

Abstract
Domain-minimized insulin receptors (IRs) have enabled crystallographic analysis of insulin-bound “micro-receptors.” In such structures, the C-terminal segment of the insulin B chain inserts between conserved IR domains, unmasking an invariant receptor-binding surface that spans both insulin A and B chains. This “open” conformation not only rationalizes the inactivity of single-chain insulin (SCI) analogs (in which the A and B chains are directly linked), but also suggests that connecting (C) domains of sufficient length will bind the IR. Here, we report the high-resolution solution structure and dynamics of such an active SCI. The hormone's closed-to-open transition is foreshadowed by segmental flexibility in the native state as probed by heteronuclear NMR spectroscopy and multiple conformer simulations of crystallographic protomers as described in the companion article. We propose a model of the SCI's IR-bound state based on molecular-dynamics simulations of a micro-receptor complex. In this model, a loop defined by the SCI's B and C domains encircles the C-terminal segment of the IR α-subunit. This binding mode predicts a conformational transition between an ultra-stable closed state (in the free hormone) and an active open state (on receptor binding). Optimization of this switch within an ultra-stable SCI promises to circumvent insulin's complex global cold chain. The analog's biphasic activity, which serendipitously resembles current premixed formulations of soluble insulin and microcrystalline suspension, may be of particular utility in the developing world.
Keywords: diabetes, hormone, protein engineering, protein structure, receptor tyrosine kinase
Introduction
Insulin is a small globular protein that regulates metabolic homeostasis in vertebrates. A classical model for studies of protein structure and long a mainstay of therapy for diabetes mellitus (DM),4 insulin was the first recombinant protein in clinical use and a pioneering target of protein engineering (1). Whereas design of first-generation analogs sought to optimize pharmacokinetic (PK) properties (i.e. rapid- or long-acting therapeutic formulations), recent efforts have focused on protein stability (2–4). Ultra-stable insulin analogs promise to circumvent a costly “cold chain” underlying its global distribution (2). The salience of this issue has been sharpened by an emerging pandemic of DM in the developing world (5).
Insulin contains two chains:5 an A chain (21 residues) and B chain (30 residues) (Fig. 1A) (6). The mature hormone nonetheless belongs to a metazoan superfamily of single-chain proteins (7) and itself derives from a single-chain precursor (proinsulin (8)). In the accompanying article (9), we described an ultra-stable single-chain insulin analog (labeled SCI-a; Fig. 1C) with appropriate biological activity (including duration of signaling) in a rat model (9). The crystal structure of SCI-a, determined as a non-canonical zinc-free hexamer, revealed native-like component dimers with apparent disorder in the engineered C domains and adjoining B domain residues. Because of this structure's limited resolution (2.8 Å) and incomplete electron density, we sought a high-resolution solution structure and its dynamic assessment. This study thus focused on a monomeric variant of SCI-a, viz. SCI-b, that is amenable to heteronuclear 3D/4D-NMR spectroscopy (10, 11). SCI-b differed from its parent at residues B28 and B29, recapitulating features of rapid-acting insulin analogs (Fig. 1B) (12, 13). Intravenous (i.v.) bolus injection of SCI-b led, for unknown reasons, to protracted insulin action (9), similar to that of ultra-stable two-chain insulin analog cross-linked by an additional disulfide bridge (3). The monophasic pharmacodynamics (PD) profile of SCI-a by contrast resembled that of wild-type (WT) insulin (9).
Figure 1.

Structural overview and insulin analog sequences. A, crystal structure of WT porcine insulin (PDB code 4INS (6)) from two perspectives showing A (green) and B (blue) chains and disulfide bridges (yellow sticks): A20–B19, A7–B7, and A6–A11 (black boxed labels). Side chains of ThrA8 and TyrA14 are highlighted as red sticks. The C-terminal B chain segment is labeled. B, protein sequences of WT insulin, insulin aspart, and insulin lispro. C, protein sequences of novel single chains SCI-a and SCI-b as well as progenitor SCI-c (4). SCIs contain a six-residue C domain; peptide bonds connecting the A and B domains to the C domain are shown as red lines. Substitutions relative to WT insulin in B and C are shown in red. Gold lines in sequences indicate disulfide bonds. Black arrows at bottom of C highlight residues B10, B28, B29, and A14.
Insulin binds as a monomer to a receptor tyrosine kinase (TK), the insulin receptor (IR). The product of a single gene, the IR precursor is processed in the trans-Golgi network to form a disulfide-linked (αβ)2 homodimer (14). The extracellular α subunits bind insulin, and the transmembrane β subunits contain the intracellular TK domains (15). Whereas the structure of the holoreceptor has been visualized only at low resolution (>20 Å (16)), its domain dissection has enabled stepwise crystallographic analysis. (i) Structures of the intracellular TK domains and their mode of interaction have been determined at a resolution of 1.9 Å (17, 18). (ii) The structure of the N-terminal three domains of the α subunit (Leu-rich domain 1, Cys-rich domain, and Leu-rich domain 2; L1-CR-L2) has been determined at 2.3 Å (19). (iii) The structure of the dimeric ectodomain ((αβΔ)2, containing the entire α subunit and the extracellular portion of the β subunit, βΔ) has been determined at 3.3 Å (20), wherein its conformation resembles an inverted V. Although co-crystals of an insulin–ectodomain complex have not been obtained, a variety of evidence indicates that insulin binds within the crux of the dimer (15, 21, 22).
Construction of domain-minimized models of the IR α subunit has enabled crystallization of “micro-receptor” (μIR) complexes (15, 21). Such complexes contain, at a minimum, the primary insulin-binding elements L1 and αCT stabilized by an Fab antibody fragment. One such co-crystal structure, determined at 3.5 Å resolution, depicted a ternary complex between insulin, an L1-CR fragment, and a synthetic αCT peptide spanning residues 704–719 of IR isoform A (15). In this structure the C-terminal segment of the insulin B chain pivots from the hormone's α-helical core to enable its insertion between the conserved surfaces of L1 and the αCT peptide. This mode of binding, anticipated based on studies of anomalous insulin analogs (23, 24) and residue-specific photo-cross-linking (25), defined the binding surfaces in the μIR for insulin's conserved triplet of aromatic residues: PheB24, PheB25, and TyrB26. The side chain of TyrB26, although important for insulin self-assembly (6), packs at a solvated edge of the μIR (15) and can be substituted by Ala, Ser, or Glu without loss of affinity (26).
The solution structure of SCI-b presented here contains a well-ordered α-helical domain whose overall structure is similar to corresponding portions of the crystallographic protomers of SCI-a. The latter resemble T-state protomers as observed in T6 crystal structures of WT insulin (9). The B chain β-strand (residues B24–B28) exhibits local order in the absence of a dimer-related β-sheet, but its precise positioning relative to the A and B domain α-helices differs from crystallographic dimers (9). The C domain is less well-ordered than the globular domain, but its ensemble of conformations is not random; non-polar and electrostatic contacts with residues in the N-terminal α-helix of the A domain are on average maintained. Order parameters derived from 13C chemical shifts and 1H-15N relaxation studies are in accordance with these trends.
NMR-derived structures of proteins are ordinarily based on the assumption that all restraints (as derived from nuclear Overhauser effects (NOEs), J-coupling constants, and patterns of chemical shifts (27)) are enforced simultaneously (28). This assumption may not be valid in the presence of local or segmental mobility (29). Accordingly, we have extended such modeling through use of time-averaged molecular-dynamics (MD) simulations in which all or part of the protein satisfies the NMR-derived restraints only as time-averaged in an MD trajectory (30, 31). This dynamic view was extended by simulated annealing (SA) and multiconformer simulation within the SCI-a crystal lattice to obtain an ensemble of possible states. A third and more distantly related ensemble was obtained through MD simulations of SCI-b as docked within a μIR complex (15, 21). Comparison of these ensembles highlights the role of segmental mobility in the mechanism of receptor binding.
Together, our results imply a functional transition between an ultra-stable closed state in the free hormone and an active open state on receptor binding. A model of an SCI–μIR complex is proposed in which the critical IR αCT element “threads” within a loop opened by displacement of the B domain C-terminal segment and bounded by a disordered C domain. Beyond these mechanistic implications, the biphasic PD features of SCI-b in vivo closely matched the dual-action profile of premixed soluble and microcrystalline insulin formulations (32): regimens with potential benefits among minority communities in the West (33) and of broad current use in Africa (34) and Asia (35). The biphasic action profile of the present SCI together with its extraordinary resistance to thermal degradation (9) may enhance access to insulin among underprivileged patients in the developing world (2, 5).
Results
SCI-a and SCI-b are described in the accompanying article (9). Designed based on prior studies of two-chain insulin analogs (12, 13), chemically cross-linked analogs (36), and an SCI prototype (4), these proteins contain sequence elements intended to co-optimize specific biochemical and biophysical properties (Fig. 1C). Whereas SCI-a (amenable to protective self-assembly) was envisaged as a therapeutic candidate, SCI-b provides a monomeric model amenable to high-resolution NMR study. Paired substitutions AspB28 and ProB29 combine features of two rapid-acting analogs in current clinical use (insulins lispro and aspart) (Fig. 1B) (12, 13). Dimerization and in turn higher-order assembly are impaired by (i) the absence of proline at B28, whose pyrrolidine ring engages GlyB23′ across the dimer interface (37), and (ii) the negative charge of AspB28 itself (1). The tractability of the one-dimensional 1H NMR spectrum of SCI-b at high protein concentration (9) motivated its present uniform 15N/13C-labeling in yeast Pichia pastoris; heteronuclear resonances assignments were obtained by standard methods (10, 27).
Solution structure resembles crystallographic protomers
The solution structure of SCI-b was calculated on the basis of 1370 NOE distance restraints (average of 24 per residue; Table S1) and 75 dihedral angle restraints. An ensemble, based on simultaneous enforcement of these restraints, contains a native-like insulin fold (A and B domains; green and blue in Fig. 2, A and B) connected by the 6-residue C domain (red in Fig. 2, A and B). Statistics are given in Table S1. The SCI-b core is similar to that of WT insulin (gray in Fig. 2C) and our previous SCI prototype (gray ensemble in Fig. 2D). In the SCI-b monomer residues, B23–B28 appear less well-ordered and displaced relative to the corresponding segments at a classical dimer interface in SCI-a (9), wherein B24–B28 and its dimer-related partner form an anti-parallel β-sheet (6). Following helical alignment of the SCI-b ensemble to all protomers of SCI-a (Fig. 3A), the mean root-mean-square deviations (r.m.s.d.) of respective residues B24–B26 are 1.2(±0.3), 1.3(±0.3), and 2.0(±0.5) Å, respectively. SCI-b also differs from crystallographic protomers of SCI-a in the presumed orientation of the B27-C6 segment, although well-defined electron density in the latter structure was not observed (9).
Figure 2.

SCI-b structural ensemble. A, 30 best-fit structures of SCI-b at two views rotated by 180°. B, expanded view of the C domain. C and D, overlay of WT porcine insulin (gray; PDB code 4INS) (C) and published SCI-c NMR ensemble (gray; PDB code 2JZQ) relative to SCI-b (red, green, blue) (D). Superpositions were aligned according to the main-chain atoms of the A1–A8, A13–A19, and B9–B19 segments. A representative model in E–G was selected to demonstrate close proximity of ProC4-Hγ2 and ValA3-Hγ2 extending the A1–A8 helix (E), the salt bridge between ArgC6 guanidinium nitrogen and GluA4 carboxylate (F), and putative π-cation interactions between ArgC5 and TyrA19 or PheB25 (G). Color code: A domain (green); B domain (blue); C domain linker (red); disulfide bridges (yellow sticks); oxygen (red); nitrogen (cyan); carbon (black); and hydrogen (white). Distance measurements are indicated by red dashed lines; π-cation distances were measured from centers of the aromatic rings (small black spheres) to the terminal nitrogen atoms of ArgC5.
Figure 3.
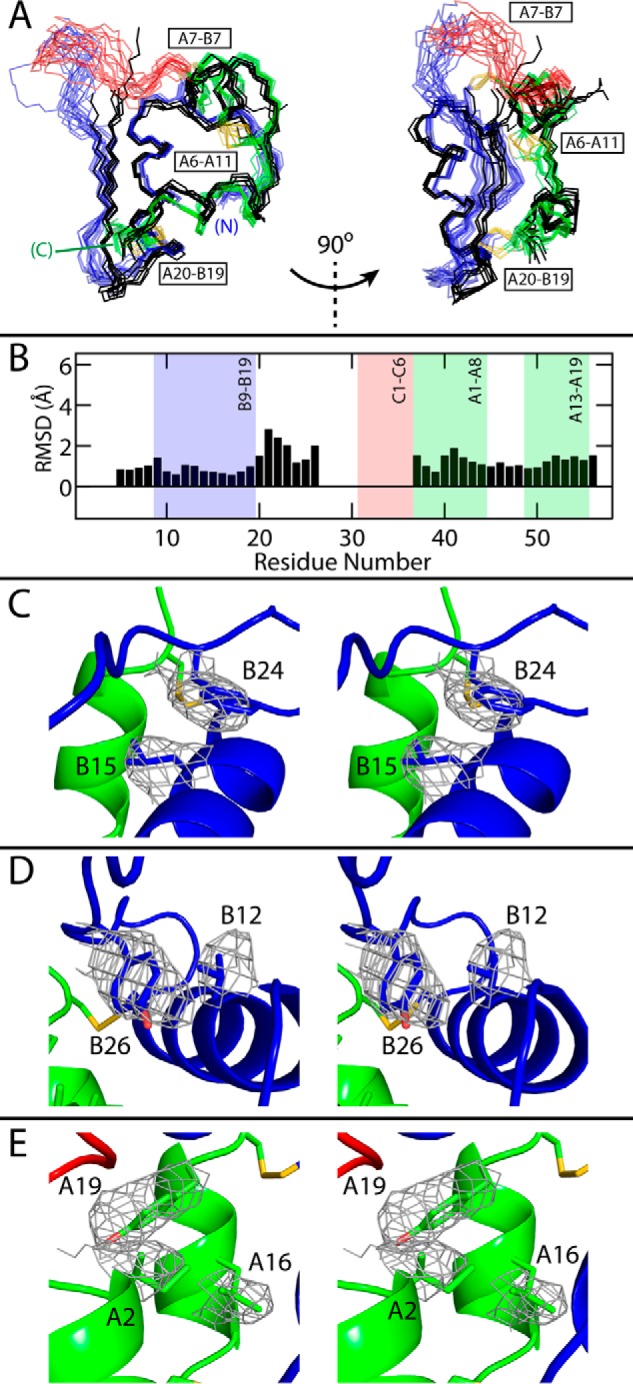
Comparison of SCI structures. A, backbone traces of 18 SCI-b NMR structures (with the A domain in green; B domain in blue; C domain in red; and disulfide bridges in yellow) with all six monomers of SCI-a (black, crystal refinement from accompanying article (9)); the structures were aligned as in Fig. 2. Two views are given at relative angle 90° to illustrate differences among B24–B30 segments. For clarity, SCI-b structures are at low opacity. B, average pairwise backbone r.m.s.d. comparing all monomers of SCI-a to 18 SCI-b structures. Absent r.m.s.d. values reflect residues with indeterminate electron densities. C–E, stereo models illustrating side-chain packing of residues B15 and B24 (C), B12 and B26 (D), and A2, A16, and A19 (E) within the mean SCI-b structure overlaid with the side-chain electron densities of SCI-a monomer D; structures were aligned as in Fig. 2 (SCI-a structure is hidden for clarity). Disulfide bridges are shown in yellow, and atoms otherwise colored by domain: A domain, green; B domain, blue; C domain, red; and atom colors: oxygen, light red; and nitrogen, light blue.
The C domain is neither well-ordered nor disordered. Its ensemble of trajectories spans a crevice between A and B domains. Segmental imprecision reflected a lower density of NOE restraints; evidence of underlying dynamic disorder is presented below. Apparently, stabilizing interactions were introduced by the arginine guanidinium groups as follows (Fig. 2, E–G). (i) The N-terminal portion of the A1–A8 α-helix extends to include residues C4–C6 (sequence PRR) as demonstrated by the proximity of ProC4 and ValA3 (Fig. 2E; corresponding 4D NOESY cross-peaks in Fig. S1). (ii) A salt bridge is formed between ArgC6 and GluA4 (Fig. 2F). (iii) Favorable π-cation interactions (38) are inferred between TyrA19–ArgC5 and possibly PheB25–ArgC5 (Fig. 2G). The latter is observed in some but not all members of the ensemble due to imprecision at residue B25. All residues within the insulin moiety mutated relative to WT (B28, B29, A8 and A14; Fig. S2) are solvent-exposed. Whereas the A8 and A14 side chain positions are well-defined within the ensemble, AspB28 and ProB29 side chains are imprecise, residing proximal to the flexible BC junction.
Segmental imprecision in the NMR-derived ensemble mirrors pattern of order parameters
To probe protein dynamics, 15N relaxation studies were undertaken to measure residue-specific R1 and R2 relaxation rates and heteronuclear NOE intensities (Fig. 4A). These data, probing motions on the picosecond to nanosecond time scale, enabled calculation of generalized order parameters (S2) (Fig. 4B, top). Such parameters distinguish between rigid (S2 ≥ 0.65) and flexible (S2 < 0.65) elements (39). The latter correlated with sites of fast 1H–2H amide-proton exchange (red squares) as described below. These residues were fit best by an extended model-free analysis where S2fast = S2 − S2slow (see “Experimental procedures”).
Figure 4.

Fast time scale dynamical studies. A, 15N spin-lattice (R1, black) and spin-spin (R2, blue) relaxation rates and heteronuclear NOEs (red). Data were obtained using a 13C,15N-SCI-b sample. Vertical error bars in each plot result from input spectral noise in each T1 or T2 experiment and subsequent error analysis by the Relax NMR software package (90). B, top, generalized (S2, black squares) and fast (S2fast, red squares) order parameters calculated from experimental 1H-15N T1, T2, and heteronuclear NOE data with the DYNAMICS software package (91). Predicted generalized order parameter (S2′; middle plot) and secondary structure prediction scores (bottom plot) were calculated by the TALOS+ software program (60) based on chemical shifts. In all plots, blue-, red-, and green-shaded areas, respectively, highlight the B9–B19 helix, the C1–C6 segment, A1–A8 helix, and the A13–A19 helix. For SSP probabilities (bottom), positive values are associated with α-helix, whereas negative values suggest β-strand; an SSP value of zero implies disorder.
Regions of flexibility or order, as distinguished by the above 1H-15N relaxation studies, correlated with patterns of 13C NMR chemical shifts. Shown in Fig. 4B are secondary-structure probabilities (SSP; bottom) and predicted order parameters (S2′; middle). Ordered regions (S2 and S2′ > 0.65 and SSP ≠ 0) correspond to the three canonical α-helices. SSP scores also suggest β-strand features within the B23–B30 segment. The latter segment exhibited increasing flexibility (0.45 < S2 < 0.65) from B23 to B30 with maximal flexibility within the C domain (residues 31–36 or C1–C6). Values of S2 and S2′ were consistently <0.6 in the C domain. Although relaxation parameters could not be obtained at ProC4 (which lacks an amide proton), its chemical shift-derived S2′ suggested less disorder than at flanking sites. These dynamic probes may be taken into account to generate alternative ensembles of protein structures based on time-averaged MD simulations (30, 31) (see “Discussion”).
SCI-b exhibits enhanced thermodynamic stability
As expected, based on its native-like solution structure, far-UV circular dichroism (CD) spectra of SCI-b and insulin lispro implied similar helix contents at 4 °C (Fig. 5, A and B). Thermal unfolding was monitored by mean residue ellipticity at 222 nm in the range 4–88 °C (Fig. 5C). Whereas insulin lispro exhibited a marked and irreversible loss of helix content with increasing temperature, SCI-b displayed only a small and limited attenuation of ellipticity on heating. On cooling from 88 to 4 °C, the far-UV CD spectrum of SCI-b was indistinguishable from the original spectrum, although lispro did not refold. Dynamic light scattering (DLS) revealed that lispro forms large aggregates following 10 min of incubation at 88 °C, whereas these conditions do not affect the size distribution of SCI-b (Fig. S3). Thermodynamic stability was likewise assessed by CD-monitored guanidine denaturation with application of a two-state model (Fig. 5D and Table 1). The SCI's free energy of unfolding (ΔGCD 4.7(±0.1) kcal/mol) was larger than that of insulin lispro (3.1(±0.1) kcal/mol). Respective titration midpoints (Cmid) were 5.7(±0.1) m (SCI-b) and 4.7(±0.1) m (insulin lispro). In such modeling the m-value was higher for SCI-b (0.82(±0.01) kcal/(mol·m)) than for insulin lispro (0.67(±0.01) kcal/(mol·m)), suggesting that hydrophobic surfaces are more effectively desolvated in SCI-b. These estimates depend on the assumption of two-state unfolding and on fitting of post-transition baselines (40).
Figure 5.
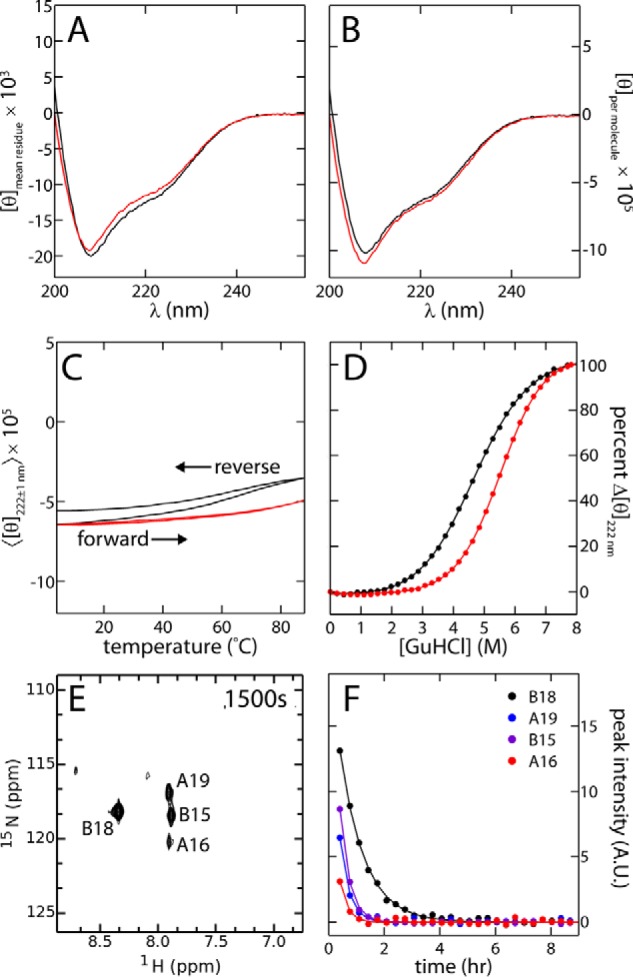
Protein folding and thermodynamic stability. A and B, CD spectra of SCI-b (red) and insulin lispro (black) acquired at 25 °C with molar ellipticities calculated and presented as per residue (A) and per molecule (B). C, forward (4 → 88 °C) and reverse (88 → 4 °C) temperature scans of SCI-b (red) and insulin lispro (black) presented as 〈[θ]222 ± 1 nm〉, an average of the ellipticities at helix-sensitive wavelengths of 222(±1) nm. Distinguishable forward and reverse traces for insulin lispro are labeled. D, comparative CD-monitored guanidine denaturation studies of SCI-b (red) and lispro (black) acquired at 25 °C (solid lines are fits; results given in Table 1). E, 1H-15N-HSQC of 15N-SCI-b ∼1500 s after placement of dried protein sample in 100% D2O potassium phosphate buffer. F, exponential decay profiles generated by plotting HSQC cross-peak intensities as a function of time. Only B15, B18, A16, and A19 resonances were present in first and subsequent HSQCs due to rapid baseline base-catalyzed 1H-2H exchange at pH 7.4. Solid lines show single-exponential fits; parameters are given in Table 2.
Table 1.
CD denaturation studies at pH 7.4 and 25 °C
| Analog | ΔGCDa | m-valueb | Cmidc |
|---|---|---|---|
| kcal/mol | kcal/(mol·m) | m | |
| lispro | 3.0 ± 0.1 | 0.63 ± 0.01 | 4.7 ± 0.1 |
| SCI-b | 4.7 ± 0.1 | 0.82 ± 0.01 | 5.7 ± 0.1 |
a Data are from two-state modeling of CD-monitored denaturation studies (25 °C and pH 7.4).
b The m-value is the slope of unfolding free energy ΔGU versus molar concentration of denaturant and is in units of kcal/(mol·m).
c Cmid is the guanidine denaturant concentration at which 50% of the protein is unfolded.
To obtain estimates of thermodynamic stability without reference to two-state modeling, we investigated amide–proton exchange kinetics in D2O at pD 7.0 and 25 °C. These studies employed protein samples uniformly enriched in 15N. The first 15N-HSQC spectrum (Fig. 5E; obtained ∼10 min following dissolution in D2O) exhibited only four 1H-15N cross-peaks: B15, B18, A16, and A19. Their progressive 1H-2H exchange in subsequent HSQCs yielded monoexponential kinetics (Fig. 5F). Comparison of observed (kobs) and intrinsic exchange rates (kint; calculated using SPHERE (41)) yielded protection factors (PF = kint/kobs) and in turn provided an estimate of thermodynamic stability (ΔGpH 7.4): 4.63(±0.08) kcal/mol (averaged over sites B15, B18, and A16; Table 2).6 This estimate, in striking accord with the results of guanidine denaturation studies, assumes global-exchange kinetics, i.e. sites where 1H-2H exchange requires complete unfolding (42, 43).
Table 2.
NMR-based stability estimates of SCI-b at pH 7.4 (pD 7.0) and 25 °C
| Residue | kobs | kint | PF | ΔGpH 7.4 |
|---|---|---|---|---|
| s−1 × 10−6 | s−1 × 10−3 | kcal/mol | ||
| B15 | 818 ± 18 | 1800 | 2201 ± 49 | 4.55 ± 0.01 |
| B18 | 314 ± 7 | 842 | 2681 ± 61 | 4.67 ± 0.01 |
| A16 | 1040 ± 142 | 2850 | 2740 ± 373 | 4.68 ± 0.08 |
| A19a | 850 ± 34 | 7670 | 9028 ± 361 | 5.39 ± 0.02 |
a The overestimate of ΔGpH 7.4 for A19 is ascribed to its proximity to cystine A20–B19, which may confound calculation of kint, possibly due to residual structure in the unfolded state under these conditions.
Subglobal conformational fluctuations are similar to those in a two-chain analog
Analysis of presumed global 1H-2H exchange at pD 2.4 (Fig. 6A; residues B18, A16, and A19) yielded estimates of ΔGpH 2.8 (Table S2) similar to those at neutral pH. Amide-proton exchange is markedly slower under these conditions as intrinsic exchange rates are slower than at neutral pD (41). Despite such global stabilization, the residue-specific pattern of subglobal and local PFs (Fig. 6B) was in accordance with a previous analysis of insulin lispro at pD 3.0 (43). Such similarities provide evidence that the C domain does not significantly damp subglobal motions on this prolonged time scale (>1 min).
Figure 6.

Long time scale dynamics probed by amide-proton exchange kinetics at pD 2.8. A, successive 1H-15N-HSQCs of SCI-b at the stated time points on dissolution of the protein in 100% D2O at pD 2.4 (pH 2.8). B, plots of PFs against residue number: all observed PFs (top) and an enlarged view of non-global sites of exchange (bottom). Color code: putative sites of global 1H-2H exchange (red); sites of local exchange (green); sites of subglobal exchange (blue); and unobserved or absent amide resonances (filled black circles).
Modeling of receptor-bound SCI-b predicted a novel open conformation
Models of the complex (Fig. 7) between SCI-b and μIR resulted in αCT being threaded through the loop formed by the C domain and B24–B30 segment. The proximity of αCT to the C terminus of the A domain precluded all attempts to form an un-threaded model. Removing αCT residue Phe-714 from the alignment with the template allowed αCT to disengage all contact with SCI-b, resulting in models with αCT no longer threaded through the loop; these models also no longer had Phe-714 occupying its usual site bounded by IleA2, ValB12, LeuB15, and μIR residues Leu-37 and Phe-64.
Figure 7.

Model of SCI-b bound to μIR. A, stereo view of bound state of SCI-b (A domain, yellow; B1–B23, gray) in complex with μIR L1 domain (white) and αCT peptide (green) as obtained from MD simulations. The open state of SCI-b enables detachment of the B24–B30 segment from the protein core as in WT insulin with the B24-C6 segment wrapping around the αCT (green) in a thread-like motif. B, overlays of 20 structures from the 30 structure best-fit SCI-b ensemble (red, green, blue) and 20 receptor-bound SCI-b structures (gray), as aligned according to the main-chain atoms of the B9–B19, A1–A8, and A13–A19 helices. Black double arrows highlight open → closed transition in B24–C6 segment. Disulfides are yellow and labeled by black boxes. N and C termini are as labeled. C, per-residue average Cα–Cα distances (〈RCα-Cα〉) between all possible pairwise Cα comparisons across models in an 18-structure free SCI-b selectively time-averaged ensemble (from A, right) and 18 structures from the bound SCI-b simulation, and D, among all models in an 18 structure SCI-a monomer “D” ensemble (from B, right) and the bound SCI-b ensemble. Error bars in C and D represent standard deviations; error in D is systematically larger than in C due to averaging over the six independent SCI-a crystallographic protomers. The B9–B19, A1–A8, and A13–A19 helices and C1–C6 segment are shown as shaded boxes.
Interactions between SCI-b and μIR over the 300-ns simulation (Fig. 7A) mirrored those observed in the co-crystal structure of the WT insulin complex, with notable key conserved interactions; in particular, PheB24 occupied the space within the hydrophobic pocket generated by B domain residues ValB12, LeuB15, TyrB16, and CysB19 and IR residues Asn-15, Leu-37, Phe-39, and Phe-714. PheB25 projects away from L1–β2 and inserts between αCT residues Val-715 and Pro-718 in the same way as observed in the WT complex. Numerous transient intramolecular electrostatic interactions were observed: between TyrB26 and AspB28, between C domain residues GluC1 and GluC2 with ArgC5 and ArgC6, and between GluA4 and ArgC5. The C domain of SCI-b contacted the flexible loop on the CR domain of the μIR between residues 264 and 277, predicting numerous nonspecific interactions. Superposition of bound SCI-b from this simulation to free SCI-b obtained by NMR (Fig. 7B) illustrates the closed → open conformational change on binding the receptor. Residual α-carbon distances from this overlay (Fig. 7C) and from a superposition of bound structures to all monomers from the SCI-a crystal structure (Fig. 7D; structure from Ref. 9) highlight that the B23–C6 segment primarily contributes to the closed → open conformational transition.
SCI-b resembles a premixed soluble-microcrystalline insulin formulation
The biological activity of SCI-b was tested on SQ injection in Lewis rats rendered diabetic by streptozotocin (Fig. 8, A and B) (44). Because its intrinsic PD profile (as defined on i.v. bolus injection; see our accompanying article (9)) exhibits fast and delayed phases, our SQ studies were conducted in relation to a premixed clinical formulation containing soluble and microcrystalline components (Humalog Mix 75/25®; Lilly) (32). Soluble insulin lispro provided a rapid-acting control (red squares in Fig. 8). Whereas standard premixed formulations were developed to provide effective post-prandial bolus (rapid) and basal (delayed) insulin activity via differential absorption rates in the SQ depot (black and aquamarine in Fig. 8), a monocomponent solution of SCI-b provided a similar action profile (green triangles in Fig. 8). The potential implications of this finding for global health are discussed below.
Figure 8.

PD profile of SCI-b resembled that of a 75/25 premixed NPH/regular insulin analog formulation. Shown are rat studies of insulin action following SQ injection of stated dose (in nanomoles) per 300-g rat of the specified SCI or control. A, blood-glucose concentration, and B, percent change relative to initial blood-glucose concentration. Symbols and doses are defined as inset in (A): data pertaining to SCI-b are shown as green triangles. Two doses of the premixed formulation (Humalog Mix75/25®; Lilly) are provided (black and aquarmarine). A solution of insulin lispro provided a rapid-acting control (red). A 360-min time point for 75/25 at 3.4-nmol dose was not acquired. Samples sizes: (diluent control) n = 4; (insulin lispro) n = 21; (SCI-b) n = 22; (75/25 2.6 nmol) n = 7; and (75/25 3.4 nmol) n = 6. Error bars represent standard error. C, schematic of a pancreatic β-cell and its response to increased plasma [glucose]. The glucose transporter (brown) regulates glucose entry, whereupon its metabolism generates ATP as an intracellular ligand for the ligand-gated K+ channel (blue). Binding of ATP closes K+ channels and thereby depolarizes the cell membrane, which in turn opens voltage-gated Ca2+ channels (green). Entry of Ca2+ triggers first-phase exocytosis of secretory vesicles followed by mobilization of storage granules (orange with red borders). D, plasma insulin concentration curve after subcutaneous injection of stated clinical analogs. The isolated soluble insulin and microcrystalline are indicated as green and blue curves, respectively. Premixed soluble and microcrystalline insulin is shown in red. D is adapted from http://watcut.uwaterloo.ca/webnotes/Metabolism/Diabetes.html.
Discussion
Protein dynamics is integral to a molecular understanding of structure and function. Segmental flexibility, for example, may underlie conformational change and in turn facilitate protein–protein recognition (45). Furthermore, such dynamic features may accelerate evolution of novel functions (46). In this and our accompanying article (9), we have sought to apply such biophysical principles to the engineering of ultra-stable protein therapeutics. We first discuss our biophysical findings and then their translational implications.
SCIs provide an ultra-stable platform for protein engineering
This study focused on an ancestral folding motif: the insulin-related superfamily (7). The high-resolution NMR structure of an active SCI, designed as an engineered monomer in solution, provided a foundation for analysis of protein dynamics. Our findings were extended by use of SA/MD simulations to gain insight into the dynamics of the parent protein in a crystal lattice and in a model receptor complex.
In the accompanying article in this issue (9), we presented the design rationale for SCI-a and SCI-b (Fig. 1C). The crystal structure of SCI-a was determined at moderate resolution (2.8 Å) as a zinc-free hexamer. Because an entire hexamer lies within the lattice's asymmetric unit, each protomer was crystallographically independent. Despite the limited resolution of the diffraction data, the six insulin moieties (i.e. the A- and B domains) exhibited canonical structures with native dimerization. Root mean square deviation (r.m.s.d.) values among the six crystallographic protomers were consistent with the structural variability of WT insulin in different crystalline environments (47). Incomplete electron density was observed (to a varying extent among the six protomers) in the respective C domains and preceding residues of the B domain (B27–B30). Precise analysis of side-chain conformations and interactions in the hydrophobic core was limited by the crystallographic resolution.
The crystal structure of SCI-a exhibited a novel overall feature: in the absence of axial zinc ions, the trimer interfaces were displaced relative to classical zinc hexamers (9). We speculated that this novel zinc-free SCI hexamer pertains in β-cells to self-assembly of proinsulin in the endoplasmic reticulum and/or Golgi apparatus (in which zinc ion concentrations are typically less than 1 pm (48)). Such native assembly might mitigate aggregation-coupled misfolding en route to prohormone processing and storage in glucose-regulated secretory granules. Subsequent formation of zinc-insulin hexamers and their microcrystallization within these granules are made possible by a β-cell-specific zinc transporter ZnT8 (49).
Interrelation of stability, dynamics, and function
Interest in SCIs was stimulated by classical studies in which bifunctional chemical reagents were employed to tether the C terminus of the B chain to the N terminus of the A chain (50, 51). Such non-standard linkers most often connected the ϵ-amino group of LysB29 to the α-amino group of GlyA1, thereby mimicking a connecting peptide. Because d-amino acid substitutions at A1 are well-tolerated (52), an alternative approach employed a d-LysA1 ϵ-amino group, extending the effective linker length (51). The relative receptor-binding affinities of the resulting B29-A1 tethered insulin analogs reflected the number of atoms within the cross-link: the longer the tether, the stronger the insulin analog–IR binding interaction.
The relationship between linker length and IR affinity suggested that conformational “play” between LysB29 and GlyA1 is required for high-affinity hormone binding. Biophysical studies of NαA1, NϵB29-ethylene glycol-bis-succinoyl-insulin, whose tether contains 22 connecting atoms inclusive of the B29 side chain, demonstrated increased thermodynamic stability (ΔΔGu 1.9 kcal/mol at 23 °C) and decreased protein flexibility, as indicated by an ∼103-fold retardation in overall rate of amide–proton exchange in D2O (36). To our knowledge, the latter study provided the first evidence for a biophysical linkage between inter-chain dynamics and protein stability in the insulin molecule. The present amide 1H-2H exchange studies of SCI-b similarly revealed increased global thermodynamic stability through significantly delayed global and subglobal exchange in D2O at pD 2.4 (pH 2.8) relative to analogous studies of two-chain WT insulin (53).
SCI-b exhibits a combination of structural order and dynamic disorder
The structure of SCI-b was determined as an engineered monomer in solution by heteronuclear multidimensional NMR methods (10, 11). Whereas SCI-a contained ProB28–GluB29, a feature of clinical analog insulin glulisine compatible with a native mode of dimerization (54), paired substitutions AspB28 and ProB29 in SCI-b impaired dimerization; this design combined features of insulin aspart and lispro (12, 13). The 1H NMR spectrum of SCI-b was found to exhibit sharp resonances even at a protein concentration of 1 mm (9). The pattern of secondary 1H NMR chemical shifts was consistent with stable folding of a globular domain. Ring-current shifts associated with formation of classical dimer and trimer interfaces (55) were not observed.
The mean NMR structure of SCI-b is similar to but not identical to crystallographic protomers of SCI-a (Fig. 3A) or WT insulin (6, 56). Although similarities in core side-chain packing were observed between the average SCI-b solution structure and the crystallographic electron density of SCI-a (Fig. 3, C–E), minor differences occur in segment B24–B28 (backbone r.m.s.d. in Fig. 3B). The latter is a “naked” β-strand in the monomer (57) but stabilized as an anti-parallel β-sheet at the dimer interface of the hexamer (6, 56). In accordance with past NMR studies of two-chain analogs (24, 57, 58), the aromatic side chains of PheB24 and TyrB26 pack against the α-helical core even in the absence of its partner strand.
Also congruent between solution and crystal structures was the partial disorder of the C domain. Four types of NMR observations provide evidence for its reduced structural organization. First, the C domain exhibited more limited 1H, 13C, and 15N chemical-shift dispersion than do the A and B domains. Such attenuated dispersion predicted lower order parameters relative to those of the insulin moiety (with the exception of frayed N-terminal residues B1–B3) (59). Second, C domain resonances exhibited motional narrowing (57, 58) as evidenced by greater carbonyl carbon peak intensities (relative to rigid core residues) in 3D HNCO spectra (Fig. S4). Third, the density of inter-residue NOEs in the C domain (i.e. contacts either (a) between residues in the C domain or (b) from the C domain to the insulin moiety) was lower than the density of NOEs among the α-helical segments; reduced NOE density led to greater imprecision in a canonical distance-restrained SA ensemble. Finally, explicit interrogation of main-chain order parameters (based on 1H-15N T1, T2, and hetNOE measurements at 700 MHz) verified that nanosecond-scale fluctuations are more marked in the C domain than in the A or B domains. Notably, regions of structural order (S2 ≥ 0.65) correlated well with the three helices, whereas segmental flexibilities in the B1–B4 and B27–C6 segments were identified by S2 < 0.65 (Fig. 4).
Dynamical information may also be inferred from patterns of 13C and 1H chemical shifts (60). Comparison of SCI-b and a reference two-chain analog (two-chain insulin analog containing AspB10, LysB28, and ProB29-insulin (61)) (Fig. S5) suggested that the B25–B30 segment is better ordered in SCI-b. Predicted S2′ parameters (where the prime indicates chemical shift-derived values) provides evidence that conformational fluctuations in C-terminal segment of the B domain are damped by the presence of the C domain, despite the latter's partial disorder (Fig. S5, top). Furthermore, the stabilizing ThrA8 → His substitution in SCI-b was associated with enhanced secondary-structure propensity (SSP) scores within the A1–A8 helix (Fig. S5, bottom). These conclusions were strengthened by analysis of the methyl proton secondary shifts of Ile, Leu, Val, and Ala residues (probes of aromatic ring currents related to tertiary structure Table S3) (55).
Complementary evidence for C domain disorder was provided by qualitative features of the electron-density maps of the variant ProB28–GluB29 SCI-a hexamer (9). In this crystal form the asymmetric unit was the hexamer itself, and so three independent views of the SCI-a dimer were obtained, in turn providing six views of the C domain. Each dimer exhibited a canonical B domain interface with discontinuous electron density between B27 and A1. In most monomers, density in this region was not interpretable; the most complete (monomer D) included limited density for residues B28–B29 and C6. Although static disorder in the lattice might in principle have contributed to such attenuated or discontinuous electron density, the consistency of these findings and their coherence with the NMR data strongly suggest that these incomplete features of the crystal structure represent dynamic disorder.
Ensemble perspective as a thought experiment
Despite evidence of segmental flexibility, both the NMR (Fig. 2) and X-ray-based (see accompanying article (9)) structural techniques are limited in their ability to visualize internal protein dynamics. To provide a depiction of SCI-b that a “Maxwell's Demon” might observe on the nanoscale, we recalculated the SCI-b NMR ensemble (Fig. 9A, left) using the Torda-Scheek-van Gunsteren time-averaged (TA) distance restraint protocol (30, 31) on either all restraints (Fig. 9A, middle) or in a modified protocol, wherein time-averaging is applied only to regions known to be dynamic (Fig. 9A, right). Whereas time averaging for all restraints increases imprecision globally, selective time-averaging essentially reproduced the ensemble calculated using conventional static distance restraints, thereby raising confidence in the latter's physical accuracy. Notably, segmental flexibility is readily apparent via increased r.m.s.d. in the B28–C5 segment (Fig. 9, C and D). As a corollary to these alternative ensembles of the solution structure, an ensemble model of hexameric SCI-a was generated. First, restraints derived from the carbon–carbon distances in the SCI-a crystallographic refinement (see accompanying article (9)) were utilized in a conventional SA protocol to generate an ensemble of 60 structures (Fig. 9B, left). Each of these structures was then subjected to multiconformer simulation wherein the B-factors of all atoms were fixed (B = 2 Å2) either without (Fig. 9B, middle) or with (Fig. 9B, right) constraining motion of the B5–B27 and A1–A21 segments, which had well-defined electron density in the original refinement. The protocol for these multiconformer simulations, distinct from standard ensemble X-ray refinement,7 is described in Fig. S6A. Ensemble-averaged R-factor calculations (REns, see Equations 2 and 3) yielded REns = 0.37 (“B = 2 Å2 crystal simulation”) and REns = 0.56 (“B = 2 Å2 core constrained”); the former was slightly higher than RFree = 0.31 for the single-structure SCI-a refinement (9). Notably, the REns of the most imprecise ensemble (Fig. 9B, middle) was lower than that of the core-constrained ensemble (Fig. 9B, right). This was a consequence of more efficient sampling of atom positions relative to the electron density by the ensemble as a whole (Fig. S6B). Although this would in principle also result in a lower REns than RFree of the single-structure refinement, the ensemble models of SCI-a contain C domain residues with no corresponding electron density, resulting in REns > RFree. We emphasize that over-parametrization in our protocol precludes use of REns as a comparative refinement parameter; its value instead corroborates the physical plausibility of the ensemble.
Figure 9.

Alternative structural ensembles of SCI-b and SCI-a. A, time-averaged NOE-based ensemble of 98 structures of SCI-b using a standard SA protocol (original SA; left). Each structure was run through a TA distance-restrained MD simulation wherein MD time-averaging was activated for all restraints (All Residues TA; middle). A separate simulation was then performed that enforced time-averaged restraints only for residues residing within flexible regions (predicted S2 < 0.65; right). B, each of the 35 hexameric structures generated using carbon–carbon distance restraints derived from the single-structure SCI-a crystallographic refinement (Hexamer Rebuild SA; left) were subjected to multiconformer simulation (see Fig. S6A for schematic description of the simulation procedure) with all thermal B-factors set to 2 Å2 and either no residues constrained (B = 2 Å2 crystal simulation; middle) or with the positions of B5–B26 and A1–A21 constrained (B = 2 Å2 with core constrained; right). The main-chain (C) and heavy-atom side-chain (D) r.m.s.d. per residue for SCI-b (black) or SCI-a (gray) were calculated from ensembles in A (right) and B (right), respectively. The r.m.s.d. for the SCI-a hexamer ensemble are averaged over all monomers. The B9–B19, A1–A8, and A13–A19 helices and C1–C6 segment are shown as shaded boxes. Gly residues at positions B8, B20, B23, C3, and A1 were excluded from heavy-atom side-chain r.m.s.d. calculations.
The above ensemble exhibits a striking overall similarity to the NMR-derived models in Fig. 9A and provides a nanoscale perspective of the hexamer as it might exist in the crystal lattice. Here, the B1–B3 and B28–C6 segments are flexible (r.m.s.d. in Fig. 9, C and D), but are constrained to lie within specific channels defined by neighboring hexamers within the lattice. The per residue r.m.s.d. (Fig. S7) of other alternative ensembles (from Fig. 9) highlight the same trends of structural order and flexibility in the solution and in this ensemble-based visualization of the crystal structure.
The ensemble perspectives of SCI-a and SCI-b highlight the segmental flexibility that foreshadows our MD-based model of SCI binding to the IR (Fig. 7). We envisage these as models of what a Maxwell's Demon, operating on the nanoscale, might observe. Such a Demon might play an active role in conformational selection, akin to Maxwell's mechanism by which environmental information is ostensibly extracted to change local order while still obeying the 2nd law of thermodynamics (62, 63). As the SCI samples conformational space, the Demon would be charged with selecting an open conformation to enable receptor binding, a process that likely occurs on the micro- to millisecond time scale inaccessible to our multiconformer and TA-NOE simulations. Nevertheless, the above ensemble perspective provides insight into the process of conformational selection by highlighting flexible sites (i.e. B24–C6) that must undergo closed → open conformational fluctuation before IR binding. We cannot exclude a mechanism by which an initial encounter complex by the closed state facilitates such opening as envisaged in classical notions of induced fit; here the Demon would be the receptor itself, coupled to the regulatory intelligence of the cell.
Model of an SCI–μIR complex
In accordance with an extensive prior biochemical literature (23, 24, 64), recent crystallographic studies of insulin bound to a domain-minimized insulin micro-receptor (μIR, composed of the L1 and αCT domains of IR) demonstrated that the B24–B27 segment of insulin is displaced from its classical packing against the A1–A8 and B9–B19 α-helices (15, 21). Effected by changes in Ramachandran dihedral angles flanking PheB24, this “unhinging” (by ∼60° relative to classical crystal structures of the free hormone dimer or hexamer (6)) enables conserved non-polar surface (spanned by the side chains of IleA2, ValA3, PheB24, and PheB25) to engage the receptor (15, 21). An extended B24–B27 segment lies in a groove between receptor elements L1 and αCT (15, 21). Residues B28–B30, not well-conserved among vertebrate insulins and dispensable for activity, were presumed to be disordered in the μIR complex. Accordingly, these three residues would contribute to the C domain of biologically active SCIs to provide a flexible tether between ThrB27 and GlyA1. Thus, the present SCI would effectively contain a 9-residue connecting peptide between the displaced B24–B27 segment and the A1–A8 α-helix. Our MD simulations of such an SCI–μIR complex (Fig. 7) have demonstrated the plausibility of this connection. The model is notable for breakage of C domain interactions with the insulin moiety, including packing of its central Pro against ValA3; distances between ValA3 Cγ2 and ProC4 Cγ are 6.1(±1.5) Å in the free NMR ensemble and 16.0(±1.9) Å in the predicted SCI–μIR complex. Close packing of these side chains in the predicted complex is precluded in the μIR complex by αCT. The bound SCI conformation thus differs from its free structure (as a monomer in solution and as a zinc-free hexamer in a crystal lattice) with respect to both the position of the B24–B27 β-strand and displacement of the C domain.
Ultimately, segmental flexibilities in SCI-b and SCI-a foreshadow the closed to open conformational transition necessary to bind and activate the IR. MD modeling of SCI-b and μIR (Fig. 7A) illustrates that the C domain, although relatively ordered in the solution-state monomer, opens up to wrap around the αCT segment of the IR-A receptor. In these threaded models, the interactions between insulin A and B domain residues with L1 and αCT were native-like, maintaining all interactions observed in the crystal structure of native insulin in complex with μIR (15, 21). Significant flexibility within the C domain was observed, with glutamic acids (GluC1 and GluC2) and arginine residues (ArgC5 and ArgC6) forming transient intramolecular electrostatic interactions as well as intermolecular interactions with the N-terminal αCT helical residues, ostensibly stabilizing the bound conformation. Models in which αCT did not thread the C domain resulted in SCI disengagement from the μIR during MD simulations, suggesting an inherent instability with these models.
The open state of SCI-b thus emulates that of two-chain insulin through the liberation of the B24–B30 segment from the protein core. This binding mode justifies SCI-b's retained activity in vivo while also rationalizing the biological inactivity of SCIs with shorter C domain sequences that would sterically hinder αCT-SCI “threading.” The striking difference between the open and closed states is highlighted by an overlay (Fig. 7B) of 18 best-fit SCI-b NMR structures (from Fig. 2) with 18 models of the receptor-bound SCI-b from the MD simulation. The average per-residue Cα–Cα distances across a helix-aligned ensemble composed of 18 structures each of the bound SCI-b and free SCI-b quantifies the magnitude of the closed → open transition (Fig. 7C), which is localized primarily from B24 to C6. A similar trend is seen when comparing the bound SCI-b ensemble to one monomer of the core-constrained SCI-a multiconformer simulation ensemble (Fig. 7D). The dynamic properties of the C domain thus appear to facilitate the conformational switch from the closed to open (receptor-bound) state and are thus crucial for SCI function.
The MD simulations of SCI-b in complex with μIR recapitulate the flexibility observed experimentally in the SCI monomer and provide evidence of the requirement for these residues to remain intrinsically dynamic both when unbound and in complex with the receptor. The requirement for αCT to thread through the open C domain would be difficult to characterize experimentally; future attempts to determine the crystal structures of these SCIs in complex with μIR may not resolve the positions of the dynamic C domain residues. The ability for MD to probe the time-dependent interactions made by these flexible domains will be integral for the future design of novel insulin analogs.
Relationship of SCI-b to IGFs
Solution structures of insulin-like growth factors (IGF-I and IGF-II) have previously been described (65, 66). IGF-I and IGF-II are homologous to SCI-b but with respective C domain lengths of 12 and 8 residues. Although similar in overall structure, there is one striking difference; whereas free SCI-b exhibits a well-defined native state, IGFs are well-organized only as ligand complexes or on specific binding to carrier proteins (IGF-binding proteins) (67). Indeed, in the absence of a bound detergent molecule or a bound phage-display peptide (68), NMR spectra of IGFs exhibit partial aggregation and conformational broadening on the millisecond time scale (65, 66). Presumably, ligand-free IGF proteins are in part molten (66).
As with SCI-b, main-chain dynamic studies of an extended variant of IGF-I (long-[Arg3]IGF-1) provided evidence of C domain flexibility with attenuation of corresponding hetNOE values (65). As a consequence, NMR-based modeling of IGF-I and IGF-II is remarkable for the imprecision of their respective C domains (Fig. S8).
Also unlike insulin and proinsulin, free IGF-I undergoes slow disulfide rearrangement to form an equilibrium between iso-energetic native and non-native (“swapped”) pairing schemes (69). By contrast, insulin has a unique ground state (70). Studies of insulin variants suggest that its native disulfide pairing is maintained, at least in part, by favorable contacts between HisB5 and A chain residues (including CysA6, CysA7, and CysA11); these long-range interactions have no counterpart in IGFs wherein HisB5 is substituted by Thr (70, 71).
Translational implications as the fruit of serendipity
In our accompanying article (9), we observed that subtle sequence variation among SCIs was associated with changes in their PD profiles as evaluated in rats on i.v. bolus injection. These findings were serendipitous and not predicted on the basis of structural modeling. SCI-a, for example, exhibited a PD profile akin to that of insulin lispro (or WT insulin), whereas SCI-b exhibited similar initial activity but a subsequent tail of activity; this tail was even more pronounced in i.v. studies of SCI-c. Because the plasma half-lives of SCI-a and SCI-c are similar (each ∼17 min (72)),8 we presume that these PD differences arise at the level of the responding target (73). Evidently, subtle differences in the structure or dynamics of the hormone–receptor complex can be associated with biological differences in the duration or time course of attenuation of the insulin signal.
The biphasic PD profile of SCI-b on i.v. injection (9) motivated the present SQ studies. Biphasic release of insulin by pancreatic β-cells is a general physiological feature of metabolic homeostasis (Fig. 8C and Fig. S9A) (74). The two phases of insulin release correspond to immediate and delayed continuous glucose-responsive exocytosis of insulin-containing secretory vesicles. The latter requires the ATP-dependent mobilization of a storage pool of vesicles (orange in Fig. S9A). Such biphasic secretion, recapitulated in isolated perfused islets (Fig. S9B) (75), provides a rapid bolus of insulin in response to a meal followed by prolonged basal release. Whereas both phases of insulin release are lost in type 1 DM (T1D; due to autoimmune β-cell destruction), early stages of type 2 DM are characterized by preferential blunting of first-phase release (76). In the natural history of T2D, eventual β-cell exhaustion also leads to progressive attenuation of second-phase insulin secretion (76).
The goal of insulin replacement therapy in T1D is to recapitulate the biphasic physiological pattern of post-prandial pancreatic insulin secretion and thereby prevent either hyperglycemic or hypoglycemic excursions. This goal has stimulated the development of rapid and basal insulin analogs (77) and sophisticated insulin delivery devices, including an “artificial pancreas” in which the output of a continuous glucose monitor controls (via a predictive algorithm) a pump as a closed-loop system (77). Rigorous glycemic control in T1D has been shown to delay or prevent microvascular complications and may also reduce the risk of macrovascular disease (78). The associated technologies primarily pertain to patients in affluent societies.
To an increasing degree over the next two decades, the majority of new cases of T2D are predicted to be in the developing world (5). This reflects an intersection between genetic factors and cultural changes, including urbanization, Western diet, and level of physical exertion (5). For unknown reasons, the incidence of T1D (classically a syndrome of northern latitudes and so more prevalent among Caucasians) is also increasing toward the equator (5). The predominant form of insulin prescribed in such populations is a premixed formulation for twice-a-day injection. Such products contain (a) a short-acting soluble component (as zinc insulin analog hexamers) and (b) a long-acting microcrystalline insulin suspension; the latter consists of zinc-stabilized protamine–insulin analog complexes (neutral protamine Hagedorn; NPH (77)). An example is provided by Humalog® Mix75/25 (Lilly), in which 75% of the hormone (insulin lispro) is contained within the NPH microcrystals and 25% is in the soluble phase (32); a similar product in broad clinical use contains an insulin aspart mixture of 70% NPH and 30% soluble analog (Novolog® Mix 70/30 from Novo-Nordisk).
The essential idea underlying design of premixed formulations exploits the combined PK properties of the soluble and microcrystalline insulin components within the SQ depot (Fig. 8D). The PD properties of the insulin analog are identical once in the bloodstream. Biphasic absorption of the rapid and delayed components following breakfast and dinner provides reasonable glycemic control in patients for whom these are the major meals (i.e. lunches are small). Disadvantages derive from the cost and complexity of manufacture and from the susceptibility of insulin component in NPH microcrystals to thermal degradation.
We have observed that the biphasic PD properties of SCI-b fortuitously recapitulated the biphasic PK-based profile of standard premixed formulation but in a solution that contains a single phase. The striking resistance of SCI-b to thermal degradation (predicting an extended shelf life) could be of clinical advantage in regions of the developing world lacking access to refrigeration (2, 5).
Concluding remarks
This study has described the high-resolution solution structure and dynamics of an active monomeric SCI. Ensembles generated by alternative SA protocols and multiconformer simulations extended our findings, providing alternative depictions of protein dynamics. Segmental flexibility in the foreshortened C domain of the SCI foreshadows a closed → open transition on binding to the IR: an SCI-αCT threading model was supported by MD simulation. Whereas this model rationalizes a wealth of prior biochemical data pertaining to smaller, inactive SCIs, optimization of this structural switch in a therapeutic formulation promises to extend the unrefrigerated shelf-life of insulin pharmaceuticals and enhance their global access in the face of an emerging DM pandemic (5).
It would be of future interest to test our MD-based model of an SCI–μIR complex through crystallographic studies. If observed in such model complexes and relevant to holoreceptor binding, the proposed threading mechanism predicts an abrupt transition between inactive SCIs (whose C domains contain three or fewer residues) and active SCIs (four or more residues). A systematic series of SCIs might therefore provide a molecular ruler by which to probe the coupling between conformational change and SCI–αCT threading. The same SCI series might exhibit an analogous yet uncorrelated transition from fibrillation resistance to susceptibility. Because cross-β assembly entails a more marked conformational change than is predicted on receptor binding, we imagine that longer C domains would be needed in a fibril to span the gap between B30 and A1. Comparison between these independent rulers thus promises to define the sweet spot for ultra-stable insulin analog design.
Experimental procedures
Construction of SCI precursor expression vectors
A set of synthetic plasmids (pPICZα) was designed and constructed for expression in yeast P. pastoris to enable direct secretion of SCI precursors (SCI-a, SCI-b, and SCI-c; Fig. 1C) into the media (79, 80). These plasmids were constructed as described in the accompanying article (9).
Biosynthesis of SCI precursor
SCI precursor biosynthesis followed a method adapted for insulin production in P. pastoris yeast (80) as described in the accompanying article (9). This study also adapted a protocol (81) for isotopic labeling of the proteins. Cells from the most expression-optimized colony were inoculated in 10 ml of sterile YPG + Zx1 (1% yeast extract, 2% peptone, 2% glycerol, 100 μg/ml Zeocin) in a 50-ml Falcon tube and grown overnight at 30 °C and a stirring speed 225 rpm (C25KC Incubator/Shaker; New Brunswick Scientific, Edison, NJ). The 10-ml culture was then added to 100 ml of YPG + Zx1 in a sterile 500-ml Erlenmeyer flask and grown another 24 h at 30 °C and 225 rpm. Cells were then pelleted and resuspended into 1 liter of sterile YPS (1% yeast extract, 2% peptone, 0.1 m sorbitol) in 2-liter flasks and incubated 24 h at 30 °C and 225 rpm. Cells centrifuged in sterile 400-ml bottles were then resuspended in media containing 0.17% yeast nitrogen base without amino acids with 0.5% [15N](NH4)2SO4 and 1% [13C]methanol (both from Cambridge Isotopes, Tewksbury, MA). For 15N-only labeling, unlabeled methanol was used for induction. For non-isotopically labeled protein, both unlabeled (NH4)2SO4 and methanol were used. Cells were incubated at 20 °C and 225 rpm for 48 h with addition of methanol to 0.5% after the first 24 h. Cells were pelleted in 400-ml centrifuge bottles (4500 × g for 20 min), and the supernatant was filtered through 0.22-μm Stericup filters (Merck Millipore, Billerica, MA).
Purification of yeast product
For purification of 13C,15N-SCI-b precursor, hydrophobic interaction chromatography (butyl-Sepharose 4 Fast Flow resin; GE Healthcare) was first used to capture protein from the filtered supernatant. All reverse-phase HPLC (rp-HPLC) purifications were performed using aqueous 0.1% trifluoroacetic acid (TFA) as Buffer A and 0.1% TFA in acetonitrile as the organic modifier (Buffer B). Preparative purifications utilized a Waters 2545 quaternary pumping system with FlexInject (Waters). The precursor was isolated by preparative C4 rp-HPLC (C4 Proto 300 Å, 10-μm, 250 × 20 mm; Higgins Analytical Inc., Mountain View, CA). For purification of all other precursors (unlabeled and 15N-only labeled), a minimized rp-HPLC-only protocol was used. To the filtered cell media, acetonitrile and TFA were added to 20 and 0.1%, respectively, followed by preparative C4 rp-HPLC (as above). In either case, eluted SCI was collected and lyophilized.
Enzymatic cleavage and SCI purification
The Achromobacter protease I (ACP-1) gene from Achromobacter lyticus (82) was expressed in Escherichia coli and purified as described in the accompanying article (9). Enzymatic conversion of SCI precursor to SCI (via cleavage of the EEAEAEAEPK spacer peptide at the C terminus of lysine) was also performed as described (9). Converted 13C,15N-SCI-b was first purified by preparative C8 rp-HPLC (C8 Kromasil 100 Å, 10-μm, 250 × 20 mm; Akzo Nobel, Amsterdam, Netherlands). Final purification utilized a C18 column (C18 Proto 300 Å, 10-μm, 250 × 20 mm; Higgins Analytical Inc.). SCI purity was verified to be ≥98% using analytical C18 rp-HPLC (C18 Discovery BIOwide 300 Å, 5-μm, 250 × 4.6 mm; Supelco, Bellefonte, PA). For unlabeled and 15N-SCI-b, the digestion reaction was purified by preparative C4 rp-HPLC (as above). Eluted SCI was lyophilized and the purity assessed to be ≥98% by C4 analytical rp-HPLC.
Optical spectroscopy
Far ultraviolet (255–200 nm) CD spectra were obtained at high signal-to-noise for insulin analogs using a CD spectropolarimeter (Aviv Biomedical Inc., Lakewood, NJ) equipped with temperature control and an automated titration unit. CD spectra (255–200 nm) of buffer were first obtained using degassed potassium/phosphate buffer (10 mm KH2PO4/K2HPO4 (pH 7.4) buffer with 50 mm KCl) at 4, 25, and 37 °C using a 30-s averaging time. As these spectra did not vary with temperature, the 25 °C KPi dataset was subtracted from all protein spectra. Samples were prepared at a concentration of 20–70 μm protein in KPi buffer, brought to pH 7.4 with KOH, and placed in a parafilm-sealed 1-mm path-length quartz cuvette. Full CD spectra (255–200 nm) were acquired at 4, 25, and 37 °C with 0.5 nm wavelength resolution and a 15-s photocount averaging time. All CD samples were quantitated via reference-subtracted UV-visible spectra acquired in KPi buffer with a UV-visible spectrometer (Aviv Biomedical Inc.) and a 3-mm quartz cuvette. Protein concentrations in potassium/phosphate buffer were calculated using absorbance at λ = 280 nm and estimated extinction coefficients predicted by the on-line ExPASy ProtParam tool.
CD studies of protein unfolding
To assess temperature dependence and reversibility of protein folding, a thermal scan (monitored at helix sensitive wavelengths 222(±1) nm) was performed on all CD protein samples (see above) from 4 to 88 °C followed by a reverse gradient from 88 to 4 °C (all in 4 °C steps). A full CD spectrum (255–200 nm) was then acquired at 4 °C to verify reversible folding (or lack thereof). Thermodynamic stabilities in KPi buffer at 25 °C were determined by monitoring guanidine-induced unfolding at the helix-sensitive wavelength 222 nm as described (83).
Heat-induced aggregation probed by DLS
Aqueous aliquots of insulin lispro or SCI-b necessary to obtain 0.6 mm protein solutions in 30-μl final volumes were pipetted into separate Eppendorf tubes and lyophilized. The powder was then dissolved in “Tris diluent” (25 mm Tris-HCl (pH 7.4) containing 16 mg/ml glycerin, 1.6 mg/ml meta-cresol, 0.65 mg/ml phenol, and ZnCl2 at a ratio of three Zn2+ ions per insulin hexamer). All buffer components (i.e. deionized water, 4× Tris-HCl diluent (pH 7.4), and 10 mm ZnCl2) were degassed via nitrogen bubbling for 15 min and then syringe-filtered (0.02-μm Whatman® Anton® 10 filters; Sigma), followed by centrifugation (5 min at 16,100 × g). Gentle mixing of the filtered components with lyophilized protein was performed by repeated pipetting.
Unheated samples were probed by DLS as described (9) following 30 min of incubation at room temperature. Heated samples were placed on a heat block set to 88 °C for 10 min to replicate the high temperatures encountered during the CD-monitored thermal scan (see above) before DLS measurement.
NMR spectroscopy
All spectra were acquired at a protein concentration of 0.6 mm at 25 °C with a Bruker AVANCE 700 MHz spectrometer equipped with a triple-resonance cryoprobe (Bruker Biospin Corp, Billerica, MA) as described (83). All chemical shifts were calibrated in parts/million (ppm) relative to 4,4-dimethyl-4-silapentane-1-sulfonic acid as an internal standard. All raw NMR spectra (except four-dimensional data) were processed using Bruker's TopSpin software before analysis using third party software packages, as described below.
NMR structure determination
A 4D time-shared 13C,13C/13C,15N/15N,15N NOESY was acquired as described (11), followed by a suite of 2D and triple-resonance 3D spectra, including 15N HSQC, 13C HSQC, aromatic 13C HSQC, HNCA, CBCA(CO)NH, HN(CO)CA, H(CCCO)NH, (H)CC(CO)NH, HNCACB, HCC-TOCSY, and 13C NOESY-HSQC (10). Because of a large total acquisition time, 15N HSQC spectra were acquired periodically to verify sample consistency. After processing 2D and 3D spectra in TopSpin, resonance assignment was performed using Sparky (84). The 4D time-shared NOESY dataset was processed using NMRPipe (85) into three separate 4D spectra consisting of a 4D 13C,13C NOESY, 4D 13C,15N NOESY, and 4D 15N,15N NOESY, which were subsequently imported into Sparky for assignment.
Custom routines written in R-script and Bash unix shell performed automated NOE distance restraint generation and structural refinement according to established protocols (28). Distance restraints were obtained from 4D NOESY data; dihedral-angle restraints for all non-dynamic residues were predicted by TALOS+ (60, 86) using chemical shifts. An SA protocol (28) performed by XPLOR-NIH (87) generated 98 structures; refinement involved iterative identification of best-fit structures followed by relaxation of distance restraints (or removal, if restraint became ≥6 Å) and ensemble recalculation until no restraint violations ≥0.4 Å were found. The final ensemble (generated using 1370 NOE and 75 dihedral angle restraints) was consistent with observed 4D NOESY data. All structural figures were generated in PyMOL (Schrödinger, LLC, New York).
1H-2H amide–proton exchange
To avoid potential artifacts in the SCI-b CD-monitored guanidine titration, 1H-2H amide exchange was performed in 100% D2O (Cambridge Isotopes) containing 10 mm KH2PO4/K2HPO4 (pH 7.4) and 50 mm KCl (100% D2O-KPi) to replicate the salt conditions of the guanidine titration studies. KPi buffer was first added to lyophilized 15N-SCI-b (to 0.6 mm protein), adjusted to pH 7.4 with KOH, and the sample lyophilized. Following this, the NMR spectrometer was shimmed, the 1H/15N/13C probe channels tuned, and the 90° 1H pulse calibrated using a separate SCI-b sample in 100% D2O-KPi. A 90° 1H pulse of 15.94 μs (1 dB power level) was used. The lyophilized 15N-SCI-b sample (containing KPi salts at pH 7.4) was placed on ice followed by addition of chilled 100% D2O with gentle mixing and placement in a Shigemi tube. The sample was shimmed at 25 °C and ∼30 successive 2D 15N-HSQC spectra were acquired and processed in batch.
For pH 2.8 amide exchange studies, lyophilized protein was directly mixed with 100% D2O, followed by pH measurement (using an electrode rinsed with 100% D2O) verifying the sample at pD 2.43 (pH 2.83). This sample was used for spectrometer 90° pulse and probe calibration. A multirun was set up to acquire 2D 15N-HSQC spectra once every 3 h for about 1 week at 25 °C. Resonance assignments needed to extract kobs from pH 2.8 amide exchange were obtained from a separate analog identical to 13C,15N-SCI-b but with AlaC4 instead of ProC4. Despite this mutation, the pH 3 and pH 7.4 15N-HSQCs of the two analogs are essentially identical.
The observed rate of amide exchange, kobs, for each resonance was determined using the Sparky “rh” extension. The intrinsic rate of exchange for each residue, kint, was calculated using the SCI-b sequence, pH 2.8 or 7.4, and 100% D2O content as input parameters in the on-line SPHERE program (41). Calculating kobs/kint gives the PF, which can be inserted into Equation 1,
| (Eq. 1) |
to obtain a model-free estimate of thermodynamic stability, ΔGAmEx. Here, R is the ideal gas law constant, and T is the absolute temperature. ΔGAmEx values obtained from pH 2.8 amide exchange studies are defined as ΔGpH 2.8, whereas ΔGpH 7.4 indicates an amide exchange result obtained at pH 7.4. The 1H-15N cross-peak of LeuB15 was omitted from pH 2.8 analysis due to overlap with that of IleA10 (Fig. 6A). Very slow exchange at pH 2.8 led to significant error in ΔGpH 2.8 from these resonances.
1H-15N NMR relaxation studies
2D spin-lattice (T1), spin-spin (T2), and hetNOE 15N-HSQC spectra were acquired at 700 MHz as described (88, 89). HetNOE spectra, which probe sub-nanosecond motions, used a relaxation delay of 6 s. T1 and T2 relaxation experiments yield information about internal protein dynamics; these experiments used a relaxation delay of 2 s. Spectra were processed in TopSpin and assigned in Sparky. Relaxation rates (R1 and R2) and NOE values were calculated by Relax NMR (90); errors in these values were derived from spectral noise. DYNAMICS generated by-residue generalized order parameters, S2, as described (91), using a rotational correlation time of 4.8 ns (isotropic diffusion model) and fixed chemical shift anisotropy of −240 ppm. This analysis detected sites of fast chemical exchange by fitting data for some residues to an extended model-free model that also presents the fast dynamics order parameter S2fast = S2 − S2slow.
TALOS+ was used to predict both secondary structure propensity (SSP) scores and generalized order parameters (S2′, which do not account for chemical exchange) from chemical shifts as described (60).
Alternative ensemble models of SCI-a and SCI-b
Using XPLOR-NIH, the refined 98 structure ensemble of SCI-b generated by distance-restrained SA was subjected to Verlet equilibration at 300 K for 10 ps, followed by another 25-ps simulation where NOE distance restraints were applied using the Torda-Sheek-van Gunsteren TA NOE protocol (30, 31) and a decay constant of 0.625 ps. Time averaging was applied to either all residues or, in a separate simulation, to only those with predicted order parameter lower than 0.65 (i.e. dynamic regions).
Because of incomplete electron density in the original SCI-a crystal refinement (see accompanying article in this issue (9)), a model of the “complete” structure of the hexamer was artificially generated. First, a custom MATLAB script calculated all possible carbon–carbon distances in the SCI-a structure and generated a restraint for any distance within the range 35–40 Å. These ∼200 restraints were used in the XPLOR-NIH SA protocol to bring a starting structure of six independent SCI-a monomers (generated in PyMOL using an SCI-b structure as a template) together, resulting in a coarse hexameric structure. To this structure, restraints derived from all carbon–carbon distances within 1.8–30 Å (∼670,000 restraints) in the SCI-a crystal refinement were applied to generate an ensemble of 60 hexameric structures (labeled Hexamer Rebuild SA in Fig. 9B). A multiconformer simulation (all B-factors fixed to B = 2 Å2) utilizing reflection data from SCI-a was performed on each individual structure from Hexamer Rebuild SA. Here, B-factors of 2 Å2 were chosen so that the mean square displacement (u, obtained from the equation B = 8π2u2) of carbon atoms would be just under 0.17 Å or ∼10% the van der Waals radius of carbon (1.7A). A separate simulation was performed wherein any residue that was well-resolved in the SCI-a crystal refinement was spatially fixed, whereas all others retained B-factors of B = 2 Å2, allowing only dynamic regions to move, paralleling the TA NOE ensembles generated for SCI-b. The protocol for our multiconformer simulation, modifications of slow cooling crystallographic SA (2000 → 300 K; 25 K reduction per 5-fs time step), is given in schematic form in Fig. S6. R-factors for each structure in the multiconformer ensemble, Ri, were calculated using the standard Equation 2,
| (Eq. 2) |
where FObs are observed structure factors, a function of Miller indices h⃗. Fcalc and k are calculated structure factors and the weighting coefficient as obtained during the B = 2 Å2 simulations. From here, the ensemble averaged R-factor, REns, is calculated using Equation 3,
| (Eq. 3) |
where n = 60, the number of structures in the ensemble. Note that for clarity only 35 structures of each hexameric ensemble are shown in Fig. 9.
Homology modeling of SCI-b bound to insulin micro-receptor
Models of SCI-b in complex with the IR isoform A (IR) L1-CR (μIR), including the IR αCT domain, were generated using the MODELLER (92) program utilizing the crystal structure of native insulin in complex with the micro-receptor and αCT as a template. To account for glycosylation a single N-linked N-acetyl-d-glucosamine carbohydrate at each of the IR residues Asn-16, Asn-25, Asn-111, Asn-215, and Asn-255 was included. From 25 models created, the model with the lowest MODELLER objective function was used in subsequent MD simulations.
MD simulations were performed using the GROMACS (version 5.1.2) (93) suite of programs and the CHARMM36 (94, 95) force field. Briefly, the simulation consisted of an initial steepest decent minimization, a short 50-ps positionally restrained MD holding the protein fixed, and finally unrestrained MD for 300 ns. The system was solvated using the TIP3P water model in a cubic box extending 10 Å beyond all atoms and utilized periodic boundary conditions. The system was made electrically neutral to a final ionic strength of 0.1 m with the addition of sodium and chloride ions; ionizable residues were assumed to be in their charged state at a pH of 7. The temperature of the system was maintained by coupling the protein and solvent independently to a velocity rescaling (96) thermostat at 300 K with a time constant of 0.1 ps. Pressure was controlled to 1 bar using a Berendsen barostat (97) with a coupling of 0.5 ps. A cutoff of 12 Å was used to account for non-bonded interactions and the particle-mesh Ewald method (98) to account for long-range electrostatics applying a grid width of 1.2 Å and a sixth-order spline interpolation. Neighbor searching applied a Verlet grid cutoff scheme with a neighbor-list update frequency of 40 steps and a time step of 2 fs. All bond lengths were constrained with the P-LINCS algorithm. Coordinates of the model were archived every 15 ns after an initial 45 ns equilibration.
Biological testing in diabetic rats
Humalog Mix75/25® (Lilly) provided a biphasic standard on SQ injection. Insulin lispro or SCI-b was dissolved at concentrations for which each 100-μl volume contained the doses (in nanomoles) specified in Fig. 8. These proteins were dissolved in Lilly® diluent (a product containing 3.8 mg/ml sodium phosphate (pH 7.4), 16 mg/ml glycerin, 1.6 mg/ml meta-cresol, 0.65 mg/ml phenol). The formulations each contained ZnCl2 at a ratio of three Zn2+/1 insulin hexamer. Insulin purity was verified by C4 analytical rp-HPLC as above. Protein-free Lilly® diluent was used as a negative control (buffer only). Male Lewis rats (mean body mass ∼300 g) were rendered diabetic by streptozotocin (44) and studied as described in our accompanying article (9). Because of day-to-day variation (between rats and in the same rat on different days), 4–8 rats were injected per analog formulation on each test day, and the experiments were repeated on one or more non-consecutive days to obtain a larger sample size.
Author contributions
NMR studies were performed by M. D. G., Y. Y., and N. P. W.; MD simulations were performed by M. D. G., N. A. S., and B. J. S.; biochemical and biophysical assays were performed by M. D. G., N. B. P., and N. P. W.; molecular modeling of the variant hormone-μIR interface was undertaken by N. A. S., B. J. S. with the assistance of M. C. L. and M. A. W.; rat studies were performed by M. D. G., K. C., under the guidance of F .I.-B. with data analysis by M. D. G. and F. I.-B; and therapeutic issues were considered by F. I.-B. and M. A. W. The overall program of research was guided by M. A. W. Each of the authors contributed to the manuscript.
Supplementary Material
Acknowledgments
We thank Y. Liu, L. Broadwater, T. Gray, T. Hattier, and T. Cardon for assistance with yeast fermentation, isotopic labeling, purification, and enzymatic processing. We also thank Y.-S. Chen for assistance with figures and B. Dhayalan for assistance with DLS.
This work, a contribution of the Cleveland Center for Membrane and Structural Biology and CWRU Institute for the Science of Origins, was supported in part by National Institutes of Health Grants R01 DK040949 and R01 DK069764 (to M. A. W.), by Australian National Health and Medical Research Council Project Grant APP1058233 (to M. C. L.), Victorian State Government Operational Infrastructure Support, Australian NHMRC Independent Research Institutes Infrastructure Support Scheme, both to his institution, and by the National Computational Infrastructure (NCI), which is supported by the Australian Government. This work was also supported by an Australian Government Research Training Program Scholarship (to N. A. S.). M. A. W. has equity in Thermalin Diabetes, LLC (Cleveland, OH), where he serves as Chief Innovation Officer; he has also been a consultant to Merck Research Laboratories and DEKA Research & Development Corp. N. B. P. and F. I.-B. are consultants to Thermalin Diabetes, LLC. Part of M.C.L.'s research is funded by Sanofi (Germany). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S9, Tables S1–S3, and supporting Refs. 1–8.
The atomic coordinates and structure factors (code 5WBT) have been deposited in the Protein Data Bank (http://wwpdb.org/).
SCI residues are denoted by residue type (in standard three-letter code) followed by the domain and position (e.g. TyrB16 designates a tyrosine at the 16th position of the B domain). Because the B, C, and A domains have lengths of 30, 6, and 21 residues, respectively, ProC4 is SCI residue 34, and IleA2 is SCI residue 38.
TyrA19 (residue 55 in SCI-b) exhibited an anomalously large PF, likely to reflect its proximity to cysteine A20-B19. SPHERE relies on primary structure and does not take into account disulfide bonding and possible residual local structure in the unfolded state (41).
Multiconformer approaches to X-ray structure refinement have been described that probe thermal motion and disorder without over-parametrization (99–101).
Whereas commercial insulin RIA/ELISA kits enabled detection of SCI-a and SCI-c (following establishment of a standard curve), the available antisera did not efficiently recognize SCI-b (72).
- DM
- diabetes mellitus
- αCT
- C-terminal domain of insulin receptor
- μIR
- domain-minimized insulin micro-receptor complex composed of the L1 and αCT domains of IR
- DLS
- dynamic light scattering
- hetNOE
- heteronuclear 1H-15N NOE
- HSQC
- heteronuclear single-quantum coherence
- IGF
- insulin-like growth factor
- IR
- insulin receptor
- L1
- first leucine-rich-repeat domain of IR
- MD
- molecular dynamics
- NPH
- neutral protamine Hagedorn
- PDB
- Protein Data Bank
- PF
- protection factor
- r.m.s.d.
- root-mean-square deviation
- PK
- pharmacokinetic
- rp-HPLC
- reverse-phase high-performance liquid chromatography
- SA
- simulated annealing
- SCI
- single-chain insulin
- SQ
- subcutaneous
- TA
- time-averaged
- TK
- tyrosine kinase
- PD
- pharmacodynamics
- SSP
- secondary-structure probability
- T1D
- type 1 DM
- T2D
- type 2 DM.
References
- 1. Brange J., Ribel U., Hansen J. F., Dodson G., Hansen M. T., Havelund S., Melberg S. G., Norris F., Norris K., and Snel L. (1988) Monomeric insulins obtained by protein engineering and their medical implications. Nature 333, 679–682 [DOI] [PubMed] [Google Scholar]
- 2. Weiss M. A. (2013) Design of ultra-stable insulin analogues for the developing world. J. Health Spec. 1, 59–70 10.4103/1658-600X.114683 [DOI] [Google Scholar]
- 3. Vinther T. N., Norrman M., Ribel U., Huus K., Schlein M., Steensgaard D. B., Pedersen T. Å., Pettersson I., Ludvigsen S., Kjeldsen T., Jensen K. J., and Hubálek F. (2013) Insulin analog with additional disulfide bond has increased stability and preserved activity. Protein Sci. 22, 296–305 10.1002/pro.2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hua Q. X., Nakagawa S. H., Jia W., Huang K., Phillips N. B., Hu S. Q., and Weiss M. A. (2008) Design of an active ultrastable single-chain insulin analog: synthesis, structure, and therapeutic implications. J. Biol. Chem. 283, 14703–14716 10.1074/jbc.M800313200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shaw J. E., Sicree R. A., and Zimmet P. Z. (2010) Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 87, 4–14 10.1016/j.diabres.2009.10.007 [DOI] [PubMed] [Google Scholar]
- 6. Baker E. N., Blundell T. L., Cutfield J. F., Cutfield S. M., Dodson E. J., Dodson G. G., Hodgkin D. M., Hubbard R. E., Isaacs N. W., and Reynolds C. D. (1988) The structure of 2Zn pig insulin crystals at 1.5 Å resolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 319, 369–456 10.1098/rstb.1988.0058 [DOI] [PubMed] [Google Scholar]
- 7. Shabanpoor F., Separovic F., and Wade J. D. (2009) The human insulin superfamily of polypeptide hormones. Vitam. Horm. 80, 1–31 10.1016/S0083-6729(08)00601-8 [DOI] [PubMed] [Google Scholar]
- 8. Steiner D. F. (1967) Evidence for a precursor in the biosynthesis of insulin. Trans. N. Y. Acad. Sci. 30, 60–68 10.1111/j.2164-0947.1967.tb02452.x [DOI] [PubMed] [Google Scholar]
- 9. Glidden M. D., Aldabbagh K., Phillips N. B., Carr K., Chen Y.-S., Whittaker J., Phillips M., Wickramasinghe N. P., Rege N., Swain M., Peng Y., Yang Y., Lawrence M. C., Yee V. C., Ismail-Beigi F., and Weiss M. A. (2017) An ultra-stable single-chain insulin analog resists thermal inactivation and exhibits biological signaling duration equivalent to the native protein. J. Biol. Chem. 293, 47–68 10.1074/jbc.M117.808626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bax A. (1994) Multidimensional nuclear magnetic resonance methods for protein studies. Curr. Opin. Struct. Biol. 4, 738–744 10.1016/S0959-440X(94)90173-2 [DOI] [Google Scholar]
- 11. Xu Y., Long D., and Yang D. (2007) Rapid data collection for protein structure determination by NMR spectroscopy. J. Am. Chem. Soc. 129, 7722–7723 10.1021/ja071442e [DOI] [PubMed] [Google Scholar]
- 12. DeFelippis M. R., Chance R. E., and Frank B. H. (2001) Insulin self-association and the relationship to pharmacokinetics and pharmacodynamics. Crit. Rev. Ther. Drug. Carrier. Syst. 18, 201–264 [PubMed] [Google Scholar]
- 13. Setter S. M., Corbett C. F., Campbell R. K., and White J. R. (2000) Insulin aspart: a new rapid-acting insulin analog. Ann. Pharmacother. 34, 1423–1431 [DOI] [PubMed] [Google Scholar]
- 14. Boucher J., Kleinridders A., and Kahn C. R. (2014) Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 6, a009191 10.1101/cshperspect.a009191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Menting J. G., Yang Y., Chan S. J., Phillips N. B., Smith B. J., Whittaker J., Wickramasinghe N. P., Whittaker L., Pandyarajan V., Wan Z., Yadav S. P., Carroll J. M., Srokes N., Roberts C. T., Ismail-Beigi F., et al. (2014) Protective hinge in insulin opens to enable its receptor engagement. Proc. Natl. Acad. Sci. U.S.A. 111, 1–48 10.1073/iti0114111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Meyts P., and Whittaker J. (2002) Structural biology of insulin and IGF1 receptors: implications for drug design. Nat. Rev. Drug Discov. 1, 769–783 10.1038/nrd917 [DOI] [PubMed] [Google Scholar]
- 17. Hubbard S. R. (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581 10.1093/emboj/16.18.5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cabail M. Z., Li S., Lemmon E., Bowen M. E., Hubbard S. R., and Miller W. T. (2015) The insulin and IGF1 receptor kinase domains are functional dimers in the activated state. Nat. Commun. 6, 6406 10.1038/ncomms7406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lou M., Garrett T. P., McKern N. M., Hoyne P. A., Epa V. C., Bentley J. D., Lovrecz G. O., Cosgrove L. J., Frenkel M. J., and Ward C. W. (2006) The first three domains of the insulin receptor differ structurally from the insulin-like growth factor 1 receptor in the regions governing ligand specificity. Proc. Natl. Acad. Sci. U.S.A. 103, 12429–12434 10.1073/pnas.0605395103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Croll T. I., Smith B. J., Margetts M. B., Whittaker J., Weiss M. A., Ward C. W., and Lawrence M. C. (2016) Higher-resolution structure of the human insulin receptor ectodomain: multi-modal inclusion of the insert domain. Structure 24, 469–476 10.1016/j.str.2015.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Menting J. G., Whittaker J., Margetts M. B., Whittaker L. J., Kong G. K., Smith B. J., Watson C. J., Záková L., Kletvíková E., Jiráček J., Chan S. J., Steiner D. F., Dodson G. G., Brzozowski A. M., Weiss M. A., et al. (2013) How insulin engages its primary binding site on the insulin receptor. Nature 493, 241–245 10.1038/nature11781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Q., Wong Y. L., and Kang C. (2014) Solution structure of the transmembrane domain of the insulin receptor in detergent micelles. Biochim. Biophys. Acta 1838, 1313–1321 10.1016/j.bbamem.2014.01.005 [DOI] [PubMed] [Google Scholar]
- 23. Mirmira R. G., Nakagawa S. H., and Tager H. S. (1991) Importance of the character and configuration of residues B24, B25, and B26 in insulin-receptor interactions. J. Biol. Chem. 266, 1428–1436 [PubMed] [Google Scholar]
- 24. Hua Q. X., Shoelson S. E., Kochoyan M., and Weiss M. A. (1991) Receptor binding redefined by a structural switch in a mutant human insulin. Nature 354, 238–241 10.1038/354238a0 [DOI] [PubMed] [Google Scholar]
- 25. Xu B., Huang K., Chu Y. C., Hu S. Q., Nakagawa S., Wang S., Wang R. Y., Whittaker J., Katsoyannis P. G., and Weiss M. A. (2009) Decoding the cryptic active conformation of a protein by synthetic photoscanning: Insulin inserts a detachable arm between receptor domains. J. Biol. Chem. 284, 14597–14608 10.1074/jbc.M900087200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pandyarajan V., Phillips N. B., Rege N., Lawrence M. C., Whittaker J., and Weiss M. A. (2016) Contribution of TyrB26 to the function and stability of insulin: structure-activity relationships at a conserved hormone-receptor interface. J. Biol. Chem. 291, 12978–12990 10.1074/jbc.M115.708347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clore G. M., Nilges M., Brünger A., and Gronenborn A. M. (1988) Determination of the backbone conformation of secretin by restrained molecular dynamics on the basis of interproton distance data. Eur. J. Biochem. 171, 479–484 10.1111/j.1432-1033.1988.tb13814.x [DOI] [PubMed] [Google Scholar]
- 28. Yang Y., Hua Q.-x., Liu J., Shimizu E. H., Choquette M. H., Mackin R. B., and Weiss M. A. (2010) Solution structure of proinsulin: connecting domain flexibility and prohormone processing. J. Biol. Chem. 285, 7847–7851 10.1074/jbc.C109.084921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ángyán A. F., and Gaspári Z. (2013) Ensemble-based interpretations of NMR structural data to describe protein internal dynamics. Molecules 18, 10548–10567 10.3390/molecules180910548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Torda A. E., Scheek R. M., and van Gunsteren W. F. (1989) Time-dependent distance restraints in molecular dynamics simulations. Chem. Phys. Lett. 157, 289–294 10.1016/0009-2614(89)87249-5 [DOI] [Google Scholar]
- 31. Torda A. E., Scheek R. M., and van Gunsteren W. F. (1990) Time-averaged nuclear overhauser effect distance restraints applied to tendamistat. J. Mol. Biol. 214, 223–235 10.1016/0022-2836(90)90157-H [DOI] [PubMed] [Google Scholar]
- 32. Heise T., Weyer C., Serwas A., Heinrichs S., Osinga J., Roach P., Woodworth J., Gudat U., and Heinemann L. (1998) Time-action profiles of novel premixed preparations of insulin lispro and NPL insulin. Diabetes Care 21, 800–803 10.2337/diacare.21.5.800 [DOI] [PubMed] [Google Scholar]
- 33. Shi L. X., Li P. F., and Hou J. N. (2017) Differential treatment response to insulin intensification therapy: a post hoc analysis of a randomized trial comparing premixed and basal-bolus insulin regimens. Diabetes Ther. 8, 915–928 10.1007/s13300-017-0286-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wambui C. K., Kumar A. M. V., Hinderaker S. G., Chinnakali P., Pastakia S. D., and Kamano J. (2016) Do diabetes mellitus patients adhere to self-monitoring of blood glucose (SMBG) and is this associated with glycemic control? Experiences from a SMBG program in western Kenya. Diabetes Res. Clin. Pract. 112, 37–43 10.1016/j.diabres.2015.11.006 [DOI] [PubMed] [Google Scholar]
- 35. Kalra S., Balhara Y. P., Sahay B. K., Ganapathy B., and Das A. K. (2013) Why is premixed insulin the preferred insulin? Novel answers to a decade-old question. J. Assoc. Physicians India 61, 9–11 [PubMed] [Google Scholar]
- 36. Brems D. N., Brown P. L., Nakagawa S. H., and Tager H. S. (1991) The conformational stability and flexibility of insulin with an additional intramolecular cross-link. J. Biol. Chem. 266, 1611–1615 [PubMed] [Google Scholar]
- 37. Ciszak E., Beals J. M., Frank B. H., Baker J. C., Carter N. D., and Smith G. D. (1995) Role of C-terminal B-chain residues in insulin assembly: the structure of hexameric LysB28ProB29-human insulin. Structure 3, 615–622 10.1016/S0969-2126(01)00195-2 [DOI] [PubMed] [Google Scholar]
- 38. Gallivan J. P., and Dougherty D. A. (1999) Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. U.S.A. 96, 9459–9464 10.1073/pnas.96.17.9459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lipari G., and Szabo A. (1982) Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104, 4546–4559 10.1021/ja00381a009 [DOI] [Google Scholar]
- 40. Sosnick T. R., Fang X., and Shelton V. M. (2000) Application of circular dichroism to study RNA folding transitions. Methods Enzymol. 317, 393–409 10.1016/S0076-6879(00)17026-0 [DOI] [PubMed] [Google Scholar]
- 41. Zhang Y. Z. (1995) Protein and Peptide Structure and Interactions Studied by Hydrogen Exchange and NMR. PhD thesis, University of Pennsylvania, Philadelphia [Google Scholar]
- 42. Englander S. W., Sosnick T. R., Englander J. J., and Mayne L. (1996) Mechanisms and uses of hydrogen exchange. Curr. Opin. Struct. Biol. 6, 18–23 10.1016/S0959-440X(96)80090-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hua Q.-X., Jia W., and Weiss M. A. (2011) Conformational dynamics of insulin. Front. Endocrinol. 2, 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saker F., Ybarra J., Leahy P., Hanson R. W., Kalhan S. C., and Ismail-Beigi F. (1998) Glycemia-lowering effect of cobalt chloride in the diabetic rat: role of decreased gluconeogenesis. Am. J. Physiol. 274, E984–E991 [DOI] [PubMed] [Google Scholar]
- 45. Mittag T., Kay L. E., and Forman-Kay J. D. (2010) Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 23, 105–116 [DOI] [PubMed] [Google Scholar]
- 46. Campbell E., Kaltenbach M., Correy G. J., Carr P. D., Porebski B. T., Livingstone E. K., Afriat-Jurnou L., Buckle A. M., Weik M., Hollfelder F., Tokuriki N., and Jackson C. J. (2016) The role of protein dynamics in the evolution of new enzyme function. Nat. Chem. Biol. 12, 944–950 10.1038/nchembio.2175 [DOI] [PubMed] [Google Scholar]
- 47. Badger J. (1992) Flexibility in crystalline insulins. Biophys. J. 61, 816–819 10.1016/S0006-3495(92)81886-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qin Y., Dittmer P. J., Park J. G., Jansen K. B., and Palmer A. E. (2011) Measuring steady-state and dynamic endoplasmic reticulum and Golgi Zn2+ with genetically encoded sensors. Proc. Natl. Acad. Sci. U.S.A. 108, 7351–7356 10.1073/pnas.1015686108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wijesekara N., Dai F. F., Hardy A. B., Giglou P. R., Bhattacharjee A., Koshkin V., Chimienti F., Gaisano H. Y., Rutter G. A., and Wheeler M. B. (2010) Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia 53, 1656–1668 10.1007/s00125-010-1733-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cutfield J., Cutfield S., Dodson E., Dodson G., Hodgkin D., and Reynolds C. (1981) Evidence concerning insulin activity from the structure of a cross-linked derivative. Hoppe Seylers Z Physiol. Chem. 362, 755–761 10.1515/bchm2.1981.362.1.755 [DOI] [PubMed] [Google Scholar]
- 51. Nakagawa S. H., and Tager H. S. (1989) Perturbation of insulin-receptor interactions by intramolecular hormone cross-linking. Analysis of relative movement among residues A1, B1, and B29. J. Biol. Chem. 264, 272–279 [PubMed] [Google Scholar]
- 52. Cosmatos A., Cheng K., Okada Y., and Katsoyannis P. G. (1978) The chemical synthesis and biological evaluation of [1-l-alanine-A]-and [1-d-alanine-A]insulins. J. Biol. Chem. 253, 6586–6590 [PubMed] [Google Scholar]
- 53. Hua Q. X., Jia W., and Weiss M. A. (2011) Conformational dynamics of insulin. Front. Endocrinol. (Lausanne) 2, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Becker R. H. (2007) Insulin glulisine complementing basal insulin: a review of structure and activity. Diabetes Technol. Ther. 9, 109–121 10.1089/dia.2006.0035 [DOI] [PubMed] [Google Scholar]
- 55. Jacoby E., Hua Q. X., Stern A. S., Frank B. H., and Weiss M. A. (1996) Structure and dynamics of a protein assembly. 1H-NMR studies of the 36 kDa R6 insulin hexamer. J. Mol. Biol. 258, 136–157 10.1006/jmbi.1996.0239 [DOI] [PubMed] [Google Scholar]
- 56. Smith G. D., Pangborn W. A., and Blessing R. H. (2003) The structure of T6 human insulin at 1.0 A resolution. Acta Crystallogr. D Biol. Crystallogr. 59, 474–482 10.1107/S0907444902023685 [DOI] [PubMed] [Google Scholar]
- 57. Hua Q. X., Hu S. Q., Frank B. H., Jia W., Chu Y. C., Wang S. H., Burke G. T., Katsoyannis P. G., and Weiss M. A. (1996) Mapping the functional surface of insulin by design: structure and function of a novel A-chain analogue. J. Mol. Biol. 264, 390–403 10.1006/jmbi.1996.0648 [DOI] [PubMed] [Google Scholar]
- 58. Olsen H. B., Ludvigsen S., and Kaarsholm N. C. (1996) Solution structure of an engineered insulin monomer at neutral pH. Biochemistry 35, 8836–8845 10.1021/bi960292+ [DOI] [PubMed] [Google Scholar]
- 59. Hua Q. X., Ladbury J. E., and Weiss M. A. (1993) Dynamics of a monomeric insulin analogue: testing the molten-globule hypothesis. Biochemistry 32, 1433–1442 10.1021/bi00057a006 [DOI] [PubMed] [Google Scholar]
- 60. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 10.1007/s10858-009-9333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang Y., Petkova A., Huang K., Xu B., Hua Q.-X., Ye I.-J., Chu Y.-C., Hu S.-Q., Phillips N. B., Whittaker J., Ismail-Beigi F., Mackin R. B., Katsoyannis P. G., Tycko R., and Weiss M. A. (2010) An Achilles' Heel in an amyloidogenic protein and its repair: insulin fibrillation and therapeutic design. J. Biol. Chem. 285, 10806–10821 10.1074/jbc.M109.067850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Binder P. M., and Danchin A. (2011) Life's demons: information and order in biology. EMBO Reports 12, 495–499 10.1038/embor.2011.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sanbonmatsu K. Y. (2012) Computational studies of molecular machines: the ribosome. Curr. Opin. Struct. Biol. 22, 168–174 10.1016/j.sbi.2012.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Derewenda U., Derewenda Z., Dodson E. J., Dodson G. G., Bing X., and Markussen J. (1991) X-ray analysis of the single chain B29-A1 peptide-linked insulin molecule. A completely inactive analogue. J. Mol. Biol. 220, 425–433 10.1016/0022-2836(91)90022-X [DOI] [PubMed] [Google Scholar]
- 65. Laajoki L. G., Francis G. L., Wallace J. C., Carver J. A., and Keniry M. A. (2000) Solution structure and backbone dynamics of long-[Arg(3)]insulin-like growth factor-I. J. Biol. Chem. 275, 10009–10015 [DOI] [PubMed] [Google Scholar]
- 66. Torres A. M., Forbes B. E., Aplin S. E., Wallace J. C., Francis G. L., and Norton R. S. (1995) Solution structure of human insulin-like growth factor II: relationship to receptor and binding protein interactions. J. Mol. Biol. 248, 385–401 10.1016/S0022-2836(95)80058-1,10.1006/jmbi.1995.0229 [DOI] [PubMed] [Google Scholar]
- 67. Brzozowski A. M., Dodson E. J., Dodson G. G., Murshudov G. N., Verma C., Turkenburg J. P., de Bree F. M., and Dauter Z. (2002) Structural origins of the functional divergence of human insulin-like growth factor-I and insulin. Biochemistry 41, 9389–9397 10.1021/bi020084j [DOI] [PubMed] [Google Scholar]
- 68. Schaffer M. L., Deshayes K., Nakamura G., Sidhu S., and Skelton N. J. (2003) Complex with a phage display-derived peptide provides insight into the function of insulin-like growth factor I. Biochemistry 42, 9324–9334 10.1021/bi034386c [DOI] [PubMed] [Google Scholar]
- 69. Miller J. A., Narhi L. O., Hua Q. X., Rosenfeld R., Arakawa T., Rohde M., Prestrelski S., Lauren S., Stoney K. S., and Tsai L. (1993) Oxidative refolding of IGF-1 yields two products of similar thermodynamic stability: a bifurcating protein-folding pathway. Biochemistry 32, 5203–5213 10.1021/bi00070a032 [DOI] [PubMed] [Google Scholar]
- 70. Hua Q. X., Jia W., Frank B. H., Phillips N. F., and Weiss M. A. (2002) A protein caught in a kinetic trap: structures and stabilities of insulin disulfide isomers. Biochemistry 41, 14700–14715 10.1021/bi0202981 [DOI] [PubMed] [Google Scholar]
- 71. Sohma Y., Hua Q.-X., Liu M., Phillips N. B., Hu S.-Q., Whittaker J., Whittaker L. J., Ng A., Roberts C. T. Jr., Arvan P., Kent S. B., and Weiss M. A. (2010) Contribution of residue B5 to the folding and function of insulin and IGF-I: constraints and fine-tuning in the evolution of a protein family. J. Biol. Chem. 285, 5040–5055 10.1074/jbc.M109.062992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Carr K. (2017) The Mechanism of the Prolonged Action of the Single-chain Insulin, 70-01. Masters thesis, Case Western Reserve University, Cleveland, OH [Google Scholar]
- 73. Rask-Madsen C., and Kahn C. R. (2012) Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 32, 2052–2059 10.1161/ATVBAHA.111.241919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang Z., and Thurmond D. C. (2009) Mechanisms of biphasic insulin-granule exocytosis – roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci. 122, 893–903 10.1242/jcs.034355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dror V., Nguyen V., Walia P., Kalynyak T. B., Hill J. A., and Johnson J. D. (2007) Notch signalling suppresses apoptosis in adult human and mouse pancreatic islet cells. Diabetologia 50, 2504–2515 10.1007/s00125-007-0835-5 [DOI] [PubMed] [Google Scholar]
- 76. Guillausseau P. J., Meas T., Virally M., Laloi-Michelin M., Médeau V., and Kevorkian J. P. (2008) Abnormalities in insulin secretion in type 2 diabetes mellitus. Diabetes Metab. 34, (Suppl. 2), S43–S48 [DOI] [PubMed] [Google Scholar]
- 77. Zaykov A. N., Mayer J. P., and DiMarchi R. D. (2016) Pursuit of a perfect insulin. Nat. Rev. Drug Discov. 15, 425–439 10.1038/nrd.2015.36 [DOI] [PubMed] [Google Scholar]
- 78. Nathan D. M., and DCCT/EDIC Research Group (2014) The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study at 30 years: overview. Diabetes Care 37, 9–16 10.2337/dc13-2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang Y., Liang Z. H., Zhang Y. S., Yao S. Y., Xu Y. G., Tang Y. H., Zhu S. Q., Cui D. F., and Feng Y. M. (2001) Human insulin from a precursor overexpressed in the methylotrophic yeast Pichia pastoris and a simple procedure for purifying the expression product. Biotechnol. Bioeng. 73, 74–79 10.1002/1097-0290(20010405)73:1%3C74::AID-BIT1038%3E3.0.CO%3B2-V [DOI] [PubMed] [Google Scholar]
- 80. Kjeldsen T., Pettersson A. F., and Hach M. (1999) Secretory expression and characterization of insulin in Pichia pastoris. Biotechnol. Appl. Biochem. 29, 79–86 [PubMed] [Google Scholar]
- 81. Sugiki T., Ichikawa O., Miyazawa-Onami M., Shimada I., and Takahashi H. (2012) Isotopic labeling of heterologous proteins in the yeast Pichia pastoris and Kluyveromyces lactis. Methods Mol. Biol. 831, 19–36 10.1007/978-1-61779-480-3_2 [DOI] [PubMed] [Google Scholar]
- 82. Ohara T., Makino K., Shinagawa H., Nakata A., Norioka S., and Sakiyama F. (1989) Cloning, nucleotide sequence, and expression of Achromobacter protease I gene. J. Biol. Chem. 264, 20625–20631 [PubMed] [Google Scholar]
- 83. Pandyarajan V., Phillips N. B., Cox G. P., Yang Y., Whittaker J., Ismail-Beigi F., and Weiss M. A. (2014) Biophysical optimization of a therapeutic protein by non-standard mutagenesis. studies of an iodo-insulin derivative. J. Biol. Chem. 289, 23367–23381 10.1074/jbc.M114.588277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Goddard T. D., and Kneller D. G. (2008) SPARKY 3. University of California, San Francisco [Google Scholar]
- 85. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 86. Cornilescu G., Delaglio F., and Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302 10.1023/A:1008392405740 [DOI] [PubMed] [Google Scholar]
- 87. Schwieters C., Kuszewski J., and Clore G. M. (2006) Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 48, 47–62 10.1016/j.pnmrs.2005.10.001 [DOI] [Google Scholar]
- 88. Kay L. E., Torchia D. A., and Bax A. (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28, 8972–8979 10.1021/bi00449a003 [DOI] [PubMed] [Google Scholar]
- 89. Stone M. J., Fairbrother W. J., Palmer A. G. 3rd, Reizer J., Saier M. H. Jr., and Wright P. E. (1992) Backbone dynamics of the Bacillus subtilis glucose permease IIA domain determined from nitrogen-15 NMR relaxation measurements. Biochemistry 31, 4394–4406 10.1021/bi00133a003 [DOI] [PubMed] [Google Scholar]
- 90. d'Auvergne E. J., and Gooley P. R. (2008) Optimisation of NMR dynamic models I. Minimisation algorithms and their performance within the model-free and Brownian rotational diffusion spaces. J. Biomol. NMR 40, 107–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Fushman D. (2012) Determining protein dynamics from (15)N relaxation data by using DYNAMICS. Methods Mol. Biol. 831, 485–511 10.1007/978-1-61779-480-3_24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Webb B., and Sali A. (2016) Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 54, 5.6.1–5.6.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., and Lindahl E. (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 [Google Scholar]
- 94. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and MacKerell A. D. Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 10.1021/ct300400x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Guvench O., Mallajosyula S. S., Raman E. P., Hatcher E., Vanommeslaeghe K., Foster T. J., Jamison F. W. 2nd, and MacKerell A. D. Jr. (2011) CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate–protein modeling. J. Chem. Theory Comput. 7, 3162–3180 10.1021/ct200328p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- 97. Berendsen H. J. C., Postma J. P. M., Gunsteren W. F. v., DiNola A., and Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 10.1063/1.448118 [DOI] [Google Scholar]
- 98. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 10.1063/1.470117 [DOI] [Google Scholar]
- 99. Wilson M. A., and Brünger A. T. (2000) The 1.0 Å crystal structure of Ca(2+)-bound calmodulin: an analysis of disorder and implications for functionally relevant plasticity. J. Mol. Biol. 301, 1237–1256 10.1006/jmbi.2000.4029 [DOI] [PubMed] [Google Scholar]
- 100. Burling F. T., and Brünger A. T. (1994) Thermal motion and conformational disorder in protein crystal structures: comparison of multi-conformer and time-averaging models. Isr. J. Chem. 34, 165–175 10.1002/ijch.199400022 [DOI] [Google Scholar]
- 101. Levin E. J., Kondrashov D. A., Wesenberg G. E., and Phillips G. N. (2007) Ensemble refinement of protein crystal structures: validation and application. Structure 15, 1040–1052 10.1016/j.str.2007.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.