

Abstract
The bacterial second messenger cyclic di-GMP (c-di-GMP) has emerged as a prominent mediator of bacterial physiology, motility, and pathogenicity. c-di-GMP often regulates the function of its protein targets through a unique mechanism that involves a discrete PilZ adaptor protein. However, the molecular mechanism for PilZ protein–mediated protein regulation is unclear. Here, we present the structure of the PilZ adaptor protein MapZ cocrystallized in complex with c-di-GMP and its protein target CheR1, a chemotaxis-regulating methyltransferase in Pseudomonas aeruginosa. This cocrystal structure, together with the structure of free CheR1, revealed that the binding of c-di-GMP induces dramatic structural changes in MapZ that are crucial for CheR1 binding. Importantly, we found that restructuring and repositioning of two C-terminal helices enable MapZ to disrupt the CheR1 active site by dislodging a structural domain. The crystallographic observations are reinforced by protein–protein binding and single cell–based flagellar motor switching analyses. Our studies further suggest that the regulation of chemotaxis by c-di-GMP through MapZ orthologs/homologs is widespread in proteobacteria and that the use of allosterically regulated C-terminal motifs could be a common mechanism for PilZ adaptor proteins. Together, the findings provide detailed structural insights into how c-di-GMP controls the activity of an enzyme target indirectly through a PilZ adaptor protein.
Keywords: chemotaxis, crystal structure, cyclic di-GMP (c-di-GMP), enzyme inhibitor, Pseudomonas aeruginosa (P. aeruginosa), PilZ adaptor, methyltransferases
Introduction
c-di-GMP4 is a widespread second messenger that controls a multitude of cellular functions and bacterial behaviors (1–3). In human pathogenic bacteria such as Pseudomonas aeruginosa, Salmonella enterica, and Vibrio cholerae, c-di-GMP plays a prominent role regulating the motile–sessile transition and formation of antibiotic-resistant biofilm communities. c-di-GMP exerts its control on cellular functions by binding to a variety of protein and riboswitch targets (4, 5). Among the known c-di-GMP–binding proteins, discrete single-domain PilZ proteins are widespread and by far the most prevalent (6–12). As the single-domain PilZ proteins only contain 80–130 amino acids and do not exhibit enzymatic or other activities, their physiological roles often remain elusive. Emerging evidence suggests that the single-domain PilZ proteins function as adaptor proteins to enable c-di-GMP to control its protein targets indirectly (8, 9, 13, 14). However, the detailed molecular mechanism of how c-di-GMP regulates the activity of its protein targets through the PilZ adaptor proteins remains unknown. How specificity in c-di-GMP signaling, which is crucial for preventing cross-talk among different pathways, is achieved through the PilZ adaptor proteins also remains a key question.
We recently discovered that c-di-GMP controls the enzymatic activity of the methyltransferase CheR1 in P. aeruginosa through the PilZ adaptor protein MapZ (14). High cellular c-di-GMP concentrations lead to the formation of the MapZ–c-di-GMP–CheR1 ternary complex and subsequently inhibit the methyltransferase activity of CheR1. Inhibition of CheR1 results in a decrease in the methylation level of methyl-accepting chemoreceptor proteins (MCPs), which ultimately leads to the suppression of flagellar motor switching and a decrease of swimming reversal frequency. Because cellular c-di-GMP concentration fluctuates with metabolic and extracellular stimuli, P. aeruginosa can fine-tune its chemotactic response and motility in different environments using the MapZ-dependent regulatory mechanism. Although it is known that the protein-protein interaction between CheR1 and MapZ is highly specific and dependent on c-di-GMP, the structural basis for the c-di-GMP–mediated specific protein-protein interaction is unknown. Here, we report the crystal structures of the free CheR1 and the MapZ–c-di-GMP–CheR1 ternary complex along with other experimental data to support the physiological relevance of the structural observations. The results reveal an exquisite allosteric mechanism that is potentially conserved in many PilZ adaptor proteins.
Results and discussion
Overall structure of the MapZ–c-di-GMP–CheR1 ternary complex
We determined the crystal structure of CheR1 in complex with MapZ and c-di-GMP at 2.3-Å resolution (Fig. 1A and Fig. S1) with the crystallographic statistics listed in Table 1. The difference Fourier map (Fo − Fc) clearly shows the ligand c-di-GMP (Fig. S2). The c-di-GMP–bound MapZ is composed of a β-barrel core consisting of eight antiparallel β-strands, an N-terminal loop, and two C-terminal α-helices as seen in other PilZ proteins (15–19) (Fig. 1B). Observed as an intercalated dimer in the electron density map, c-di-GMP is held in position by residues stemming from the N-terminal loop and strands β4, β5, β7, and β8. The methyltransferase CheR1 is composed of two domains (Fig. 1C). The N-terminal domain of CheR1 is a relatively small domain consisting of four perpendicularly packed α-helices, whereas the C-terminal domain contains a large α/β fold core and a small β-subdomain (Fig. 1C). The core of the C-terminal domain adopts a Rossmann fold consisting of a mixed seven-stranded β-sheet flanked by α-helices. The architecture of the C-terminal domain core is similar to those of other class I methyltransferases (20). In addition to the α/β fold core, the C-terminal domain also contains a β-subdomain consisting of two short helices and three antiparallel strands (Fig. 1C). The β-subdomain, which is inserted between α7 and α10, is unique to chemotaxis methyltransferases (21–23) (Fig. S3). The N- and C-terminal domains are connected by a flexible domain linker (aa 70–80) with a cleft formed between the two domains. In the ternary complex structure, the β-barrel core of MapZ sits on the top of CheR1 with the two C-terminal helices α1 and α2 wedged into the central cleft of CheR1, resulting in extensive contacts between the two proteins that are likely to account for the formation of a stable MapZ–c-di-GMP–CheR1 ternary complex.
Figure 1.

Crystal structure of MapZ–c-di-GMP–CheR1 ternary complex. A, overall view of the ternary complex. MapZ and CheR1 are shown in salmon and green, respectively. The intercalated c-di-GMP dimer is bound by MapZ and shown as sticks with carbon, oxygen, and nitrogen atoms colored cyan, red, and blue, respectively. B, an enlarged view of c-di-GMP–bound MapZ. The view is rotated by 90° along the vertical axis in A. C, schematic diagram of CheR1. The N-terminal domain, domain linker, C-terminal domain, and β-subdomain are colored red, blue, light blue, and yellow, respectively. The protein secondary structures are numbered in sequential order from the N to C terminus.
Table 1.
Summary of crystallographic data and refinement statistics
ASU, asymmetric unit; r.m.s., root mean square; CC1/2, Pearson's correlation coefficient.
| ApoCheR1 | MapZ–c-di-GMP–CheR1 | |
|---|---|---|
| Data collection | ||
| Space group | I41 | P212121 |
| Protein Data Bank code | 5Y4S | 5Y4R |
| Wavelength (Å) | 0.9779 | 0.9537 |
| Cell dimensions | ||
| a, b, c (Å) | 279.13, 279.13, 138.58 | 88.44, 98.82, 110.66 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Molecules/ASU | 10 | 2 |
| Resolution (Å)a | 50–3.41 (3.61–3.41) | 50–2.30 (2.44–2.30) |
| CC1/2 | 99.8 (53.1) | 99.8 (56.8) |
| I/σ(I) | 13.82 (1.42) | 12.64 (1.43) |
| Completeness (%)a | 99.9 (99.8) | 99.8 (99.0) |
| Redundancya | 14.5 (14.7) | 6.75 (6.91) |
| Refinement | ||
| Resolution (Å) | 49.42–3.41 | 48.29–2.30 |
| No. reflections | 143,720 | 83,410 |
| Rwork (Rfree) (%) | 25.7 (29.5) | 19.4 (23.9) |
| No. atoms | ||
| Protein | 20,244 | 6,230 |
| Ligand/ion | c-di-GMP, 184; SO4, 60 | |
| Water | 163 | |
| B-factors (Å2) | ||
| Protein | 246.3 | 59.4 |
| Ligand/ion | 60.7 | |
| Water | 50.9 | |
| r.m.s. deviations | ||
| Bond lengths (Å) | 0.004 | 0.011 |
| Bond angles (°) | 1.19 | 1.44 |
| Ramachandran plot | ||
| Favored (outlier) (%) | 90.0 (0.56) | 95.0 (0.26) |
a Values for the highest-resolution shell are in parentheses.
Specific interactions between MapZ and CheR1
In the ternary complex, MapZ makes direct contacts with CheR1 through its C-terminal motifs, the β6-β7 loop, and a small segment of the N-terminal loop. The C-terminal motifs of MapZ encompass the two helices α1 (aa 92–106) and α2 (aa 109–121) and the adjacent loops. Helix α1 seems to play a central role in mediating the MapZ-CheR1 interaction by situating in the central cleft of CheR1 with several highly specific polar interactions formed between α1 and CheR1. In particular, Arg99 interacts with Asp67 and Thr70 of CheR1 through salt bridges and hydrogen bonds, and Arg100 forms a salt bridge with Asp144 (Fig. 2A). The side chains of Asp93, Ser96, His97, and Asn105 and the main chain oxygen of Leu104 also interact with various CheR1 residues through hydrogen bonds. The side chains of several residues from α1, including Leu92, Ile95, Leu101, and Leu104, as well as Arg99, Arg100, and Glu103 interact with various CheR1 residues through hydrophobic forces. In addtition, α2 seems to interact with CheR1 mainly through hydrophobic interaction via residues Leu112, Val116, Ala117, Leu119, and Val120 (Fig. 2B). The only noticeable direct polar interaction that involves α2 seems to be the hydrogen bond formed between Glu109 (α2) and Ser146 (CheR1). Apart from the residues in α1 and α2, the main chain groups of Leu92 and Ile90 from the loop prior to α1 form three hydrogen bonds with Val22 and Gly20 of CheR1 (Fig. 2C), whereas His123 and Asp124 from the loop downstream of α2 interact with Thr18 and Arg62 of CheR1 through a hydrogen bond and salt bridge, respectively.
Figure 2.
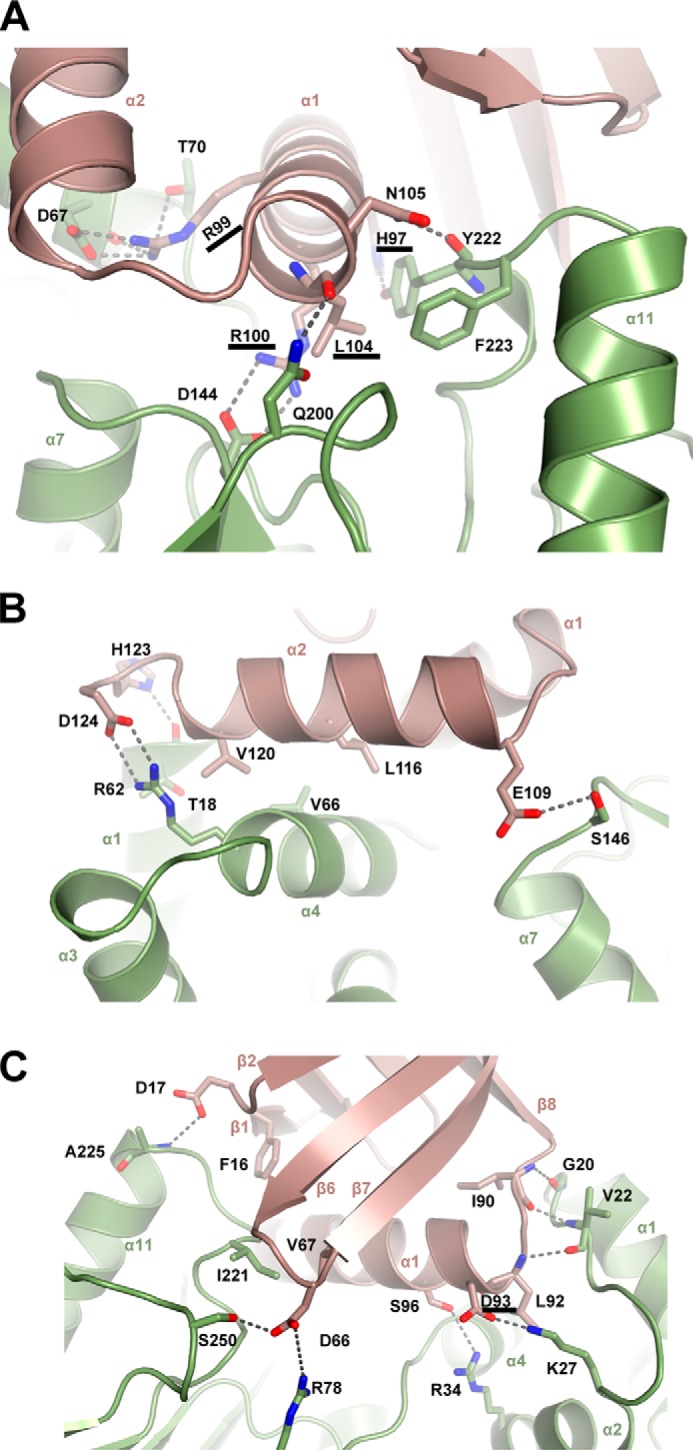
Specific interactions that stabilize the MapZ–c-di-GMP–CheR1 ternary complex. A, specific interactions involving residues from the helix α1 of MapZ. Helix α1 (aa 92–106) is positioned in the central cleft of CheR1 to form extensive contacts, including salt bridges for Arg99-Asp67 and Arg100-Asp144; hydrogen bonds for His97-Tyr222, Arg99-Thr70, Leu104-Asn200, and Asn105-Tyr222; and hydrophobic interactions for Leu104-Tyr222 and -Phe223 (the former residues in the pairs are from MapZ; same as below). B, specific interactions involving helix α2 and adjacent loops of MapZ. The residues in α2 and the adjacent loops in MapZ mainly interact with the N-terminal domain (α1 and α4) of CheR1. C, specific interactions involving residues from the non-C-terminal regions of MapZ. The N-terminal β1-β2 loop and β6-β7 loop in MapZ contact the C-terminal domain of CheR1, and the loop residues Ile90 and Leu92 between β8 and α1 engage in hydrogen-bonding interactions with the N-terminal loop (α1-α2) residues Gly20 and Val22 in CheR1. For clarity, the residues Asp93 and Ser96 from α1 of MapZ interacting with Lys27 and Arg34 of CheR1 are shown in this panel instead of in A. Note that residues involved in the above interactions are shown as sticks with oxygen and nitrogen atom colored red and blue, respectively. Hydrogen bonds and salt bridges are shown as dashed lines. The residues involved in critical interactions are underlined.
In addition to the residues from the C-terminal motifs, several residues from other non-C-terminal regions of MapZ are also involved in direct binding of CheR1. Asp66 and Val67 from the β6-β7 loop interact with Arg78, Ser250, and Ile221 of CheR1 through hydrogen bond and hydrophobic interactions, whereas Phe16 and Asp17 from the N-terminal loop interact with CheR1 residues through hydrophobic and hydrogen bond interactions, respectively (Fig. 2C).
Binding affinity measurements reinforce the key role of the MapZ C-terminal residues in binding CheR1
The extensive contacts between the two proteins are consistent with the high thermodynamic binding affinity (Kd = 28.6 ± 17.7 nm) between the two proteins. c-di-GMP is essential for MapZ-CheR1 interaction because no binding could be detected in the absence of c-di-GMP. To evaluate the contribution of the aforementioned residues of α1 and α2 to CheR1 binding, we measured the binding affinity of several MapZ mutants for CheR1. The results (Fig. 3, A and B) show that Arg99 is the most crucial residue as the substitution of Arg99 by Ala reduced the binding affinity to below the detection limit, consistent with the structural observation that the residue Arg99 engages in multiple interactions with CheR1 residues (Fig. 2A). The substitution of other α1 residues, His97, Arg100, and Leu104, by Ala reduced the Kd by 7.1-, 6-, and 2.4-fold, respectively. Replacement of Asp93, which positions His97 for interacting with CheR1 residues, caused a 10.4-fold decrease in Kd. In comparison, removal of the entire α2 helix (MapZ(Δα2)) decreased the Kd by 14.6-fold. These observations support the relevance of the crystal structure and reinforce the central role of α1 in mediating the MapZ-CheR1 interaction.
Figure 3.

Assessment of the contribution of the residues from helices α1 and α2 to protein-protein interaction by binding affinity measurement. A, binding isotherm (top panels) and data-fitting curves (bottom panels) obtained from ITC assay. Solution containing CheR1 was titrated into solution containing MapZ (or a mutant) and saturating c-di-GMP (250 μm). The experimental conditions are described under “Experimental procedures.” The binding measurements were performed in two independent experiments, and the representative data set is presented here. DP, differential power. B, apparent dissociation constant (Kd) values obtained by fitting the isotherm data to a 1:1 binding model.
The C-terminal binding residues are important for the function of MapZ in suppressing flagellar motor switching
To corroborate the structural and binding data and further validate the importance of the two C-terminal helices for the in vivo function of MapZ, we investigated how mutations in MapZ impact flagellar motor switching in P. aeruginosa cells by cell tethering assay (14, 24). We previously observed that the overexpression of MapZ in P. aeruginosa cells dramatically suppressed flagellar motor switching, which is similar to the effect caused by the deletion of cheR1 (14). Most of the MapZ-overexpressing cells cannot switch motor direction and exhibit unidirectional flagellar rotation (14). The suppression is attributed to the ability of MapZ to inhibit CheR1 and decrease MCP methylation at high c-di-GMP levels. Hence, it was expected that overexpression of defective MapZ mutants that lack key residues for c-di-GMP or CheR1 binding would exhibit reduced suppression on flagellar motor switching. Indeed, the cell tethering assays showed that overexpression of MapZ mutants that lack binding residues for CheR1 suppressed motor switching to a lesser extent than MapZ. MapZ(R99A) did not seem to suppress motor switching at all, whereas MapZ(D93A), MapZ(H97A), MapZ(R110A), MapZ(L104A), and MapZ(Δα2) exhibited less suppression than MapZ to various extents (Fig. 4A and Movies S1–S8). Overexpression of MapZ(R13A), which is defective in c-di-GMP binding (14), also exhibited a much weaker suppression effect. As supported by Western blotting assays, the different suppression effects observed for the mutant proteins are not caused by a reduction in protein expression level (Fig. 4B and Fig. S4). Taken together, the results provide strong support for the physiological relevance of the MapZ–c-di-GMP–CheR1 ternary structure and establish the pivotal role of the C-terminal motifs in mediating MapZ-CheR1 interaction and suppressing flagellar motor switching.
Figure 4.
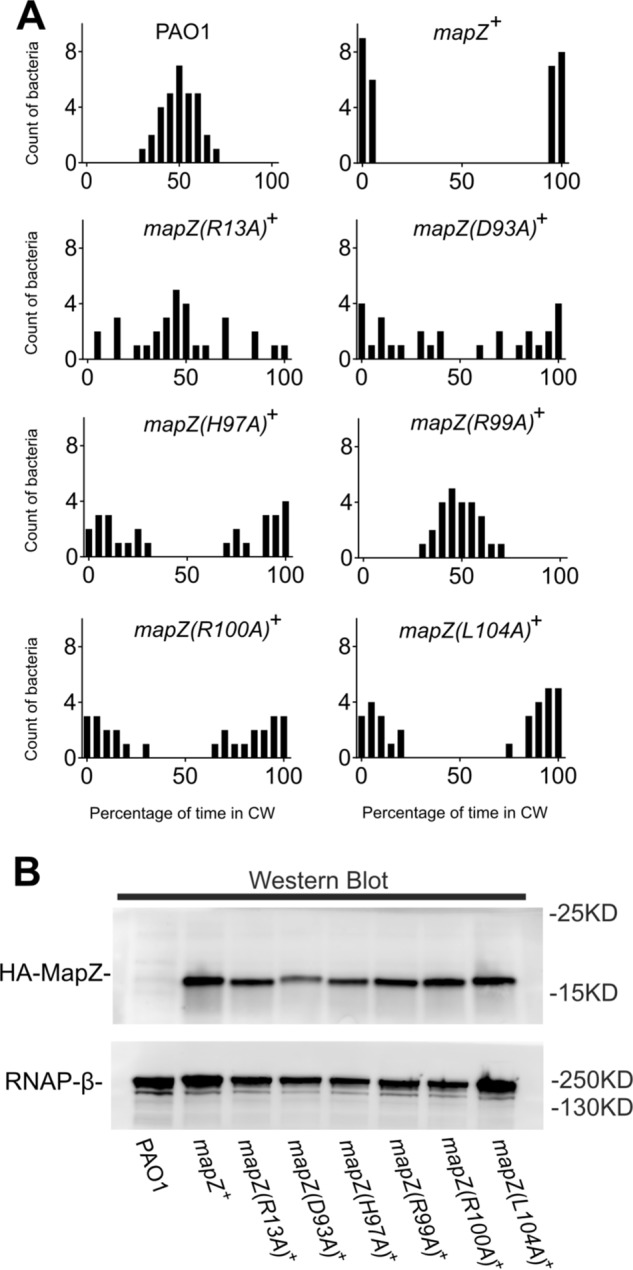
Residues from helices α1 and α2 are important for the function of MapZ in suppressing flagellar motor switching. A, bar graphs showing the percentage of time that P. aeruginosa PAO1 and overexpression mutant strains underwent clockwise (CW) rotation. The sample sizes are 32, 30, 30, 28, 27, 25, 27, and 31 cells for PAO1, mapZ+, mapZ(R13A)+, mapZ(D93A)+, mapZ(H97A)+, mapZ(R99A)+, mapZ(R100A)+, and mapZ(L104A)+, respectively. The corresponding videos for representative rotating cells can be found in Movies S1–S8. Data are representative of two independent experiments. B, protein expression levels of MapZ and MapZ mutants in P. aeruginosa by Western blotting using HA-specific antibody. RNAP-β served as a loading control and was visualized using RNAP-β–specific antibody. Blots are representative of two independent experiments.
Structural rearrangement in MapZ induced by the binding of c-di-GMP and CheR1
The structures of MapZ with and without c-di-GMP bound were determined previously by NMR spectroscopy (25, 26). It was noted that MapZ undergoes large structural rearrangements upon the binding of c-di-GMP. Comparing the ternary complex structure with the NMR structures (Protein Data Bank code 1YWU (26)) reveals how the structural rearrangement induced by c-di-GMP binding prepares MapZ for CheR1 binding. Upon the binding of c-di-GMP, the N-terminal loop becomes organized by wrapping itself around the intercalated c-di-GMP dimer (Fig. 5A). Without the c-di-GMP–induced reorganization, the N-terminal loop would hinder the binding of CheR1 sterically (Fig. 5A). In the C-terminal region, one of the most notable changes is that the binding of c-di-GMP results in the straightening of the initially bent α2 and concomitant dissociation of α2 from the β-barrel core (Fig. 5A). The restructuring and displacement of α2 is crucial for setting up MapZ for the interaction with CheR1.
Figure 5.

Conformational changes in MapZ and CheR1 induced by c-di-GMP binding and protein complex formation. A, superimposition of the structures of MapZ–c-di-GMP–CheR1 complex and free MapZ (gray; Protein Data Bank code 1YWU). The c-di-GMP–binding pocket and CheR1 are represented by the cyan and green surfaces. The N-terminal loop and C-terminal helices (α1 and α2) of the free MapZ adopt conformations that prohibit the binding of CheR1 due to steric hindrance. B, superimposition of CheR1-bound MapZ (salmon) and CheR1-free MapZ (gray; Protein Data Bank code 2L74) in the presence of c-di-GMP. Structures are aligned according to the β-barrel cores. The residue Phe11 and c-di-GMP in both structures are shown in sticks. In the case of CheR1 binding, carbon, oxygen, and nitrogen atoms in c-di-GMP are colored cyan, red, and blue, respectively. Phe11 interacts with α2 in the two structures. Upon CheR1 binding, conformational changes in α2 cause rearrangement of the N-terminal loop of MapZ through their interactions. Given the interaction of the N-terminal loop with c-di-GMP, such a rearrangement would thus cause deformation of the c-di-GMP–binding pocket. The conformational changes of α2 helix, residue Phe11, and c-di-GMP that occur upon CheR1 binding are indicated by arrows. C, coordination of dimeric c-di-GMP in the ternary complex. Three additional residues (Glu7, Phe11, and Arg79) are involved in the binding of c-di-GMP in the ternary complex. D, coordination of dimeric c-di-GMP in the binary complex based on the NMR structure (Protein Data Bank code 2L74). His12 is involved in binding c-di-GMP in the binary complex but not in the ternary complex. Hydrogen bonds are shown as dashed lines. E, superimposition of the structures of free CheR1 and CheR1 from the ternary complex. The most notable differences are seen in the position of the N-terminal domain and the conformation adopted by the domain linker. F, superimposition of the structures of MapZ-bound CheR1 and SAH-bound BsCheR (Protein Data Bank code 5FTW). The two methyltransferase (BsCheR in gray) structures are aligned according to their C-terminal domains. SAH is shown as a stick with carbon, oxygen, nitrogen, and sulfur atoms colored yellow, red, blue, and brown, respectively. In the enlarged view of the cofactor-binding site, it can be seen that Arg100 and Leu104 from α1 of MapZ occupy the SAH-binding site to prevent the binding of the cofactor AdoMet. The highly conserved catalytic residue Asp144/Asp130 (CheR1/BsCheR) that interacts with Arg100 (MapZ) is also highlighted as a stick.
Unexpectedly, the c-di-GMP binding mode in the ternary complex differs to some extent from that in the c-di-GMP–MapZ binary complex (Protein Data Bank code 2L74) (25). The N-terminal loop adopts a rather dissimilar conformation as characterized by the different positions and orientations of several loop residues (e.g. Glu7, Phe11, and His12) in the ternary complex (Fig. 5B). Also, noticeably, the helix α1 from the C terminus is tilted toward the β-barrel core by ∼2 Å, and α2 is pushed toward α1 by as much as 12 Å in the ternary complex. Although residues Arg8, Arg9, Arg10, Arg13, Asp35, and Trp77 are seen to interact with c-di-GMP directly in both the binary and ternary complexes, three additional residues (Glu7, Phe11, and Arg79) are directly involved in the binding of c-di-GMP only in the ternary complex (Fig. 5, C and D). It should be noted that although Glu7 and Phe11 are conserved in most single-domain PilZ proteins, their functions remain unknown. The above observations suggest that the two residues are conserved because they play a direct role in c-di-GMP binding. On the contrary, His12, which is not a conserved residue and seen to form a hydrogen bond with c-di-GMP in the binary complex, does not participate in c-di-GMP binding in the ternary complex. The discrepancies in c-di-GMP binding between the binary and ternary complexes highlight the malleability of the c-di-GMP–binding pocket and call for caution when interpreting the structures of PilZ proteins determined in the absence of their protein partners.
Binding of MapZ to CheR1 resulting in disruption of the S-adenosylmethionine (AdoMet)-binding pocket and inhibition of methyltransferase activity
We also determined the crystal structure of free CheR1 protein to determine how the binding of MapZ impacts the structure of CheR1. Crystals of free CheR1 belong to the tetragonal space group I41 with 10 copies of the molecule (chains A–J) in an asymmetric unit. Superposition of crystallographic independent chains B–J onto chain A gives an average root mean square deviation of 0.76 Å over main-chain atoms, indicating almost identical conformations and a limited influence of crystal packing on the individual chain. The structure was refined to 3.4-Å resolution, and the simulated annealing 2Fo − Fc composite omit map clearly demonstrates that the two domains and the domain linker are nicely modeled (Fig. S5). Comparison of the structures of the free CheR1 and the ternary complex shows that although the C-terminal domains are almost superimposable the N-terminal domains adopt very different conformations in the two structures (Fig. 5E). The spatial positioning of the N-terminal domain relative to the C-terminal domain is also different from that of BsCheR (29) (Fig. S6) despite the fact that the structures of the N-terminal domains are highly similar (Fig. S7). The N-terminal domain and the domain linker of CheR1 have relatively higher B-factors (Fig. S8), suggesting that they are intrinsically flexible. The most important implication from the comparison of the structures of free CheR1 and the ternary complex is that the binding of MapZ induces a relocation of the N-terminal domain by a translocation of more than 10 Å and a rotation of ∼105° as well as a noticeable conformational change in the domain linker (Fig. 5E and see Movie S9).
Based on the studies on other chemotaxis methyltransferases (22, 23), the C-terminal domain and the domain linker are likely involved in the binding of the cofactor AdoMet. In the free CheR1 structure, an AdoMet-binding pocket can be readily identified by overlaying the structure with that of the S-adenosyl-l-homocysteine (SAH)-bound BsCheR (Protein Data Bank code 5FTW; Fig. 5F). Two highly conserved catalytic residues (Asp144 and Tyr222) that are essential for AdoMet binding can also be identified. In the ternary complex, the AdoMet-binding pocket is completely occupied by MapZ upon dislodging of the N-terminal domain and conformational changes in the domain linker. The AdoMet-binding pocket is filled by residues from the α1 of MapZ with Asp144 and Tyr222 engaging in multiple interactions with residues from α1 (Figs. 2A and 5F). These observations strongly suggest that the C-terminal motifs of MapZ, particularly α1, have evolved to interact specifically with the conserved residues in the CheR1 active site to inhibit its enzymatic activity. MapZ only interacts with CheR1 but not the other three chemotaxis methyltransferases of P. aeruginosa (27, 28). Sequence alignment of the four methyltransferases suggests that this is likely because some of the MapZ-binding residues in CheR1 are absent in the other three CheR proteins (Fig. S9).
MapZ-mediated regulation of chemotaxis by c-di-GMP is conserved in many proteobacteria
Together with our previous study (14), the current study firmly establishes the regulation of chemotaxis by c-di-GMP through a MapZ-mediated mechanism in P. aeruginosa. We further determined whether this regulatory mechanism is conserved in the genus Pseudomonas and other non-Pseudomonas species by searching MapZ orthologs/homologs encoded by bacterial genomes. We first identified and collected the sequences of a total of 11,781 single-domain PilZ proteins from the InterPro protein database with most sequences belonging to the IPR009875 family (29). From this collection, we identified 1,063 potential functional orthologs/homologs of MapZ according to the following two criteria. First, the proteins must share high sequence similarity (>60% similarity) with MapZ. Second, the proteins must contain two highly similar C-terminal helices with the CheR1-binding residues conserved. As shown by the sequence logo (Fig. 6A), the critical CheR1-interacting residues are highly conserved in the putative orthologs/homologs.
Figure 6.

MapZ homologs are widespread in proteobacteria. A, sequence log for MapZ and its 1,063 functional homologs. The two essential c-di-GMP–binding motifs (RXXXR and DXSXXG) and the C-terminal α1 and α2 helices are highlighted by bars. The residues from α1 and α2 that are involved in direct binding of CheR1 are highlighted by red residue numbers. B, phylogenetic tree analysis of MapZ homologs. The clades for the two major Pseudomonadales and Vibrionales orders are colored. Most of the remaining homologs belong to other orders from the Proteobacteria phylum, but a small number of homologs belong to unclassified species whose sequences were obtained by metagenomics projects. A FASTA file that contains the 1,063 sequences can be found in the supporting information.
A taxonomic and phylogenetic relationship analysis for MapZ and the 1,063 proteins suggests that most MapZ orthologs/homologs come from Pseudomonas, Vibrio, and other proteobacteria, in particular γ-proteobacteria (Fig. 6B). A small percentage of the orthologs/homologs were encoded by genes from unclassified bacterial species uncovered by metagenomics projects. PlzA, one of the PilZ proteins from the pathogenic bacterium V. cholerae (9) with unknown cellular function was also identified as an ortholog/homolog of MapZ. This hints that PlzA is likely to play a similar role in V. cholerae by binding to a chemotactic methyltransferase to regulate chemotaxis. Furthermore, for the MapZ homolog/ortholog-containing bacterial species whose genomes have been sequenced, we could identify genes encoding chemotaxis methyltransferases that are highly similar to CheR1. Altogether, the bioinformatics analysis suggests that the MapZ-mediated mechanism for fine-tuning chemotactic response and bacterial motility is not a unique mechanism restricted to P. aeruginosa but a widely distributed mechanism adapted by many proteobacteria.
In summary, the studies described here establish the structural basis for the regulation of the methyltransferase CheR1 by c-di-GMP through the PilZ protein MapZ. The data support a model whereby MapZ, at high cellular c-di-GMP concentrations, undergoes dramatic conformational changes and occupies the CheR1 active site to inhibit the enzyme activity (Fig. 7). Inhibition of CheR1 decreases the methylation level of MCPs and ultimately results in the suppression of flagellar motor switching. Further analysis of the sequences of about 12,000 single-domain PilZ proteins encoded by sequenced bacterial genomes revealed that most of them contain C-terminal motifs that vary in sequence length and secondary structure. Although the variation in sequence and structure may reflect the diverse protein partners with which the PilZ proteins interact, the mechanism of allosteric regulation of C-terminal motifs for controlling protein-protein interaction observed in MapZ could be a common theme for the PilZ adaptor proteins.
Figure 7.

A model depicting the c-di-GMP–regulated methylation of MCP through the PilZ adaptor protein MapZ. M, methyl group; CW, clockwise; CCW, counterclockwise.
Experimental procedures
Materials
All bacterial strains, plasmids, and primers used in this study are listed in Tables S1 and S2.
Protein expression and purification
For the recombinant proteins used for crystallization, the CheR1- and MapZ-encoding genes PA3348 and PA4608 were amplified by polymerase chain reaction (PCR) using the genomic DNA of P. aeruginosa PAO1 as template. The PCR fragments were cloned into pACYCDuet-1 expression vector (Novagen), which provides a hexahistidine tag at the N terminus of recombinant CheR1 and MapZ, respectively. In brief, the expression plasmid carrying cheR1 or mapZ gene was transformed into Escherichia coli BL21/DE3 strain using a heat-shock method. Protein expression was induced by 0.2 mm isopropyl β-d-thiogalactoside at 16 °C overnight. Cells were harvested by centrifugation, and the pellet was resuspended and homogenized five times using an EmulsiFlex-C3 (Avestin). The lysate was centrifuged at 40,000 rpm for 1 h to remove insoluble cell debris. The supernatant was applied to a nickel-nitrilotriacetic acid affinity column, sequentially washed, and eluted by a gradient of increasing imidazole concentration in buffer. The pooled fractions were treated with tobacco etch virus protease and dialyzed against a buffer containing 20 mm Tris-HCl, pH 7.5, 100 mm NaCl. After removal of tobacco etch virus protease and uncleaved protein by the second affinity column, the protein samples were further purified by size-exclusion chromatography. The target protein was concentrated to ∼15 mg/ml for subsequent crystallization and biochemical analysis. The mutant MapZ proteins were generated using a QuikChange kit (Stratagene) and prepared following the aforementioned procedure except the final buffer contained20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine hydrochloride (Sigma).
Crystallization and data collection
The initial crystal of the isolated CheR1 was achieved at 20 °C by mixing 1.0 μl of protein with 1.0 μl of reservoir solution containing 1.5 m sodium phosphate monobasic/potassium phosphate dibasic, pH 5.0. For further optimization, the crystallization condition was supplemented with 1% (v/v) glycerol and 3% (v/v) dextran sulfate sodium salt (Mr 5,000). The crystals were flash frozen (100 K) in the reservoir solution supplemented with 30% (v/v) glycerol. The native data set (total of 3,600 images with 0.2 oscillations) was collected at PXIII, Swiss Light Source.
For the MapZ–c-di-GMP–CheR1 complex, the molar ratio between the two proteins was determined to be 1:1 by analytical gel filtration. The MapZ–c-di-GMP–CheR1 complex was prepared by incubating 0.3 mm CheR1 with 0.3 mm MapZ and 1 mm c-di-GMP (Sigma-Aldrich). Crystals of this ternary complex were grown at 20 °C by mixing 2.0 μl of protein with 1.0 μl of reservoir solution containing 0.1 mm Bis-Tris, pH 5.5, 19% PEG 3350, and 0.2 m lithium sulfate. The crystals were flash frozen (100 K) in the reservoir solution with a final concentration of 40% (v/v) PEG 3350. The data set (total of 2,000 images with 0.1 oscillations) for MapZ–c-di-GMP–CheR1 complex was collected at the Swiss Light Source.
Data processing, structure determination, and refinement
All data were processed using the XDS package (30). The structure of MapZ–c-di-GMP–CheR1 ternary complex was determined by molecular replacement with Phenix combined with Phaser (31) using BsCheR (Protein Data Bank code 5FTW) and MapZ (Protein Data Bank code 2L74) as search models. The structure of apoCheR1 was determined by molecular replacement (Phaser/Phenix) (31) using the CheR1 model from our ternary complex as a search model. The models were improved by manual model building with Coot (32) and refined using Phenix (31). Crystallographic statistics of the structures are listed in Table 1, and all structural figures were generated with PyMOL (Schrödinger, LLC).
Preparation of MapZ overexpression mutant strains and Western blotting assay
The DNA fragments of MapZ mutants, including MapZ(D93A), MapZ(H97A), MapZ(R99A), MapZ(R100A), and MapZ(L104A) were synthesized by Integrated DNA Technologies, Inc. Synthesized DNA fragments were digested and ligated into the pUCP18 overexpression vector (Novagen) (33). The constructs were verified by sequencing and transformed into P. aeruginosa mPAO1 strain following the established procedure (34). Single colonies of overexpression strains were used to inoculate 10 ml of LB medium that was kept at 37 °C with shaking overnight. One milliliter of culture was centrifuged at 11,325 × g for 10 min at 4 °C. Cells were resuspended with 100 μl of 2× loading dye and heated at 95 °C for 10 min. Samples were analyzed by SDS-PAGE and Western blotting using THETM HA tag antibody (HRP) (GenScript) and RNA polymerase β-subunit (RNAP-β) (loading control) antibody (a kind gift from Dr. Wang).
Binding affinity measurement by isothermal titration calorimetry (ITC)
ITC measurements were performed at 25 °C using a MicroCal ITC200 (MicroCal Inc.). Protein samples were dialyzed into a buffer containing 20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine hydrochloride. A sample syringe with stirring speed of 800 rpm was used to titrate the CheR1 (250–320 μm) into a cell containing 25–30 μm MapZ or MapZ mutant in the presence of c-di-GMP (250 μm), which was prepared enzymatically as described previously (35, 36). The titration comprised 19 injections of 2 μl each separated by 150-s equilibration time. The isothermal data were analyzed using the Origin 7.0 program, fitting to a single-site binding model.
Cell tethering assay for the analysis of the flagellar rotation
Single colonies of P. aeruginosa PAO1 MapZ and mutants overexpression strains were used to inoculate 10 ml of LB broth (BD Biosciences) that was kept at 37 °C with shaking at 250 rpm overnight. Cultures were diluted to an A600 of 0.2 using M9 medium and grown at 37 °C with shaking at 250 rpm until the cells reached the late-exponential growth phase at A600 of 0.8. Cell cultures were then diluted 10-fold before the microfluidic experiments. Cell tethering assays were performed by using a microfluidic stagnation flow device precoated with flagellar antibodies (37). Flagella were sheared off by passing the bacterial cells through a 34-gauge blunt-end needle four times. Cells were loaded into the cell chamber of the flow device, and non-tethered cells were washed away using M9 medium. Cells were visualized using an inverted microscope (Nikon TE2000-U) under a 40× objective. Videos of tethered bacteria were taken at 40 frames/s for 1–5 min using a complementary metal-oxide semiconductor camera (Thorlabs DCC1645). The rotation of the cells was monitored and recorded using a procedure reported previously (38). The time spent by each cell in clockwise, counterclockwise, or pause phases was tallied in 20-s intervals. Image processing of the tethered cells was carried out as described previously (39) with videos of the tethered cells converted to grayscale and the contrast of the cells adjusted (saturated pixels, 0.4; histogram stack) using NIH ImageJ. Images of the cells were binarized to isolate them from the background for every frame in the video. The center of mass coordinates of the rotating cell bodies of each cell were measured from the obtained binarized image stack and imported into our in-house analysis program BTAP (bacterial tethering analysis program) in MATLAB. Student's two-tailed t test was used to test the statistical significance of the data. The normality assumption was considered to be sound because, by the central limit theorem, sample means of moderately large samples are well-approximated by a normal distribution.
Genome mining and phylogenetic relationship analysis
Single-domain PilZ proteins were identified by mining PilZ protein sequences deposited in the InterPro database (http://www.ebi.ac.uk/interpro/)5 (29). A total of 11,781 sequences of single-domain PilZ proteins were then submitted to the EFI/EST sequence similarity network server (http://efi.igb.illinois.edu/efi-est/tutorial_startscreen.php)5 (40) for analysis and generation of a sequence similarity network by using an alignment score of 16. The clusters formed in the network were analyzed and visualized using Cytoscape v3.5. MapZ was found to be clustered with a group of 1,023 proteins in the network. The sequences of the 1,023 proteins were curated manually to verify whether they contain the crucial C-terminal helices and key residues for CheR1 binding.
For the generation of the sequence logo, the 1,024 protein sequences contained in a FASTA file were first aligned using ClustalW. The FASTA file containing the aligned sequences is included in the supporting information and can be visualized using the free software UGENE and MEGA. ClustalW was also used to generate the phylogenetic tree file (.ph file) to allow visualization of the tree using the web-based tool interactive Tree Of Life (iTOL) (41).
Author contributions
Z.-X. L. and Y.-G. G. conceived and oversaw the design and implementation of the research project. X.-F. Y., J. T. Y., and S. J. crystallized the proteins and solved the structures. L. X., and R. A. C. Y. F. performed gene cloning, protein purification, and binding assays and prepared mutant strains. Z.-X. L. and Q. W. C. identified and analyzed the MapZ homologs by database mining. Y. Z. and K.-H. C. performed the cell tethering assay and data analysis. Y.-G. G. and Z.-X. L. wrote the manuscript with input from the other authors.
Supplementary Material
This work was supported by Ministry of Education of Singapore Tier I Grants RG40/17 (to Z.-X. L.) and RG43/15 (to Y.-G. G.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S9, Tables S1 and S2, and Movies S1–S9.
The atomic coordinates and structure factors (codes 5Y4R and 5Y4S) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- c-di-GMP
- cyclic di-GMP
- MCP
- methyl-accepting chemoreceptor protein
- aa
- amino acids
- Bs
- Bacillus subtilis
- AdoMet
- S-adenosylmethionine
- SAH
- S-adenosyl-l-homocysteine
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- RNAP-β
- RNA polymerase β-subunit
- ITC
- isothermal titration calorimetry.
References
- 1. Römling U., Galperin M. Y., and Gomelsky M. (2013) Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol. Mol. Biol. Rev. 77, 1–52 10.1128/MMBR.00043-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jenal U., Reinders A., and Lori C. (2017) Cyclic di-GMP: second messenger extraordinaire. Nat. Rev. Microbiol. 15, 271–284 10.1038/nrmicro.2016.190 [DOI] [PubMed] [Google Scholar]
- 3. Liang Z. X. (2015) The expanding roles of c-di-GMP in the biosynthesis of exopolysaccharides and secondary metabolites. Nat. Prod. Rep. 32, 663–683 10.1039/C4NP00086B [DOI] [PubMed] [Google Scholar]
- 4. Ryan R. P., Tolker-Nielsen T., and Dow J. M. (2012) When the PilZ don't work: effectors for cyclic di-GMP action in bacteria. Trends Microbiol. 20, 235–242 10.1016/j.tim.2012.02.008 [DOI] [PubMed] [Google Scholar]
- 5. Nelson J. W., Sudarsan N., Furukawa K., Weinberg Z., Wang J. X., and Breaker R. R. (2013) Riboswitches in eubacteria sense the second messenger c-di-AMP. Nat. Chem. Biol. 9, 834–839 10.1038/nchembio.1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amikam D., and Galperin M. Y. (2006) PilZ domain is part of the bacterial c-di-GMP binding protein. Bioinformatics 22, 3–6 10.1093/bioinformatics/bti739 [DOI] [PubMed] [Google Scholar]
- 7. Ryjenkov D. A., Simm R., Römling U., and Gomelsky M. (2006) The PilZ domain is a receptor for the second messenger c-di-GMP: the PilZ protein YcgR controls motility in enterobacteria. J. Biol. Chem. 281, 30310–30314 10.1074/jbc.C600179200 [DOI] [PubMed] [Google Scholar]
- 8. Christen M., Christen B., Allan M. G., Folcher M., Jenö P., Grzesiek S., and Jenal U. (2007) DgrA is a member of a new family of cyclic diguanosine monophosphate receptors and controls flagellar motor function in Caulobacter crescentus. Proc. Natl. Acad. Sci. U.S.A. 104, 4112–4117 10.1073/pnas.0607738104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pratt J. T., Tamayo R., Tischler A. D., and Camilli A. (2007) PilZ domain proteins bind cyclic diguanylate and regulate diverse processes in Vibrio cholerae. J. Biol. Chem. 282, 12860–12870 10.1074/jbc.M611593200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McCarthy Y., Ryan R. P., O'Donovan K., He Y. Q., Jiang B. L., Feng J. X., Tang J. L., and Dow J. M. (2008) The role of PilZ domain proteins in the virulence of Xanthomonas campestris pv. campestris. Mol. Plant Pathol. 9, 819–824 10.1111/j.1364-3703.2008.00495.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pitzer J. E., Sultan S. Z., Hayakawa Y., Hobbs G., Miller M. R., and Motaleb M. A. (2011) Analysis of the Borrelia burgdorferi cyclic-di-GMP-binding protein PlzA reveals a role in motility and virulence. Infect. Immun. 79, 1815–1825 10.1128/IAI.00075-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pultz I. S., Christen M., Kulasekara H. D., Kennard A., Kulasekara B., and Miller S. I. (2012) The response threshold of Salmonella PilZ domain proteins is determined by their binding affinities for c-di-GMP. Mol. Microbiol. 86, 1424–1440 10.1111/mmi.12066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu L., Venkataramani P., Ding Y., Liu Y., Deng Y., Yong G. L., Xin L., Ye R., Zhang L., Yang L., and Liang Z.-X. (2016) A cyclic di-GMP-binding adaptor protein interacts with histidine kinase to regulate two-component signaling. J. Biol. Chem. 291, 16112–16123 10.1074/jbc.M116.730887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu L., Xin L., Zeng Y., Yam J. K. H., Ding Y., Venkataramani P., Cheang Q. W., Yang X., Tang X., Zhang L.-H., Chiam K.-H., Yang L., and Liang Z.-X. (2016) A cyclic di-GMP–binding adaptor protein interacts with a chemotaxis methyltransferase to control flagellar motor switching. Sci. Signal. 9, ra102 10.1126/scisignal.aaf7584 [DOI] [PubMed] [Google Scholar]
- 15. Whitney J. C., Whitfield G. B., Marmont L. S., Yip P., Neculai A. M., Lobsanov Y. D., Robinson H., Ohman D. E., and Howell P. L. (2015) Dimeric c-di-GMP is required for post-translational regulation of alginate production in Pseudomonas aeruginosa. J. Biol. Chem. 290, 12451–12462 10.1074/jbc.M115.645051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ko J., Ryu K.-S., Kim H., Shin J.-S., Lee J.-O., Cheong C., and Choi B.-S. (2010) Structure of PP4397 reveals the molecular basis for different c-di-GMP binding modes by PilZ domain proteins. J. Mol. Biol. 398, 97–110 10.1016/j.jmb.2010.03.007 [DOI] [PubMed] [Google Scholar]
- 17. Shin J. S., Ryu K. S., Ko J., Lee A., and Choi B. S. (2011) Structural characterization reveals that a PilZ domain protein undergoes substantial conformational change upon binding to cyclic dimeric guanosine monophosphate. Protein Sci. 20, 270–277 10.1002/pro.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fujiwara T., Komoda K., Sakurai N., Tajima K., Tanaka I., and Yao M. (2013) The c-di-GMP recognition mechanism of the PilZ domain of bacterial cellulose synthase subunit A. Biochem. Biophys. Res. Commun. 431, 802–807 10.1016/j.bbrc.2012.12.103 [DOI] [PubMed] [Google Scholar]
- 19. Schumacher M. A., and Zeng W. (2016) Structures of the activator of K. pneumonia biofilm formation, MrkH, indicates PilZ domains involved in c-di-GMP and DNA binding. Proc. Natl. Acad. Sci. U.S.A. 113, 10067–10072 10.1073/pnas.1607503113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boriack-Sjodin P. A., and Swinger K. K. (2016) Protein methyltransferases: a distinct, diverse, and dynamic family of enzymes. Biochemistry 55, 1557–1569 10.1021/acs.biochem.5b01129 [DOI] [PubMed] [Google Scholar]
- 21. Djordjevic S., Goudreau P. N., Xu Q., Stock A. M., and West A. H. (1998) Structural basis for methylesterase CheB regulation by a phosphorylation-activated domain. Proc. Natl. Acad. Sci. U.S.A. 95, 1381–1386 10.1073/pnas.95.4.1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Batra M., Sharma R., Malik A., Dhindwal S., Kumar P., and Tomar S. (2016) Crystal structure of pentapeptide-independent chemotaxis receptor methyltransferase (CheR) reveals idiosyncratic structural determinants for receptor recognition. J. Struct. Biol. 196, 364–374 10.1016/j.jsb.2016.08.005 [DOI] [PubMed] [Google Scholar]
- 23. Djordjevic S., and Stock A. M. (1997) Crystal structure of the chemotaxis receptor methyltransferase CheR suggests a conserved structural motif for binding S-adenosylmethionine. Structure 5, 545–558 10.1016/S0969-2126(97)00210-4 [DOI] [PubMed] [Google Scholar]
- 24. Qian C., Wong C. C., Swarup S., and Chiam K.-H. (2013) Bacterial tethering analysis reveals a “run-reverse-turn” mechanism for Pseudomonas species motility. Appl. Environ. Microbiol. 79, 4734–4743 10.1128/AEM.01027-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Habazettl J., Allan M. G., Jenal U., and Grzesiek S. (2011) Solution structure of the PilZ domain protein PA4608 complex with cyclic di-GMP identifies charge clustering as molecular readout. J. Biol. Chem. 286, 14304–14314 10.1074/jbc.M110.209007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramelot T. A., Yee A., Cort J. R., Semesi A., Arrowsmith C. H., and Kennedy M. A. (2007) NMR structure and binding studies confirm that PA4608 from Pseudomonas aeruginosa is a PilZ domain and a c-di-GMP binding protein. Proteins 66, 266–271 [DOI] [PubMed] [Google Scholar]
- 27. Ferrández A., Hawkins A. C., Summerfield D. T., and Harwood C. S. (2002) Cluster II che genes from Pseudomonas aeruginosa are required for an optimal chemotactic response. J. Bacteriol. 184, 4374–4383 10.1128/JB.184.16.4374-4383.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. García-Fontana C., Reyes-Darias J. A., Muñoz-Martínez F., Alfonso C., Morel B., Ramos J. L., and Krell T. (2013) High specificity in CheR methyltransferase function. CheR2 of Pseudomonas putida is essential for chemotaxis, whereas CheR1 is involved in biofilm formation. J. Biol. Chem. 288, 18987–18999 10.1074/jbc.M113.472605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hunter S., Apweiler R., Attwood T. K., Bairoch A., Bateman A., Binns D., Bork P., Das U., Daugherty L., Duquenne L., Finn R. D., Gough J., Haft D., Hulo N., Kahn D., et al. (2009) InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 10.1093/nar/gkn785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 12 10.1107/S09074449090473375–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu L., Xin L., Zeng Y., Yam J. K., Ding Y., Venkataramani P., Cheang Q. W., Yang X., Tang X., Zhang L. H., Chiam K. H., Yang L., and Liang Z. X. (2016) A cyclic di-GMP-binding adaptor protein interacts with a chemotaxis methyltransferase to control flagellar motor switching. Sci. Signal. 9, ra102 10.1126/scisignal.aaf7584 [DOI] [PubMed] [Google Scholar]
- 34. Choi K. H., Kumar A., and Schweizer H. P. (2006) A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64, 391–397 10.1016/j.mimet.2005.06.001 [DOI] [PubMed] [Google Scholar]
- 35. Venkataramani P., and Liang Z. X. (2017) Enzymatic production of c-di-GMP using a thermophilic diguanylate cyclase. Methods Mol. Biol. 1657, 11–22 10.1007/978-1-4939-7240-1_2 [DOI] [PubMed] [Google Scholar]
- 36. Rao F., Pasunooti S., Ng Y., Zhuo W., Lim L., Liu W., and Liang Z.-X. (2009) Enzymatic synthesis of c-di-GMP using a thermophilic diguanylate cyclase. Anal. Biochem. 389, 138–142 10.1016/j.ab.2009.03.031 [DOI] [PubMed] [Google Scholar]
- 37. Alicia T. G., Yang C., Wang Z., and Nguyen N. T. (2016) Combinational concentration gradient confinement through stagnation flow. Lab Chip 16, 368–376 10.1039/C5LC01137J [DOI] [PubMed] [Google Scholar]
- 38. Wang C. J., Bergmann A., Lin B., Kim K., and Levchenko A. (2012) Diverse sensitivity thresholds in dynamic signaling responses by social amoebae. Sci. Signal. 5, ra17. 10.1126/scisignal.2002449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Long Z., Olliver A., Brambilla E., Sclavi B., Lagomarsino M. C., and Dorfman K. D. (2014) Measuring bacterial adaptation dynamics at the single-cell level using a microfluidic chemostat and time-lapse fluorescence microscopy. Analyst 139, 5254–5262 10.1039/C4AN00877D [DOI] [PubMed] [Google Scholar]
- 40. Gerlt J. A., Bouvier J. T., Davidson D. B., Imker H. J., Sadkhin B., Slater D. R., and Whalen K. L. (2015) Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): a web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 1854, 1019–1037 10.1016/j.bbapap.2015.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Letunic I., and Bork P. (2007) Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128 10.1093/bioinformatics/btl529 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.