

Abstract
UBIAD1 (UbiA prenyltransferase domain–containing protein-1) utilizes geranylgeranyl pyrophosphate (GGpp) to synthesize vitamin K2. We previously reported that sterols stimulate binding of UBIAD1 to endoplasmic reticulum (ER)–localized 3-hydroxy-3-methylglutaryl (HMG) CoA reductase. UBIAD1 binding inhibits sterol-accelerated, ER-associated degradation (ERAD) of reductase, one of several mechanisms for feedback control of this rate-limiting enzyme in the branched pathway that produces cholesterol and nonsterol isoprenoids such as GGpp. Accumulation of GGpp in ER membranes triggers release of UBIAD1 from reductase, permitting its maximal ERAD and ER-to-Golgi transport of UBIAD1. Mutant UBIAD1 variants associated with Schnyder corneal dystrophy (SCD), a human disorder characterized by corneal accumulation of cholesterol, resist GGpp-induced release from reductase and remain sequestered in the ER to block reductase ERAD. Using lines of genetically manipulated cells, we now examine consequences of UBIAD1 deficiency and SCD-associated UBIAD1 on reductase ERAD and cholesterol synthesis. Our results indicated that reductase becomes destabilized in the absence of UBIAD1, resulting in reduced cholesterol synthesis and intracellular accumulation. In contrast, an SCD-associated UBIAD1 variant inhibited reductase ERAD, thereby stabilizing the enzyme and contributing to enhanced synthesis and intracellular accumulation of cholesterol. Finally, we present evidence that GGpp-regulated, ER-to-Golgi transport enables UBIAD1 to modulate reductase ERAD such that synthesis of nonsterol isoprenoids is maintained in sterol-replete cells. These findings further establish UBIAD1 as a central player in the reductase ERAD pathway and regulation of isoprenoid synthesis. They also indicate that UBIAD1-mediated inhibition of reductase ERAD underlies cholesterol accumulation associated with SCD.
Keywords: endoplasmic reticulum (ER), endoplasmic reticulum–associated protein degradation (ERAD), Golgi, cholesterol metabolism, isoprenoid
Introduction
Mevalonate, which is formed by the action of the endoplasmic reticulum (ER)–localized2 enzyme 3-hydroxy-3-methylglutaryl CoA reductase, is a crucial intermediate in the branched pathway that produces cholesterol and the nonsterol isoprenoids farnesyl pyrophosphate (Fpp) and geranylgeranyl pyrophosphate (GGpp) (1, 2). These two isoprenoids are transferred to many cellular proteins and utilized in the synthesis of other nonsterol isoprenoids such as ubiquinone, vitamin K2, heme, and dolichol. Nonsterol isoprenoids are essential for numerous cellular processes ranging from electron transport (ubiquinone and heme), asparagine-linked protein glycosylation (dolichol), and coagulation (vitamin K2) to signal transduction, cell growth, and migration (Fpp and GGpp) (2). Sterol and nonsterol end-products of mevalonate metabolism combine to exert stringent control on reductase through multiple feedback mechanisms (3). This complex, multivalent regulatory system ensures that cells maintain constant production of nonsterol isoprenoids while avoiding the potentially toxic overaccumulation of cholesterol or one of its sterol precursors.
One mechanism for the multivalent feedback regulation of reductase involves its sterol-accelerated, ER-associated degradation (ERAD) from membranes. This ERAD is initiated by the accumulation of sterols in ER membranes, which stimulates binding of reductase to ER membrane proteins called Insig-1 and Insig-2 (4, 5). Insig binding is mediated by the membrane domain of reductase, which contains eight transmembrane helices and precedes the cytosolic catalytic domain (6, 7). Insig-associated ubiquitin ligases facilitate ubiquitination of reductase (8, 9), marking it for extraction across the ER membrane and subsequent release into the cytosol for proteasomal degradation (10, 11). Maximal ERAD of reductase requires the addition to cells of geranylgeraniol, the alcohol derivative of GGpp (5). We postulate that geranylgeraniol becomes converted to GGpp, which in turn augments reductase ERAD by enhancing its membrane extraction (11).
We recently discovered that sterols also cause a subset of reductase molecules to bind to UBIAD1 (UbiA prenyltransferase domain–containing protein-1) (12), an integral membrane prenyltransferase that utilizes GGpp to synthesize the vitamin K2 subtype menaquinone-4 (MK-4) (13). GGpp triggers release of UBIAD1 from reductase, allowing for the maximal ERAD of reductase and ER-to-Golgi transport of UBIAD1. Eliminating expression of UBIAD1 relieves the GGpp requirement for reductase ERAD, indicating that the reaction is inhibited by the prenyltransferase. Missense mutations in the UBIAD1 gene cause Schnyder corneal dystrophy (SCD), an autosomal eye disease in humans caused by abnormal accumulation of cholesterol in the cornea (14, 15). SCD-associated mutants of UBIAD1 resist GGpp-induced release from reductase and remain sequestered in the ER where they inhibit reductase ERAD in a dominant-negative fashion (12, 16). Utilizing cells genetically manipulated using CRISPR/Cas9 techniques, we determine in the current studies the consequences of UBIAD1 deficiency and SCD-associated UBIAD1 on regulation of the mevalonate pathway. The results of these studies not only further establish a key role for UBIAD1 in regulation of reductase ERAD and mevalonate metabolism, but they also indicate that inhibition of reductase ERAD significantly contributes to accumulation of cholesterol associated with SCD.
Results
In the experiment of Fig. 1A, we compared the level of reductase protein in wild-type and genetically manipulated SV-589 cells. The results show that when wild-type SV-589 cells were cultured in sterol-replete medium containing FCS, a low but detectable level of reductase protein was observed by immunoblot analysis of isolated membrane fractions (Fig. 1A, lane 1). The level of reductase protein was reduced in SV-589 (ΔUBIAD1) cells (lane 2), which harbor a CRISPR-Cas9-induced deletion in the endogenous UBIAD1 gene. SV-589 (ΔUBIAD1)/pMyc-UBIAD1 cells are SV-589 (ΔUBIAD1) cells stably transfected with UBIAD1 containing an N-terminal Myc tag. Immunoblot analysis of membrane fractions from SV-589 (ΔUBIAD1)/pMyc-UBIAD1 cells revealed that expression of Myc-UBIAD1 restored expression of reductase protein to approximately wild-type levels (compare lanes 1 and 3). A marked accumulation of reductase was observed in SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells (lane 4), which express the SCD-associated variant of UBIAD1 that harbors substitution of serine for asparagine-102 (N102S). No significant changes were observed in the level of reductase mRNA in SV-589, SV-589 (ΔUBIAD1), SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (WT), and SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells (Fig. 1B). Considering these results together with the previous finding that overexpression of UBIAD1 (N102S) and other SCD-associated UBIAD1 variants blocks ERAD of endogenous reductase (16), we conclude that changes in levels of reductase protein observed here were attributable to modulation of its sterol-accelerated ERAD. When SV-589 and SV-589 (ΔUBIAD1) cells were cultured in sterol-depleted lipoprotein-deficient serum (LPDS)-containing medium, a condition that enhances transcription of the reductase gene and slows reductase ERAD (1), similar levels of reductase protein were observed (lanes 5 and 6). However, it should be noted that reductase continued to accumulate in SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells compared with that in SV-589 (ΔUBIAD1)/pMyc-UBIAD1 cells when grown in LPDS (compare lanes 7 and 8). The immunofluorescence experiment of Fig. 1C shows that despite their overexpression, Myc-UBIAD1 (WT) and (N102S) were localized to the Golgi and ER, respectively, of isoprenoid-replete cells as previously observed (12, 16).
Figure 1.

Expression of HMG CoA reductase protein is reduced in absence of UBIAD1 and enhanced in presence of SCD-associated UBIAD1 (N102S). A and B, SV-589, SV-589(ΔUBIAD1), SV-589(ΔUBIAD1)/pMyc-UBIAD1 (WT), and SV-589(ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells were set up on day 0 at 4 × 105 cells/60-mm dish in medium A containing 10% FCS and 1 mm mevalonate. On day 1, the cells were refed medium A supplemented with either 10% FCS or LPDS as indicated. After 16 h at 37 °C, the cells were harvested for subcellular fractionation (A) or preparation of total RNA (B) as described under “Experimental procedures.” A, aliquots of membrane fractions (20 μg protein/lane) were subjected to SDS-PAGE, and immunoblot analysis was carried with IgG-A9 (against HMG CoA reductase), IgG-H8 (against endogenous UBIAD1), IgG-D1 (against ACAT-1), and anti-calnexin IgG. B, total RNA was subjected to quantitative RT-PCR using primers against human reductase; the human 36B4 mRNA was used as an invariant control. Each value represents the amount of reductase mRNA relative to that in SV-589 cells, which is arbitrarily defined as 1. C, SV-589(ΔUBIAD1)/pMyc-UBIAD1 (WT) and SV-589(ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells were set up on day 0 at 3 × 105 cells/60-mm dish in medium A containing 10% FCS. On day 1, the cells were refed identical medium and incubated for 16 h at 37 °C. The cells were then fixed and analyzed by immunofluorescence microscopy using IgG-9E10 (against Myc-UBIAD1) using a Zeiss Axio Observer Epifluorescence microscope as described under “Experimental procedures.” D, SV-589, SV-589(ΔUBIAD1), SV-589(ΔUBIAD1)/pMyc-UBIAD1 (WT), and SV-589(ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells were set up on day 0 at 5 × 105 cells/100-mm dish in medium A containing 10% FCS and 1 mm mevalonate. On day 1, the cells were depleted of sterols through incubation in medium A supplemented with 10% LPDS, 10 μm compactin, and 50 μm mevalonate. After 16 h at 37 °C, the cells were refed the identical medium containing 10 μm MG-132, and the indicated concentration of 25-HC. Following incubation for 1 h at 37 °C, the cells were harvested, lysed in detergent-containing buffer, and immunoprecipitated with 30 μg of polyclonal IgG-839 (against reductase) as described under “Experimental procedures.” Aliquots of the resulting immunoprecipitates were then subjected to SDS-PAGE, followed by immunoblot analysis with IgG-A9 (against reductase) and IgG-P4D1 (against ubiquitin). The results in this and other figures are representative of at least two independent experiments. MW, molecular mass.
Although UBIAD1 (N102S) inhibits sterol-accelerated ERAD of reductase, the experiment of Fig. 1D shows that sterols continue to trigger ubiquitination of reductase in the presence of the mutant vitamin K2. SV-589, SV-589 (ΔUBIAD1), SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (WT), and SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells were first depleted of isoprenoids through incubation for 16 h in medium containing LPDS and the statin compactin to trigger accumulation of reductase. The cells were then treated with the proteasome inhibitor MG-132 (to block degradation of ubiquitinated reductase) in the presence of various concentrations of the oxysterol 25-hydroxycholesterol (25-HC). After 1 h, the cells were harvested for preparation of detergent lysates that were immunoprecipitated with polyclonal anti-reductase. In a dose-dependent manner, 25-HC caused reductase to become ubiquitinated in SV-589 cells, as indicated by smears of reactivity in anti-ubiquitin immunoblots of the reductase immunoprecipitates (Fig. 1D, lanes a--d). Similar results were observed in SV-589 (ΔUBIAD1) (lanes e–h), SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (WT) (lanes i–l), and SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells (lanes m–p). It is notable that reductase became ubiquitinated in SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells, even though its ERAD was inhibited (Fig. 1A, lane 4).
We next sought to determine the consequence of UBIAD1 deficiency and reduced expression of reductase on synthesis of cholesterol from the two-carbon precursor acetate. As shown in the experiment of Fig. 2A, SV-589 (ΔUBIAD1) cells incorporated significantly less [14C]acetate into cholesterol than parental SV-589 cells when cultured in FCS-containing medium. To determine whether exogenous cholesterol from FCS-derived lipoproteins contributed to reduced synthesis of cholesterol in SV-589 (ΔUBIAD1) cells, we measured incorporation of [3H]mevalonate into cholesterol in wild-type and UBIAD1-deficient cells. The results show that incorporation of [3H]mevalonate into cholesterol was reduced 3–4-fold in SV-589 (ΔUBIAD1) cells cultured in FCS compared with that in SV-589 cells (Fig. 2B). However, this difference was ablated when the cells were cultured in LPDS. This result indicates that reduced expression of reductase renders SV-589 (ΔUBIAD1) cells reliant on exogenous lipoproteins to satisfy cellular demands for cholesterol. The uptake of exogenous lipoproteins likely reduced incorporation of [3H]mevalonate into cholesterol because of modulation of the activation of sterol regulatory element-binding protein (SREBP)-2, a membrane-bound transcription factor that controls expression of genes encoding reductase and other enzymes of the mevalonate pathway (17).
Figure 2.
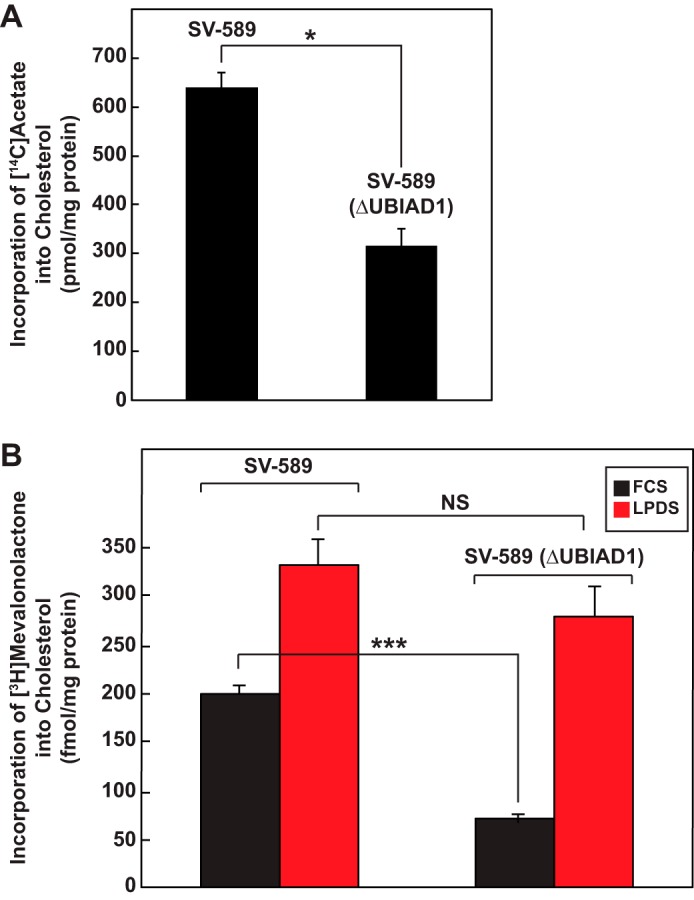
Reduced synthesis of cholesterol in SV-589 cells lacking UBIAD1. SV-589 and SV-589(ΔUBIAD1) cells were set up on day 0 at a density of 4 × 105 cells/60-mm dish in medium A containing 10% FCS and 1 mm mevalonate. On day 1, the cells were switched to medium A containing 10% FCS. A, on day 2, the cells were refed the identical medium supplemented with 15 μCi/ml [14C]acetate; cold acetate was to obtain a final concentration of 0.5 mm. Following incubation for 4 h at 37 °C, the cells were harvested for preparation of lysates from which lipids were extracted and analyzed by TLC as described under “Experimental procedures.” Incorporation of [14C]acetate into [14C]cholesterol was determined by scintillation counting. A blank value, representing the amount of [14C]acetate incorporated into [14C]cholesterol in each cell line that was chilled to 4 °C, refed [14C]acetate-containing medium and immediately washed and extracted, was subtracted from each value. Each value is the mean of triplicate incubations (± standard error). B, on day 2, the cells were refed medium A supplemented with 10 μCi/ml [3H]mevalonolactone and either 10% FCS or LPDS. Following incubation for 4 h at 37 °C, the cells were harvested and lysed for lipid extraction; incorporation of [3H]mevalonolactone into [3H]cholesterol was determined as described in A. Each value is the mean of triplicate incubations (± standard error). The p values were calculated using Student's t test: NS, not significant; *, p ≤ 0.05; ***, p ≤ 0.001.
Fig. 3A compares synthesis of cholesterol from acetate in SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (WT) and (N102S) cells cultured in FCS. The incorporation of [14C]acetate was markedly enhanced (>5-fold) in SV-589 (ΔUBIAD1) cells expressing UBIAD1 (N102S) compared with that in cells expressing wild-type UBIAD1. In contrast, the synthesis of cholesterol from [3H]mevalonate in SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (WT) and (N102S) cells was similar, regardless of culture in FCS or LPDS (Fig. 3B). Thus, enhanced synthesis of cholesterol from acetate in SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells likely resulted from accumulation of reductase caused by UBIAD1 (N102S)-mediated inhibition of its ERAD. It should be noted that cholesterol synthesis from mevalonate was not enhanced when SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (WT) cells were cultured in LPDS (Fig. 3B). Moreover, we noticed that cholesterol synthesis is reduced in SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (WT) cells compared that observed in parental SV-589 (ΔUBIAD1) cells. We reason that when UBIAD1 is overexpressed, mevalonate metabolites are diverted away from cholesterol and become incorporated into nonsterol isoprenoids.
Figure 3.
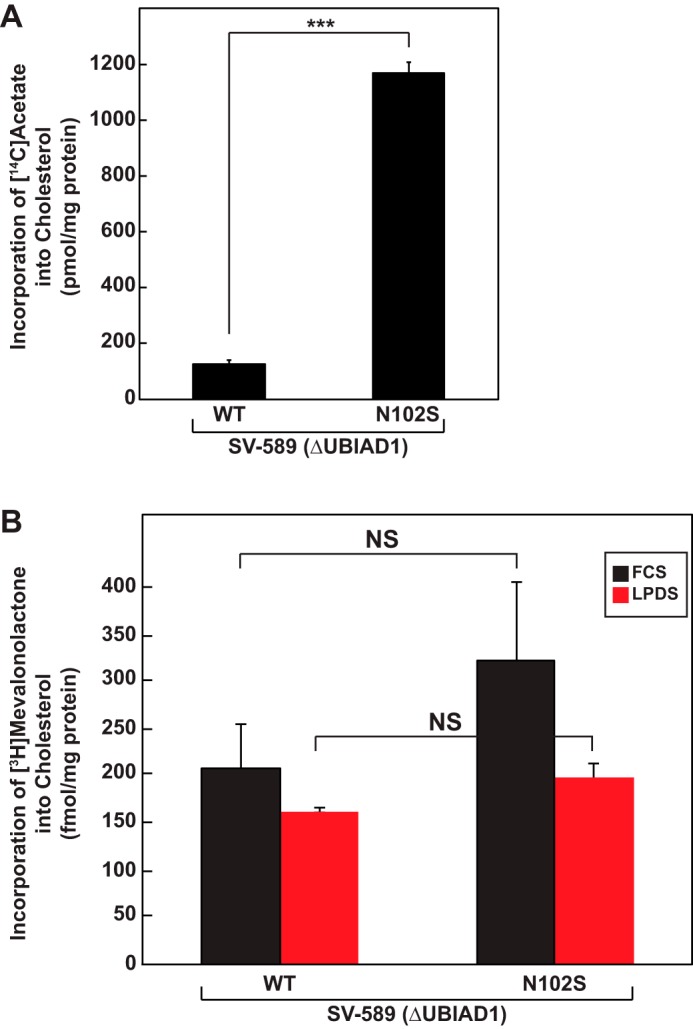
Expression of SCD-associated UBIAD1 (N102S) leads to enhanced synthesis of cholesterol in SV-589 cells. SV589(ΔUBIAD1)/pMyc-UBIAD1 (WT) and SV589(ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells were set up on day 0 at a density of 4 × 105 cells/60-mm dish in medium A containing 10% FCS and 1 mm mevalonate. On day 1, the cells were switched to medium A containing 10% FCS. On day 2, the cells received the identical medium supplemented with either 15 μCi/ml [14C]acetate (A) or 10 μCi/ml [3H]mevalonolactone (B) and incubated for 4 h at 37 °C. Incorporation of [14C]acetate and [3H]mevalonolactone into [3H]cholesterol was determined by TLC and scintillation counting as described in the legend to Fig. 2. Each value is the mean of triplicate incubations (± standard error). The p values were calculated using the Student's t test: NS, not significant; ***, p ≤ 0.001.
The synthesis of cholesteryl esters, the major storage form of cellular cholesterol, was next measured in wild-type and UBIAD1-deficient SV-589 cells (Fig. 4A). Similar to results obtained for cholesterol synthesis (Fig. 3A), incorporation of [3H]oleate into cholesteryl esters was reduced in the absence of UBIAD1 (Fig. 4A). Overexpression of UBIAD1 (N102S) in SV-589 (ΔUBIAD1) cells led to enhanced synthesis of cholesteryl esters compared with that in SV-589 (ΔUBIAD1) cells expressing the wild-type prenyltransferase (Fig. 4B). Notably, we did not observe significant differences in the amounts of triglycerides produced by SV-589 (ΔUBIAD1), SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (WT), and SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells (data not shown). In Fig. 4C, SV-589 (ΔUBIAD1), SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (WT), and SV-589 (ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells were cultured in FCS and subsequently stained with oil red O, a dye that stains neutral lipids such as triglycerides and cholesteryl esters. The results show that oil red O staining was reduced by almost 50% in SV-589 (ΔUBIAD1) cells compared with parental SV-589 cells (Fig. 4C, compare panels 1 and 2, and D). The level of oil red O staining was rescued by overexpression of wild-type UBIAD1 in SV-589 (ΔUBIAD1) cells (Fig. 4C, panel 3, and D) and neutral lipids overaccumulated ∼2-fold upon the overexpression of UBIAD1 (N102S) (Fig. 4C, panel 4, and D).
Figure 4.

Cholesterol ester synthesis and neutral lipid accumulation in SV-589 cells expressing wild-type and SCD-associated UBIAD1. A and B, SV589(ΔUBIAD1)/pMyc-UBIAD1 (WT) and SV589(ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells were set up on day 0 as described in the legend to Fig. 2. On day 1, the cells were refed medium A containing 10% DFCS. On day 2, the cells were refed the identical medium supplemented with 0.5 μCi/ml [3H]oleate and incubated for the indicated periods of time at 37 °C. Following incubations, the cells were harvested for preparation of lysates from which lipids were extracted and analyzed by TLC as described under “Experimental procedures.” Incorporation of [3H]oleate into [3H]cholesterol esters was determined by scintillation counting. The values were corrected for background as described in the legend to Fig. 2. Each value is the mean of triplicate incubations (± standard error). C, SV589(ΔUBIAD1)/pMyc-UBIAD1 (WT) and SV589(ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells were set up on day 0 at 3 × 105 cells/60-mm dish with glass coverslips in medium A containing 5% FCS and 1 mm mevalonate. On day 1, the medium was switched to medium A supplemented with 10% FCS. On day 2, the cells were fixed and double-stained with 4′6-diamino-phenylindole for nuclei (blue) and with oil red O for neutral lipids (red) as described under “Experimental procedures.” D, quantification of the oil red O signal was performed using ImageJ. The values are the average of 10 images (± standard error). Scale bar, 20 μm. The p values were calculated using the Student's t test: ***, p ≤ 0.001.
Considering our previous results that implicated UBIAD1 as a novel sensor of GGpp embedded in membranes (16) and the reciprocal synthesis of sterol and nonsterol isoprenoids in sterol-replete cells (3, 18), we next compared the ability of mevalonate to restore Golgi localization of endogenous UBIAD1 in statin-treated cells cultured in LPDS and FCS. When SV-589 cells were depleted of exogenous sterols through incubation in LPDS-containing medium, UBIAD1 localized to the Golgi as expected (Fig. 5A, panel 1). Treatment of the cells with compactin to deplete nonsterol isoprenoids disrupted Golgi localization of UBIAD1, causing it to accumulate in the ER (panel 2). A high concentration of mevalonate (10 mm) was required to stimulate transport of UBIAD1 from the ER to Golgi in the sterol and nonsterol isoprenoid-depleted cells (panels 3-6). The Golgi localization of UBIAD1 was also disrupted by compactin when SV-589 cells were grown in lipoprotein-rich FCS-containing medium (Fig. 5A, compare panels 7 and 8). However, Golgi transport of UBIAD1 was triggered by lower concentrations of mevalonate (1–3 mm) when SV-589 cells were cultured under sterol-replete conditions (panels 10-12). Similar results were obtained when isoprenoid-depleted SV-589 cells were treated with 25-HC rather than FCS for repletion of sterols. As shown in Fig. 5B, Golgi localization of UBIAD1 was restored by lower concentrations of mevalonate when cells were incubated with 25-HC (panels 10-12) compared with those required in the absence of the oxysterol (panels 4-6).
Figure 5.

Mevalonate restores Golgi localization of UBIAD1 in compactin-treated SV-589 cells. SV-589 cells were set up on day 0 at 8 × 104 cells/well of a 6-well dish containing glass coverslips in medium A supplemented with 5% FCS. On day 1, the cells were refed medium A supplemented with either 10% FCS or 10% LPDS, 10 μm compactin, and 0.05–10 mm mevalonate (A) or 0.3 μg/ml 25-HC (B) as indicated. Following incubation for ∼16 h at 37 °C, the cells were fixed and analyzed by immunofluorescence microscopy using IgG-1H12 (against human UBIAD1) and 4′6-diamino-phenylindole as described under “Experimental procedures.” Images were acquired as described in the legend to Fig. 4. Scale bars, 10 μm.
We next measured the synthesis of MK-4 in cells using a recently established assay in our group. SV-589 cells were deprived of isoprenoids through incubation in medium containing LPDS and compactin. In addition, the cells received [3H]menadione in the presence or absence of various concentrations of mevalonate (to provide substrates for synthesis of nonsterol isoprenoids). Following incubation for 16 h, the cells were lysed, and incorporation of [3H]menadione into MK-4 was determined by TLC and scintillation counting. The results show that the addition to cells of mevalonate led to a dose-dependent increase in the amount of [3H]menadione incorporated into MK-4 (Fig. 6A, white circles). Incorporation of [3H]menadione into MK-4 was enhanced at lower concentrations of mevalonate (>0.3 mm) when cells were treated with 25-HC (red circles).
Figure 6.

The oxysterol 25-hydroxycholesterol enhances synthesis of MK-4. A, SV-589 cells were set up on day 0 at 2.5 × 105 cells/60-mm dish in medium A supplemented with 10% FCS. On day 1, the cells were refed medium A supplemented with 10% LPDS, 10 μm compactin, 0.2 μCi/ml [3H]menadione, and the indicated concentration of mevalonate in the absence or presence of 0.3 μg/ml 25-HC. On day 2, the cells were harvested, the lipids were extracted, and the amount of [3H]menadione incorporated into MK-4 was determined by TLC and scintillation counting as described under “Experimental procedures.” The values are the means of triplicate samples (± standard error). B and C, SV-589 cells were set up on day 0 at 2.6 × 104 cells/60-mm dish (for RNA isolation), 7 × 105 cells/100-mm dish (subcellular fractionation) in medium A containing 10% FCS. On day 1, the cells were switched to medium A supplemented with 10% LPDS in the absence or presence of 10 μm compactin, 0.3 μg/ml 25-HC, and various concentrations of mevalonate as indicated. Following incubation for 16 h at 37 °C, the cells were harvested for isolation of total RNA (B) or subcellular fractionation (C) as described under “Experimental procedures.” B, total RNA from each condition was subjected to quantitative RT-PCR using primers against the indicated gene; 36B4 was used as an invariant control. Each value represents the amount of mRNA relative to that in untreated cells, which is arbitrarily defined as 1. The error bars represent ± standard error of triplicate samples. HMGCR, HMG CoA reductase; FPPS, farnesyl pyrophosphate synthase. C, resulting cytosolic, nuclear, and membrane fractions were subjected to SDS-PAGE (14–20 μg total protein/lane), followed by immunoblot analysis with antibodies against HMGCS, GGPPS, SREBP-2, nucleoporin, HMG CoA reductase, SQS, UBIAD1, actin, and calnexin.
In sterol-deprived cells, transcriptionally active fragments of SREBP-2 become proteolytically released from Golgi membranes (17, 19). Once released, these fragments migrate to the nucleus, where they modulate transcription of genes required for the synthesis of cholesterol (17). Sterol accumulation inhibits proteolytic release of SREBP-2 from membranes, which results in the decline in transcription of SREBP-2 target genes and the rate of cholesterol synthesis. Quantitative RT-PCR reveals that expression of mRNAs encoding reductase and other cholesterol biosynthetic enzymes including HMG CoA synthase (HMGCS), Fpp synthase, and squalene synthase (SQS) were markedly reduced by 25-HC (Fig. 6B). In contrast, levels of mRNAs for geranylgeranyl pyrophosphate synthase (GGPPS) and UBIAD1 remained constant regardless of the absence or presence of compactin, mevalonate, or 25-HC. Immunoblot analysis revealed a reduction in the protein levels for HMGCS, transcriptionally active nuclear SREBP-2, reductase, and SQS in 25-HC treated cells, whereas GGPPS and UBIAD1 protein levels were unchanged (Fig. 6C).
Discussion
The current results provide further evidence for a pivotal role of UBIAD1 in the regulation of reductase ERAD and metabolism of mevalonate. This evidence includes results showing that in the absence of UBIAD1, ERAD of reductase was accelerated (Fig. 1A), causing a fall in the synthesis of cholesterol (Fig. 2A) and cholesteryl esters (Fig. 4A). As a result, UBIAD1-deficient cells exhibited reduced storage of cholesteryl esters and other neutral lipids compared with wild-type cells as indicated by oil red O staining (Fig. 4, C and D). Nickerson et al. (20) reported an association of UBIAD1 with the ER enzyme acyl CoA cholesterol acyltransferase-1 (ACAT), which mediates synthesis of cholesteryl esters. However, we did not observe changes in the level of ACAT-1 in UBIAD1-deficient cells (Fig. 1A). Thus, reduced synthesis of cholesteryl esters in SV-589 (ΔUBIAD1) cells can be attributed to enhanced synthesis of cholesterol. In contrast, expression of SCD-associated UBIAD1 (N102S) led to the overaccumulation of neutral lipids (Fig. 4, C and D), because of the inhibition of reductase ERAD (Fig. 1A) that led to enhanced synthesis of cholesterol (Fig. 3A) and cholesteryl esters (Fig. 4B). These findings indicate that UBIAD1 (N102S)-mediated inhibition of reductase ERAD significantly contributes to enhanced synthesis and accumulation of cholesterol in cultured cells. Previously, we found that all 20 SCD-associated mutants of UBIAD1 are trapped in ER membranes and confer resistance of cells to growth in medium containing SR-12813 (16), a 1,1-bisphosphonate ester that mimics sterols in accelerating reductase ERAD (21). Thus, we feel the current results with UBIAD1 (N102S) are applicable to the remaining 19 SCD-associated UBIAD1 variants. Finally, it should be noted that although UBIAD1 (N102S) blocks sterol-accelerated ERAD of reductase, sterols continued to stimulate its ubiquitination (Fig. 1D). This result is consistent with our previous hypothesis that UBIAD1 inhibits post-ubiquitination steps in ERAD of reductase (12), the molecular basis of which is currently under investigation.
In contrast to our findings summarized above, previous studies reported reduced levels of cholesterol not only in cells overexpressing wild-type, full-length UBIAD1, but also in those expressing truncated and SCD-associated variants (including N102S) of UBIAD1 (22–24). In addition, a mild accumulation of intracellular cholesterol was observed upon RNAi-mediated knockdown of UBIAD1. It should be noted that all of the previous studies utilized an indirect method employing the enzyme cholesterol oxidase to measure intracellular cholesterol levels, whereas we directly measured synthesis of cholesterol (Figs. 2A and 3A) and cholesteryl esters (Fig. 4, A and B), as well as intracellular accumulation of neutral lipids (Fig. 4, C and D). Direct measurement of these parameters revealed that reduced synthesis and accumulation of cholesterol/cholesteryl esters correlated with reduced levels of reductase in UBIAD1-deficient cells, whereas enhanced synthesis and accumulation of cholesterol/cholesteryl esters in cells expressing SCD-associated UBIAD1 correlated with enhanced levels of reductase (Fig. 1A).
In 1979, Faust, Brown, and Goldstein (18) found that when cells were deprived of both sterol and nonsterol isoprenoids, reductase accumulated so as to produce mevalonate that was primarily converted into cholesterol (Fig. 7A). The addition of LDL to the cells satisfied their requirements for cholesterol and partially suppressed reductase, thereby limiting synthesis of mevalonate. As a result, incorporation of mevalonate into cholesterol was reduced, but paradoxically, its incorporation into ubiquinone-10 (and presumably other nonsterol isoprenoids) was enhanced (Fig. 7B). The results of Fig. 5 show that the Golgi localization of UBIAD1 parallels synthesis of nonsterol isoprenoids as reported by Faust et al. (18). Depleting cells of sterol and nonsterol isoprenoids through incubation in LPDS and compactin triggered the accumulation of reductase (Fig. 6C), which was be attributed to enhanced transcription of the reductase gene (Fig. 6B), and slowed ERAD of reductase protein (Fig. 6C). Under these conditions, a high concentration of mevalonate (10 mm) was required to stimulate the ER-to-Golgi transport of UBIAD1 (Fig. 5A). However, considerably less mevalonate (3–10-fold) caused UBIAD1 to appear in the Golgi when statin-treated cells were cultured in lipoprotein-rich FCS or LPDS plus 25-HC (Fig. 5B). Importantly, synthesis of MK-4 (used as a surrogate for production of GGpp) followed a similar pattern in that the reaction was enhanced in cells replete with sterols (Fig. 6A). These observations indicate that subcellular localization of UBIAD1 is a gauge for flux through the nonsterol branch of the mevalonate pathway.
Figure 7.

Flux through the mevalonate pathway in cells depleted of sterol and/or nonsterol isoprenoids. Shown is a schematic representation of flux through the mevalonate pathway when cells are depleted of both sterol and nonsterol isoprenoids (A) or replete with sterols and depleted of nonsterol isoprenoids (B) as determined by Faust et al. (18).
Based on previous (12, 16) and current results, we conclude that GGpp-regulated, ER-to-Golgi transport allows UBIAD1 to modulate reductase ERAD such that synthesis of nonsterol isoprenoids continues in sterol-replete cells. When cells are replete with sterols, reductase levels are suppressed by 1) reduced transcription of the reductase gene, caused by inhibition of SREBP-2, and 2) accelerated ERAD of reductase protein. However, sterols also trigger binding of UBIAD1 to a subset of reductase molecules (12). UBIAD1 binding inhibits ERAD of reductase, as indicated by reduced levels of the protein in UBIAD1-deficient cells cultured under sterol-replete conditions (Fig. 1A). This inhibition of reductase ERAD allows for production of small amounts of mevalonate that are diverted into GGpp and other nonsterol isoprenoids (i.e. dolichol, ubiquinone-10, etc.). In 1980, Brown and Goldstein (3) postulated that this diversion is facilitated by the high substrate affinity of enzymes in the nonsterol branch of the mevalonate pathway compared with that of enzymes in the sterol branch of the pathway. When GGpp accumulates to appropriate levels in ER membranes, the nonsterol isoprenoid binds to UBIAD1, causing its release from reductase. The GGpp-induced release permits transport of UBIAD1 from ER to Golgi and ERAD of reductase. The importance of this release is illustrated by the finding that GGpp-resistant UBIAD1 (N102S) is sequestered in the ER (Fig. S1) where it blocks reductase ERAD, which leads to enhanced synthesis and intracellular accumulation of cholesterol (Figs. 1, 3, and 4). Considering our recent discovery that sterol-accelerated ERAD plays a significant role in regulation of reductase and cholesterol homeostasis in whole animals (25, 26), we are now poised to establish a role for UBIAD1 in reductase ERAD in vivo and determine whether UBIAD1 (N102S)-mediated inhibition of the reaction contributes to the accumulation of cholesterol that characterizes SCD.
Experimental procedures
Materials
We obtained geranylgeraniol from Santa Cruz Biotechnology; 25-hydroxycholesterol was from Steraloids (Newport, RI); menadione was obtained from Sigma–Aldrich; and [3H]menadione, [3H]mevalonolactone, and [14C]acetate were obtained from American Radiolabeled Chemicals (St. Louis, MO). The expression plasmids, pCMV-Myc-UBIAD1, which encodes human UBIAD1 containing a single copy of a Myc epitope at the N terminus under transcriptional control of the cytomegalovirus promoter, and pCMV-Myc-UBIAD1 (N102S) encoding Myc-tagged human UBIAD1 harboring the substitution of serine for asparagine-102 (N102S) were previously described (12). Other reagents, including FCS, newborn calf LPDS (d > 1.215 g/ml), delipidated FCS (DFCS), sodium compactin, and sodium mevalonate, were prepared or obtained as previously described (11, 27, 28).
Cell culture
Monolayers of SV-589 cells, an immortalized line of human fibroblasts expressing the SV40 large T antigen (29), were maintained at 37 °C, 5% CO2 in medium A (Dulbecco's modified Eagle's medium containing 100 units/ml penicillin and 100 μg/ml streptomycin sulfate) supplemented with 10% (v/v) FCS. SV-589(ΔUBIAD1) cells, a line of UBIAD1-deficient SV-589 cells (12), were grown in medium A supplemented with 10% FCS and 1 mm mevalonate. SV-589(ΔUBIAD1)/pMyc-UBIAD1 (WT) and SV-589(ΔUBIAD1)/pMyc-UBIAD1 (N102S), lines of SV-589(ΔUBIAD1) cells that stably express wild-type (WT) or N102S Myc-UBIAD1, were generated by transfection of cells with 3 μg of pCMV-Myc-UBIAD1 (WT) or (N102S) as described previously (16), followed by 2 weeks of selection in medium A supplemented with 10% FCS, 1 mm mevalonate, and 700 μg/ml G418. Individual colonies were isolated using cloning cylinders, and expression of Myc-UBIAD1 was determined by immunoblot analysis. Cells from a single colony were cloned by limiting dilution; clones expressing similar levels of wild-type and N102S Myc-UBIAD1 (as determined by immunoblot analysis) were selected and maintained in medium A supplemented with 10% FCS, 1 mm mevalonate, and 700 μg/ml G418.
Subcellular fractionation, immunoprecipitation, and immunoblot analysis
The cells were set up for experiments on day 0 as described in the figure legends. Following incubations described in the figure legends, triplicate dishes of cells for each variable were harvested and pooled for analysis. Subcellular fractionation of cells by differential centrifugation was performed as described previously (12). For immunoprecipitations, detergent lysates were prepared and subjected to immunoprecipitation with polyclonal antibodies against the 60-kDa C-terminal domain of human reductase as described previously (21). Aliquots of detergent lysates and pellet fractions from immunoprecipitation and membrane fractions from subcellular fractionations were subjected to SDS-PAGE and immunoblot analysis. Primary antibodies used for immunoblot analysis included IgG-A9, a mouse monoclonal antibody against the catalytic domain of reductase (30); rabbit polyclonal anti-calnexin IgG (Novus Biologicals, Littleton, CO); IgG-9E10, a mouse monoclonal antibody against c-Myc purified from the culture medium of hybridoma clone 9E10 (American Type Culture Collection); mouse monoclonal anti-SOAT-1 IgG (Santa Cruz Biotechnology); IgG-P4D1, a mouse monoclonal antibody against bovine ubiquitin (Santa Cruz Biotechnology); IgG-1H12, a mouse monoclonal antibody against human UBIAD1; IgG-A6, a mouse monoclonal antibody against human cytosolic HMG CoA synthase (Santa Cruz Biotechnology); IgG-B2, a mouse monoclonal antibody against human geranylgeranyl pyrophosphate synthase (Santa Cruz Biotechnology); IgG-C4, a mouse monoclonal antibody against chicken actin (BD Biosciences); IgG-53, a mouse monoclonal antibody against human nucleoporin (BD Biosciences); IgG-22D5, a rabbit polyclonal antibody against SREBP-2; and anti-SQS (Santa Cruz Biotechnology).
Immunofluorescence
SV-589, SV-589(ΔUBIAD1)/pMyc-UBIAD1 (WT), and SV-589(ΔUBIAD1)/pMyc-UBIAD1 (N102S) cells were set up for experiments on day 0 as described in the figure legends. Following incubations described in figure legends, the cells were washed with PBS and subsequently fixed and permeabilized for 15 min in methanol at −20 °C. Upon blocking with 1 mg/ml BSA in PBS, coverslips were incubated for 30 min at 37 °C with primary antibodies (IgG-1H12, a mouse monoclonal antibody against human UBIAD1, or IgG-9E10, a mouse monoclonal antibody against c-Myc) diluted in PBS containing 1 mg/ml BSA. Bound antibodies were visualized with goat anti-mouse IgG conjugated to Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 594 (Life Technologies) as described in the figure legends. In addition, coverslips were stained for 5 min with 300 nm 4′,6-diamidino-2-phenylindole (Life Technologies) to visualize nuclei. The coverslips were then mounted with Fluoromount G (Southern Biotech, Birmingham, AL), and fluorescence analysis was performed using a Plan-Apochromat 63×/1.4 objective (Zeiss, Peabody, MA), an Axio Observer microscope (Zeiss), an), an Axiocam (Zeiss) color digital camera in black and white mode, and ImageJ software (National Institutes of Health, Bethesda, MD).
Metabolic labeling studies
Incorporation of [14C]acetate and [3H]mevalonate into cholesterol and that of [3H]oleate into cholesteryl esters was determined as described (27, 31) with minor modifications. For cholesterol synthesis experiments, the cells were set up on day 0 as described in the figure legends. On day 2, the cells received medium A supplemented with 10% FCS or LPDS and either 15 μCi/ml [14C]acetate (cold acetate was added to obtain a final concentration of 0.5 mm) or 10 μCi/ml [3H]mevalonate. Following incubation for 4 h at 37 °C, the cell lysates were prepared, and lipids were extracted and separated by TLC on plastic-backed silica gel TLC plates (Macherey–Nagel) developed in 100% chloroform as described (32). The amount of radioactivity incorporated into cholesterol was determined by scintillation counting. To establish background radioactivity, the cells were chilled to 4 °C, refed with radiolabeled medium and immediately washed and extracted. An aliquot of each sample was taken for protein determination using the BCA protein assay reagent (Thermo Scientific).
For cholesteryl ester synthesis experiments, the cell lines were cultured on day 0 as described in the figure legends. On day 1, the cells were switched to medium A containing 10% DFCS. On day 2, the cells were refed identical medium containing 0.5 μCi/ml [3H]oleate and incubated for various periods of times at 37 °C. The cells were subsequently washed twice with PBS + 2% BSA at room temperature, followed by two washes with PBS. The lipids were then extracted with hexane/isopropanol (3:2) for 30 min at room temperature. Extracted lipids were transferred to glass tubes and mixed with recovery solution containing 0.25 μCi/ml [14C]triolein plus 6.8 mg/ml cold triolein and 10 mg/ml cholesteryl ester (chloroform/methanol, 2:1 solvent). The lipids were dried down and resuspended in 40 μl of heptane and spotted on TLC plates that were developed in heptane:diethlyether:glacial acid (90:30:1) solvent. The lipids were visualized by staining in iodine vapor, and incorporation of [3H]oleate into CE was determined by scintillation counting. The values were corrected for background as described above; they were also corrected for recovery as judged by the percent of [14C]triolein radioactivity recovered in each sample. An aliquot of each sample was taken for protein determination using the BCA protein assay reagent (Thermo Scientific Pierce).
In studies measuring incorporation of [3H]menadione into MK-4, SV-589 cells were set up on day 0 as described in figure legends. On day 1, the cells were refed medium A containing 10% LPDS, 10 μm compactin, and 0.2 μCi/ml [3H]menadione (cold menadione was added to achieve a final concentration of 50 nm). After 16 h at 37 °C, the cells were washed twice with PBS + 2% BSA, followed by an additional wash with PBS. The cells were then lysed with 0.1 n NaOH; the resulting lysates were mixed with recovery solution containing 16 μg/ml MK-4, 0.025 μCi/ml [14C]cholesterol, and 16 μg/ml unlabeled cholesterol and extracted with dicholoromethane:methanol (2:1). The lipids were dried down, resuspended in dicholoromethane, and spotted on TLC plates that were developed in chloroform. The lipids were visualized by staining in iodine vapor and incorporation of [3H]menadione into MK-4 was determined by scintillation counting. The values were corrected for recovery as judged by the percent of [14C]cholesterol recovered in each sample. An aliquot of each sample was taken for protein determination.
Neutral lipid staining of cells with oil red O
The cells were set up for experiments on day 0 as described in figure legends. On day 2, the cells were washed with PBS and then fixed in 4% paraformaldehyde for 20 min at room temperature. The fixed cells were then stained with oil red O solution for 10 min. Following multiple washes with PBS, the coverslips were stained with 300 nm DAPI in PBS for 5 min at room temperature. After additional washes with PBS, the coverslips were mounted with Fluoromount G. Images were acquired using a Zeiss Axio Observer Epifluorescence microscope using a 63×/1.4 oil Plan-Apochromat objective and Zeiss Axiocam color digital camera (Zeiss, Peabody, MA) in black and white mode.
Quantitative real-time PCR
The protocol for quantitative RT-PCR was similar to that described previously (33). Total RNA was prepared from SV-589 cells using Tri-Reagent (Molecular Research Center, Cincinnati, OH). Equal amounts of RNA were treated with DNase I (DNA-freeTM; ThermoFisher Scientific). First strand cDNA was synthesized from 2 μg of DNase I-treated total RNA with random hexamer primers using TaqMan reverse transcription reagents (Applied Biosystems/Roche Applied Science). Specific primers for each gene were designed using Primer Express software (Life Technologies). Triplicate RT-PCRs were set up in a final volume of 20 μl containing 20 ng of reverse-transcribed total RNA, 167 nm forward and reverse primers, and 10 μl of 2× SYBR Green PCR Master Mix (Life Technologies). The relative amount of all mRNAs was calculated using the comparative threshold cycle (CT) method. Human 36B4 mRNA were used as the invariant control. Sequences of the primers for RT-PCR used in the current study are listed in Table 1.
Table 1.
Primers used for real-time PCR analysis
| Gene | Primers |
|---|---|
| 36B4 (control) | |
| Forward | 5′-GGCCTGAGCTCCCTGTCTCT-3′ |
| Reverse | 5′-GCGGTGCGTCAGGGATT-3′ |
| HMGCS | |
| Forward | 5′-GACTTGTGCATTCAAACATAGCAA-3′ |
| Reverse | 5′-GCTGTAGCAGGGAGTCTTGGTACT-3′ |
| HMG CoA reductase | |
| Forward | 5′-CAAGGAGCATGCAAAGATAATCC-3′ |
| Reverse | 5′-GCCATTACGGTCCCACACA-3′ |
| Fpp synthase | |
| Forward | 5′-GCCCCCAGCCGTCTTT-3′ |
| Reverse | 5′-CCTTGCAATCTCTAGGTCACTTTCT-3′ |
| GGPPS | |
| Forward | 5′-TCCGACGTGGCTTTCCA-3′ |
| Reverse | 5′-CGTAATTGGCAGAATTGATGACA-3′ |
| SQS | |
| Forward | 5′-ATGACCATCAGTGTGGAAAAGAAG-3′ |
| Reverse | 5′-CCGCCAGTCTGGTTGGTAA-3′ |
| SREBP-2 | |
| Forward | 5′-CGGTAATGATCACGCCAACAT-3′ |
| Reverse | 5′-TGGTATATCAAAGGCTGCTGGAT-3′ |
| UBIAD1 | |
| Forward | 5′-ACTTGTGGACCGAATCTTGGA-3′ |
| Reverse | 5′-CGCAGCCCAACGTGTAGAG-3′ |
Author contributions
M. M. S. and R. A. D.-B. conceptualization; M. M. S., D.-J. J., and B. M. J. data curation; M. M. S., D.-J. J., B. M. J., and R. A. D.-B. formal analysis; M. M. S., D.-J. J., B. M. J., and R. A. D.-B. validation; M. M. S. and R. A. D.-B. investigation; M. M. S., D.-J. J., and B. M. J. methodology; M. M. S. and R. A. D.-B. writing-original draft; M. M. S., D.-J. J., B. M. J., and R. A. D.-B. writing-review and editing; R. A. D.-B. supervision; R. A. D.-B. funding acquisition; R. A. D.-B. project administration.
Supplementary Material
Acknowledgments
We thank Kristi Garland-Brasher and Gennipher Young-Smith for excellent technical assistance and Lisa Beatty, Shomanike Head, and Ijeoma Onweneme for help with tissue culture.
This work was supported by National Institutes of Health Grants HL020948 and GM112409 (to R. D. B.) The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- Fpp
- farnesyl pyrophosphate
- GGpp
- geranylgeranyl pyrophosphate
- SCD
- Schnyder corneal dystrophy
- HMG
- 3-hydroxy-3-methylglutaryl
- MK-4
- menaquinone-4
- LPDS
- lipoprotein-deficient serum
- 25-HC
- 25-hydroxycholesterol
- SREBP
- sterol regulatory element-binding protein
- HMGCS
- HMG CoA synthase
- SQS
- squalene synthase
- GGPPS
- geranylgeranyl pyrophosphate synthase
- ACAT
- acyl CoA cholesterol acyltransferase
- DFCS
- delipidated FCS.
References
- 1. Goldstein J. L., and Brown M. S. (1990) Regulation of the mevalonate pathway. Nature 343, 425–430 10.1038/343425a0 [DOI] [PubMed] [Google Scholar]
- 2. Edwards P. A., and Ericsson J. (1999) Sterols and isoprenoids: signaling molecules derived from the cholesterol biosynthetic pathway 63. Annu. Rev. Biochem. 68, 157–185 10.1146/annurev.biochem.68.1.157 [DOI] [PubMed] [Google Scholar]
- 3. Brown M. S., and Goldstein J. L. (1980) Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J. Lipid Res. 21, 505–517 [PubMed] [Google Scholar]
- 4. Sever N., Yang T., Brown M. S., Goldstein J. L., and DeBose-Boyd R. A. (2003) Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol. Cell 11, 25–33 10.1016/S1097-2765(02)00822-5 [DOI] [PubMed] [Google Scholar]
- 5. Sever N., Song B. L., Yabe D., Goldstein J. L., Brown M. S., and DeBose-Boyd R. A. (2003) Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem. 278, 52479–52490 10.1074/jbc.M310053200 [DOI] [PubMed] [Google Scholar]
- 6. Roitelman J., Olender E. H., Bar-Nun S., Dunn W. A. Jr, and Simoni R. D. (1992) Immunological evidence for eight spans in the membrane domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase: implications for enzyme degradation in the endoplasmic reticulum. J. Cell Biol. 117, 959–973 10.1083/jcb.117.5.959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liscum L., Finer-Moore J., Stroud R. M., Luskey K. L., Brown M. S., and Goldstein J. L. (1985) Domain structure of 3-hydroxy-3-methylglutaryl coenzyme A reductase, a glycoprotein of the endoplasmic reticulum. J. Biol. Chem. 260, 522–530 [PubMed] [Google Scholar]
- 8. Song B. L., Sever N., and DeBose-Boyd R. A. (2005) Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 19, 829–840 10.1016/j.molcel.2005.08.009 [DOI] [PubMed] [Google Scholar]
- 9. Jo Y., Lee P. C., Sguigna P. V., and DeBose-Boyd R. A. (2011) Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc. Natl. Acad. Sci. U.S.A. 108, 20503–20508 10.1073/pnas.1112831108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morris L. L., Hartman I. Z., Jun D. J., Seemann J., and DeBose-Boyd R. A. (2014) Sequential actions of the AAA-ATPase valosin-containing protein (VCP)/p97 and the proteasome 19 S regulatory particle in sterol-accelerated, endoplasmic reticulum (ER)-associated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 289, 19053–19066 10.1074/jbc.M114.576652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elsabrouty R., Jo Y., Dinh T. T., and DeBose-Boyd R. A. (2013) Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from membranes of permeabilized cells. Mol. Biol. Cell 24, 3300–3308 10.1091/mbc.E13-03-0157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schumacher M. M., Elsabrouty R., Seemann J., Jo Y., and DeBose-Boyd R. A. (2015) The prenyltransferase UBIAD1 is the target of geranylgeraniol in degradation of HMG CoA reductase. eLife 4, 10.7554/eLife.05560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakagawa K., Hirota Y., Sawada N., Yuge N., Watanabe M., Uchino Y., Okuda N., Shimomura Y., Suhara Y., and Okano T. (2010) Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature 468, 117–121 10.1038/nature09464 [DOI] [PubMed] [Google Scholar]
- 14. Orr A., Dubé M. P., Marcadier J., Jiang H., Federico A., George S., Seamone C., Andrews D., Dubord P., Holland S., Provost S., Mongrain V., Evans S., Higgins B., Bowman S., et al. (2007) Mutations in the UBIAD1 gene, encoding a potential prenyltransferase, are causal for Schnyder crystalline corneal dystrophy. PLoS One 2, e685 10.1371/journal.pone.0000685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weiss J. S., Kruth H. S., Kuivaniemi H., Tromp G., White P. S., Winters R. S., Lisch W., Henn W., Denninger E., Krause M., Wasson P., Ebenezer N., Mahurkar S., and Nickerson M. L. (2007) Mutations in the UBIAD1 gene on chromosome short arm 1, region 36, cause Schnyder crystalline corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 48, 5007–5012 10.1167/iovs.07-0845 [DOI] [PubMed] [Google Scholar]
- 16. Schumacher M. M., Jun D. J., Jo Y., Seemann J., and DeBose-Boyd R. A. (2016) Geranylgeranyl-regulated transport of the prenyltransferase UBIAD1 between membranes of the ER and Golgi. J. Lipid Res. 57, 1286–1299 10.1194/jlr.M068759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Horton J. D., Shah N. A., Warrington J. A., Anderson N. N., Park S. W., Brown M. S., and Goldstein J. L. (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. U.S.A. 100, 12027–12032 10.1073/pnas.1534923100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Faust J. R., Goldstein J. L., and Brown M. S. (1979) Synthesis of ubiquinone and cholesterol in human fibroblasts: regulation of a branched pathway. Arch. Biochem. Biophys. 192, 86–99 10.1016/0003-9861(79)90074-2 [DOI] [PubMed] [Google Scholar]
- 19. Goldstein J. L., DeBose-Boyd R. A., and Brown M. S. (2006) Protein sensors for membrane sterols. Cell 124, 35–46 10.1016/j.cell.2005.12.022 [DOI] [PubMed] [Google Scholar]
- 20. Nickerson M. L., Bosley A. D., Weiss J. S., Kostiha B. N., Hirota Y., Brandt W., Esposito D., Kinoshita S., Wessjohann L., Morham S. G., Andresson T., Kruth H. S., Okano T., and Dean M. (2013) The UBIAD1 prenyltransferase links menaquinone-4 [corrected] synthesis to cholesterol metabolic enzymes. Hum. Mutat. 34, 317–329 10.1002/humu.22230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sever N., Lee P. C., Song B. L., Rawson R. B., and DeBose-Boyd R. A. (2004) Isolation of mutant cells lacking Insig-1 through selection with SR-12813, an agent that stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 279, 43136–43147 10.1074/jbc.M406406200 [DOI] [PubMed] [Google Scholar]
- 22. Fredericks W. J., McGarvey T., Wang H., Lal P., Puthiyaveettil R., Tomaszewski J., Sepulveda J., Labelle E., Weiss J. S., Nickerson M. L., Kruth H. S., Brandt W., Wessjohann L. A., and Malkowicz S. B. (2011) The bladder tumor suppressor protein TERE1 (UBIAD1) modulates cell cholesterol: implications for tumor progression. DNA Cell Biol. 30, 851–864 10.1089/dna.2011.1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fredericks W. J., Yin H., Lal P., Puthiyaveettil R., Malkowicz S. B., Fredericks N. J., Tomaszewski J., Rauscher F. J. 3rd, Malkowicz S. B. (2013) Ectopic expression of the TERE1 (UBIAD1) protein inhibits growth of renal clear cell carcinoma cells: altered metabolic phenotype associated with reactive oxygen species, nitric oxide and SXR target genes involved in cholesterol and lipid metabolism. Int. J. Oncol. 43, 638–652 10.3892/ijo.2013.1985 [DOI] [PubMed] [Google Scholar]
- 24. Liu S., Guo W., Han X., Dai W., Diao Z., and Liu W. (2016) Role of UBIAD1 in intracellular cholesterol metabolism and vascular cell calcification. PLoS One 11, e0149639 10.1371/journal.pone.0149639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hwang S., Hartman I. Z., Calhoun L. N., Garland K., Young G. A., Mitsche M. A., McDonald J., Xu F., Engelking L., and DeBose-Boyd R. A. (2016) Contribution of accelerated degradation to feedback regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase and cholesterol metabolism in the liver. J. Biol. Chem. 291, 13479–13494 10.1074/jbc.M116.728469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hwang S., Nguyen A. D., Jo Y., Engelking L. J., Brugarolas J., and DeBose-Boyd R. A. (2017) Hypoxia-inducible factor 1alpha activates insulin-induced gene 2 (Insig-2) transcription for degradation of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase in the liver. J. Biol. Chem. 292, 9382–9393 10.1074/jbc.M117.788562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goldstein J. L., Basu S. K., and Brown M. S. (1983) Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 98, 241–260 10.1016/0076-6879(83)98152-1 [DOI] [PubMed] [Google Scholar]
- 28. Hannah V. C., Ou J., Luong A., Goldstein J. L., and Brown M. S. (2001) Unsaturated fatty acids down-regulate srebp isoforms 1a and 1c by two mechanisms in HEK-293 cells. J. Biol. Chem. 276, 4365–4372 10.1074/jbc.M007273200 [DOI] [PubMed] [Google Scholar]
- 29. Yamamoto T., Davis C. G., Brown M. S., Schneider W. J., Casey M. L., Goldstein J. L., and Russell D. W. (1984) The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell 39, 27–38 10.1016/0092-8674(84)90188-0 [DOI] [PubMed] [Google Scholar]
- 30. Liscum L., Luskey K. L., Chin D. J., Ho Y. K., Goldstein J. L., and Brown M. S. (1983) Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase and its mRNA in rat liver as studied with a monoclonal antibody and a cDNA probe. J. Biol. Chem. 258, 8450–8455 [PubMed] [Google Scholar]
- 31. Brown M. S., Faust J. R., Goldstein J. L., Kaneko I., and Endo A. (1978) Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J. Biol. Chem. 253, 1121–1128 [PubMed] [Google Scholar]
- 32. Jo Y., Hartman I. Z., and DeBose-Boyd R. A. (2013) Ancient ubiquitous protein-1 mediates sterol-induced ubiquitination of 3-hydroxy-3-methylglutaryl CoA reductase in lipid droplet-associated endoplasmic reticulum membranes. Mol. Biol. Cell 24, 169–183 10.1091/mbc.E12-07-0564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liang G., Yang J., Horton J. D., Hammer R. E., Goldstein J. L., and Brown M. S. (2002) Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 277, 9520–9528 10.1074/jbc.M111421200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.