inflammation, RAGE, and neurodegenerative signaling may be related to the systemic origin of inflammation, which may affect CNS structures at different extents depending on their localization, contact area with blood vessels, and relative constitution of different cell types. However, LPS-induced systemic inflammation was reported to cause loss of neurons in both hippocampus and prefrontal cortex, with substantial loss of cholinergic innervations, resembling a neurodegenerative process; and different types of cognitive deficits were observed in this model (70). Previous works demonstrated several different cognitive deficits in sepsis survivors, and these deficits were associated with the prefrontal cortex and hippocampus inflammation (6, 13, 71). Here, it was demonstrated that blocking RAGE signaling in the hippocampus inhibited long-term cognitive dysfunction. All these data taken together suggest that systemic inflammation induces long-term hippocampal and prefrontal alterations that are somehow similar to a neurodegenerative process. These molecular and structural alterations are linked to a phenotype of cognitive dysfunction that is dependent on hippocampus and prefrontal connections.
Antagonism of RAGE signaling using RAGE antibodies or sequestering RAGE ligands with sRAGE have been previously undertaken in models of inflammation and sepsis. The use of monoclonal, polyclonal, or Fab fragments has been extensively applied as a pharmacological inhibitor was available only very recently. RAGE inhibition by these approaches has a significant impact on sepsis, mainly through attenuation of pro-inflammatory signaling (24). Utilization of Fab fragments or monoclonal antibodies instead of polyclonal antibodies used here could represent a practical advantage as they would not bind to other scavenger receptors sharing similar epitopes. A necessary step for development of a clinical protocol based on immune neutralization of RAGE, in this context, is the rational design of a selective RAGE antibody for human utilization in a clinical trial. Besides, the delivery of an anti-RAGE molecule to specific brain regions should be considered another essential step, which could demand a surgical approach. A clinical trial with a synthetic RAGE inhibitor, PF-04494700 (Azeliragon or TTP488), indicated no consistent effect on plasma levels of Aβ, inflammatory biomarkers, or secondary cognitive outcomes in phase II (72). However, a follow-up examination, conducted after treatment was suspended, suggested a possible clinical benefit for a low dose, but as these data became evident only long after discontinuation of the treatment based on preliminary results, they remain inconclusive (73). Currently, a phase III clinical trial is ongoing for efficacy and safety in mild AD (NCT02080364). Nonetheless, in an experimental context, blocking of RAGE with polyclonal antibodies has been successfully demonstrated in different rodent models of systemic inflammation, and effects caused by unspecific binding have not been observed (74–76). Systemic blocking of RAGE previous to sepsis attenuated the effects of endotoxemic shock (23, 25, 77). Here, RAGEab was used to evaluate the role of RAGE in brain dysfunction occurring 30 days after CLP. Importantly, the hippocampal administration of RAGEab was performed from 15 to 19 days after CLP, to establish the importance of CLP-induced up-regulation of RAGE in changes emerging after the acute pro-inflammatory phase of sepsis. Blocking RAGE prior to CLP, or using constitutive RAGE knock-out models, would result in an overall inhibition of systemic inflammation, as demonstrated previously (77), whereas the main goal of the current approach was to identify a molecular link connecting systemic inflammation and brain dysfunction that may be involved in long-term brain impairment caused by sepsis. This allowed identifying RAGE as an important molecular component in the course of CNS signaling events often associated with the onset and progression of neurodegeneration, such as Aβ accumulation, Tau phosphorylation, astrocyte/microglia activation, and modulation of Akt and mTOR. Also, the participation of RAGE in cognitive impairment as animals recover from an episode of sepsis could also be identified.
In conclusion, the results presented here demonstrate a prominent role of RAGE in the development of biochemical and behavioral changes commonly associated with the onset of neurodegenerative processes. Although the limitations of the current model do not allow an extrapolation of a more important role for RAGE in the evolution of characteristic neurodegenerative conditions (such as AD and other tauopathies, for instance), this study provided valuable insight to the understanding of mechanistic links that could associate episodes of acute systemic inflammation to the activation of neurodegenerative-associated signaling in the CNS in later periods of life. Besides, the development of a clinical protocol for RAGE immune neutralization in a specific brain region could be designed to be applied in early steps of neurodegenerative diseases, such as AD, in future clinical studies. The results presented here indicate that inhibition of RAGE in the hippocampus and prefrontal cortex by injection of RAGEab in hippocampus is able to counteract the degenerative process triggered by sepsis. Clinical strategies that treat the cause of neuronal death in neurodegenerative diseases are currently scarce, as most therapeutic approaches focus on alleviating or retarding the symptoms of neurodegeneration.
Experimental procedures
Chemicals
Electrophoresis and immunoblot reagents were from Bio-Rad, Thermo Fisher Scientific Pierce Protein Biology Products, and GE Healthcare Brazilian Headquarter (São Paulo, Brazil). Polyclonal and monoclonal antibodies from Abcam (Cambridge, UK) were as follows: TNF-α (ab6671); IL-1β (ab9722); IL-6 (ab9324); CML (ab27684); nitrotyrosine (ab7048); RAGE (ab37647); and TLR4 (ab22048). Antibodies against S100B (catalog no. 9550), HSP70 (catalog no. 4872), HMGB1 (catalog no. 3935), GFAP (catalog no. 3670), p-Tau (Ser-202) (catalog no. 11834), Tau (catalog no. 4019), Aβ (catalog no. 2454), p-ERK-44/42 (Thr-202/Tyr-204) (catalog no. 9101), ERK-44/42 (catalog no. 9102), RAGE (catalog no. 4679), p-Akt (Ser-308) (catalog no. 13038), Akt (catalog no. 9272), p-mTOR (catalog no. 2971), and mTOR (catalog no. 4517) were from Cell Signaling Technology. Polyclonal antibody against nNOS (AB5380) and NeuN (MAB377) was from Merck Millipore. β-Actin (A1978) monoclonal antibody was from Sigma. Anti-Iba-1 (catalog no. 019-19741) was from Wako Chemicals. Antibody concentrations were used as indicated by the manufacturers. Biotinylated ladder protein marker was from Cell Signaling Technology. Anti-rabbit IgG peroxidase-conjugated (catalog no. AP132P) and anti-mouse IgG peroxidase-conjugated (catalog no. AP124P) were from Merck Millipore. Immunoblot chemiluminescence detection was carried out with the West Pico detection kit from Thermo Fisher Scientific Pierce Protein Biology Products (Rockford, IL). MilliQ-purified H2O was used for preparing solutions. For RAGE immune neutralization, anti-RAGE rat IgG (SC5563) from Santa Cruz Biotechnology, Inc. (Dallas, TX), was used. All other reagents used in this study were of analytical or HPLC grade.
CLP
Adult (60-day-old) male Wistar rats were used in this study. Animals were subjected to CLP or sham-surgery as described previously (78). In brief, rats were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg) given intraperitoneally. A 3-cm midline laparotomy was performed to expose the cecum with adjoining intestine, under aseptic conditions. The cecum was tightly ligated with a 3.0 silk suture at its base, below the ileocecal valve, and perforated with a 14-gauge needle. The cecum was then gently squeezed to extrude a small amount of feces from the perforation site. The cecum was then returned to the peritoneal cavity, and the laparotomy was closed with 4.0 silk sutures. A sham operation (laparotomy and cecal exposure without further manipulation) was performed as control. After surgery, animals were resuscitated with normal saline (50 ml/kg b.w. subcutaneously as bolus injection) and received 30 mg/kg ceftriaxone and 25 mg/kg clindamycin in saline (50 ml/kg b.w.) subcutaneously every 6 h for a total of 3 days. Sodium dipyrone was used as analgesic due to its high gastric tolerability and to avoid opioid analgesics that could have any possible influence in behavior tests. Besides, there are no reported effects of sodium dipyrone over sepsis aggravation to date. Administration of 80 mg/kg was performed at the end of surgery, and 0.5% flavored dipyrone suspension was provided in the water for 3 days after surgery. All these drugs were equally administered to sham and CLP animals. It is relevant to cite that even flavored dipyrone could alter water taste, although evident changes in water consumption were not observed. To minimize variability between different experiments, the CLP procedure was always performed by the same investigator. For each experiment, every group had an initial number of 22 animals, with a survival rate of 100% in the sham group and around 40% in the sepsis group, which was in accordance with our previous observations (30, 79). A minimum of six animals per group (sham versus CLP) on each day (1, 15, or 30 days after surgery) was used for individual biochemical and behavioral analyses. For behavioral analyses, animals that were unable to complete tasks within a maximum time were excluded. To collect samples for biochemical analyses, animals were euthanized by decapitation, and samples (tissues and blood) were collected immediately. The whole blood was collected in a centrifuge tube, and the serum was immediately separated by centrifugation while the brain was dissected. Brain was dissected in a Petri's dish containing isotonic PBS placed on ice. After surgical isolation of the hippocampus and prefrontal cortex, these samples were immediately homogenized in ice-cold RIPA buffer containing a complete protease and phosphatase inhibitor mixture (CST catalog no. 5872). For histology preparation, the procedure is provided below. All experimental procedures were performed in accordance with the National Institutes of Health, Guide for the Care and Use of Laboratory Animals (NRC80), and the Brazilian Society for Neuroscience and Behavior recommendations for animal care (80). The experimental protocols were approved by Committee of Ethics for the Use of Animals under protocol number 068/2014-2 (Universidade do Extremo Sul Catarinense) and the Universidade Federal do Rio Grande do Sul Committee of Ethics for the Use of Animals under protocol number 22629.
RAGE immune neutralization
Rats were bilaterally implanted with guide cannulas under anesthesia with isoflurane (1.0–2.0% delivered in O2). Fourteen days after CLP, the rat was anesthetized with 2.0% isoflurane and placed in a Kopfe stereotaxic frame and a stainless steel guide cannula (27 gauge, 9-mm length) aimed 1 mm above the hippocampus coordinates: anteroposterior, 2.2 mm, lateral ± 2.0 mm, and ventral −3.7 (81) was inserted. Isoflurane (1.0–2.0%) was delivered during the entire procedure. Anchoring screws and dental acrylic secured the guide cannula to the skull. The skin was approximated around the implant and sutured in place. The solutions were infused through an injection needle of 10 mm length, thus extending 1 mm beyond the length of guide cannula. The needle had a flat extremity and was connected to a 10-μl Hamilton syringe, and infusions were carried out over 30 s, first on one side and then on the other. In each case the infusion cannula was left in place for 15 s after the infusion had been completed. Rat polyclonal RAGEab was administered bilaterally into the hippocampus at 100 μg/kg saline at days 15, 17, and 19 after CLP. Control animals received 100 μg/kg of isotype IgG.
Behavior tests
Animals were subjected to inhibitory avoidance tasks 30 days after CLP. The behavioral procedures were conducted between 13:00 and 16:00 h in a sound-isolated room. All behavioral tests were performed by the same person who was blind to the animal group. The inhibitory avoidance procedure was described in a previous report (82). Training sessions were performed 30 days after surgery. Immediately after stepping down on the grid, animals received a foot shock of 0.3 mA for 2 s. In test sessions carried out 24 h after training, no foot shock was given, and the step-down latency (maximum of 180 s) was used as a measure of retention. Animals that failed to step down on the grid or presented any signs of unusual locomotor activity were excluded. During object recognition habituation, session animals were allowed to explore an open field. Training was conducted by placing rats in the field, in which two identical objects (objects A1 and A2; both being cubes) were positioned. Twenty four hours after training, animals were allowed to explore the field in the presence of the familiar object A and a novel object B (a sphere with a square-shaped base). A recognition index was calculated and was reported as the ratio TB/(TA + TB) (TA = time spent exploring the familiar object A; TB = time spent exploring the novel object B). Animals that presented unusual locomotor activity during training session were excluded.
ELISA measurements
To determine TNF-α, IL1-β, IL-6, and CML concentrations in serum and TNF-α, IL-1β, IL-6, S100B, CML, HSP70, and HMGB1 in tissues, we used an indirect enzyme-linked immunosorbent assay (ELISA) procedure. Samples were normalized according to protein content (83) and added to ELISA microplates. The antigen was incubated for 24 h at room temperature. The immunoreactivity (1:1) was detected using a spectrophotometric detection kit from BD Biosciences. The reaction was stopped with sulfuric acid (2 m), and samples were read at 450 nm. Purified proteins were used for standard curve calculation.
Western blotting
To perform immunoblot experiments, the tissue was homogenized with 1× RIPA buffer, centrifuged 14,000 × g for 10 min at 4 °C, and supernatant proteins were measured by the Bradford method (83). Laemmli sample buffer was added to complete volume according to the protein content of each sample, and equal amounts of cell protein (30 μg/well) were fractionated by SDS-PAGE and electroblotted onto nitrocellulose membranes with Trans-Blot® SD Semi-Dry Electrophoretic Transfer Cell, Bio-Rad. Membranes were incubated for 20 min at room temperature in SNAP i.d.® 2.0 Protein Detection System, Merck Millipore, with each primary antibody. Anti-rabbit or -mouse IgG peroxidase-linked secondary antibody was incubated with membranes for an additional 20 min in SNAP (1:2000 dilution range), and the immunoreactivity was detected by enhanced chemiluminescence using Supersignal West Pico Chemiluminescent kit from Thermo Fisher Scientific. Molecular weight was accessed with a biotinylated protein ladder (5 μl per well). Densitometric analysis of the images was performed with ImageJ software (ImageJ version 1.49, National Institutes of Health). Blots were developed to be linear in the range used for densitometry.
Perfusion fixation of tissue for histology
Animals (eight per group) were anesthetized with thiopental and lidocaine (120 and 10 mg/kg i.p.). Once the animal was unresponsive to toe pinch-response (anesthesia validation test), it was placed on the operating table with its back down. A scalpel was used to make an incision through the abdomen at the length of the diaphragm, followed by a cut of the rib cage up to the collarbone on both sides of the ribs providing a clear view of the heart. A small incision was made in the posterior end of the left ventricle and an olive-tipped perfusion needle was inserted through the ventricle to extend straight up about 5 mm. An incision to the rat right atrium was made to create an outlet for free flow of the solution. A hemostat was used to stabilize the needle and to clamp the descendent aorta to optimize perfusion in the CNS. Eight animals per group were perfused with 0.9% sterile saline during 10 min (flow rate 20 ml/min) followed by 10 min with paraformaldehyde solution 4% in PBS (pH 7.4) (flow rate 20 ml/min). The brains were then carefully extracted and maintained in 4% paraformaldehyde for 24 h at 4 °C, then placed in sucrose 15% for 24 h at 4 °C, and placed in sucrose 30% for 24 h at 4 °C. Brains were slightly dried and frozen in −20 °C. After 24 h the prefrontal cortex and the hippocampus were sectioned in slices of 15 μm on the coronal plane using a cryostat at −20 °C (Jung Histoslide 2000R; Leica; Heidelberg, Germany). A total of 20–30 slices per rat containing the structures was collected in PBS containing 0.1% Triton X-100 (PBS-0.1%). The free-floating sections were incubated with 5% albumin during 2 h to block non-specific binding sites. The antibody was incubated during 48 h at 4 °C. Anti-RAGE, anti-Iba-1, anti-GFAP, anti-Aβ, and anti-phospho-Tau were used at 1:500 dilution. DAPI was used for nucleic acid staining (1:500; D9542, Sigma). Antibodies were diluted in PBS containing bovine serum albumin (2%). After washing four times with PBS-0.1%, tissue sections were incubated with secondary antibody according to reactive species (anti-rabbit or mouse Alexa 488 or 555 from Cell Signaling Technology), all diluted 1:500 in PBS and 2% BSA. After 1 h at room temperature, the slices were washed several times in PBS-0.1%, transferred to gelatinized slides, mounted with FluorSaveTM (345789, Merck Millipore), and covered with coverslips. The images were obtained with a Microscopy EVOS® FL Auto Imaging System (AMAFD1000, Thermo Fisher Scientific). Quantitative analysis of immunofluorescence staining was performed with ImageJ software (ImageJ version 1.49, National Institutes of Health). Results are expressed as fold relative to control (sham) group.
Statistical analysis
Statistical analysis was performed with GraphPad 5.0 software (GraphPad Software Inc., San Diego). Student's t test (one-tailed) was applied for simple comparisons between sham and CLP animals in each assay and one-way analysis of variance (ANOVA) followed by Tukey's post hoc test in assays with more groups. For behavioral analyses, individual groups were compared by the Wilcoxon tests, and comparisons among groups in inhibitory avoidance tasks were performed using the Mann-Whitney U test. Differences were considered significant when p < 0.05.
Author contributions
J. G. performed experiments, analyzed all data, elaborated the figures, and wrote the majority of the manuscript. C. S. G., N. S., and C. T. R. performed Western blottings and ELISA measurements. M. M., B. S., M. R., and A. V. S. performed surgical procedures (CLP and hippocampal cannulation), RAGEab administration, and behavioral tests plus analyses. J. C. F. M. designed experiments, discussed data, and helped to write the manuscript. T. B. and J. Q. designed behavioral experiments, analyzed and discussed data, and helped to write the manuscript. F. D. P. and D. P. G. provided the aims, design, and setting of the study, analyzed data, and wrote and reviewed the manuscript. All authors read and approved the final manuscript.
Acknowledgments
The Translational Psychiatry Program is funded by the Department of Psychiatry and Behavioral Sciences, McGovern Medical School, University of Texas Health Science Center at Houston. Laboratory of Neurosciences and Fisiopatologia Experimental (Brazil) are one of the members of the Center of Excellence in Applied Neurosciences of Santa Catarina (NENASC).
This work was supported by grants from Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (to D. P. G., J. C. F. M., J. Q., and F. D.-P.), Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (to D. P. G. and J. C. F. M.), Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina (to J. Q. and F. D.-P.), Propesq/UFRGS (to D. P.G. and J. C. F. M.), Fundação CAPES, Instituto Cérebro e Mente (to J. Q.) and UNESC (to T. B., J. Q., and F. D.-P.). The authors declare that they have no conflicts of interest with the contents of this article.
- CLP
- cecal ligation and perforation
- RAGE
- receptor for advanced glycation end product
- sRAGE
- soluble form of RAGE
- RAGEab
- RAGE antibody
- AGE
- advanced glycation end product
- Aβ
- amyloid-β
- AβPP
- amyloid precursor protein
- BBB
- blood–brain barrier
- GFAP
- glial fibrillary acidic protein
- mTOR
- mechanistic target of rapamycin
- nNOS
- neuronal nitric-oxide synthase
- AD
- Alzheimer's disease
- ANOVA
- analysis of variance
- CML
- Nϵ-(carboxymethyl)lysine
- b.w.
- body weight
- TBI
- traumatic brain injury.
References
- 1. Singer M., Deutschman C. S., Seymour C. W., Shankar-Hari M., Annane D., Bauer M., Bellomo R., Bernard G. R., Chiche J. D., Coopersmith C. M., Hotchkiss R. S., Levy M. M., Marshall J. C., Martin G. S., Opal S. M., et al. (2016) The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315, 801–810 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilson J. X., and Young G. B. (2003) Progress in clinical neurosciences: sepsis-associated encephalopathy: evolving concepts. Can. J. Neurol. Sci. 30, 98–105 10.1017/S031716710005335X [DOI] [PubMed] [Google Scholar]
- 3. Widmann C. N., and Heneka M. T. (2014) Long-term cerebral consequences of sepsis. Lancet Neurol. 13, 630–636 10.1016/S1474-4422(14)70017-1 [DOI] [PubMed] [Google Scholar]
- 4. Semmler A., Hermann S., Mormann F., Weberpals M., Paxian S. A., Okulla T., Schäfers M., Kummer M. P., Klockgether T., and Heneka M. T. (2008) Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J. Neuroinflammation 5, 38 10.1186/1742-2094-5-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winters B. D., Eberlein M., Leung J., Needham D. M., Pronovost P. J., and Sevransky J. E. (2010) Long-term mortality and quality of life in sepsis: a systematic review. Crit. Care Med. 38, 1276–1283 10.1097/CCM.0b013e3181d8cc1d [DOI] [PubMed] [Google Scholar]
- 6. Barichello T., Martins M. R., Reinke A., Feier G., Ritter C., Quevedo J., and Dal-Pizzol F. (2005) Cognitive impairment in sepsis survivors from cecal ligation and perforation. Crit. Care Med. 33, 221–223 10.1097/01.CCM.0000150741.12906.BD [DOI] [PubMed] [Google Scholar]
- 7. Yende S., Austin S., Rhodes A., Finfer S., Opal S., Thompson T., Bozza F. A., LaRosa S. P., Ranieri V. M., and Angus D. C. (2016) Long-term quality of life among survivors of severe sepsis: analyses of two international trials. Crit. Care Med. 44, 1461–1467 10.1097/CCM.0000000000001658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iwashyna T. J., Ely E. W., Smith D. M., and Langa K. M. (2010) Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304, 1787–1794 10.1001/jama.2010.1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Annane D., and Sharshar T. (2015) Cognitive decline after sepsis. Lancet. Respir. Med. 3, 61–69 10.1016/S2213-2600(14)70246-2 [DOI] [PubMed] [Google Scholar]
- 10. Varatharaj A., and Galea I. (2017) The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 60, 1–12 [DOI] [PubMed] [Google Scholar]
- 11. Dal-Pizzol F., Rojas H. A., dos Santos E. M., Vuolo F., Constantino L., Feier G., Pasquali M., Comim C. M., Petronilho F., Gelain D. P., Quevedo J., Moreira J. C., and Ritter C. (2013) Matrix metalloproteinase-2 and metalloproteinase-9 activities are associated with blood-brain barrier dysfunction in an animal model of severe sepsis. Mol. Neurobiol. 48, 62–70 10.1007/s12035-013-8433-7 [DOI] [PubMed] [Google Scholar]
- 12. Michels M., Steckert A. V., Quevedo J., Barichello T., and Dal-Pizzol F. (2015) Mechanisms of long-term cognitive dysfunction of sepsis: from blood-borne leukocytes to glial cells. Intensive Care Med. Exp. 3, 30 10.1186/s40635-015-0066-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Michels M., Vieira A. S., Vuolo F., Zapelini H. G., Mendonça B., Mina F., Dominguini D., Steckert A., Schuck P. F., Quevedo J., Petronilho F., and Dal-Pizzol F. (2015) The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav. Immun. 43, 54–59 10.1016/j.bbi.2014.07.002 [DOI] [PubMed] [Google Scholar]
- 14. Cunningham C., and Hennessy E. (2015) Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research. Alzheimers Res. Ther. 7, 33 10.1186/s13195-015-0117-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spires-Jones T. L., Stoothoff W. H., de Calignon A., Jones P. B., and Hyman B. T. (2009) Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 32, 150–159 10.1016/j.tins.2008.11.007 [DOI] [PubMed] [Google Scholar]
- 16. Gendron T. F., and Petrucelli L. (2009) The role of Tau in neurodegeneration. Mol. Neurodegener. 4, 13 10.1186/1750-1326-4-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Multhaup G., Huber O., Buée L., and Galas M. C. (2015) Amyloid precursor protein (APP) metabolites APP intracellular fragment (AICD), Aβ42, and tau in nuclear roles. J. Biol. Chem. 290, 23515–23522 10.1074/jbc.R115.677211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nalivaeva N. N., and Turner A. J. (2013) The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett. 587, 2046–2054 10.1016/j.febslet.2013.05.010 [DOI] [PubMed] [Google Scholar]
- 19. Zuo L., Hemmelgarn B. T., Chuang C. C., and Best T. M. (2015) The role of oxidative stress-induced epigenetic alterations in amyloid-β production in Alzheimer's disease. Oxid. Med. Cell. Longev. 2015, 604658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Creagh-Brown B. C., Quinlan G. J., Evans T. W., and Burke-Gaffney A. (2010) The RAGE axis in systemic inflammation, acute lung injury and myocardial dysfunction: an important therapeutic target? Intensive Care Med. 36, 1644–1656 10.1007/s00134-010-1952-z [DOI] [PubMed] [Google Scholar]
- 21. Batkulwar K. B., Bansode S. B., Patil G. V., Godbole R. K., Kazi R. S., Chinnathambi S., Shanmugam D., and Kulkarni M. J. (2015) Investigation of phosphoproteome in RAGE signaling. Proteomics 15, 245–259 10.1002/pmic.201400169 [DOI] [PubMed] [Google Scholar]
- 22. Tóbon-Velasco J. C., Cuevas E., and Torres-Ramos M. A. (2014) Receptor for AGEs (RAGE) as mediator of NF-κB pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug Targets 13, 1615–1626 10.2174/1871527313666140806144831 [DOI] [PubMed] [Google Scholar]
- 23. Lutterloh E. C., Opal S. M., Pittman D. D., Keith J. C. Jr, Tan X. Y., Clancy B. M., Palmer H., Milarski K., Sun Y., Palardy J. E., Parejo N. A., and Kessimian N. (2007) Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit. Care 11, R122 10.1186/cc6184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Zoelen M. A., Schmidt A. M., Florquin S., Meijers J. C., de Beer R., de Vos A. F., Nawroth P. P., Bierhaus A., and van der Poll T. (2009) Receptor for advanced glycation end products facilitates host defense during Escherichia coli-induced abdominal sepsis in mice. J. Infect. Dis. 200, 765–773 10.1086/604730 [DOI] [PubMed] [Google Scholar]
- 25. van Zoelen M. A., and van der Poll T. (2008) Targeting RAGE in sepsis. Crit. Care 12, 103 10.1186/cc6187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Srikanth V., Maczurek A., Phan T., Steele M., Westcott B., Juskiw D., and Münch G. (2011) Advanced glycation end products and their receptor RAGE in Alzheimer's disease. Neurobiol. Aging 32, 763–777 [DOI] [PubMed] [Google Scholar]
- 27. Li X. H., Lv B. L., Xie J. Z., Liu J., Zhou X. W., and Wang J. Z. (2012) AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol. Aging 33, 1400–1410 [DOI] [PubMed] [Google Scholar]
- 28. Chavan S. S., Huerta P. T., Robbiati S., Valdes-Ferrer S. I., Ochani M., Dancho M., Frankfurt M., Volpe B. T., Tracey K. J., and Diamond B. (2012) HMGB1 mediates cognitive impairment in sepsis survivors. Mol. Med. 18, 930–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dal-Pizzol F., Pasquali M., Quevedo J., Gelain D. P., and Moreira J. C. (2012) Is there a role for high mobility group box 1 and the receptor for advanced glycation end products in the genesis of long-term cognitive impairment in sepsis survivors? Mol. Med. 18, 1357–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schwalm M. T., Pasquali M., Miguel S. P., Dos Santos J. P., Vuolo F., Comim C. M., Petronilho F., Quevedo J., Gelain D. P., Moreira J. C., Ritter C., and Dal-Pizzol F. (2014) Acute brain inflammation and oxidative damage are related to long-term cognitive deficits and markers of neurodegeneration in sepsis-survivor rats. Mol. Neurobiol. 49, 380–385 10.1007/s12035-013-8526-3 [DOI] [PubMed] [Google Scholar]
- 31. Deane R., Singh I., Sagare A. P., Bell R. D., Ross N. T., LaRue B., Love R., Perry S., Paquette N., Deane R. J., Thiyagarajan M., Zarcone T., Fritz G., Friedman A. E., Miller B. L., and Zlokovic B. V. (2012) A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Invest. 122, 1377–1392 10.1172/JCI58642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu Y., and Ye R. D. (2015) Microglial Aβ receptors in Alzheimer's disease. Cell. Mol. Neurobiol. 35, 71–83 10.1007/s10571-014-0101-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barichello T., Martins M. R., Reinke A., Constantino L. S., Machado R. A., Valvassori S. S., Moreira J. C., Quevedo J., and Dal-Pizzol F. (2007) Behavioral deficits in sepsis-surviving rats induced by cecal ligation and perforation. Braz. J. Med. Biol. Res. 40, 831–837 10.1590/S0100-879X2007000600013 [DOI] [PubMed] [Google Scholar]
- 34. Gutierrez E. G., Banks W. A., and Kastin A. J. (1993) Murine tumor necrosis factor α is transported from blood to brain in the mouse. J. Neuroimmunol. 47, 169–176 10.1016/0165-5728(93)90027-V [DOI] [PubMed] [Google Scholar]
- 35. Yarlagadda A., Alfson E., and Clayton A. H. (2009) The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry 6, 18–22 [PMC free article] [PubMed] [Google Scholar]
- 36. Pan W., Stone K. P., Hsuchou H., Manda V. K., Zhang Y., and Kastin A. J. (2011) Cytokine signaling modulates blood-brain barrier function. Curr. Pharm. Des. 17, 3729–3740 10.2174/138161211798220918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dal-Pizzol F., Di Leone L. P., Ritter C., Martins M. R., Reinke A., Pens Gelain D., Zanotto-Filho A., de Souza L. F., Andrades M., Barbeiro D. F., Bernard E. A., Cammarota M., Bevilaqua L. R., Soriano F. G., Cláudio J., et al. (2006) Gastrin-releasing peptide receptor antagonist effects on an animal model of sepsis. Am. J. Respir. Crit. Care Med. 173, 84–90 10.1164/rccm.200507-1118OC [DOI] [PubMed] [Google Scholar]
- 38. Ritter C., Andrades M. E., Reinke A., Menna-Barreto S., Moreira J. C., and Dal-Pizzol F. (2004) Treatment with N-acetylcysteine plus deferoxamine protects rats against oxidative stress and improves survival in sepsis. Crit. Care Med. 32, 342–349 10.1097/01.CCM.0000109454.13145.CA [DOI] [PubMed] [Google Scholar]
- 39. du Moulin G. C., Paterson D., Hedley-Whyte J., and Broitman S. A. (1985) E. coli peritonitis and bacteremia cause increased blood-brain barrier permeability. Brain Res. 340, 261–268 10.1016/0006-8993(85)90922-9 [DOI] [PubMed] [Google Scholar]
- 40. Lee J. Y., Lee J. D., Phipps S., Noakes P. G., and Woodruff T. M. (2015) Absence of toll-like receptor 4 (TLR4) extends survival in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuroinflammation 12:90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang J. Z., Xia Y. Y., Grundke-Iqbal I., and Iqbal K. (2013) Abnormal hyperphosphorylation of Tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimers Dis. 33, S123 [DOI] [PubMed] [Google Scholar]
- 42. Goedert M., Jakes R., Crowther R. A., Six J., Lübke U., Vandermeeren M., Cras P., Trojanowski J. Q., and Lee V. M. (1993) The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc. Natl. Acad. Sci. U.S.A. 90, 5066–5070 10.1073/pnas.90.11.5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goedert M., Jakes R., Crowther R. A., Hasegawa M., Smith M. J., and Spillantini M. G. (1998) Intraneuronal filamentous tau protein and α-synuclein deposits in neurodegenerative diseases. Biochem. Soc. Trans. 26, 463–471 10.1042/bst0260463 [DOI] [PubMed] [Google Scholar]
- 44. Šimič G., Babič Leko M., Wray S., Harrington C., Delalle I., Jovanov-Milošević N., Bažadona D., Buée L., de Silva R., Di Giovanni G., Wischik C., and Hof P. R. (2016) Tau protein hyperphosphorylation and aggregation in Alzheimer's disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6, 6 10.3390/biom6010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharshar T., Gray F., Lorin de la Grandmaison G., Hopkinson N. S., Ross E., Dorandeu A., Orlikowski D., Raphael J. C., Gajdos P., and Annane D. (2003) Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric-oxide synthase after death from septic shock. Lancet 362, 1799–1805 10.1016/S0140-6736(03)14899-4 [DOI] [PubMed] [Google Scholar]
- 46. Barichello T., Fortunato J. J., Vitali A. M., Feier G., Reinke A., Moreira J. C., Quevedo J., and Dal-Pizzol F. (2006) Oxidative variables in the rat brain after sepsis induced by cecal ligation and perforation. Crit. Care Med. 34, 886–889 10.1097/01.CCM.0000201880.50116.12 [DOI] [PubMed] [Google Scholar]
- 47. Zilka N., Kazmerova Z., Jadhav S., Neradil P., Madari A., Obetkova D., Bugos O., and Novak M. (2012) Who fans the flames of Alzheimer's disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J. Neuroinflammation 9, 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Šimić G., Babić Leko M., Wray S., Harrington C., Delalle I., Jovanov-Milošević N., Bažadona D., Buée L., de Silva R., Di Giovanni G., Wischik C., and Hof P. R. (2016) Tau protein hyperphosphorylation and aggregation in Alzheimer's disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6, 6 10.3390/biom6010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Simic G. (2002) Pathological tau proteins in argyrophilic grain disease. Lancet Neurol. 1, 276 10.1016/S1474-4422(02)00130-8 [DOI] [PubMed] [Google Scholar]
- 50. O'Brien R. J., and Wong P. C. (2011) Amyloid precursor protein processing and Alzheimer's disease. Annu. Rev. Neurosci. 34, 185–204 10.1146/annurev-neuro-061010-113613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Edwards G. 3rd, Moreno-Gonzalez I., and Soto C. (2017) Amyloid-β and tau pathology following repetitive mild traumatic brain injury. Biochem. Biophys. Res. Commun. 483, 1137–1142 10.1016/j.bbrc.2016.07.123 [DOI] [PubMed] [Google Scholar]
- 52. Cho H. J., Son S. M., Jin S. M., Hong H. S., Shin D. H., Kim S. J., Huh K., and Mook-Jung I. (2009) RAGE regulates BACE1 and Aβ generation via NFAT1 activation in Alzheimer's disease animal model. FASEB J. 23, 2639–2649 10.1096/fj.08-126383 [DOI] [PubMed] [Google Scholar]
- 53. Takuma K., Fang F., Zhang W., Yan S., Fukuzaki E., Du H., Sosunov A., McKhann G., Funatsu Y., Nakamichi N., Nagai T., Mizoguchi H., Ibi D., Hori O., Ogawa S., et al. (2009) RAGE-mediated signaling contributes to intraneuronal transport of amyloid-β and neuronal dysfunction. Proc. Natl. Acad. Sci. U.S.A. 106, 20021–20026 10.1073/pnas.0905686106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Candela P., Gosselet F., Saint-Pol J., Sevin E., Boucau M. C., Boulanger E., Cecchelli R., and Fenart L. (2010) Apical-to-basolateral transport of amyloid-β peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J. Alzheimers Dis. 22, 849–859 10.3233/JAD-2010-100462 [DOI] [PubMed] [Google Scholar]
- 55. Hong Y., Shen C., Yin Q., Sun M., Ma Y., and Liu X. (2016) Effects of RAGE-specific inhibitor FPS-ZM1 on amyloid-β metabolism and AGEs-induced inflammation and oxidative stress in rat hippocampus. Neurochem. Res. 41, 1192–1199 10.1007/s11064-015-1814-8 [DOI] [PubMed] [Google Scholar]
- 56. Johnson V. E., Stewart W., and Smith D. H. (2010) Traumatic brain injury and amyloid-β pathology: a link to Alzheimer's disease? Nat. Rev. Neurosci. 11, 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gouras G. K., Willén K., and Faideau M. (2014) The inside-out amyloid hypothesis and synapse pathology in Alzheimer's disease. Neurodegener. Dis. 13, 142–146 [DOI] [PubMed] [Google Scholar]
- 58. Takahashi R. H., Almeida C. G., Kearney P. F., Yu F., Lin M. T., Milner T. A., and Gouras G. K. (2004) Oligomerization of Alzheimer's β-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 24, 3592–3599 10.1523/JNEUROSCI.5167-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Capetillo-Zarate E., Gracia L., Yu F., Banfelder J. R., Lin M. T., Tampellini D., and Gouras G. K. (2011) High-resolution 3D reconstruction reveals intra-synaptic amyloid fibrils. Am. J. Pathol. 179, 2551–2558 10.1016/j.ajpath.2011.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Opp M. R., George A., Ringgold K. M., Hansen K. M., Bullock K. M., and Banks W. A. (2015) Sleep fragmentation and sepsis differentially impact blood-brain barrier integrity and transport of tumor necrosis factor-α in aging. Brain Behav. Immun. 50, 259–265 10.1016/j.bbi.2015.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dent P. (2014) Crosstalk between ERK, AKT, and cell survival. Cancer Biol. Ther. 15, 245–246 10.4161/cbt.27541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kamat P. K., Rai S., Swarnkar S., Shukla R., and Nath C. (2014) Molecular and cellular mechanism of okadaic acid (OKA)-induced neurotoxicity: a novel tool for Alzheimer's disease therapeutic application. Mol. Neurobiol. 50, 852–865 10.1007/s12035-014-8699-4 [DOI] [PubMed] [Google Scholar]
- 63. Shi S., Liang D., Bao M., Xie Y., Xu W., Wang L., Wang Z., and Qiao Z. (2016) Gx-50 inhibits neuroinflammation via α7 nAChR activation of the JAK2/STAT3 and PI3K/AKT pathways. J. Alzheimers Dis. 50, 859–871 10.3233/JAD-150963 [DOI] [PubMed] [Google Scholar]
- 64. Heras-Sandoval D., Pérez-Rojas J. M., Hernández-Damián J., and Pedraza-Chaverri J. (2014) The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 26, 2694–2701 10.1016/j.cellsig.2014.08.019 [DOI] [PubMed] [Google Scholar]
- 65. Guillot F., Kemppainen S., Lavasseur G., Miettinen P. O., Laroche S., Tanila H., and Davis S. (2016) Brain-specific basal and novelty-induced alternations in PI3K-Akt and MAPK/ERK signaling in a middle-aged AβPP/PS1 mouse model of Alzheimer's disease. J. Alzheimers Dis. 2016;51(4):1157–1173 10.3233/JAD-150926 [DOI] [PubMed] [Google Scholar]
- 66. Maiese K. (2016) Targeting molecules to medicine with mTOR, autophagy, and neurodegenerative disorders. Br. J. Clin. Pharmacol. 82, 1245–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Asati V., Mahapatra D. K., and Bharti S. K. (2016) PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 109, 314–341 10.1016/j.ejmech.2016.01.012 [DOI] [PubMed] [Google Scholar]
- 68. Chiang G. G., and Abraham R. T. (2005) Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 280, 25485–25490 10.1074/jbc.M501707200 [DOI] [PubMed] [Google Scholar]
- 69. Lukic I. K., Humpert P. M., Nawroth P. P., and Bierhaus A. (2008) The RAGE pathway: activation and perpetuation in the pathogenesis of diabetic neuropathy. Ann. N.Y. Acad. Sci. 1126, 76–80 10.1196/annals.1433.059 [DOI] [PubMed] [Google Scholar]
- 70. Semmler A., Frisch C., Debeir T., Ramanathan M., Okulla T., Klockgether T., and Heneka M. T. (2007) Long-term cognitive impairment, neuronal loss and reduced cortical cholinergic innervation after recovery from sepsis in a rodent model. Exp. Neurol. 204, 733–740 10.1016/j.expneurol.2007.01.003 [DOI] [PubMed] [Google Scholar]
- 71. Barichello T., Martins M. R., Reinke A., Feier G., Ritter C., Quevedo J., and Dal-Pizzol F. (2005) Long-term cognitive impairment in sepsis survivors. Crit. Care Med. 33, 1671 10.1097/01.CCM.0000170192.54682.C1 [DOI] [PubMed] [Google Scholar]
- 72. Sabbagh M. N., Agro A., Bell J., Aisen P. S., Schweizer E., and Galasko D. (2011) PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 25, 206–212 10.1097/WAD.0b013e318204b550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Galasko D., Bell J., Mancuso J. Y., Kupiec J. W., Sabbagh M. N., van Dyck C., Thomas R. G., Aisen P. S., and Alzheimer's Disease Cooperative Study. (2014) Clinical trial of an inhibitor of RAGE-Aβ interactions in Alzheimer disease. Neurology 82, 1536–1542 10.1212/WNL.0000000000000364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Origlia N., Righi M., Capsoni S., Cattaneo A., Fang F., Stern D. M., Chen J. X., Schmidt A. M., Arancio O., Yan S. D., and Domenici L. (2008) Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-β-mediated cortical synaptic dysfunction. J. Neurosci. 28, 3521–3530 10.1523/JNEUROSCI.0204-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kuhla A., Norden J., Abshagen K., Menger M. D., and Vollmar B. (2013) RAGE blockade and hepatic microcirculation in experimental endotoxaemic liver failure. Br. J. Surg. 100, 1229–1239 10.1002/bjs.9188 [DOI] [PubMed] [Google Scholar]
- 76. Xia P., Deng Q., Gao J., Yu X., Zhang Y., Li J., Guan W., Hu J., Tan Q., Zhou L., Han W., Yuan Y., and Yu Y. (2016) Therapeutic effects of antigen affinity-purified polyclonal anti-receptor of advanced glycation end-product (RAGE) antibodies on cholestasis-induced liver injury in rats. Eur. J. Pharmacol. 779, 102–110 10.1016/j.ejphar.2016.03.017 [DOI] [PubMed] [Google Scholar]
- 77. Lutterloh E. C., and Opal S. M. (2007) Antibodies against RAGE in sepsis and inflammation: implications for therapy. Expert Opin. Pharmacother. 8, 1193–1196 10.1517/14656566.8.9.1193 [DOI] [PubMed] [Google Scholar]
- 78. Ritter C., Andrades M., Frota Júnior M. L., Bonatto F., Pinho R. A., Polydoro M., Klamt F., Pinheiro C. T., Menna-Barreto S. S., Moreira J. C., and Dal-Pizzol F. (2003) Oxidative parameters and mortality in sepsis induced by cecal ligation and perforation. Intensive Care Med. 29, 1782–1789 10.1007/s00134-003-1789-9 [DOI] [PubMed] [Google Scholar]
- 79. Barichello T., Machado R. A., Constantino L., Valvassori S. S., Réus G. Z., Martins M. R., Petronilho F., Ritter C., Quevedo J., and Dal-Pizzol F. (2007) Antioxidant treatment prevented late memory impairment in an animal model of sepsis. Crit. Care Med. 35, 2186–2190 10.1097/01.CCM.0000281452.60683.96 [DOI] [PubMed] [Google Scholar]
- 80. National Research Council (2011) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. 8th Ed., National Academies Press, Washington, D. C. [Google Scholar]
- 81. Paxinos G., and Watson C. (2005) The Rat Brain in Stereotaxic Coordinates, 5th Ed., pp. 181–207, Elsevier Academic Press, Amsterdam [Google Scholar]
- 82. Roesler R., Vianna M. R., de-Paris F., and Quevedo J. (1999) Memory-enhancing treatments do not reverse the impairment of inhibitory avoidance retention induced by NMDA receptor blockade. Neurobiol. Learn. Mem. 72, 252–258 10.1006/nlme.1999.3910 [DOI] [PubMed] [Google Scholar]
- 83. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]