

Abstract
C-type lectin domain family 3 member A (CLEC3A) is a poorly characterized protein belonging to the superfamily of C-type lectins. Its closest homologue tetranectin binds to the kringle 4 domain of plasminogen and enhances its association with tissue plasminogen activator (tPA) thereby enhancing plasmin production, but whether CLEC3A contributes to plasminogen activation is unknown. Here, we recombinantly expressed murine and human full-length CLEC3As as well as truncated forms of CLEC3A in HEK-293 Epstein-Barr nuclear antigen (EBNA) cells. We analyzed the structure of recombinant CLEC3A by SDS-PAGE and immunoblot, glycan analysis, matrix-assisted laser desorption ionization time-of-flight mass spectrometry, size-exclusion chromatography, circular dichroism spectroscopy, and electron microscopy; compared the properties of the recombinant protein with those of CLEC3A extracted from cartilage; and investigated its tissue distribution and extracellular assembly by immunohistochemistry and immunofluorescence microscopy. We found that CLEC3A mainly occurs as a monomer, but also forms dimers and trimers, potentially via a coiled-coil α-helix. We also noted that CLEC3A can be modified with chondroitin/dermatan sulfate side chains and tends to oligomerize to form higher aggregates. We show that CLEC3A is present in resting, proliferating, and hypertrophic growth-plate cartilage and assembles into an extended extracellular network in cultures of rat chondrosarcoma cells. Further, we found that CLEC3A specifically binds to plasminogen and enhances tPA-mediated plasminogen activation. In summary, we have determined the structure, tissue distribution, and molecular function of the cartilage-specific lectin CLEC3A and show that CLEC3A binds to plasminogen and participates in tPA-mediated plasminogen activation.
Keywords: cartilage, cartilage biology, extracellular matrix, plasminogen, plasminogen regulation, tissue plasminogen activator (tPA), C-type lectin domain family 3 member A, C-type lectin, CLEC3A
Introduction
The C-type lectins form a diverse protein family with many different functions across species. Most members are found extracellularly and carry C-type carbohydrate recognition domains (CRD)2 that in some cases specifically recognize or bind proteins, lipids, or carbohydrates in a Ca2+-dependent manner, whereas those in other proteins only form a structural motif. The protein family is classified into 17 subgroups, but the classification criteria are not consistent with regard to function, phylogenesis criteria, or gene structure (1). C-type lectin domain family 3 member A (CLEC3A, originally called CLECSF1) was first described as a cartilage-derived member of the C-type lectin superfamily and according to its domain structure assigned to the tetranectin IX group, together with tetranectin (CLEC3B) and stem cell growth factor (SCGF) with α and β forms (CLEC11A) (1, 2). Whereas tetranectin binds to the plasminogen kringle 4 (3) and is thought to enhance tissue plasminogen activator (tPA)-mediated plasminogen activation (4), SCGF (CLEC11A) has been described to be a growth factor stimulating hematopoietic progenitor cells (5). In cartilage, the large proteoglycan aggrecan possesses a C-type lectin domain (CLD) and interacts via the CLD with other extracellular matrix proteins, e.g. fibulins and tenascins, and with glycosaminoglycans on the cell surface (6). Missense mutations in the aggrecan CLD repeat have been described to cause the human disorders spondyloepimetaphyseal dysplasia (autosomal recessive aggrecan-type) and familial osteochondritis dissecans (7).
The murine and human CLEC3A gene consists of three exons (Fig. 1). The first exon codes for a potential signal peptide with 22 amino acids and the subsequent 16 amino acids (2), 8 of which are positively charged. The second exon encodes 27 amino acids and the third a CRD domain of 130 amino acids. The CRD domain contains six cysteine residues which form disulfide bonds in the pattern 1 + 2, 3 + 6, and 4 + 5 (2). Based on the sequence homology with its closest relative tetranectin (Fig. 1), CLEC3A could potentially occur as an oligomer that forms trimers via an N-terminal coiled-coil domain (2). Northern blot analysis of human CLEC3A showed an expression restricted to cartilage (2) which was up-regulated in osteoarthritis (8). Human CLEC3A mRNA has been detected in normal breast and breast cancer tissue as well as in two colon cancer cell lines (9).
Figure 1.

Alignment of CLEC3A and tetranectin and schematic illustration of CLEC3A. A, alignment of the amino acid sequence of mouse CLEC3A (NM_001007223.3) and mouse tetranectin (NM_011606.2). The potential signal peptide cleavage site is marked by an arrow, exon/intron borders of exons 1, 2, and 3 by vertical lines (2), and MMP-7 cleavage sites by arrowheads (9). The amino acid sequences of mouse and human CLEC3A show a homology of 87%. Three amino acids that are involved in binding the plasminogen kringle 4 domain are conserved in mouse and human CLEC3A (asterisks). Hash mark, predicted O-glycosylation sites; the illustration is based on Ref. 2. B, alignment of the murine CLEC3A and murine tetranectin sequences encoded by exon 2. The positions “a” and “d” that are preferentially occupied by hydrophobic amino acids in heptad repeats are indicated (15). C, schematic depiction of CLEC3A. OD, potential oligomerization (coiled-coil) domain. CRD, carbohydrate recognition domain. D, schematic illustration of recombinant full-length CLEC3A and truncated forms of CLEC3A.
Although the exact function of CLEC3A is not known, tetranectin enhances the association between tissue plasminogen activator and plasminogen 10-fold and is thought to regulate proteolytic processes (3, 4). Three specific amino acid residues that are involved in plasminogen binding were identified and are conserved in human and murine CLEC3A (Fig. 1) (2) but not in SCGF (CLEC11A). CLEC3A-expressing HT1080 cells adhere with higher efficiency to laminin and fibronectin than mock-transfected HT1080 cells, and CLEC3A also binds heparin (9). In WiDr human colon carcinoma cells CLEC3A is a membrane-associated substrate for matrix metalloproteinase-7 (MMP-7) (9), and it was speculated that cleavage of CLEC3A by MMP-7 in the tumor microenvironment may affect tumor cell invasion and metastasis by modulating cell adhesion and the plasminogen/plasminogen-activator system (9).
In this study, we used mouse CLEC3A for structural characterization of CLEC3A, and human CLEC3A for functional studies. We recombinantly expressed murine and human full-length CLEC3A as well as truncated forms comprising the potential α-helical oligomerization domain (OD) and the CRD (CLEC3A-Ex23) or only the CRD (CLEC3A-Ex3) in HEK-293 EBNA cells and generated specific antisera. We analyzed the structure of murine CLEC3A by a variety of biochemical and biophysical methods and compared the properties of the recombinant protein with those of CLEC3A extracted from cartilage. Further, we investigated its tissue distribution and extracellular assembly by immunohistochemistry and immunofluorescence microscopy. Finally, we studied the interaction of human CLEC3A and plasminogen by enzyme-linked immunosorbent binding assay (ELISA-style binding assay) and by surface plasmon resonance and analyzed the tPA-mediated activation of plasminogen in the presence of human CLEC3A by plasminogen conversion assay.
Results
Recombinant expression of CLEC3A
cDNAs encoding human and murine full-length CLEC3A (CLEC3A-flc) as well as truncated forms comprising the OD and CRD (CLEC3A-Ex23), or only the CRD (CLEC3A-Ex3) were cloned into modified pCEP-Pu vectors utilizing either the BM40 signal peptide (10) or the endogenous CLEC3A signal peptide. The recombinant vectors were used to transfect HEK-293 EBNA cells. Interestingly, only the vector containing the endogenous CLEC3A signal peptide yielded an appropriate protein expression of full-length CLEC3A, whereas those containing the BM40 signal peptide gave very low secretion. Transfection of CLEC3A-Ex3 carrying the BM40 signal peptide gave adequate secretion. The identity of recombinant murine full-length CLEC3A and CLEC3A-Ex3 was verified by peptide mass fingerprint analysis (not shown).
Antibody generation
Murine CLEC3A-Ex3 and human full-length CLEC3A were used to immunize a rabbit. The resulting antisera were Affinity-purified by binding to a column coupled with human full-length CLEC3A or murine CLEC3A-Ex3. In immunoblot analysis the eluted antibodies reacted strongly with CLEC3A and not with tetranectin (Fig. S1).
Structure of CLEC3A
We used recombinant mouse CLEC3A for structural characterization and recombinant human CLEC3A for functional studies. Recombinant mouse and human full-length CLEC3A as well as truncated forms of mouse and human CLEC3A were purified, and analyzed by SDS-PAGE and immunoblotting (Fig. 2, A, B, F, and G). Under nonreducing conditions full-length CLEC3A (Fig. 2, A and G) mainly migrates as distinct bands with a molecular mass of ∼20–25 kDa, in good agreement with the calculated mass of 23.4 kDa for mouse and human CLEC3A. This shows that mouse and human CLEC3A occur mainly as monomers under denaturing conditions. After reduction of recombinant CLEC3A additional bands appeared below 20 kDa (Fig. 2A), presumably representing proteolytic fragments released upon cleavage of intrachain disulfide bonds. Interestingly, Coomassie Blue staining of mouse full-length CLEC3A revealed an additional diffuse band migrating between 35 and 100 kDa (Fig. 2A). By sequence analysis of mouse CLEC3A (www.expasy.org)3 we could identify two potential O-glycosylation sites (Fig. 1A) at positions Ser-25 and Ser-33. Upon treatment of mouse CLEC3A with chondroitinase ABC the diffuse band largely disappeared, demonstrating substitution with chondroitin and/or dermatan sulfate, and an additional band was detected at 46 kDa, which may represent dimeric CLEC3A (Fig. 2C).
Figure 2.

Immunoblot analysis of mouse and human CLEC3A. A–G, recombinant mouse and human full-length CLEC3A (CLEC3A-flc), CLEC3A-Ex23, CLEC3A-Ex3, chondroitinase ABC–treated recombinant full-length CLEC3A, tissue extracts from a mixture of forelegs and ribs of a newborn mouse (newborn) and from the tail of a 2-year-old mouse (adult), and human cartilage extracts were separated by SDS-PAGE under reducing (+SH) and nonreducing (−SH) conditions and visualized by Coomassie staining or by immunoblot with Affinity-purified antibodies against murine CLEC3A-Ex3 (A–D) or human full-length CLEC3A (flc) (E–G). Both recombinant and tissue CLEC3A occur mainly as monomer. Faint bands with a molecular mass around 40 kDa most likely represent dimeric CLEC3A. Note that recombinant CLEC3A possesses a 3.5-kDa strepII-tag. C, incubation of CLEC3A with chondroitinase ABC (+) led to the loss of the higher diffuse band, indicating that CLEC3A carries chondroitin sulfate and/or dermatan sulfate side chains. Additional bands in the Coomassie Blue–stained gel of chondroitinase ABC–treated CLEC3A originate from the enzyme preparation and from albumin which was used as a carrier (all samples +SH). E, human cartilage extracts were separated by SDS-PAGE followed by immunoblot with Affinity-purified antibodies against human CLEC3A. 1–4, extraction buffer 1–4 (see “Experimental procedures”). Please note that the human antibodies react only weakly with nonreduced monomeric CLEC3A.
Extracts of cartilage from forelegs and ribs of a newborn mouse and from the tail of a 2-year-old mouse were analyzed by immunoblotting (Fig. 2D). Mouse tissue–derived CLEC3A migrates as a distinct band at 20 kDa, showing that also tissue-derived CLEC3A occurs mainly as a monomer in SDS-PAGE. Further, a weak band with a mass of about 38 kDa was detected, possibly representing dimeric CLEC3A. In contrast with recombinant mouse CLEC3A, we did not find evidence for glycosylation of tissue-derived CLEC3A (Fig. 2D).
In addition, we analyzed sequential extracts from human osteoarthritic cartilage (Fig. 2E). We found the highest amounts of CLEC3A in extracts obtained with 2 m urea. Also in human cartilage CLEC3A occurs mainly as a monomer. Furthermore, a minor amount of dimeric CLEC3A as well as proteolytic cleavage products was detected (Fig. 2E).
Recombinant mouse CLEC3A was submitted to MALDI-TOF mass spectrometry with or without prior reduction (Fig. 3). Under nonreducing conditions, a major molecule ion peak was detected at 21.9 kDa, representing the CLEC3A monomer, and a second very faint peak at 44.6 kDa, probably originating from dimers. The calculated mass of full-length mouse CLEC3A is 23.4 kDa and the lower mass indicates that the protein is proteolytically processed. Peptide mass fingerprint analysis gave evidence for a processing at the N terminus (not shown). Similar processing at the positively charged N-terminal end was seen for matrilin-3 that had been recombinantly expressed in HEK-293 EBNA cells (11). Analysis of CLEC3A-Ex3 showed that it is not cleaved at either the N or the C terminus. Reduced full-length CLEC3A yielded additional molecule ion peaks with masses between 10 and 17 kDa, in accordance with the immunoblotting results.
Figure 3.

Analysis of mouse CLEC3A by mass spectrometry. Purified recombinant mouse CLEC3A was investigated by MALDI-TOF mass spectrometry prior to (−SH) and after (+SH) reduction. Under nonreducing conditions a main molecule ion peak with a mass of 21949 Da and a faint peak with a mass of 44640 Da could be detected, and after reduction a main peak with a mass of 21949 Da.
Size-exclusion chromatography and circular dichroism (CD) spectroscopy
We characterized the oligomeric state of mouse CLEC3A, CLEC3A-Ex3, and CLEC3A-Ex2 by calibrated size-exclusion chromatography (Fig. 4A). Full-length CLEC3A eluted in a broad manner at positions corresponding to molecular masses ranging from more than 756 kDa to less than 43 kDa. This indicates that full-length CLEC3A forms higher order complexes under native conditions. Such complexes were also seen in electron microscopy.4 Some complex formation could also be detected in agarose-acrylamide composite (12) native gels separating very large protein complexes and in native polyacrylamide gel electrophoresis (Fig. S2, A and B). CLEC3A-Ex3 eluted close to the total volume of the size-exclusion column, which reflects that this protein lacks the potential coiled-coil oligomerization domain. The 29 amino acid–long synthetic peptide CLEC3A-Ex2 with a calculated mass of 3387 Da also eluted close to the total volume.
Figure 4.

Size-exclusion chromatography and CD spectroscopy analysis of mouse CLEC3A. A, full-length CLEC3A (CLEC3A-flc), CLEC3A-Ex3, or CLEC3A-Ex2 were analyzed on a size-exclusion column and the elution positions compared with molecular mass standards. Elution fractions were collected and analyzed by SDS-PAGE followed by immunoblot with the Affinity-purified antiserum against mouse CLEC3A-Ex3 in case of CLEC3A and CLEC3A-Ex3 and by silver staining in case of CLEC3A-Ex2. V0, void volume; Vt, total volume. B, full-length CLEC3A (CLEC3A-flc), CLEC3A-Ex3, and CLEC3A-Ex2 were analyzed by CD spectroscopy. All spectra showed minima at 207 nm and 227 nm, characteristic for α-helical structure.
The secondary structure of recombinant murine full-length CLEC3A, CLEC3A-Ex3, and CLEC3A-Ex2 was analyzed by CD spectroscopy (Fig. 3B). The CLEC3A spectra indicated a structurally intact protein with about 90% α-helical conformation and some β-turns or disordered structures, and also the spectra of CLEC3A-Ex3 showed mainly an α-helical structure. The spectra for CLEC3A-Ex2 showed clear minima at 207 nm and 227 nm, characteristic for an α-helix (about 87%). Indeed, the second exon codes for 3.5 heptad repeats of 27 amino acids' residues (2) that are predicted to form a coiled-coil α-helix (http://embnet.vital-it.ch/software/COILS_form.html)3 (Fig. 1B), which may explain the oligomerization of CLEC3A seen in size-exclusion chromatography.
Electron microscopy of recombinant murine full-length CLEC3A
We also investigated the oligomeric structure of mouse full-length CLEC3A by electron microscopy (Fig. 5). The stained particles were heterogeneous in size and although monomers predominated, dimers and trimers also could be observed. At higher magnification the monomeric molecules show a globular domain, that most likely represents the CRD (exon 3), and a filamentous tail domain, probably representing the N-terminal part (exon 1) and the putative coiled-coil domain (exon 2). Dimers and trimers are joined at a single point via the filamentous domain, reminiscent of the structures seen for some oligomeric proteins held together by a coiled-coil, e.g. the matrilins (11).
Figure 5.

Negative stain electron microscopy of purified recombinant mouse full-length CLEC3A. A, the overview shows monomeric and oligomeric forms of CLEC3A. B, selected particles that represent monomeric, dimeric, and trimeric CLEC3A. Scale bars: 100 nm.
Tissue distribution and extracellular assembly of CLEC3A
When investigating the tissue distribution of CLEC3A by extraction of mouse tissues followed by SDS-PAGE and immunoblotting we could only detect the protein in cartilage (Fig. 2D). By immunohistochemistry on a section of a 14.25-day-old mouse embryo, CLEC3A could be seen in the primordial skeleton, e.g. in the vertebrae and ribs, in the cartilage primordium of the legs, and in the hyoid bone of the tongue (Fig. 6A). In the head of a newborn mouse, CLEC3A was present in the developing occipital bones and in cartilages of the nasal cavity (Fig. 6B). Also after birth, strong expression of CLEC3A could be detected only in cartilage. In knee joint cartilage of a newborn mouse, CLEC3A was present in the resting, proliferating, and hypertrophic zones of the growth plate (Fig. 6C). The staining was more prominent in the territorial matrix, but also seen in the interterritorial matrix. Close to the surface of the developing articular cartilage the signal was faint. CLEC3A was also detected in the cartilage remnants of growing bones, in costal and sternal cartilage, and in the cartilage plates of the sternum (Fig. 6D). The extracellular assembly of CLEC3A was studied by immunofluorescence microscopy of the extracellular matrix formed by Swarm rat chondrosarcoma (RCS) cells (Fig. 6, E and F). Here CLEC3A was part of an extensive extracellular network, reminiscent of the network staining seen for matrilin-3 (11) (Fig. 6F), and is also in the pericellular matrix (Fig. 6E).
Figure 6.
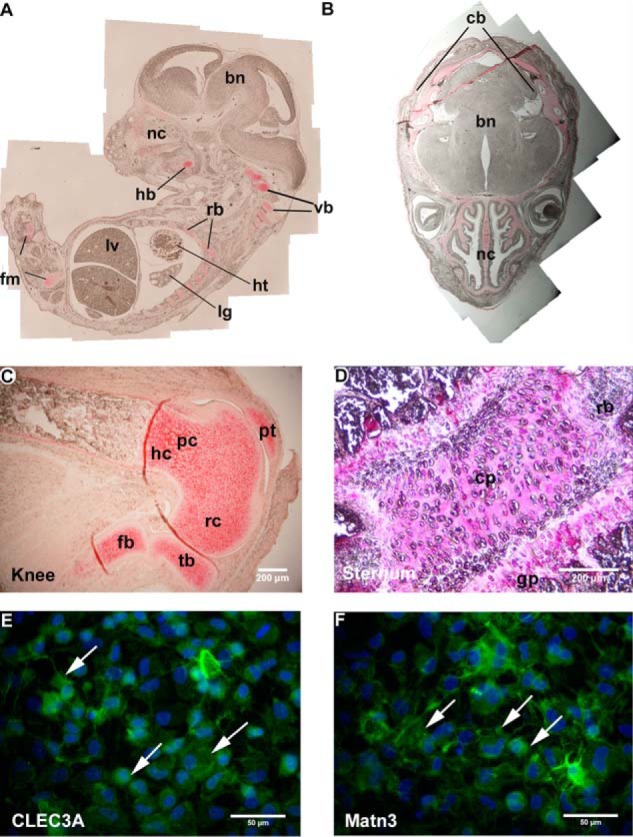
Tissue distribution (A–D) and extracellular assembly (E and F) of CLEC3A. A–D, immunohistochemistry was performed on paraffin-embedded tissue sections of a 14.25-day-old mouse embryo (A), a newborn mouse head (B), a knee of a newborn (C), and the sternum of a 6-week-old (D) mouse using the Affinity-purified murine CLEC3A-Ex3 antibodies. A, in a 14.25-day-old mouse embryo, CLEC3A was found in cartilage primordiae of the legs and the vertebrae (vb), in rib cartilage (rb), and in cartilages of the nasal cavity (nc). lv, liver; ht, heart; lg, lung; fm, femur; hb, hyoid bone. B, in the head, CLEC3A was present in the cartilage primordium of the developing cranial bones (cb) and in cartilages of the nasal cavity (nc). bn, brain. C, in a knee, CLEC3A was detected in the resting (rc), proliferating (pc), and hypertrophic (hc) zones of cartilage. tb, tibia; pt, patella; fb, fibula. D, in the sternum CLEC3A was detected in the cartilage plates (cp) and the growth plates (gp) and rib (rb). C and D, scale bars: 200 μm. E and F, extracellular assembly of CLEC3A (E) and matrilin-3 (F) was studied by immunofluorescence microscopy on cultures of RCS cells. CLEC3A is part of an extensive extracellular network (E) and is also present in the pericelluar matrix reminiscent of the network staining seen for matrilin-3 (F). E and F, scale bars: 50 μm. Arrows indicate CLEC3A or matrilin-3 staining.
CLEC3A-plasminogen interaction
Three specific amino acid residues that are involved in binding of tetranectin to plasminogen are conserved in CLEC3A (Fig. 1). By SDS-PAGE and immunoblot analysis we found evidence for plasminogen/plasmin in cartilage extracts (not shown). We therefore investigated a potential CLEC3A-plasminogen interaction by ELISA-style binding assay (Fig. 7A). Human full-length CLEC3A was coated on a 96-well plate and after incubation with serial dilutions of plasminogen, bound plasminogen was detected with a polyclonal antibody. A concentration-dependent binding of plasminogen to human full-length CLEC3A was seen which did not, however, reach saturation (Fig. 7A).
Figure 7.

Binding of CLEC3A to plasminogen. A, for ELISA-style binding assay, human CLEC3A was coated on 96-well plates at 1 μg/well followed by incubation with human plasminogen in serial dilutions. Error bars indicate ±S.D. B, for surface plasmon resonance analysis, human plasminogen was immobilized on a sensor chip and serial dilutions of human full-length CLEC3A (0.315–5 μm) were passed over the chip. Both methods showed a concentration-dependent binding of plasminogen to CLEC3A.
We further studied the binding of CLEC3A to plasminogen by surface plasmon resonance (Fig. 7B). Plasminogen was immobilized on a sensor chip and serial dilutions of human full-length CLEC3A (0.315–5 μm) were passed over the chip. The results showed a concentration-dependent binding of human full-length CLEC3A to plasminogen (KD 230 nm) (Fig. 7B).
CLEC3A-enhanced plasminogen activation
To determine whether CLEC3A is involved in tPA-mediated plasminogen activation, we assessed the ability of human tPA to convert human plasminogen to plasmin and measured the hydrolysis of the substrate S-2251 (H-D-Val-Leu-Lys-pNA) by plasmin in the presence of human full-length and truncated CLEC3A (Fig. 8A) at different concentrations (Fig. S3). Both full-length CLEC3A and to a lesser extent CLEC3A-Ex23 enhanced tPA-mediated plasminogen activation. The enzymatic activity detected in the presence of the various proteins was full-length CLEC3A, 52 units/liter; CLEC3A-Ex23, 31 units/liter; CLEC3A-Ex3, 10 units/liter; CLEC3A-Ex12, 7 units/liter; and BSA, 7 units/liter.
Figure 8.

Plasminogen conversion assay. A, plasminogen activation was studied in an assay based on the ability of tPA to convert plasminogen to plasmin, and the hydrolysis of the substrate S-2251 by plasmin. The cleavage of the substrate leads to the release of p-nitroaniline which can be detected at 405 nm. The plasminogen activation assay was performed with 2 μm Glu-plasminogen, 10 nm tPA, and 5 μg of human full-length CLEC3A (CLEC3A-flc), CLEC3A-Ex23, CLEC3A-Ex3, CLEC3A-Ex12, or BSA, as indicated. Full-length CLEC3A and to a lesser extent CLEC3A-Ex23 enhance tPA-mediated plasminogen activation. B, CLEC3A (flc) was incubated with tPA in the presence (+) or absence (−) of plasminogen (Plg). Cleavage of CLEC3A by plasmin was studied by immunoblotting under reducing (+SH) and nonreducing (−SH) conditions. After incubation of CLEC3A with tPA and plasminogen, CLEC3A could no longer be detected, showing that CLEC3A is a plasmin substrate.
CLEC3A a plasmin substrate
We incubated human full-length CLEC3A with tPA in the presence or absence of plasminogen for 30 min at 37 °C. Subsequently, cleavage of CLEC3A was analyzed by SDS-PAGE followed by immunoblotting. After 30-min incubation of CLEC3A with tPA and plasminogen, CLEC3A could no longer be detected, which shows that CLEC3A is a plasmin substrate (Fig. 8B).
Discussion
We investigated the structure, tissue distribution, and extracellular assembly of CLEC3A, and its role in tPA-mediated plasminogen activation. CLEC3A mainly occurs as a monomer but can also form oligomers, presumably via a coiled-coil α-helix. Tetranectin has been shown to form non-covalent homotrimers (13). The genes encoding murine CLEC3A and tetranectin are orthologous and their amino acid sequence is 42% identical (Fig. 1). The mouse CLEC3A gene consists of three exons and the second exon codes for 3.5 heptad repeats with a length of 27 amino acids (2) that are predicted to form a trimeric (2) coiled-coil α-helix (http://embnet.vital-it.ch/software/COILS_form.html)3 (Fig. 1B). However, the coiled coil of CLEC3A was predicted to be rather unstable, which is reflected in the rather low abundance of oligomers detected. A reason for this could be the relatively short heptad repeat region. By database analysis, it was predicted that at least four to five heptad repeats are necessary to form a stable coiled-coil domain (14). Interestingly, tetranectin is known to form a trimeric α-helical coiled coil (15) although the amino acid residues in the “a” and “d” positions of the heptad repeat are mostly identical of those in CLEC3A (Fig. 1B). Furthermore, CLEC3A possesses six cysteine residues within the CRD domain that is encoded by CLEC3A exon 3. Cysteines frequently form disulfide bridges to stabilize the structure of multimeric proteins that are assembled via a coiled-coil domain, e.g. in matrilin 1 (16). However, for CLEC3A it has been predicted that the six cysteine residues within the CRD will form intrachain instead of interchain disulfide bridges, and stabilize the structure of the CRD rather than bridging CLEC3A subunits (2). This is in full agreement with the findings from our biochemical analysis.
We found a distinct sharp band for CLEC3A in SDS-PAGE, but in addition a higher, diffuse band was seen as is often the case for highly glycosylated proteins such as proteoglycans and mucins. Tetranectin is not N-glycosylated but by mass spectrometry, a heterogeneous O-glycosylation was detected (17). For CLEC3A, no N-glycosylation is predicted, but potential O-glycosylation sites occur at the positively charged N terminus (http://www.cbs.dtu.dk/services/NetOGlyc/).3 By treatment of CLEC3A with chondroitinase ABC we could show that CLEC3A carries glycosaminoglycan side chains of the chondroitin sulfate/dermatan sulfate type. Interestingly, only a portion of the CLEC3A molecules carry side chains. Analysis of tissue-derived CLEC3A did not show any substitution with glycosaminoglycans, possibly because of the low amounts of protein in tissue extracts which makes the detection of diffuse bands difficult.
Immunofluorescence microscopy of the extracellular matrix formed by RCS cells reveals that CLEC3A is part of an extended filamentous extracellular network. This network is similar to that seen for matrilin-3 (11). In addition, CLEC3A shows a pericellular deposition, in agreement with the proposal that CLEC3A is a membrane-bound target of MMP-7 (9).
By Northern blot analysis of tissue extracts, CLEC3A was predicted to be cartilage specific (2). By RT-PCR CLEC3A mRNA was also detected in human colon cancer cell lines, in human breast cancer cell lines, and in normal breast and breast cancer tissue by use of a human tissue cDNA library (9). By SDS-PAGE and immunoblotting as well as by immunohistochemistry, we found CLEC3A protein only in cartilaginous tissues, e.g. newborn femur, sternum, and tail. In growth plate cartilage, CLEC3A is present in the resting, proliferating, and hypertrophic zones and the protein was also detected in cartilage remnants of the growing bone. Recently, it was shown that murine CLEC3A mRNA levels are up-regulated in IL-1α–induced cartilage degradation and that human CLEC3A is significantly more highly expressed in osteoarthritic cartilage than in normal donor articular cartilage (8, 18). Taken together, the cartilage-specific protein expression and the enhanced expression in osteoarthritic cartilage makes CLEC3A an interesting biomarker candidate for the diagnosis of degenerative joint diseases, e.g. for osteoarthritis.
The function of CLEC3A in cartilage has been unknown. CLEC3A has a heparin-binding activity, which is abolished after cleavage of CLEC3A by MMP-7 (9). In addition, recombinantly expressed CLEC3A promotes cell adhesion and spreading onto laminin and fibronectin substrates. It was speculated that CLEC3A acts as a cell adhesion modulator and that cleavage of CLEC3A by MMP-7 affects tumor cell invasion and metastasis (9). CLEC3A may therefore be involved in the adhesion of chondrocytes to the extracellular matrix.
CLEC3A has two functional domains: a positively charged, heparin-binding N-terminal domain and a C-terminal CRD domain, i.e. the binding domain of the C-type lectins (1). We could show that CLEC3A predominantly occurs as a monomer, but can also form dimers and trimers. As an oligomer, CLEC3A could simultaneously bind multiple ligand molecules via its CRD domain. Tetranectin was shown to enhance the association between tissue plasminogen activator and plasminogen by 10-fold (4). The enhanced activation was suggested to be because of tetranectin's ability to bind and accumulate tPA in an active conformation (4). Tetranectin is thought to regulate proteolytic processes by binding to plasminogen (3, 19) and may be involved in the regulation of tissue remodeling, osteogenesis, myogenesis, wound repair, and regeneration (20–23). We could show also that the cartilage-specific homologue CLEC3A binds specifically to plasminogen and enhances tPA-mediated plasminogen activation. Furthermore, we could show that CLEC3A can form oligomers, and only full-length CLEC3A and CLEC3A-Ex23, that also possesses the potential oligomerization domain, gave an enhanced tPA-mediated plasminogen activation. Therefore, we conclude that CLEC3A may bind both plasminogen and tPA and in this way accelerate tPA-mediated plasminogen activation.
Plasmin is essential for fibrinolysis and plasminogen activation after traumatic bleeding results in enhanced fibrinolysis and clearance. In addition, plasmin activates various pro-MMPs that are involved in cartilage homeostasis (24). We propose that CLEC3A is a cartilage-specific plasminogen activator involved in cartilage remodeling and degradation, i.e. during endochondral bone formation or after traumatic lesions. Furthermore, the susceptibility of CLEC3A to proteolytic cleavage by plasmin would be an ideal feedback mechanism to reduce excessive activation.
In summary, we have determined the structure, tissue distribution, and molecular function of the cartilage-specific lectin CLEC3A and show that CLEC3A binds to plasminogen and participates in tPA-mediated plasminogen activation.
Experimental procedures
Ethics
All procedures were approved by local government authorities (LANUV NRW, permit no. 87–51.04.2010.A218) and by the local ethics committee (Ethics Committee of the University Hospital Cologne, application no. 04–196).
Cloning and recombinant expression of full-length and truncated murine and human CLEC3A proteins
cDNA of murine full-length CLEC3A (NM_001007223.3) and a truncated form of murine CLEC3A (CLEC3A-Ex3) comprising only the CRD encoded by CLEC3A exon 3, and human full-length CLEC3A (NM_005752) and truncated forms of human CLEC3A comprising CLEC3A exon 2 and exon 3 (CLEC3A-Ex23), or only CLEC3A exon 3 (CLEC3A-Ex3) was generated by RT-PCR (SuperScript III, Life Technologies) on mouse or human cartilage total RNA. The primers for murine full-length CLEC3A were designed with a 5′-HindIII (5′-ACTAAGCTTATGGCAAAGAACGGACTTGTCCTTTG-3′) and a 3′-BamHI (5′-GACTGGATCCTGGGATGATAAACTCACATATGTATCGCTTGC-3′), and the primers of murine CLEC3A-Ex3 were designed with a 5′-NheI (5′-CTAGGCTAGCTGTCTGTCTCCGAGGCACCAAAG-3′) and a 3′-NotI restriction site (5′-GGCCGCGGCCGCTTATGGGATGATAAACTCAC-3′). The primers for human full-length CLEC3A were designed with a 5′-HindIII (5′-AAGCTTGGCAACATGGCTCACAGG-3′) and a 3′-BamHI (5′-GGATCCTTGAGGGATGGTGAACTCGCATATGTAT-3′) restriction site. The primers for human CLEC3A-Ex23 and CLEC3A-Ex3 were designed with a 5′-NheI (5′-GACAAGGATGGAGATCTGAAGACTCAAATTGAA-3′ or 5′-GTCTGTCTCCGAGGCACTAAAGTTC-3′) and a 3′-BamHI (5′-GGATCCCTATTGAGGGATGGTGAACTCGCATATG-3′) restriction site. Murine and human full-length CLEC3A cDNA was amplified with the endogenous signal peptide and cloned into a modified pCEP-Pu vector (10) that encodes a C-terminal strepII-tag. The amplified cDNA of murine and human CLEC3A-Ex3 and of human CLEC3A-Ex23 was ligated into a modified pCEP-Pu vector (10) encoding an N-terminal BM40 signal peptide followed by a His6-tag. Plasmid DNA was sequenced by Seqlab (Microsynth, Göttingen, Germany). The recombinant plasmids were introduced into HEK-293 EBNA cells by transfection using FuGENE® HD (Roche Applied Science). Cells were selected with puromycin (1 μg/ml) (Sigma-Aldrich) in DMEM-F12 (1:1) (Thermo Fisher Scientific) containing 10% FCS (PAN-Biotech, Aidenbach, Germany), 1% amphotericin B (Thermo Fisher Scientific), and 0.1% gentamycin (Thermo Fisher Scientific). For protein production, serum-free cell culture medium was harvested after 3 or 4 days, centrifuged, and stored at −20 °C.
Synthetic peptides
Synthetic peptides with amino acid sequences corresponding to human CLEC3A exon 1 and exon 2 (CLEC3A-Ex12, HTSRLKARKHSKRRVRDKDGDLKTQIEKLWTEVNALKEIQALQTVCL) with a calculated mass of 5543 Da, and to mouse CLEC3A exon 2 (CLEC3A-Ex2, AKDDDLKSQVEKLWREVNALKEMQALQTV) and with a calculated molecular mass of 3387 Da were generated by Biomatik (Wilmington, DE) and Peptide 2.0 Inc. (Herndon, VA), respectively. The lyophilized peptides were dissolved in Dulbecco's Phosphate-Buffered Saline (DPBS, without Ca2+ and Mg2+) (Thermo Fisher Scientific) to a concentration of 1 mg/ml.
Affinity purification
Depending on the tag used, conditioned FCS–free cell culture medium was loaded onto columns of Strep-Tactin® (IBA, Göttingen, Germany) or TALON® Metal Affinity Resin (Clontech, Mountain View, CA). Proteins were eluted from the Strep-Tactin column with 2.5 mm desthiobiotin and from the TALON column with 250 mm imidazol.
MALDI-TOF mass spectrometry
CLEC3A and CLEC3A-Ex3 were submitted to MALDI-TOF mass spectrometry for determination of total mass. Peptide mass fingerprints were generated by MALDI-TOF after trypsin digestion. The measurements were carried out by the Central Bioanalytic Core Unit at the Center for Molecular Medicine Cologne (CMMC).
Antibody generation
Purified human full-length CLEC3A and murine CLEC3A-Ex3 protein were used to immunize rabbits (Pineda Antibody Service, Berlin, Germany). The resulting antisera were Affinity-purified either with human full-length CLEC3A or murine CLEC3A-Ex3 coupled to CNBr-activated SepharoseTM 4B column (GE Healthcare Life Sciences). The specificity of the purified antibodies was tested by immunoblotting. They reacted with CLEC3A and not with tetranectin.
SDS-PAGE and immunoblotting
Samples were separated on 4–12% or 12% Bis-Tris polyacrylamide gels (Thermo Fisher Scientific) and stained with Coomassie Brilliant Blue R 250 (Merck). For immunoblotting, the proteins were transferred to a polyvinylidene fluoride (0.45 μm) membrane (Thermo Fisher Scientific). Membranes were blocked in Tris-buffered saline containing 0.01% Tween 20 (Merck) and 5% milk powder (Carl Roth GmbH & Co., Karlsruhe, Germany) (TBS-TM), incubated with the Affinity-purified polyclonal rabbit antibodies against murine CLEC3A-Ex3 or human CLEC3A in TBS-TM and incubated with horseradish peroxidase–conjugated polyclonal donkey anti-rabbit IgG (Thermo Fisher Scientific) in TBS-TM. Membranes were treated with Amersham BiosciencesTM ECLTM Prime Western Blotting Detection Reagent (Thermo Fisher Scientific) and signals were visualized on Hyperfilms (Thermo Fisher Scientific). Pre-reduced MagicMarkTM XP (Invitrogen) for immunoblot and SeeBlue® Plus2 Pre-stained Protein Standard (Invitrogen) for Coomassie Blue staining were used.
Glycan analysis
C-terminally strepII-tagged murine CLEC3A was incubated overnight at 37 °C with chondroitinase ABC (5 milliunits/μl, pH 7.7) (Seikagaku, Japan) and analyzed by SDS-PAGE followed by Coomassie Blue staining and immunoblotting as described (25).
Electron microscopy
Murine full-length CLEC3A was stained with 0.75% uranyl format and visualized by transmission electron microscopy as described (26, 27).
Circular dichroism spectroscopy
Murine full-length CLEC3A, CLEC3A-Ex3, and CLEC3A-Ex2 were dialyzed into DPBS, (without Ca2+ and Mg2+) (Thermo Fisher Scientific), followed by determination of the protein concentration with the Bradford assay (Merck). The concentrations were full-length CLEC3A 0.25 mg/ml, CLEC3A-Ex3, and CLEC3A-Ex2 0.5 mg/ml. CD spectra were recorded in a Jasco J-715 Spectropolarimeter using a thermostated 1 mm quartz cell (Hellma, Germany). Far-ultraviolet spectra (185–260 nm) were recorded at 20 °C. For data analysis and secondary structure prediction the DicroWeb server was used (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml)3 with the algorithms SELCON 3, CDSSTR, and Contin-LL (28, 29). The buffer background was subtracted and the data converted to delta ellipticity (Δ ϵ) (30, 31).
Size-exclusion chromatography
Murine full-length CLEC3A, CLEC3A-Ex3, and CLEC3A-Ex2 were analyzed by size-exclusion chromatography using a SuperoseTM 6 10/300 GL column with a 24-ml bed volume (Thermo Fisher Scientific) on an ÄKTAexplorerTM FPLC (Thermo Fisher Scientific) system at room temperature. The column was equilibrated in DPBS to a constant baseline. The samples were eluted isocratically in PBS at a flow rate of 0.5 ml/min for 80 min. Eluted proteins were detected using UV absorbance at 280 nm. Column calibration was performed with a set of molecular mass standards ranging from 14 to 756 kDa (Low Molecular Weight Gel Filtration Calibration Kit, Amersham Bioscience). Eluates were analyzed by SDS-PAGE followed by immunoblot or silver staining (SilverQuest Silver Staining Kit, Thermo Fisher Scientific).
Tissue extraction
Tissues from newborn and 2-year-old (adult) C57/Bl6 mice were crushed with a mortar in liquid nitrogen and extracted overnight at 4 °C with 4 volumes (v/w) of 100 mm NaCl, 50 mm Tris-HCl, pH 7.4, and subsequently with 4 volumes of 4 m guanidine HCl, 10 mm EDTA, 1 m NaCl, 50 mm Tris, pH 7.4. Human hip cartilage was crushed with a mortar in liquid nitrogen and proteins were sequentially extracted overnight at 4 °C with 4 volumes (v/w) of 150 mm NaCl, 50 mm Tris-HCl, pH 7.4 (buffer 1), 10 mm EDTA, 1 m NaCl, 50 mm Tris-HCl, pH 7.4 (buffer 2), 2 m urea, 10 mm EDTA, 1 m NaCl, 50 mm Tris-HCl, pH 7.4 (buffer 3), and 4 m guanidine HCl, 10 mm EDTA, 1 m NaCl, 50 mm Tris-HCl, pH 7.4 (buffer 4). Guanidine fractions were dialyzed against DPBS (Thermo Fisher Scientific).
Immunohistochemistry
Immunohistochemistry was performed on paraffin-embedded sections of embryonic, newborn, and 6-week-old mice. All tissues were pre-fixed with 3.6% formaldehyde, and those from 6-week-old mice also demineralized with HNO3 overnight at 4 °C. After demineralization, the tissues were transferred into 5% Na2SO4 for 24 h. Deparaffinization was for 30 min in Roti®Histol (Carl Roth GmbH & Co., Karlsruhe, Germany) at 52 °C. After rehydration the sections were digested with hyaluronidase (500 units/ml) (Merck) in PBS, pH 5–6, (Merck) for 30 min at 37 °C followed by proteinase K (5.5 milliunits/ml in PBS, pH 5–6) (Merck) for 5 min at 37 °C. The sections were washed with PBS, post-fixed with 4% formaldehyde in PBS for 15 min, blocked for 1 h with 1% bovine serum albumin (Merck) in TBS and incubated with Affinity-purified antibodies against CLEC3A-Ex3 overnight at 4 °C. The primary antibodies were visualized by treatment for 1 h with biotin SP–conjugated goat anti-rabbit IgG and alkaline phosphatase–conjugated streptavidin (both from Dianova, Germany). The slides were developed with Fast Red TR/Naphthol (Merck).
Immunofluorescence microscopy
RCS cells were seeded on chamber slides (1 × 105/chamber) and cultivated with DMEM-F12 (1:1), 5% FCS (PAN, Germany), 1% amphotericin B (PAN, Germany), 0.1% gentamycin (Thermo Fisher Scientific), and cOmplete protease inhibitor mini (1:5) (Roche) in the presence of 100 μg/ml L-ascorbate (Merck) for 4–7 days. The cells were fixed for 10 min with pure methanol (prechilled to −20 °C) followed by blocking for 1 h in BD diluent (BD Biosciences). Cells were subsequently incubated overnight with Affinity-purified antibodies against human CLEC3A or matrilin-3 (11), and for 1 h with Alexa Fluor 488–conjugated goat anti-rabbit IgG (Thermo Fisher Scientific) in blocking solution. Nuclei were stained with DAPI.
ELISA-style binding assay
Recombinant human full-length CLEC3A was diluted in 0.1 mm Na2HPO4, pH 9.0, and coated on 96-well plates (Nunc MaxiSorp, Thermo Fisher Scientific) with 1 μg/well at room temperature for 1h. After three washing steps with TBS Tween 20 0.01%, wells were blocked with 1% BSA in 0.01% TBS Tween 20. Next, wells were incubated with human Glu-plasminogen (Sekisui Diagnostics, lot 150603, Pfungstadt, Germany) at different concentrations (0–12 μg/ml) in TBS with 1% BSA for 1 h at room temperature. Bound plasminogen was detected using a human plasminogen HRP-conjugated antibody (1:1000, Bio-Rad Laboratories, Germany). Color reaction was achieved using 3,3′,5,5′-tetramethylbenzidine (Sigma) and stopped with 1 m HCl. Absorbance was detected at 450 nm using a TECAN microplate reader (Tecan Life Sciences, Männedorf, Switzerland). Wells were measured in duplicates.
Surface plasmon resonance
Surface plasmon resonance was performed as described before (32–34) using a Biacore 2000 system (Biacore Life Sciences, Uppsala, Sweden). Human Glu-plasminogen (1 mg/ml) (Sekisui Diagnostics) was covalently coupled to a CM-5 sensor chip (GE Healthcare Life Sciences) at 2000 response units in 10 mm sodium acetate buffer, pH 5. To investigate the interaction of plasminogen and CLEC3A, serial dilutions of human CLEC3A (0.315–5 μm) in HBS-EP buffer (0.01 m HEPES, pH 7.4, 0.15 m NaCl, 3 mm EDTA, 0.005% (v/v) surfactant P20) were pumped over the Glu-plasminogen–coupled sensor chip. The kinetic constants were calculated by nonlinear fitting (1:1 interaction model with mass transfer) to the association and dissociation curves using the BIAevaluation software (Version 4.1). The apparent equilibrium dissociation constant (KD) was calculated as the ratio of kD/kA.
Plasminogen activation assay
Plasminogen activation was assessed based on the ability of tPA to convert plasminogen to plasmin and the hydrolysis of the substrate S-2251 (H-D-Val-Leu-Lys-pNA) by plasmin (4). The cleavage of the substrate leads to the release of p-nitroaniline which can be detected at 405 nm. The plasminogen activation assay was performed in 96-well plates (Nunc MaxiSorp, Thermo Fisher Scientific) with 2 μm glu-Plasminogen (1 mg/ml) (Sekisui Diagnostics), 10 nm two-chain recombinant tPA (Sekisui Diagnostics, Lot 1000829), 0.5 mm S-2251 (Sigma), and 5 μg of human full-length CLEC3A, human CLEC3A-Ex23, human CLEC3A-Ex3, human CLEC3A-Ex12, or BSA (Sigma), respectively, in 100 μl 0.1 m Tris, pH 7.4, 0.02% Tween 20 for 30 min at 37 °C. The OD was measured every 2 min with a TECAN microplate reader (Tecan Life Sciences). For data analysis, Excel 2010 (Microsoft Corporation), GraphPad Prism 7 (GraphPad Software, Inc.), and Magellan 7 (Tecan Life Sciences) were used.
Cleavage of CLEC3A by plasmin
Human CLEC3A (5 μg) was incubated for 30 min at 37 °C in 100 μl 0.1 m Tris pH 7.4, 0.02% Tween 20 with 2 μm plasminogen and 10 nm tPA, or only with 10 nm tPA. Cleavage of CLEC3A was analyzed by SDS-PAGE followed by immunoblotting.
Author contributions
D. L. designed, performed, and analyzed all experiments and wrote the manuscript. D. E. provided proteins. M. M. performed electron microscopy. A. P. W. participated in experimental design and analysis of CD, SEC, and SPR experiments. G. H. provided technical assistance and analyzed the experiments. M. K. provided cloning vectors. U. H. participated in experimental design for GAG analysis. S. H. provided human material. R. W. and M. P. contributed to experimental design, interpretation of data, and writing and editing of the manuscript. T. S. contributed to the interpretation of data and editing of manuscript. A. R. K. conceived and supervised the study, contributed to the experimental design and interpretation of data, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank M. Kamper for providing tissue from newborn mice and B. Dengler for organs from adult mice, G. Kühn and B. Paul-Klausch for assistance with immunoblotting and tissue staining, J. Hülsmann for technical support with chondroitinase ABC digestion, S. Höning for access to the ÄKTA system, T. Bläske for technical support with size-exclusion chromatography, U. Baumann for support in interpretation of enzymatic data.
This work was supported by European Community's Seventh Framework Programme Grant 602300 (SYBIL). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
M. Mörgelin, personal communication.
- CRD
- carbohydrate recognition domains
- CLEC3A
- C-type lectin domain family 3 member A
- SCGF
- stem cell growth factor
- tPA
- tissue plasminogen activator
- CLD
- C-type lectin domain
- MMP-7
- matrix metalloproteinase-7
- OD
- oligomerization domain
- RCS
- Swarm rat chondrosarcoma
- DPBS
- Dulbecco's phosphate-buffered saline.
References
- 1. Zelensky A. N., and Gready J. E. (2005) The C-type lectin-like domain superfamily. FEBS J. 272, 6179–6217 10.1111/j.1742-4658.2005.05031.x [DOI] [PubMed] [Google Scholar]
- 2. Neame P. J., Tapp H., and Grimm D. R. (1999) The cartilage-derived, C-type lectin (CLECSF1): Structure of the gene and chromosomal location. Biochim. Biophys. Acta 1446, 193–202 10.1016/S0167-4781(99)00087-1 [DOI] [PubMed] [Google Scholar]
- 3. Clemmensen I., Petersen L. C., and Kluft C. (1986) Purification and characterization of a novel, oligomeric, plasminogen kringle 4 binding protein from human plasma: Tetranectin. Eur. J. Biochem. 156, 327–333 10.1111/j.1432-1033.1986.tb09586.x [DOI] [PubMed] [Google Scholar]
- 4. Westergaard U. B., Andersen M. H., Heegaard C. W., Fedosov S. N., and Petersen T. E. (2003) Tetranectin binds hepatocyte growth factor and tissue-type plasminogen activator. Eur. J. Biochem. 270, 1850–1854 10.1046/j.1432-1033.2003.03549.x [DOI] [PubMed] [Google Scholar]
- 5. Hiraoka A., Sugimura A., Seki T., Nagasawa T., Ohta N., Shimonishi M., Hagiya M., and Shimizu S. (1997) Cloning, expression, and characterization of a cDNA encoding a novel human growth factor for primitive hematopoietic progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 94, 7577–7582 10.1073/pnas.94.14.7577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Melin Fürst C., Mörgelin M., Vadstrup K., Heinegård D., Aspberg A., and Blom A. M. (2013) The C-type lectin of the aggrecan G3 domain activates complement. PLoS One 8, e61407 10.1371/journal.pone.0061407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aspberg A. (2012) The different roles of aggrecan interaction domains. J. Histochem. Cytochem. 60, 987–996 10.1369/0022155412464376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karlsson C., Dehne T., Lindahl A., Brittberg M., Pruss A., Sittinger M., and Ringe J. (2010) Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage 18, 581–592 10.1016/j.joca.2009.12.002 [DOI] [PubMed] [Google Scholar]
- 9. Tsunezumi J., Higashi S., and Miyazaki K. (2009) Matrilysin (MMP-7) cleaves C-type lectin domain family 3 member A (CLEC3A) on tumor cell surface and modulates its cell adhesion activity. J. Cell. Biochem. 106, 693–702 10.1002/jcb.22062 [DOI] [PubMed] [Google Scholar]
- 10. Kohfeldt E., Maurer P., Vannahme C., and Timpl R. (1997) Properties of the extracellular calcium binding module of the proteoglycan testican. FEBS Lett. 414, 557–561 10.1016/S0014-5793(97)01070-3 [DOI] [PubMed] [Google Scholar]
- 11. Klatt A. R., Nitsche D. P., Kobbe B., Mörgelin M., Paulsson M., and Wagener R. (2000) Molecular structure and tissue distribution of matrilin-3, a filament-forming extracellular matrix protein expressed during skeletal development. J. Biol. Chem. 275, 3999–4006 10.1074/jbc.275.6.3999 [DOI] [PubMed] [Google Scholar]
- 12. Gebauer J. M., Kobbe B., Paulsson M., and Wagener R. (2016) Structure, evolution and expression of collagen XXVIII: Lessons from the zebrafish. Matrix Biol. 49, 106–119 10.1016/j.matbio.2015.07.001 [DOI] [PubMed] [Google Scholar]
- 13. Holtet T. L., Graversen J. H., Clemmensen I., Thøgersen H. C., and Etzerodt M. (1997) Tetranectin, a trimeric plasminogen-binding C-type lectin. Protein Sci. 6, 1511–1515 10.1002/pro.5560060715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lupas A., Van Dyke M., and Stock J. (1991) Predicting coiled coils from protein sequences. Science 252, 1162–1164 10.1126/science.252.5009.1162 [DOI] [PubMed] [Google Scholar]
- 15. Nielsen B. B., Kastrup J. S., Rasmussen H., Holtet T. L., Graversen J. H., Etzerodt M., Thøgersen H. C., and Larsen I. K. (1997) Crystal structure of tetranectin, a trimeric plasminogen-binding protein with an alpha-helical coiled coil. FEBS Lett. 412, 388–396 10.1016/S0014-5793(97)00664-9 [DOI] [PubMed] [Google Scholar]
- 16. Haudenschild D. R., Tondravi M. M., Hofer U., Chen Q., and Goetinck P. F. (1995) The role of coiled-coil alpha-helices and disulfide bonds in the assembly and stabilization of cartilage matrix protein subunits—a mutational analysis. J. Biol. Chem. 270, 23150–23154 10.1074/jbc.270.39.23150 [DOI] [PubMed] [Google Scholar]
- 17. Jaquinod M., Holtet T. L., Etzerodt M., Clemmensen I., Thøgersen H. C., and Roepstorff P. (1999) Mass spectrometric characterisation of post-translational modification and genetic variation in human tetranectin. Biol. Chem. 380, 1307–1314 [DOI] [PubMed] [Google Scholar]
- 18. Wilson R., Golub S. B., Rowley L., Angelucci C., Karpievitch Y. V., Bateman J. F., and Fosang A. J. (2016) Novel elements of the chondrocyte stress response identified using an in vitro model of mouse cartilage degradation. J. Proteome Res. 15, 1033–1050 10.1021/acs.jproteome.5b01115 [DOI] [PubMed] [Google Scholar]
- 19. Obrist P., Spizzo G., Ensinger C., Fong D., Brunhuber T., Schäfer G., Varga M., Margreiter R., Amberger A., Gastl G., and Christiansen M. (2004) Aberrant tetranectin expression in human breast carcinomas as a predictor of survival. J. Clin. Pathol. 57, 417–421 10.1136/jcp.2003.010058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wewer U. M., Iba K., Durkin M. E., Nielsen F. C., Loechel F., Gilpin B. J., Kuang W., Engvall E., and Albrechtsen R. (1998) Tetranectin is a novel marker for myogenesis during embryonic development, muscle regeneration, and muscle cell differentiation in vitro. Dev. Biol. 200, 247–259 10.1006/dbio.1998.8962 [DOI] [PubMed] [Google Scholar]
- 21. Wewer U. M., Ibaraki K., Schjørring P., Durkin M. E., Young M. F., and Albrechtsen R. (1994) A potential role for tetranectin in mineralization during osteogenesis. J. Cell Biol. 127, 1767–1775 10.1083/jcb.127.6.1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iba K., Sawada N., Chiba H., Wewer U. M., Ishii S., and Mori M. (1995) Transforming growth factor-beta 1 downregulates dexamethasone-induced tetranectin gene expression during the in vitro mineralization of the human osteoblastic cell line SV-HFO. FEBS Lett. 373, 1–4 10.1016/0014-5793(95)00992-I [DOI] [PubMed] [Google Scholar]
- 23. Iba K., Hatakeyama N., Kojima T., Murata M., Matsumura T., Wewer U. M., Wada T., Sawada N., and Yamashita T. (2009) Impaired cutaneous wound healing in mice lacking tetranectin. Wound Repair Regen. 17, 108–112 10.1111/j.1524-475X.2008.00447.x [DOI] [PubMed] [Google Scholar]
- 24. Salo J., Mackiewicz Z., Indahl A., Konttinen Y. T., Holm A. K., Sukura A., and Holm S. (2008) Plasmin-matrix metalloproteinase cascades in spinal response to an experimental disc lesion in pig. Spine 33, 839–844 10.1097/BRS.0b013e31816b1f1d [DOI] [PubMed] [Google Scholar]
- 25. Schnepp A., Komp Lindgren P., Hülsmann H., Kröger S., Paulsson M., and Hartmann U. (2005) Mouse testican-2. Expression, glycosylation, and effects on neurite outgrowth. J. Biol. Chem. 280, 11274–11280 10.1074/jbc.M414276200 [DOI] [PubMed] [Google Scholar]
- 26. Engel J., and Furthmayr H. (1987) Electron microscopy and other physical methods for the characterization of extracellular matrix components: laminin, fibronectin, collagen IV, collagen VI, and proteoglycans. Methods Enzymol. 145, 3–78 10.1016/0076-6879(87)45003-9 [DOI] [PubMed] [Google Scholar]
- 27. Piecha D., Muratoglu S., Mörgelin M., Hauser N., Studer D., Kiss I., Paulsson M., and Deák F. (1999) Matrilin-2, a large, oligomeric matrix protein, is expressed by a great variety of cells and forms fibrillar networks. J. Biol. Chem. 274, 13353–13361 10.1074/jbc.274.19.13353 [DOI] [PubMed] [Google Scholar]
- 28. Sreerama N., and Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN,SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260 10.1006/abio.2000.4880 [DOI] [PubMed] [Google Scholar]
- 29. Sreerama N., Venyaminov S. Y. U., and Woody R. W. (1999) Estimation of the number of α-helical and β-strand segments in proteins using CD spectroscopy. Protein Sci. 8, 370–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 89, 392–400 10.1002/bip.20853 [DOI] [PubMed] [Google Scholar]
- 31. Whitmore L., and Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 10.1093/nar/gkh371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sengle G., Ono R. N., Sasaki T., and Sakai L. Y. (2011) Prodomains of transforming growth factor beta (TGFβ) superfamily members specify different functions: Extracellular matrix interactions and growth factor bioavailability. J. Biol. Chem. 286, 5087–5099 10.1074/jbc.M110.188615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sengle G., Ono R. N., Lyons K. M., Bächinger H. P., and Sakai L. Y. (2008) A new model for growth factor activation: type II receptors compete with the prodomain for BMP-7. J. Mol. Biol. 381, 1025–1039 10.1016/j.jmb.2008.06.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wohl A. P., Troilo H., Collins R. F., Baldock C., and Sengle G. (2016) Extracellular regulation of bone morphogenetic protein activity by the microfibril component fibrillin-1. J. Biol. Chem. 291, 12732–12746 10.1074/jbc.M115.704734 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.