

Abstract
Mast cells (MCs) are involved in host defenses against pathogens and inflammation. Stimulated MCs release substances stored in their granules via regulated exocytosis. In other cell types, Munc13 (mammalian homolog of Caenorhabditis elegans uncoordinated gene 13) proteins play essential roles in regulated exocytosis. Here, we found that MCs express Munc13-2 and -4, and we studied their roles using global and conditional knock-out (KO) mice. In a model of systemic anaphylaxis, we found no difference between WT and Munc13-2 KO mice, but global and MC-specific Munc13-4 KO mice developed less hypothermia. This protection correlated with lower plasma histamine levels and with histological evidence of defective MC degranulation but not with changes in MC development, distribution, numbers, or morphology. In vitro assays revealed that the defective response in Munc13-4-deficient MCs was limited to regulated exocytosis, leaving other MC secretory effector responses intact. Single cell capacitance measurements in MCs from mouse mutants differing in Munc13-4 expression levels in their MCs revealed that as levels of Munc13-4 decrease, the rate of exocytosis declines first, and then the total amount of exocytosis decreases. A requirement for Munc13-2 in MC exocytosis was revealed only in the absence of Munc13-4. Electrophysiology and EM studies uncovered that the number of multigranular compound events (i.e. granule-to-granule homotypic fusion) was severely reduced in the absence of Munc13-4. We conclude that although Munc13-2 plays a minor role, Munc13-4 is essential for regulated exocytosis in MCs, and that this MC effector response is required for a full anaphylactic response.
Keywords: allergy, exocytosis, immunoglobulin E (IgE), mast cell, SNARE proteins, Munc13, anaphylaxis
Introduction
During exocytosis, the membrane of a secretory vesicle fuses with the plasma membrane, allowing the release of vesicular contents into the extracellular space and the incorporation of vesicle membrane components into the plasma membrane (1). Exocytosis can be constitutive or regulated (2). In constitutive exocytosis, newly formed products are secreted as they are synthesized, and the amount of secreted product is controlled by the rate of expression of the vesicular cargo. In contrast, in regulated exocytosis the formed products are stored in secretory vesicles (e.g. MC2 granules) and released upon stimulation using diacylglycerol (DAG) and Ca2+ as second messengers (3, 4). The amount of secreted product is controlled by the rate and number of vesicles to plasma membrane fusion events. Regulated exocytosis can adopt various forms. In single-vesicle exocytosis, individual secretory granules fuse with the plasma membrane. In sequential compound exocytosis, these primary fused vesicles become targets for secondary fusion events with vesicles lying deeper in the cell. In multigranular compound exocytosis, secretory vesicles fuse homotypically with each other inside the cell before fusing heterotypically with the plasma membrane (5). Some cells (e.g. MCs) use all three forms of regulated exocytosis (6).
Regulated exocytosis involves the generation of secretory vesicles and their transport toward the plasma membrane. Then, tethering and docking establish physical proximity between the vesicle and plasma membrane. The final event involves the fusion of both membranes (1), which requires the assembly of complexes between the SNARE (soluble N-ethylmaleimide-sensitive factor-activated protein receptor) domains of proteins on the vesicular (vesicle-associated membrane protein (VAMP)) and target membranes (syntaxin (Stx) and synaptosome-associated protein 25 (SNAP25)) (7) and is highly regulated by complexin and synaptotagmin (8). The physical proximity established by docking is not sufficient to drive fusion, and additional biochemical events are required to render the vesicles competent for Ca2+-triggered fusion in an intermediate step known as priming (9, 10).
The mammalian homologs of Caenorhabditis elegans uncoordinated gene 13 (Munc13) are essential for docking (11) and priming (12, 13). All isoforms contain a MUN domain, which in Munc13-1 is required for making the SNARE domain of Stx available to interact with those of VAMP and SNAP25 (14, 15) in the correct configuration (16). Munc13 proteins also contain two or three C2 domains. The C2A domain regulates the function of Munc13 through homodimerization or binding to RIM (Rab3–interacting molecule) (17, 18). C2B and C2C help to bridge the vesicular and plasma membranes (18, 19). Some Munc13 isoforms also have a C1 domain (18, 20), and binding of DAG to C1 and Ca2+ to C2B regulates the final assembly of the SNARE complex (21). Neuronal loss of Munc13-1 severely impairs neurotransmitter release and drastically reduces the number of fusion-ready vesicles (22). Munc13-4 is ubiquitously expressed, and most studies have focused on lymphocytes because its absence in humans causes familial lymphohistiocytosis type 3 (23). Compared with Munc13-1 and -2, Munc13-4 lacks the C2A and C1 domains (24). Munc13-4 binds in vitro to the SNARE domains of Stx-1, -4, and -11 (25) and facilitates the fusion of Rab7+ secretory granules with Rab11+ endosomes in RBL-2H3 cells (26).
MCs can be activated by allergens, complement, cytokines, growth factors, venoms, and other secretagogues (27). One MC effector response is degranulation, in which mediators stored in their large metachromatic granules are released via regulated exocytosis (28, 29). Stimulation also activates the transcription of multiple cytokines, chemokines, and growth factors, which are synthesized in the endoplasmic reticulum and then exported via constitutive exocytosis from the Golgi to the plasma membrane. In addition, the rise in intracellular Ca2+ induces the enzymatic processing of arachidonic acid into eicosanoids, mainly PGD2 and LTC4, which are exported through membrane transporters (30, 31).
We hypothesized that the robust degranulation kinetics of MCs would allow us to test with high resolution the requirements of Munc13 proteins in non-neuronal cell exocytosis. To this end, we used single-cell, cell population, and whole-animal assays. We found that Munc13-4 regulates the amount and rate of exocytic events, that it is specifically required for MC-regulated exocytosis but not for other MC effector responses, that it mediates homotypic fusion in multigranular compound exocytosis, and that animals with a selective deficiency of Munc13-4 in their MCs could not mount a full anaphylactic response.
Results
Expression of Munc13 proteins in MCs and generation of Munc13-4-deficient mice
We found that C57BL/6J (B6) mature peritoneal MCs express Munc13-2 and -4 (Fig. 1A), and we decided to target both isoforms. We obtained Munc13-2 global KO mice (32) and generated Munc13-4 global and conditional KO lines (Fig. 1B). We flanked the exon containing the transcription initiation codon and sequence present in all reported mouse Unc13d transcripts (https://www.ncbi.nlm.nih.gov/gene/70450) with two loxP sequences (“floxed” or F allele), and we removed it in MCs (Δ allele) using mice that express Cre recombinase under the control of the Cma1 (chymase 1) locus (33). We also crossed Munc13-4F/F with CMV-Cre mice (34) to obtain germ line deletion (−allele). PCR of genomic DNA with primers surrounding exon 3 confirmed our genetic manipulations (Fig. 1C). The resulting mutant mice (Munc13-4+/− and Munc13-4−/−, Munc13-4F/F, and Munc13-4Δ/Δ) were all viable and fertile; transmission of the mutant alleles followed a Mendelian pattern, and we found no gross anatomical abnormalities or survival differences when raised in a pathogen-free facility.
Figure 1.

Expression of Munc13 proteins in MCs from WT and mutant mice. A, qPCR of B6 peritoneal MCs for all Munc13 paralogs relative to β-actin. ND, not detected; n = 4. B, generation of the Munc13-4 global and conditional KO mice. Black bars, exons; blue flags, location of FRT sites; red flags, location of loxP sites; black arrow, PGK-Neo; gray arrow, PGK-TK; colored arrowheads, location of primers for genotyping. C, PCR of genomic tail DNA amplified with the primers in B. Arrowhead, Cre+ lane. D, qPCR for Munc13-2 and Munc13-4 of peritoneal MCs from different Munc13-4 mutant mice. Values are normalized to β-actin and then to levels in Munc13-4+/+ MCs. Bar, mean; error bar, S.E. n = 4; * = p ≤ 0.001 compared with Munc13-4+/+. E, representative immunoblot of PCMC and spleen lysates for Munc13-4. β-Actin was used as loading control. Right panels, densitometry relative to β-actin and Munc13-4+/+.
The efficacy of Cre recombination in peritoneal MCs using a surrogate locus was >99% (Fig. S1), so we used peritoneal MCs in most of our studies. Quantification of transcripts from FACS-sorted peritoneal MCs demonstrated the predicted absence of Munc13-4 transcripts in MCs from Munc13-4−/− and Munc13-4Δ/Δ mice, and ∼50% reduction in those from Munc13-4+/− mice. Also, there was no compensatory overexpression of Munc13-2 in the absence of Munc13-4 (Fig. 1D). The transcripts for Munc13-2 were undetectable in peritoneal MCs from Munc13-2−/− mice. The reduction of Munc13-4 transcript levels in Munc13-4F/F MCs to ∼15% was unexpected. By 3′-rapid amplification of cDNA ends (RACE), we confirmed that the transcripts present in Munc13-4+/+ and Munc13-4F/F correspond to the ones already described for mouse Munc13-4. Immunoblots of lysates from peritoneal cell-derived MCs (PCMCs) and different organs (only spleen is shown) confirmed the reduced expression in Munc13-4F/F mice and the specificity of Cre-mediated deletion: Munc13-4Δ/Δ animals had the same levels of Munc13-4 as Munc13-4F/F mice in all tissues except in MCs, where it was absent (Fig. 1E). The fact that no bands were seen in samples from Munc13-4−/− mice in tissues known to express Munc13-2 proved the specificity of our antibody. Because Munc13-4F/F mice were hypomorphic, Munc13-4Δ/Δ mice were strictly compared with Munc13-4F/F littermates in all experiments.
Munc13-4 is required for a full anaphylactic response
In small mammals, the massive peripheral vasodilation characteristic of anaphylaxis causes rapid loss of body heat, so core body temperature accurately reflects the severity of the anaphylactic response. Hypothermia in this model of IgE-dependent anaphylaxis depends on histamine (35). Given that histamine is released by MCs via regulated exocytosis, we tested whether the global or MC-specific deletion of Munc13-4 affected this allergic response. We sensitized mice with IgE anti-2,4-dinitrophenol (DNP), challenged them with DNP-human serum albumin (DNP-HSA), and monitored their core body temperature (Fig. 2A). As negative controls, we introduced Munc13-4+/+ mice that were challenged with a sham solution. To control for anaphylactoid reactions to DNP-HSA, we used non-sensitized but challenged B6 mice, and no reactions were observed (data not shown). The lack of response in MC-deficient KitW-sh/W-sh mice confirmed the MC dependence of this model. The absence of Munc13-2 did not affect the hypothermic response. However, there was significantly less drop in temperature in Munc13-4−/− mice compared with Munc13-4+/+ littermates, suggesting that a component of this reaction depends on Munc13-4. We recorded a similar difference between Munc13-4Δ/Δ and Munc13-4F/F mice, indicating that this Munc13-4-dependent component is limited to MCs (Fig. 2C). The partial deficit of Munc13-4 expression in Munc13-4+/− and Munc13-4F/F mice did not modify the nadir or recovery phase of the anaphylactic response, but it affected the early phase (Fig. 2B). Based on these findings we decided to drop the Munc13-2−/− mice from most of our studies. Also, because we did not find a difference between Munc13-4−/− and Munc13-4Δ/Δ mice in this and most other experiments, we opted not to compare Munc13-4+/− with Munc13-4+/Δ mice.
Figure 2.

MC Munc13-4 is required for a full anaphylactic response. Mice were sensitized i.v. with anti-DNP IgE and challenged i.v. with DNP-HSA. Sham, non-sensitized but challenged B6 mice. A, change in body core temperature from baseline (ΔT). Circle, mean; error bar, S.E. C and D, ΔT 5 min (B) and 50 min (C) post-challenge. Line, mean; box, 25th–75th percentile; whiskers, 5th–95th percentile; n = number inside boxes, applies to A–C. D, plasma histamine levels 3 min post-challenge. Bar, mean; error bar, S.E.; n = number inside bars. E, representative lip sections 15 min post-challenge stained with toluidine blue to identify MC metachromatic granules. Scale bar = 40 μm. F, quantification of MC degranulation in lips and tongues based on proportion of granules outside the MC: low, <10%; moderate, 10–50%; and severe, >50%. Number of cells inside bars, number of animals in parentheses. # = p < 0.05, † = p < 0.01, * = p < 0.001; all comparisons are to Munc13-4+/+ unless otherwise indicated.
During anaphylaxis, MC proteases contained in MC granules can be detected in serum (36). We measured plasma histamine, also stored in MC granules (37), as a surrogate of MC degranulation in mice undergoing anaphylaxis. We took special care during plasma collection to prevent the release of histamine from platelets. We found decreased plasma levels of histamine in challenged Munc13-4−/− and Munc13-4Δ/Δ mice (Fig. 2D). In histological samples of connective tissues from challenged Munc13-4+/+, Munc13-4+/−, and Munc13-4F/F mice, we observed multiple metachromatic granules surrounding degranulated MCs. Conversely, most visible MC granules from challenged Munc13-4−/− and Munc13-4Δ/Δ mice remained densely packed inside the MCs, which were almost indistinguishable from those of unchallenged Munc13-4+/+ mice (Fig. 2E). When quantified with a grading system (Fig. S2), we observed almost no MCs that we would grade as severely degranulated in the absence of Munc13-4 (Fig. 2F). There was a clear correlation between Munc13-4 expression in MCs, the severity of MC degranulation, the histamine plasma levels, and the hypothermic response to anaphylaxis.
Characterization of MCs from Munc13-4 mutant mice
Abnormalities in the number, distribution, and development of MCs caused by Munc13-4 deficiency could also blunt the anaphylactic response. Consequently, we studied the baseline characteristics of the mutant mice and their MCs. As shown in Fig. 3 and Table 1, we found no differences in the density of MCs in the dermis, the distribution of MCs in other tissues (data not shown), and the proportion and absolute number of MCs in peritoneal lavages. Qualitatively, the metachromasia of granules from all MCs was almost identical. The same proportion and number of bone marrow cells underwent differentiation into BMMCs in media enriched with stem cell factor (SCF) and IL3. Finally, all MCs and BMMCs expressed similar levels of receptors important for MC development and activation on their surfaces (FcϵRIα, Kit, and IL33R). We assessed cell morphology by stereology of EM profiles of peritoneal MCs fixed immediately after harvesting. Because the area of the cell profiles and cell-surface density (Sv cell) was similar, we concluded that there was no difference in the size, shape, and surface complexity of the MCs. The fact that both granule volume density (Vv) and granule surface density (Sv granule) did not differ indicated that the number, size, and shape of the MC secretory granules at baseline are unchanged. Therefore, we found no baseline abnormality that could explain the phenotype of Munc13-4−/− and Munc13-4Δ/Δ mice or that could interfere with the interpretation of the cell assays described below.
Figure 3.
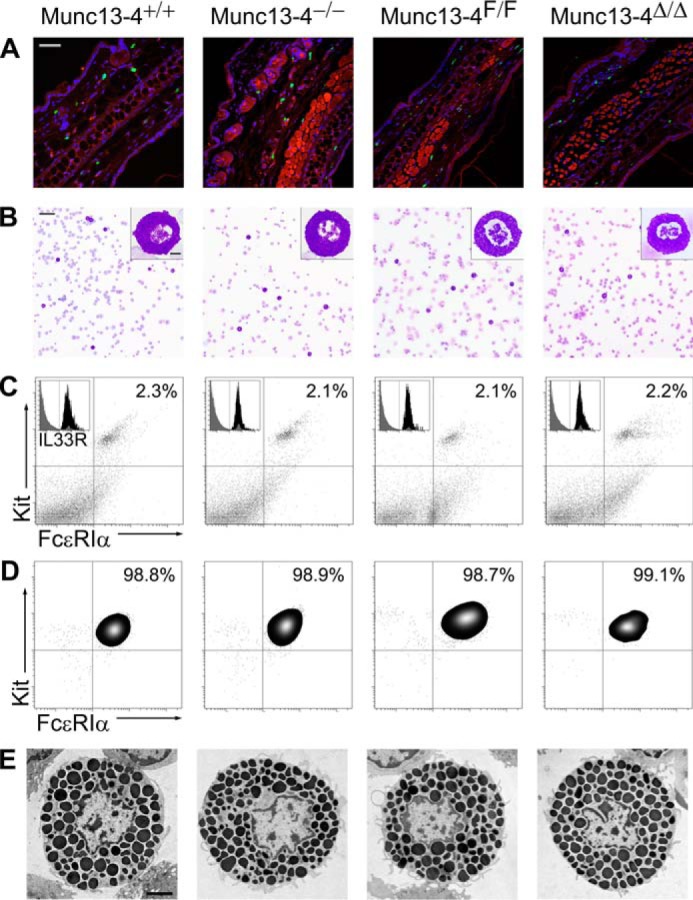
Characterization of MCs from Munc13-4 mutant mice. Representative samples are shown: for detailed quantification please refer to Table 1. A, ear sections stained with FITC-avidin (green) and Hoechst (blue). MCs are identified as FITC+ granules surrounding Hoechst+ nuclei; cartilage and epithelial autofluorescence in the red channel delimited the dermis. Scale bar = 100 μm. B, Wright-Giemsa staining of peritoneal lavages. MCs are identified by their metachromatic granules. Scale bar = 80 μm, inset scale bar = 5 μm. C, flow cytometry of peritoneal lavage cells labeled for Kit and FcϵRIα. MCs are identified as Kit+/FcϵRIα+ double-positive cells. Inset, Kit+/FcϵRIα+ double-positive cells before (gray) and after (black) adding labeled anti-IL33R antibody. D, flow cytometry of BMMCs after 6 weeks in media enriched with SCF and IL3 labeled for Kit and FcϵRIα. E, EM cell profiles of peritoneal MCs used for stereology. Scale bar = 4 μm.
Table 1.
Characterization of MCs from Munc13-4 mutant mice
Results are means ± S.E. for 10 mice of each genotype. A, area; AU, arbitrary units; BMMC, bone marrow-derived MC; DP, double-positive cells for Kit and FcϵRIα; IL33R, receptor for interleukin 33; MC, mast cell; MFI, mean fluorescence intensity; Sv, surface density; Vv, volume density.
| Munc13-4+/+ | Munc13-4−/− | Munc13-4F/F | Munc13-4Δ/Δ | |
|---|---|---|---|---|
| Dermal MCs | ||||
| Density (MCs/mm2 of dermis)a | 220.9 ± 20 | 204.9.7 ± 42 | 210.2 ± 17.6 | 209.3 ± 20.9 |
| Peritoneal MCs | ||||
| Count (MCs/μl of lavage)b | 23.67 ± 6 | 23.9 ± 6.7 | 23.3 ± 1.4 | 23.07 ± 12 |
| MCs (% of lavaged cells)b | 3.35 ± 0.6 | 3.07 ± 0.4 | 2.98 ± 0.7 | 3.1 ± 1.3 |
| Vvc | 0.51 ± 0.01 | 0.52 ± 0.01 | 0.54 ± 0.01 | 0.53 ± 0.03 |
| Sv cell (μm−1)c | 0.49 ± 0.02 | 0.50 ± 0.03 | 0.49 ± 0.02 | 0.49 ± 0.04 |
| Sv granule (μm−1)c | 6.98 ± 0.1 | 7.05 ± 0.2 | 7.01 ± 0.2 | 6.97 ± 0.4 |
| A (μm2)c | 84.61 ± 2 | 85.32 ± 1.8 | 83.75 ± 2.3 | 85.76 ± 2.4 |
| Kit+/FcϵRIα+ (% of total)d | 2.2 ± 0.2 | 1.9 ± 0.3 | 2.1 ± 0.5 | 2.2 ± 0.3 |
| IL33R (% of DP)e | 98.3 ± 0.3 | 97.1 ± 0.2 | 98.5 ± 0.3 | 97.6 ± 1.0 |
| Kit MFI (AU)f | 7.3 ± 0.8 | 8.4 ± 0.6 | 7.4 ± 0.8 | 8.7 ± 0.5 |
| FcϵRIα MFI (AU)f | 5.0 ± 0.5 | 5.0 ± 0.5 | 4.4 ± 0.4 | 4.9 ± 0.7 |
| IL33R MFI (AU)f | 8.2 ± 0.8 | 7.5 ± 0.3 | 7.6 ± 0.8 | 8.3 ± 1.1 |
| BMMCs | ||||
| Kit+/FcϵRIα+ (%)d | 97.9 ± 0.4 | 96.8 ± 1.0 | 95.2 ± 1.8 | 96.0 ± 1.2 |
| Kit MFI (AU)f | 5.0 ± 0.6 | 5.2 ± 0.6 | 5.2 ± 0.5 | 4.8 ± 0.8 |
| FcϵRIα MFI (AU)f | 4.8 ± 0.3 | 5.1 ± 0.5 | 5.4 ± 0.5 | 4.7 ± 0.7 |
a Cells with FITC+ granules and a Hoechst+ nucleus per mm2 of dermis in random ear sections (10 sections/mouse) are shown.
b Neubauer chamber counts and differentials of cytospins of peritoneal lavages are shown.
c Stereological analysis of randomly acquired EM images of peritoneal MCs (10 cell profiles/mouse) are shown.
d Fraction of Kit+/FcϵRIα+ double-positive cells by flow cytometry are shown.
e Fraction of Kit+/FcϵRIα+ double-positive cells were also IL33R-positive.
f Cell-surface expression of Kit, FcϵRIα, and IL33R was expressed as MFI of specific labeled primary antibodies.
Lack of Munc13-4 affects exclusively MC-regulated exocytosis
The defective MC degranulation in mice lacking Munc13-4 prompted us to address whether all effector responses were compromised by measuring the secretion of products that putatively depend on regulated exocytosis, constitutive exocytosis, and non-exocytic transport. PCMCs were stimulated via FcϵRI or by a combination of the DAG analog phorbol 12-myristate 13-acetate (PMA) and the Ca2+ ionophore ionomycin. We quantified the fraction of total cell histamine and β-hexosaminidase secreted after stimulation. These two compounds are preformed, stored in MC secretory granules, and secreted via regulated exocytosis. In both cases, we detected the expected bell-shaped response to increasing amounts of IgE ligand (38, 39) in Munc13-4+/+ PCMCs, whereas PCMCs from Munc13-4−/− and Munc13-4Δ/Δ mice had a flat response (Fig. 4, A and B). We could detect a small response in the hypomorphic Munc13-4F/F PCMCs, which was magnified when we used PMA/ionomycin (PI) as agonist (Fig. 4C). The almost identical results between Munc13-4−/− and Munc13-4Δ/Δ PCMCs point to the efficiency of Cre recombination in these cells.
Figure 4.

Munc13-4 controls only regulated exocytosis in MCs. PCMCs sensitized with anti-DNP IgE and then challenged with DNP-HSA (DNP) or PMA/ionomycin (PI). Color legend in A applies to all panels. Regulated exocytosis was monitored by measuring secretion of histamine (A) or β-hexosaminidase (B and C) as fraction of total cell content 30 min post-stimulation. n = 4; circle and bar, mean; error bar, S.E. Constitutive exocytosis was monitored by measuring secretion of TNFα (D) and IL4 (E) 6 h post-stimulation. n = 8. Secretion independent of exocytosis was monitored by measuring PGD2 (F) and LTC4 (G) 30 min post-stimulation. n = 4. Line, mean; box, 25th–75th percentile; whiskers, 5th–95th percentile. * = p < 0.001; all comparisons are to Munc13-4+/+ unless otherwise indicated.
We chose the dose of DNP at which we detected the largest response (10 ng/ml) and PI to interrogate other MC effector responses. To address constitutive exocytosis of newly synthesized cytokines, we measured the secretion of TNFα and IL4, and for responses independent of exocytosis, we measured the secretion of PGD2 and LTC4. As reported by others, we found that engaging IgE signaling was a weaker stimulus than other agonists for cytokine production (40). In either case, we found no differences between Munc13-4-sufficient and Munc13-4-deficient MCs (Fig. 4, D and E). Also, we observed no differences in secretion of PGD2 and LTC4 (Fig. 4, F and G). TNFα, IL4, PGD2, and LTC4 were undetectable in supernatants of unstimulated cells (data not shown). Thus, we found that only regulated exocytosis is affected by the absence of Munc13-4.
Munc13-2 and Munc13-4 regulate the kinetics of MC exocytosis
Based on our findings for regulated exocytosis and anaphylaxis, we decided to study the influence of Munc13 proteins on the dynamics of exocytosis at the single-cell level. To detect exocytosis in individual MCs, we measured changes in membrane capacitance in the whole-cell patch-clamp recording configuration. Intracellular dialysis of GTPγS and Ca2+ through the patch pipette induces almost complete MC degranulation, and the resolution of this technique is such that we can record individual granule-to-plasma membrane fusion events (41–44). Membrane capacitance (Cm) is proportional to the area of the plasma membrane, which increases by the addition of membrane from a secretory vesicle upon fusion, and is recorded as an almost instantaneous “step up” in capacitance gain (ΔCm) (43). We tested freshly isolated peritoneal MCs from all Munc13-4 genotypes, Munc13-2 KO mice, and double Munc13-2/Munc13-4 KO mice (DKO). As a negative control, we used Munc13-4+/+ MCs penetrated with a pipette loaded with an intracellular solution lacking GTPγS to demonstrate that the manipulation itself was not responsible for the changes in Cm (Fig. 5A). The baseline Cm of all the MCs studied was 6.1 ± 0.4 pF; the intracellular Ca2+ concentration achieved in all MCs as measured by ratiometry was 701 ± 44 nm (mean ± S.E. in both cases), and we found no differences in these values among all the genotypes. We recorded the ΔCm over this baseline after stimulation to quantify the total amount of exocytosis (Fig. 5, A and B). We then normalized the Cm curves (Fig. 5C) to obtain the rate of gain between 40 and 60% of total ΔCm, which corresponds to the steepest part of each curve (Fig. 5D). We used the Cm traces (Fig. 6A) to measure the interval between the beginning of stimulation and the beginning of exocytosis in each MC (Fig. 6B). Finally, we measured the sizes of steps in Cm curves from Munc13-4-sufficient and Munc13-4-deficient MCs (Fig. 6, C and D) to estimate the size of the vesicles being exocytosed.
Figure 5.
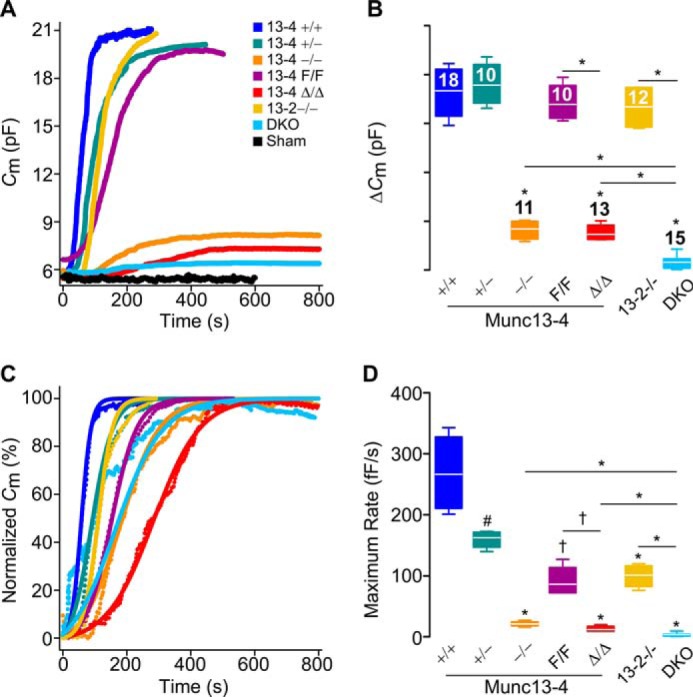
Munc13-4 regulates the amount and rate of MC exocytosis. Shown are capacitance (Cm) recordings from peritoneal MCs during exocytosis stimulated by intracellular dialysis of GTPγS and Ca2+. Access to the cell interior under the whole-cell patch clamp recording technique was achieved at time = 0. Sham, Munc13-4+/+ MCs dialyzed with a solution lacking GTPγS. DKO, double Munc13-2 and Munc13-4 KO mouse. Color legend in A applies to all panels. A, representative traces of cumulative Cm. B, total Cm change (ΔCm) above baseline. n = number inside boxes, applies to B and D. C, representative normalized Cm traces. D, maximum rate of ΔCm (rate between 40 and 60% of total ΔCm). Line, mean; box, 25th–75th percentile; whiskers, 5th–95th percentile. # = p < 0.05, † = p < 0.01, * = p < 0.001; all comparisons are to Munc13-4+/+ unless otherwise indicated.
Figure 6.
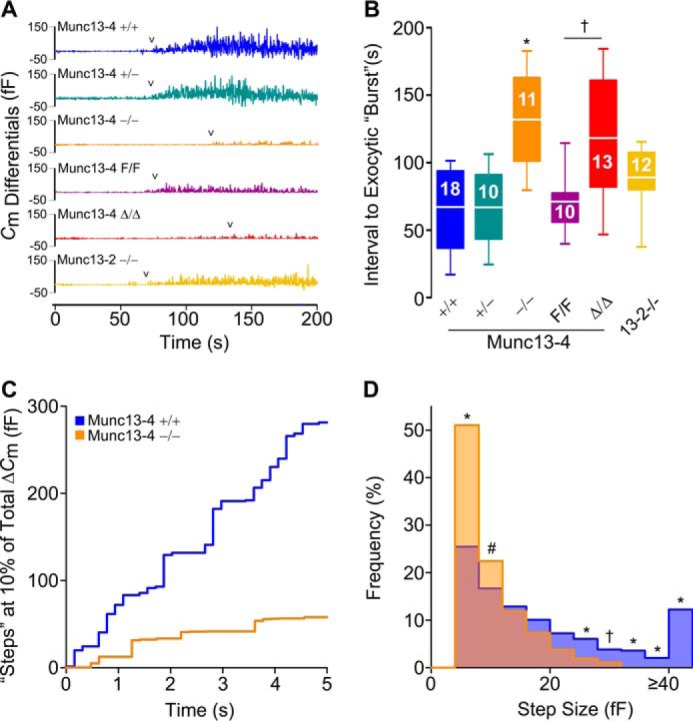
Munc13-4 regulates the start of exocytosis and the size of the steps in Cm. A, representative Cm differentials from Cm traces shown in Fig. 5A. 0, time of cell access; arrowheads, first Cm differential ≥8 fF. Color legend applies to A and B. B, interval between start of dialysis of GTPγS and Ca2+ and beginning of the exocytic burst. Line, mean; box, 25th–75th percentile; whiskers, 5th–95th percentile; n = number inside boxes. C, representative Cm traces at ∼10% of total ΔCm. Signals <0 fF have been removed for clarity. Color legend applies to C and D. D, histogram of Cm step sizes between 1 and 15% of total ΔCm. Signals <4 fF have been removed for clarity. n = numbers in B; ∼68 steps/cell. # = p < 0.05, † = p < 0.01, and * = p < 0.001; all comparisons are to Munc13-4+/+ unless otherwise indicated.
Munc13-4 deficiency (Munc13-4−/− and Munc13-4Δ/Δ) resulted in severe reductions in the total amount of exocytosis. Any residual exocytosis was very slow and delayed. The mutants with intermediate expression levels of Munc13-4 (Munc13-4+/− and Munc13-4F/F) had a normal interval to start exocytosis, and they reached a normal level of ΔCm but at a slow rate. Indeed, we found a clear dose dependence between the expression levels of Munc13-4 and the rate of exocytosis in MCs. The frequency distribution of the step sizes was very different between Munc13-4+/+ and Munc13-4−/− MCs, with a clear shift to the left and absence of large fusion events in the absence of Munc13-4.
Although deletion of Munc13-2 in isolation had no effect on the total amount of exocytosis, it partially impacted the rate of exocytosis, and any residual exocytosis we observed in Munc13-4-deficient MCs was almost completely eliminated when both Munc13-2 and Munc13-4 were deleted. The defect was so severe in the DKO MCs that we could not determine a start point of exocytosis or have enough steps to analyze. Therefore, the amount, rate, and interval between stimulation and start of exocytosis and the size of the vesicular compartments fusing with the plasma membrane all depend on Munc13-4, and a role for Munc13-2 was revealed in the absence of Munc13-4.
Lack of Munc13-4 alters the ultrastructural changes associated with exocytosis in MCs
A limitation of patch clamp is that only events at the cell surface generate a signal. Consequently, it cannot distinguish between individual vesicles fusing with the plasma membrane and sequential compound exocytosis connecting the membrane to inward granules. Also, the lack of large steps in ΔCm could reflect a failure of homotypic granule-to-granule fusion, of heterotypic fusion of large multigranular compartments to the plasma membrane, or both. To overcome this deficit, we studied stimulated peritoneal MCs under EM and stereology. Although there were marked changes between stimulated and unstimulated Munc13-4+/+ MCs, stimulated Munc13-4−/− MCs were almost indistinguishable from unstimulated controls (Fig. 7A). To quantify these qualitative differences, we based our stereological analysis on EM signs of MC activation: the granules increase in size as they become hydrated and they lose electron-density as the contents become diluted (45); the geometry of the surface of the cell increases in complexity as membrane is added during degranulation; and there is loss of the intracellular boundaries between granules as they fuse during compound exocytosis (42, 46). We show that whereas the cell profiles of stimulated Munc13-4+/+ MCs had a decrease in compactness of their granules, displayed as a right shift in relative electron lucency (Fig. 7B, blue area), the density of the granules in Munc13-4−/− cell profiles was basically unchanged (red area) compared with unstimulated controls (black area). This was confirmed when we measured the Vv of granules with a relative electron lucency of >150 (Fig. 7C). As the diameter of a sphere increases, its surface-to-volume ratio (expressed by the surface density or Sv) decreases, so the reduction in granule Sv in stimulated WT cells compared with unstimulated WT cells reflects the swelling of MC granules during activation, which did not occur in stimulated Munc13-4−/− MCs (Fig. 7D). The cell Sv increases as the surface complexity increases and the cell becomes less spherical, and again the stimulated Munc13-4−/− MCs behaved more like unstimulated Munc13-4+/+ cells than stimulated Munc13-4+/+ cells (Fig. 7E). Finally, we quantified the volume density of multigranular complexes sharing a single membrane (Fig. S3) and observed that, despite stimulation, almost none could be detected in the absence of Munc13-4 (Fig. 7F). Thus, Munc13-4 not only controls the heterotypic fusion of secretory granules with the plasma membrane, it also mediates the homotypic fusion between granules.
Figure 7.
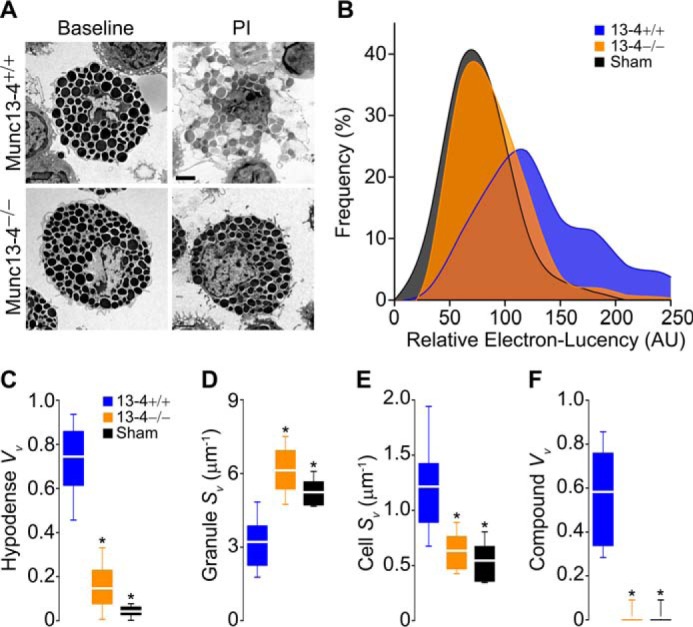
Lack of Munc13-4 alters the ultrastructural changes associated with MC exocytosis. Peritoneal MCs fixed after exposure to PI. Sham, Munc13-4+/+ MCs exposed to vehicle. A, representative cell profiles before and after stimulation. Scale bar = 4 μm. B, relative electron lucency of MC granules based on a 0 (black) to 255 (white) scale. n = ∼40 granules/cell, 30 cells/animal, four animals. C–F, stereological analysis of MC profiles. Vv, volume density; Sv, surface density; Hypodense, granules with relative electron lucency >150; Compound, multigranular profiles sharing a single delimiting membrane. n = 30 cells/animal, four animals. * = p < 0.001; all comparisons are to Munc13-4+/+ unless otherwise indicated.
Discussion
MCs store biogenic amines, lysosomal enzymes, proteases, and highly sulfated glycosaminoglycans attached to a serglycin protein backbone in their large secretory granules (29, 47), and the local and systemic release of these substances contributes to many pathological processes (48–51).
Munc13-4 has been detected in BMMCs (52, 53) and RBL-2H3 cells (26), and we found that Munc13-4 is co-expressed with Munc13-2 in mature MCs (Fig. 1). Munc13-2 has two splice variants, one expressed predominantly in brain (bMunc13-2) and another expressed ubiquitously (ubMunc13-2) (12, 54). ubMunc13-2 is the variant expressed in MCs, and its structure is very similar to that of Munc13-1 (55). The Munc13-2 KO mouse line we used lacks both splice variants (56). With this deletant mouse, we were able to detect a role for Munc13-2 in MC exocytosis (Fig. 5). It is possible that a subpopulation of MC secretory granules uses Munc13-2 for priming and that this could only be revealed in a Munc13-4-null background. Another explanation is that, similar to cytotoxic T lymphocytes where expression of Munc13-1 can supplement or supplant the function of Munc13-4 in exocytosis of lytic granules (57), Munc13-2 might partially replace the function of Munc13-4 in exocytosis of MC granules. Our findings in electrophysiology and EM that there was almost no residual compound multigranular exocytosis once Munc13-4 was removed (Figs. 6D and 7F) indicate that not all Munc13-4 functions can be replaced by Munc13-2.
While developing the Munc13-4F/F line, we disrupted the expression of Munc13-4, creating a hypomorphic mouse with expression levels between that of heterozygous and homozygous deletants (Fig. 1). In all assays where we compared Munc13-4F/F with Munc13-4Δ/Δ mice, we found a significant difference, indicating that this conditional KO line is a very useful loss-of-function model. Even more, we took advantage of this unexpected outcome, and we found a dose-dependent relationship between MC exocytosis and expression levels of Munc13-4 (Figs. 4 and 5).
By Cm measurements, we detected a 78% decrease in stimulated MC exocytosis in the absence of Munc13-4 (Fig. 5B). This defect was not due to differences in MC granules (Fig. 3 and Table 1), as is the case in other secretion anomalies (58, 59). The defect in ΔCm we found in Munc13-4-deficient mice was confirmed by the almost complete lack of evidence of degranulation in our EM stereology studies (Fig. 7). The close correlation between our electrophysiological and morphological studies indicates that stereology of EM cell profiles is a reliable method to assess exocytosis in MCs. Although a participation of Munc13-4 in exocytosis from BMMCs (52) and RBL-2H3 cells (25, 26, 60) has been described before, our findings in MCs with intermediate expression of Munc13-4 (+/− and F/F) proves that Munc13-4 is a rate-limiting factor in this process (Figs. 4C and 5D).
In most cells, only a fraction of their secretory vesicles are primed at rest, and this pool increases as more Munc13 is recruited to the plasma membrane during cell activation (61). A defect in priming could explain why Munc13-4 is rate-limiting for MC secretion. First, the longer interval between MC stimulation and response corresponds to a delayed “burst” of degranulation that we observed in the absence of Munc13-4 (Fig. 6B), indicating that this protein is a limiting factor for the number of secretory granules that may be fusion-competent before MC activation, similar to the ready releasable pool of synaptic vesicles in neurons (62). Second, Munc13-4 is required for the acceleration of exocytosis after activation (Fig. 5D), indicating that Munc13-4 might be a limiting factor for the recruitment of new secretory vesicles to the primed pool, similar to the replenishment of the ready releasable pool in neurons (63). Despite these similarities to Munc13-1-dependent neuronal priming, Munc13-4 lacks the regulatory C2A and C1 domains, implying that homodimerization, interactions with RIM, and binding to DAG are dispensable for these Munc13-4 functions (17, 18, 20, 24).
The close correlation between the maximum rate of exocytosis and the level of expression of Munc13-4 we observed (Fig. 5D) could have another explanation. Mechanistically, the transition from initial to maximum rate of exocytosis in MCs could be achieved by compound exocytosis (42). The domino-like fusion of individual granules to other granules that have already fused with the plasma membrane in sequential compound exocytosis decreases the distance between vesicle and target membranes, accelerating the rate of single-vesicle fusion. Also, the fusion between granules to create large secretory compartments before fusing with the plasma membrane in multigranular compound exocytosis adds a larger membrane surface per exocytic event. The membrane of a single MC granule has a Cm of ∼7 fF (64). We found that most fusion events in stimulated Munc13-4+/+ MCs are larger than 8 fF, indicating that multigranular compound exocytosis is common in normal MCs. In contrast, most steps recorded in Munc13-4−/− MCs were ≤8 fF (Fig. 6D), implying that most exocytosis remaining in the absence of Munc13-4 is of single vesicles. The lack of large “steps” could be due to a failure to form large multigranular compartments or a defect in fusion of these multigranular compartments with the plasma membrane. Our EM stereology studies of activated peritoneal MCs confirmed the former (Fig. 7F). This loss-of-function model and the combination of electrophysiology and EM stereology corroborate the long suspected dependence of the accelerated phase of MC exocytosis on compound multigranular exocytosis.
Our findings prove that Munc13-4 is required for both the heterotypic fusion of granule membranes with the plasma membrane and the homotypic fusion of granule membranes in mature MCs. Others have reported that the two C2 domains of Munc13-4 are required for the homotypic fusion responsible for the formation of large vacuoles in RBL-2H3 cells (26). The fact that both types of fusion are under the control of the same Munc13 protein suggests that both membrane fusion machineries may share other fundamental components.
Using populations of MCs and other stimuli we found no difference in secretion of PGD2 and LTC4 between Munc13-4+/+ and Munc13-4−/− MCs. This shows that Munc13-4 does not play a role in these exocytic-independent MC effector responses, that Munc13-4-deficient MCs can be activated, and that we are not dealing with a failure in signaling but with a pure exocytic defect. Munc13-4-deficient MCs had a dose-dependent defect in the secretion of preformed mediators stored in MC secretory granules (Fig. 4) that could not be explained by changes in size, number, or shape of MC granules (Fig. 3 and Table 1). In many systems, the presence of a synaptotagmin and a Munc13 protein defines an exocytic process as regulated (10). Similar to what we found in the synaptotagmin-2 KO mouse (38), deficiency of Munc13-4 affected only the release of MC granules, lending further support to the concept that this process is a form of regulated exocytosis.
Our manipulations of the expression of Munc13-4 did not affect the secretion of cytokines by MCs, as was the case for synaptotagmin-2 and VAMP-8 (38, 65), pointing to the existence of an alternative pathway for cytokine secretion. This is supported by studies in which MCs could be stimulated to secrete cytokines without inducing degranulation (66, 67). The most parsimonious explanation is that de novo generated mediators are secreted via constitutive exocytosis using a distinct secretory route (68–70) and that this process is not controlled by Munc13-4. Our finding that the densities of receptors on the MC surface were the same regardless of the expression levels of Munc13-4 (Fig. 3 and Table 1) supports this possibility, given that translocation of most receptors to the plasma membrane depends on constitutive exocytosis (2). Others have found TNFα inside MC granules (71), and it has been documented that vesicles containing TNFα can merge with MC secretory granules (72, 73) in a process that also requires Munc13-4 (26), suggesting that this cytokine uses regulated exocytosis as the final step in its secretion. Unfortunately, none of these studies addressed the relative contribution of this putative secretory pathway to the overall secretion of TNFα by MCs. A way to reconcile these results with our findings of normal TNFα secretion (Fig. 4D), despite a severe defect in degranulation in the absence of Munc13-4, is that the contribution of degranulation to TNFα secretion is minuscule. The existence of a discrete subpopulation of MC granules that contain TNFα but that is not controlled by Munc13-4 is incompatible with our results, unless these granules also lack β-hexosaminidase and histamine.
We did not find that the decreased or absent expression of Munc13-4 caused any alteration in the number, distribution, and differentiation of MCs in the mutant mice (Fig. 3 and Table 1). Thus, any abnormality in the animals should reflect exclusively a functional defect in MCs. There was no overall manifestation of haploinsufficiency in our anaphylaxis model. Even the hypomorphic Munc13-4F/F mice reached the same degree of hypothermia at 50 min, increases in circulating histamine and histological evidence of MC degranulation in connective tissues compared with Munc13-4+/+ mice (Fig. 2), indicating that these manifestations depend not on the rate but on the total amount of exocytosis in MCs (Fig. 5). The only significant difference was detected in the early time points (5 min after challenge), when Munc13-4+/− and Munc13-4F/F were significantly less hypothermic than Munc13-4+/+ mice. This correlates with the slower rates of exocytosis we found in the electrophysiological studies of MCs from these two mutants. These observations indicate that the earliest response engaged is MC degranulation, making the immediate anaphylactic response very sensitive to changes in the kinetics of MC-regulated exocytosis. Afterward, as the reaction progresses and MCs achieve full degranulation, and engage other effector responses, a partial deficiency in Munc13-4 has no effects.
We found a correlation between less MC degranulation, less plasma histamine, and less hypothermia (Fig. 2). Because the responses in Munc13-4−/− and Munc13-4Δ/Δ mice were indistinguishable, we conclude that the Munc13-4 dependence of this response is limited to MCs and that the failure in MC degranulation in vivo was not contingent to a Munc13-4-dependent abnormality in another cell that indirectly affected MC activation. The protection was significant but not absolute. Cma1/mMCP5 is expressed exclusively in MCs but not in all MCs; it is expressed mainly in constitutive/connective tissue MCs (74). Hence, a population of inducible/mucosal MCs that do not express Cre, and therefore do not undergo recombination in Munc13-4Δ/Δ mice, could be responsible for the residual anaphylactic response. However, that does not explain why Munc13-4−/− mice, which lack Munc13-4 in all cells, including mucosal MCs, have the same degree of hypothermia and levels of residual circulating histamine as Munc13-4Δ/Δ mice. The possibility that cells other than MCs (e.g. basophils and platelets) are responsible for the residual response is improbable, given that MC-deficient animals, which have intact numbers of other cells important in anaphylaxis (75), exhibit complete protection from hypothermia and had undetectable levels of circulating histamine. Another explanation is that the residual exocytosis we documented in our electrophysiological and cell secretion assays (Figs. 4, A–C and 5B) allows enough mediators to be secreted to precipitate a limited response in Munc13-4−/− and Munc13-4Δ/Δ mice. This could also explain why we did not detect a major difference in mice with partial expression of Munc13-4 (Munc13-4+/− and Munc13-4F/F). Finally, we think that the best explanation for the incomplete protection in Munc13-4−/− and Munc13-4Δ/Δ mice is that MC responses independent of regulated exocytosis are involved. The duration of our model is too short to be influenced by synthesis and secretion of cytokines but coincides with the time course of secretion of eicosanoids by MCs, which are well-known mediators of anaphylaxis (76) and do not require Munc13-4 for secretion (Fig. 4).
MC degranulation is always cited as one of the main culprits in the generation of the anaphylactic reaction. For the first time we can quantify the relative contribution of MC degranulation in this pathological response, and compare it with other effector responses from MCs and other cells. Based on our results, interfering with MC degranulation or blocking the effects of the multiple mediators released in this process could achieve a significant but not complete control of anaphylaxis.
Experimental procedures
Mice
All genomic portions for the Munc13-4 targeting construct were obtained from B6 mice. The 5′-homology arm (3352 bp; GRCm38:Chr11:116077882–116081234, −strand) included a portion from the 5′-flanking region to intron 2 of mouse Unc13d and the 3′-homology arm (3654 bp; GRCm38:Chr11:116073258–116076912, −strand) from exon 4 to intron 15. The vector contained an FRT-flanked phosphoglucokinase promoter-driven neomycin resistance gene (PGK-Neo) upstream from the insertion fragment (995 bp; GRCm38:Chr11:116076909–116077904, −strand) containing a loxP-flanked exon 3. PGK-herpes simplex virus thymidine kinase (PGK-TK) was placed downstream from the homology region (Fig. 1B). We electroporated the vector into 129S6:B6 ES cells and used neomycin and gancyclovir for selection. Homologous recombinant ES cells were injected into B6(Cg)-Tyrc-2J/J blastocysts and implanted into pseudopregnant B6 mice. Chimeric males were crossed with B6(Cg)-Tyrc-2J/J females, and the resulting heterozygous mutants were crossed with B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/RainJ (The Jackson Laboratory; catalog no. 009086) to remove PGK-Neo and establish our conditional KO line. This floxed mouse was crossed with B6.C-Tg(CMV-cre)1Cgn/J mice (The Jackson Laboratory; catalog no. 006054) that express Cre recombinase ubiquitously to delete the allele in the germ line (34) and to generate our global KO line, or with Tg(Cma1-cre)ARoer mice (Dr. Axel Roers, University of Cologne) for MC-specific deletion (33). We crossed our lines with B6 mice for 10 generations. All genetic manipulations were confirmed by sequencing of genomic DNA from tail biopsies and from sorted peritoneal MCs in the mice chosen as founders of our colonies. Afterward, genotypes were determined by PCR of genomic DNA with primers P1 5′-GGAAAGGTGTGTCGCCATGGTG-3′, P2 5′-ATCCCAGATCAAAATGCTCCCAC-3′, and P3 5′-CCAACATAAGGCTCTCTGAAGG-3′. P1 and P2 were used to differentiate between the conditional (F; 725 bp) and WT (+; 636 bp) alleles and P2 and P3 between the globally or MC-specifically deleted (− or Δ, respectively; 415 bp) and WT (+; 1268 bp) alleles. Genomic Cre was detected using primers 5′-ACAGTGGTATTCCCGGGGAGTGT-3′ and 5′-GTCAGTGCGTTCAAAGGCCA-3′ that give a 421-bp internal control band from the native Cma1 gene and a 555-bp band from the transgene. Littermates +/+, +/−, and −/−, and F/F and Δ/Δ were used in all experiments. We also obtained Munc13-2 KO mice (Dr. Christian Rosenmund, Charité Universitaetsmedizin) (32) and MC-deficient KitW-sh/KitW-sh mice (Dr. Stephen J. Galli, Stanford University) (75). All mice were kept in a pathogen-free facility and handled in accordance with the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center.
MC harvesting and cultures
Eight ml of PBS and 2 ml of air were injected through a 27-gauge needle into the mouse peritoneal cavity, aspirating the paramedian inguinal fat afterward to seal the puncture point. After massaging the abdomen for 5 min, the fluid was aspirated from the flanks with an 18-gauge needle. Peritoneal cells were centrifuged (380 × g, 4 °C, 5 min) and processed for different studies. For PCMCs, cells were resuspended in 10 ml of media (Iscove's modified Dulbecco's medium with glutamine, 10% FBS, 500 units/ml penicillin, 500 units/ml streptomycin, 1× vitamins, 1× non-essential amino acids, 100 μm sodium pyruvate, 50 nm 2-mercaptoethanol; Gibco) with IL3 (5 ng/ml) and SCF (50 ng/ml; both from R&D Systems), plated on 100-mm cell culture plates and incubated for 2 weeks (37 °C, 5% CO2, bi-weekly media changes). For BMMCs, we flushed out the bone marrow from the diaphyses of freshly dissected tibiae and femora with 10 ml of medium (RPMI 1640, 10% FBS, 500 units/ml penicillin, 500 units/ml streptomycin, 100 μm HEPES, pH 7.3, 1 mm sodium pyruvate, 1× nonessential amino acids, 0.1% 2-mercaptoethanol; Gibco) onto a 100-mm cell culture dish. IL3 and SCF were added as above, and cells were incubated for 6 weeks (37 °C, 5% CO2, bi-weekly media changes).
Histology
Peritoneal MC absolute and relative numbers were determined with a Neubauer chamber and differentials of cytospins stained with modified Wright-Giemsa. Ears were excised, fixed overnight at 4 °C in 4% paraformaldehyde, and embedded in paraffin. Next, 5-μm sections were stained with FITC-avidin (2 μg/ml) and Hoechst (5 μg/ml). The density of FITC+/Hoechst+ cells was reported per area of dermis, which was measured as the area between the epidermis and the subcutaneous fat and/or cartilage (77).
Flow cytometry and cell sorting
106 BMMCs, PCMCs, or peritoneal lavage cells were suspended in 250 μl of PBS, blocked with 0.5 μg/ml of anti-mouse CD16/CD32 (BD Biosciences), and incubated with 0.2 μg/ml of phycoerythrin anti-mouse CD117/kit (BD Biosciences), allophycocyanin anti-mouse FcϵRIα (eBioscience), or allophycocyanin anti-mouse IL33R (BD Biosciences) for 30 min at 4 °C (78). The cells were washed twice with PBS and resuspended in 500 μl of cold PBS for analysis in an LSRII flow cytometer. For FACS, we collected the FcϵRIα+/Kit+ double-positive cells using a FACSAria Fusion Cell Sorter (BD Biosciences).
Expression analyses
For qPCR, FACS-sorted peritoneal MCs were immersed in DNA/RNA Shield (Zymo Research) for 1 day and homogenized by vortexing for 1 min. RNA was extracted using RNeasy mini kit (Qiagen), and cDNA was obtained through reverse transcription with qScript cDNA SuperMix (Quanta Biosciences). Amplification was conducted on a ViiA 7 real-time PCR system using PerfeCTa qPCR ToughMix (Quanta Biosciences). Expression of Munc13-1 exons 3–4 (Mm01340418_m1, Life Technologies, Inc.), Munc13-2 exons 17–18 (Mm01351419_m1), Munc13-3 exons 3–4 (Mm00463432_m1), and Munc13-4 exons 7–8 (Mm01252625_m1; present in all reported mouse Unc13d splice variants) were quantified relative to that of β-actin exon 6 (Mm00607939_s1). For RACE, RNA was reverse-transcribed (SuperScript II, Life Technologies, Inc.) and amplified with the adaptor primer and 5′-AACCACCATGGCGACACACC-3′, which is common to all translated Munc13-4 splice variants (GRCm38). The gel-purified product was then sequenced. For immunoblots, mouse tissues were homogenized and sonicated on ice in 2 ml of lysis buffer (150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris, pH 8.0) with 1% protease inhibitor mixture (Sigma), or 106 PCMCs were sonicated in 100 μl of PBS with 1% protease inhibitor mixture. Protein content was estimated with BCA (Pierce). Lysates were run under denaturing conditions in a 7.5% bisacrylamide gel, transferred onto a nitrocellulose membrane (both from Bio-Rad), and incubated with a rabbit polyclonal antibody (1:4000) raised against the mouse Munc13-4 peptide N-LLESRKGDREAQAFVKLRRQRAKQASQHAP-C. β-Actin was used as loading control.
Secretion assays
3 × 104 PCMCs were incubated overnight with 100 ng/ml SPE-7 anti-DNP IgE (Sigma). They were next centrifuged (300 × g, 4 °C, 10 min), washed three times in secretion buffer (in mm: 10 HEPES, pH 7.4, 137 NaCl, 2.7 KCl, 0.4 Na2HPO4, 5.6 glucose, 1.8 CaCl2, 1.3 MgSO4), and resuspended in 90 μl of secretion buffer with 0.04% BSA (37 °C). Cells were stimulated by adding 10 μl of secretion buffer alone, with 1 ng to 10 μg of DNP-HSA, or PMA (50 ng/ml) with ionomycin (1.2 μm) (all from Sigma). After 30 min (β-hexosaminidase, histamine, LTC4, and PGD2) or 6 h (TNFα and IL4), the stimulation was halted by placing the cells on ice. Samples were centrifuged (300 × g, 4 °C, 10 min), and supernatants were collected. ELISA was used to quantify histamine, PDG2, and LTC4 (Cayman Chemical) and TNFα and IL4 (R&D Biosystems). For histamine and β-hexosaminidase, a duplicate of every sample was lysed by adding 150 μl of 0.1% Triton X-100 (10 min, 37 °C) to measure total cell content. To determine β-hexosaminidase activity, the lysates or supernatants (50 μl) were incubated with 100 μl of 3.5 mg/ml p-nitro-N-acetyl-β-d-glucosaminide (pNAG; Sigma; 90 min, 37 °C), and the reaction was stopped by adding 100 μl of 400 mm glycine, pH 10.7. Absorbance at λ = 405 nm was recorded (reference λ = 620 nm) and corrected for dilutions.
Electrophysiology
Peritoneal MCs were washed twice and plated onto glass-bottomed recording chambers filled with external recording solution (in mm: 136.89 NaCl, 2.6 KCl, 2 CaCl2, 0.493 MgCl2, 0.407 MgSO4, 0.441 KH2PO4, 0.338 Na2HPO4, 10 HEPES, and 10 glucose, pH 7.3, 310 mosm). Whole-cell recordings were performed at 21–24 °C on individual peritoneal MCs using 5–6-megohm patch pipettes coated with a silicone elastomer (Sylgard) and filled with an internal solution that both defined intracellular Ca2+ and induced degranulation (79, 80). The internal solution contained (in mm) 135 potassium gluconate, 7 MgCl2, 0.2 Na2ATP, 0.05 Li4GTPγS, 2.5 EGTA, 7.5 Ca-EGTA, 0.1 Fura-2, and 10 HEPES (pH 7.21, 0.302 osmoles). Alternatively, a similar calculated free Ca2+ concentration was achieved by replacing the EGTA and Ca-EGTA concentrations listed above with 5 mm EGTA, 7.5 mm Ca-EGTA, and 2 mm CaCl2 (MaxChelator). The free Ca2+ concentration was determined from the ratio of emitted fluorescence at 360 and 388 nm with the use of a computer-controlled monochrometer-based photometry system (79–81) and from Ca2+ calibration constants determined in vitro with the use of Fura-2 Calcium Imaging calibration kit (0–10 mm Ca-EGTA, 50 μm Fura-2; Thermo Fisher Scientific/Molecular Probes). Measurements of Cm were made with an EPC-9 patch-clamp amplifier controlled by Pulse software (HEKA Electronik). An 800-Hz sinusoidal, 30-mV peak-to-peak stimulus was applied around a holding potential of −70 mV, and the resultant signal was analyzed using the Lindau-Neher technique (80, 81) to yield Cm and the membrane (Gm) and series (Gs) conductances. For each 100-ms sweep, the average value was recorded, yielding a temporal resolution for Cm, Gm, and Gs of ∼7 Hz. Cells selected for analysis met the criteria of Gm ≤1000 pS, Gs ≥ 40 nS, and steady-state intracellular [Ca2+] ∼700 ± 100 nm. From recordings of Cm over time, we obtained the total ΔCm and from normalized curves the rates of ΔCm from 40 to 60% of total ΔCm. We logged individual changes in Cm (Cm differentials). We identified the beginning of the burst of exocytosis as successive Cm differentials ≥8 fF sustained for >4 s; this avoided baseline isolated events. Then, we recorded the time interval between cell access and the first Cm differential ≥8 fF within this 4-s region. We measured the size of the steps between 1 and 15% of total ΔCm after eliminating any signal <4 fF to decrease noise.
Electron microscopy and stereology
Peritoneal MCs were resuspended at rest or after 5 min of activation with PI and fixed in 0.1 m sodium cacodylate buffer, pH 7.2, containing 2% glutaraldehyde (2 h, room temperature). They were washed twice with PBS, resuspended in 0.1 m sodium cacodylate buffer containing 1% OsO4, washed in double-distilled H2O, pelleted, and embedded in 3% low-melting temperature agarose. The agarose pellets were dehydrated through an acetone series and then embedded in EMbed-812 epoxy resin (14120 EMS). Sections of 100 nm thickness were stained with uranyl acetate and lead citrate and viewed under a Tecnai 12 transmission electron microscope. Nucleated MC profiles were randomly photographed, and at least 30 profiles from each sample were assessed by stereology. Vv and Sv were obtained with randomly placed dot and line grids on the cell profiles (82, 83). The cell profile area was estimated selecting circumference intersections in a cycloid grid with a known perimeter value. Multigranular events were defined as granule profiles not separated by a membrane (Fig. S3). To measure the relative electron lucency of MC granules on a grayscale, a circle-cycloid stereological grid with circles of 0.0366 μm2 (diameter = 30 pixels) was randomly superimposed on cell profiles; 50 random circles that fell on granules, nuclear heterochromatin, and electron-lucent extracellular space were selected, and their grayscale value (0–255) was recorded. The values from the heterochromatin and extracellular space were used to set a linear scale to compare the values of each randomly selected granule, providing an internal scale normalized for each photograph.
Passive systemic anaphylaxis
Adult mice (15–27 weeks old) were sensitized i.v. with anti-DNP IgE (20 μg in 200 μl of PBS). A day later, they were challenged i.v. with DNP-HSA (1 mg in 200 μl of PBS). Core temperatures were recorded at baseline and at 5, 10, 30, 50, 70, and 90 min with a rectal probe. For histamine quantification, we took special care not to activate platelets because they also release significant amounts of histamine. After challenge, animals were placed in an isoflurane chamber, and 3 min later blood was drawn slowly from the inferior vena cava through a 21-gauge needle into a 1-ml syringe loaded with 110 μl of 4% sodium citrate. An equal volume of Tyrode's buffer (0.81% NaCl, 0.02% KCl, 0.09% glucose, 0.005% NaH2PO4, 0.1% NaHCO3) was added to each sample, and the plasma was separated by centrifugation (380 × g, 15 min, room temperature) and frozen at −80 °C for subsequent histamine ELISA (Cayman Biomedical). Tongues and lips of mice euthanized 15 min post-challenge were fixed in 4% paraformaldehyde (16 h, 4 °C), placed in 30% sucrose and 30% OCT (Tissue-Tek; VWR) in PBS overnight, and then embedded in OCT. Sections of 5 μm were stained with 0.1% toluidine blue, pH 0.5. MC degranulation was blindly assessed based on the fraction of granules outside the cell and described as low (0–10%), moderate (10–50%), or severe (>50%) (Fig. S2).
Statistical analysis
For continuous variables, we compared the means of all groups by one-way analysis of variance; if a significant difference was found, we applied Tukey's test for multiple pairwise comparisons or Dunnett's test for multiple comparisons against the control group. For categorical data, we used Pearson's χ2 test or Fisher's exact test. We analyzed frequency distributions with the Kolmogorov-Smirnov test. Significance was set at p < 0.05.
Author contributions
R. A. conceived the study. D. C. M., Y. P., M. J. T., B. F. D., and R. A. generated the mutant Munc13-4 lines. M. A. R. and E. I. C. performed the expression studies. E. M. R., M. A. R., D. C. M., Y. P., S. M., J. M., and R. A. characterized the MCs. D. C. M., Y. P., S. M, K. B., and E. M. R. performed in vivo assays. M. A. R. and Y. P. performed the secretion assays. A. J. D., D. S. M., A. I. R., R. H., and R. A. performed and analyzed the electrophysiology experiments. D. C. M., D. S. M., S. M, A. I. R., L. E. R., E. S., A. T., J. M., E. A. G., A. R. B., and R. A. performed the electron microscopy, histology and stereology. R. A., E. M. R., and M. A. R. analyzed the data and prepared the manuscript. All authors contributed to the discussion of the study.
Supplementary Material
Acknowledgments
We thank Evelyn S. Brown and Margaret Gondo (University of Houston) for professional assistance with the electron microscopy and Dr. Thomas C. Südhof for the backbone of the targeting vector.
This work was supported by the National Institutes of Health Grants AI093533, HL129795, CA016672, EY007551, and EY018239 and the Cancer Prevention Research Institute of Texas Grant RP110166. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- MC
- mast cell
- B6
- C57BL6/J mouse line
- BMMC
- bone marrow-derived MC
- Cm
- capacitance
- ΔCm
- capacitance gain
- ΔT
- change in body core temperature
- DAG
- diacylglycerol
- DKO
- Munc13-2/Munc13-4 double KO
- DNP
- 2,4-dinitrophenol
- F
- farad
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- HSA
- human serum albumin
- LTC4
- leukotriene C4
- Neo
- neomycin phosphotransferase
- PCMC
- peritoneal cell-derived MC
- PGD2
- prostaglandin D2
- PGK
- phosphoglucokinase promoter
- PI
- PMA/ionomycin
- PMA
- phorbol 12-myristate 13-acetate
- qPCR
- quantitative PCR
- RACE
- rapid amplification of cDNA ends
- RIM
- Rab3-interacting molecule
- S
- siemen
- SCF
- stem cell factor
- SNARE
- soluble N-ethylmaleimide-sensitive factor activated protein receptor
- Stx
- syntaxin
- Sv
- surface density
- TK
- herpes simplex virus thymidine kinase
- VAMP
- vesicle-associated membrane protein
- Vv
- volume density.
References
- 1. Jahn R. (2004) Principles of exocytosis and membrane fusion. Ann. N. Y. Acad. Sci. 1014, 170–178 10.1196/annals.1294.018 [DOI] [PubMed] [Google Scholar]
- 2. Burgess T. L., and Kelly R. B. (1987) Constitutive and regulated secretion of proteins. Annu. Rev. Cell Biol. 3, 243–293 10.1146/annurev.cb.03.110187.001331 [DOI] [PubMed] [Google Scholar]
- 3. Berridge M. J. (1984) Inositol trisphosphate and diacylglycerol as second messengers. Biochem. J. 220, 345–360 10.1042/bj2200345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pang Z. P., and Südhof T. C. (2010) Cell biology of Ca2+-triggered exocytosis. Curr. Opin. Cell Biol. 22, 496–505 10.1016/j.ceb.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pickett J. A., and Edwardson J. M. (2006) Compound exocytosis: mechanisms and functional significance. Traffic 7, 109–116 10.1111/j.1600-0854.2005.00372.x [DOI] [PubMed] [Google Scholar]
- 6. Lorentz A., Baumann A., Vitte J., and Blank U. (2012) The SNARE machinery in mast cell secretion. Front. Immunol. 3, 143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Söllner T., Whiteheart S. W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., and Rothman J. E. (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324 10.1038/362318a0 [DOI] [PubMed] [Google Scholar]
- 8. Rizo J., and Xu J. (2015) The synaptic vesicle release machinery. Annu. Rev. Biophys. 44, 339–367 10.1146/annurev-biophys-060414-034057 [DOI] [PubMed] [Google Scholar]
- 9. Klenchin V. A., and Martin T. F. (2000) Priming in exocytosis: attaining fusion-competence after vesicle docking. Biochimie 82, 399–407 10.1016/S0300-9084(00)00208-X [DOI] [PubMed] [Google Scholar]
- 10. Südhof T. C. (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690 10.1016/j.neuron.2013.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hammarlund M., Palfreyman M. T., Watanabe S., Olsen S., and Jorgensen E. M. (2007) Open syntaxin docks synaptic vesicles. PLoS Biol. 5, e198 10.1371/journal.pbio.0050198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brose N., Hofmann K., Hata Y., and Südhof T. C. (1995) Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J. Biol. Chem. 270, 25273–25280 10.1074/jbc.270.42.25273 [DOI] [PubMed] [Google Scholar]
- 13. Betz A., Okamoto M., Benseler F., and Brose N. (1997) Direct interaction of the rat unc-13 homologue Munc13-1 with the N terminus of syntaxin. J. Biol. Chem. 272, 2520–2526 10.1074/jbc.272.4.2520 [DOI] [PubMed] [Google Scholar]
- 14. Ma C., Li W., Xu Y., and Rizo J. (2011) Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat. Struct. Mol. Biol. 18, 542–549 10.1038/nsmb.2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang X., Wang S., Sheng Y., Zhang M., Zou W., Wu L., Kang L., Rizo J., Zhang R., Xu T., and Ma C. (2015) Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic-vesicle priming. Nat. Struct. Mol. Biol. 22, 547–554 10.1038/nsmb.3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lai Y., Choi U. B., Leitz J., Rhee H. J., Lee C., Altas B., Zhao M., Pfuetzner R. A., Wang A. L., Brose N., Rhee J., and Brunger A. T. (2017) Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron 95, 591–607.e10 10.1016/j.neuron.2017.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Camacho M., Basu J., Trimbuch T., Chang S., Pulido-Lozano C., Chang S. S., Duluvova I., Abo-Rady M., Rizo J., and Rosenmund C. (2017) Heterodimerization of Munc13 C2A domain with RIM regulates synaptic vesicle docking and priming. Nat. Commun. 8, 15293 10.1038/ncomms15293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu X., Seven A. B., Camacho M., Esser V., Xu J., Trimbuch T., Quade B., Su L., Ma C., Rosenmund C., and Rizo J. (2016) Functional synergy between the Munc13 C-terminal C1 and C2 domains. Elife 5, e13696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shin O. H., Lu J., Rhee J. S., Tomchick D. R., Pang Z. P., Wojcik S. M., Camacho-Perez M., Brose N., Machius M., Rizo J., Rosenmund C., and Südhof T. C. (2010) Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat. Struct. Mol. Biol. 17, 280–288 10.1038/nsmb.1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu J., Camacho M., Xu Y., Esser V., Liu X., Trimbuch T., Pan Y. Z., Ma C., Tomchick D. R., Rosenmund C., and Rizo J. (2017) Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13-1 C1C2BMUN. Elife 6, e22567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Michelassi F., Liu H., Hu Z., and Dittman J. S. (2017) A C1-C2 module in Munc13 inhibits calcium-dependent neurotransmitter release. Neuron 95, 577–590 10.1016/j.neuron.2017.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Augustin I., Rosenmund C., Südhof T. C., and Brose N. (1999) Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400, 457–461 10.1038/22768 [DOI] [PubMed] [Google Scholar]
- 23. Feldmann J., Callebaut I., Raposo G., Certain S., Bacq D., Dumont C., Lambert N., Ouachée-Chardin M., Chedeville G., Tamary H., Minard-Colin V., Vilmer E., Blanche S., Le Deist F., Fischer A., and de Saint Basile G. (2003) Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 115, 461–473 10.1016/S0092-8674(03)00855-9 [DOI] [PubMed] [Google Scholar]
- 24. Koch H., Hofmann K., and Brose N. (2000) Definition of Munc13-homology-domains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem. J. 349, 247–253 10.1042/bj3490247,10.1042/0264-6021:3490247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boswell K. L., James D. J., Esquibel J. M., Bruinsma S., Shirakawa R., Horiuchi H., and Martin T. F. (2012) Munc13-4 reconstitutes calcium-dependent SNARE-mediated membrane fusion. J. Cell Biol. 197, 301–312 10.1083/jcb.201109132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woo S. S., James D. J., and Martin T. F. (2017) Munc13-4 functions as a Ca2+ sensor for homotypic secretory granule fusion to generate endosomal exocytic vacuoles. Mol. Biol. Cell 28, 792–808 10.1091/mbc.E16-08-0617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bulfone-Paus S., Nilsson G., Draber P., Blank U., and Levi-Schaffer F. (2017) Positive and negative signals in mast cell activation. Trends Immunol. 38, 657–667 10.1016/j.it.2017.01.008 [DOI] [PubMed] [Google Scholar]
- 28. Theoharides T. C., Kempuraj D., Tagen M., Conti P., and Kalogeromitros D. (2007) Differential release of mast cell mediators and the pathogenesis of inflammation. Immunol. Rev. 217, 65–78 10.1111/j.1600-065X.2007.00519.x [DOI] [PubMed] [Google Scholar]
- 29. McNeil H. P., Adachi R., and Stevens R. L. (2007) Mast cell-restricted tryptases: structure and function in inflammation and pathogen defense. J. Biol. Chem. 282, 20785–20789 10.1074/jbc.R700017200 [DOI] [PubMed] [Google Scholar]
- 30. Reid G., Wielinga P., Zelcer N., van der Heijden I., Kuil A., de Haas M., Wijnholds J., and Borst P. (2003) The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. U.S.A. 100, 9244–9249 10.1073/pnas.1033060100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kochel T. J., and Fulton A. M. (2015) Multiple drug resistance-associated protein 4 (MRP4), prostaglandin transporter (PGT), and 15-hydroxyprostaglandin dehydrogenase (15-PGDH) as determinants of PGE2 levels in cancer. Prostaglandins Other Lipid Mediat. 116–117, 99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Varoqueaux F., Sigler A., Rhee J. S., Brose N., Enk C., Reim K., and Rosenmund C. (2002) Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. U.S.A. 99, 9037–9042 10.1073/pnas.122623799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scholten J., Hartmann K., Gerbaulet A., Krieg T., Müller W., Testa G., and Roers A. (2008) Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res. 17, 307–315 10.1007/s11248-007-9153-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schwenk F., Baron U., and Rajewsky K. (1995) A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23, 5080–5081 10.1093/nar/23.24.5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Makabe-Kobayashi Y., Hori Y., Adachi T., Ishigaki-Suzuki S., Kikuchi Y., Kagaya Y., Shirato K., Nagy A., Ujike A., Takai T., Watanabe T., and Ohtsu H. (2002) The control effect of histamine on body temperature and respiratory function in IgE-dependent systemic anaphylaxis. J. Allergy Clin. Immunol. 110, 298–303 10.1067/mai.2002.125977 [DOI] [PubMed] [Google Scholar]
- 36. Hogan A. D., and Schwartz L. B. (1997) Markers of mast cell degranulation. Methods 13, 43–52 10.1006/meth.1997.0494 [DOI] [PubMed] [Google Scholar]
- 37. Fawcett D. W. (1954) Cytological and pharmacological observations on the release of histamine by mast cells. J. Exp. Med. 100, 217–224 10.1084/jem.100.2.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Melicoff E., Sansores-Garcia L., Gomez A., Moreira D. C., Datta P., Thakur P., Petrova Y., Siddiqi T., Murthy J. N., Dickey B. F., Heidelberger R., and Adachi R. (2009) Synaptotagmin-2 controls regulated exocytosis but not other secretory responses of mast cells. J. Biol. Chem. 284, 19445–19451 10.1074/jbc.M109.002550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gimborn K., Lessmann E., Kuppig S., Krystal G., and Huber M. (2005) SHIP down-regulates FcϵR1-induced degranulation at supraoptimal IgE or antigen levels. J. Immunol. 174, 507–516 10.4049/jimmunol.174.1.507 [DOI] [PubMed] [Google Scholar]
- 40. Kulka M., Sheen C. H., Tancowny B. P., Grammer L. C., and Schleimer R. P. (2008) Neuropeptides activate human mast cell degranulation and chemokine production. Immunology 123, 398–410 10.1111/j.1365-2567.2007.02705.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fernandez J. M., Neher E., and Gomperts B. D. (1984) Capacitance measurements reveal stepwise fusion events in degranulating mast cells. Nature 312, 453–455 10.1038/312453a0 [DOI] [PubMed] [Google Scholar]
- 42. Alvarez de Toledo G., and Fernandez J. M. (1990) Compound versus multigranular exocytosis in peritoneal mast cells. J. Gen. Physiol. 95, 397–409 10.1085/jgp.95.3.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alvarez de Toledo G., and Fernandez J. M. (1990) Patch-clamp measurements reveal multimodal distribution of granule sizes in rat mast cells. J. Cell Biol. 110, 1033–1039 10.1083/jcb.110.4.1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oberhauser A. F., and Fernandez J. M. (1996) A fusion pore phenotype in mast cells of the ruby-eye mouse. Proc. Natl. Acad. Sci. U.S.A. 93, 14349–14354 10.1073/pnas.93.25.14349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Finkelstein A., Zimmerberg J., and Cohen F. S. (1986) Osmotic swelling of vesicles: its role in the fusion of vesicles with planar phospholipid bilayer membranes and its possible role in exocytosis. Annu. Rev. Physiol. 48, 163–174 10.1146/annurev.ph.48.030186.001115 [DOI] [PubMed] [Google Scholar]
- 46. Kasai H., Takahashi N., and Tokumaru H. (2012) Distinct initial SNARE configurations underlying the diversity of exocytosis. Physiol. Rev. 92, 1915–1964 10.1152/physrev.00007.2012 [DOI] [PubMed] [Google Scholar]
- 47. Stevens R. L., and Adachi R. (2007) Protease-proteoglycan complexes of mouse and human mast cells and importance of their β-tryptase-heparin complexes in inflammation and innate immunity. Immunol. Rev. 217, 155–167 10.1111/j.1600-065X.2007.00525.x [DOI] [PubMed] [Google Scholar]
- 48. Wernersson S., and Pejler G. (2014) Mast cell secretory granules: armed for battle. Nat. Rev. Immunol. 14, 478–494 10.1038/nri3690 [DOI] [PubMed] [Google Scholar]
- 49. Thakurdas S. M., Melicoff E., Sansores-Garcia L., Moreira D. C., Petrova Y., Stevens R. L., and Adachi R. (2007) The mast cell-restricted tryptase mMCP-6 has a critical immunoprotective role in bacterial infections. J. Biol. Chem. 282, 20809–20815 10.1074/jbc.M611842200 [DOI] [PubMed] [Google Scholar]
- 50. Shin K., Nigrovic P. A., Crish J., Boilard E., McNeil H. P., Larabee K. S., Adachi R., Gurish M. F., Gobezie R., Stevens R. L., and Lee D. M. (2009) Mast cells contribute to autoimmune inflammatory arthritis via their tryptase/heparin complexes. J. Immunol. 182, 647–656 10.4049/jimmunol.182.1.647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hamilton M. J., Sinnamon M. J., Lyng G. D., Glickman J. N., Wang X., Xing W., Krilis S. A., Blumberg R. S., Adachi R., Lee D. M., and Stevens R. L. (2011) Essential role for mast cell tryptase in acute experimental colitis. Proc. Natl. Acad. Sci. U.S.A. 108, 290–295 10.1073/pnas.1005758108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Singh R. K., Mizuno K., Wasmeier C., Wavre-Shapton S. T., Recchi C., Catz S. D., Futter C., Tolmachova T., Hume A. N., and Seabra M. C. (2013) Distinct and opposing roles for Rab27a/Mlph/MyoVa and Rab27b/Munc13-4 in mast cell secretion. FEBS J. 280, 892–903 [DOI] [PubMed] [Google Scholar]
- 53. Higashio H., Nishimura N., Ishizaki H., Miyoshi J., Orita S., Sakane A., and Sasaki T. (2008) Doc2α and Munc13-4 regulate Ca2+-dependent secretory lysosome exocytosis in mast cells. J. Immunol. 180, 4774–4784 10.4049/jimmunol.180.7.4774 [DOI] [PubMed] [Google Scholar]
- 54. Song Y., Ailenberg M., and Silverman M. (1998) Cloning of a novel gene in the human kidney homologous to rat munc13s: its potential role in diabetic nephropathy. Kidney Int. 53, 1689–1695 10.1046/j.1523-1755.1998.00942.x [DOI] [PubMed] [Google Scholar]
- 55. Augustin I., Betz A., Herrmann C., Jo T., and Brose N. (1999) Differential expression of two novel Munc13 proteins in rat brain. Biochem. J. 337, 363–371 10.1042/bj3370363,10.1042/0264-6021:3370363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rosenmund C., Sigler A., Augustin I., Reim K., Brose N., and Rhee J. S. (2002) Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron 33, 411–424 10.1016/S0896-6273(02)00568-8 [DOI] [PubMed] [Google Scholar]
- 57. Dudenhoffer-Pfeifer M., Schirra C., Pattu V., Halimani M., Maier-Peuschel M., Marshall M. R., Matti U., Becherer U., Dirks J., Jung M., Lipp P., Hoth M., Sester M., Krause E., and Rettig J. (2013) Different Munc13 isoforms function as priming factors in lytic granule release from murine cytotoxic T lymphocytes. Traffic 14, 798–809 10.1111/tra.12074 [DOI] [PubMed] [Google Scholar]
- 58. Barbosa M. D., Nguyen Q. A., Tchernev V. T., Ashley J. A., Detter J. C., Blaydes S. M., Brandt S. J., Chotai D., Hodgman C., Solari R. C., Lovett M., and Kingsmore S. F. (1996) Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 382, 262–265 10.1038/382262a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Henningsson F., Hergeth S., Cortelius R., Abrink M., and Pejler G. (2006) A role for serglycin proteoglycan in granular retention and processing of mast cell secretory granule components. FEBS J. 273, 4901–4912 10.1111/j.1742-4658.2006.05489.x [DOI] [PubMed] [Google Scholar]
- 60. Higashio H., Satoh Y., and Saino T. (2016) Mast cell degranulation is negatively regulated by the Munc13-4-binding small-guanosine triphosphatase Rab37. Sci. Rep. 6, 22539 10.1038/srep22539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pivot-Pajot C., Varoqueaux F., de Saint Basile G., and Bourgoin S. G. (2008) Munc13-4 regulates granule secretion in human neutrophils. J. Immunol. 180, 6786–6797 10.4049/jimmunol.180.10.6786 [DOI] [PubMed] [Google Scholar]
- 62. Kaeser P. S., and Regehr W. G. (2017) The readily releasable pool of synaptic vesicles. Curr. Opin. Neurobiol. 43, 63–70 10.1016/j.conb.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen Z., Cooper B., Kalla S., Varoqueaux F., and Young S. M. Jr. (2013) The Munc13 proteins differentially regulate readily releasable pool dynamics and calcium-dependent recovery at a central synapse. J. Neurosci. 33, 8336–8351 10.1523/JNEUROSCI.5128-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dernick G., de Toledo G. A., and Lindau M. (2007) in Electrochemical Methods for Neuroscience (Michael A. C., and Borland L. M., eds), pp. 315–336, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 65. Tiwari N., Wang C. C., Brochetta C., Ke G., Vita F., Qi Z., Rivera J., Soranzo M. R., Zabucchi G., Hong W., and Blank U. (2008) VAMP-8 segregates mast cell-preformed mediator exocytosis from cytokine trafficking pathways. Blood 111, 3665–3674 10.1182/blood-2007-07-103309 [DOI] [PubMed] [Google Scholar]
- 66. Yu Y., Huang Z., Mao Z., Zhang Y., Jin M., Chen W., Zhang W., Yu B., Zhang W., and Alaster Lau H. Y. (2016) Go is required for the release of IL-8 and TNF-α, but not degranulation in human mast cells. Eur. J. Pharmacol. 780, 115–121 10.1016/j.ejphar.2016.03.038 [DOI] [PubMed] [Google Scholar]
- 67. Gupta A. A., Leal-Berumen I., Croitoru K., and Marshall J. S. (1996) Rat peritoneal mast cells produce IFN-γ following IL-12 treatment but not in response to IgE-mediated activation. J. Immunol. 157, 2123–2128 [PubMed] [Google Scholar]
- 68. Leal-Berumen I., Conlon P., and Marshall J. S. (1994) IL-6 production by rat peritoneal mast cells is not necessarily preceded by histamine release and can be induced by bacterial lipopolysaccharide. J. Immunol. 152, 5468–5476 [PubMed] [Google Scholar]
- 69. Cairns J. A., and Walls A. F. (1996) Mast cell tryptase is a mitogen for epithelial cells. Stimulation of IL-8 production and intercellular adhesion molecule-1 expression. J. Immunol. 156, 275–283 [PubMed] [Google Scholar]
- 70. Marshall J. S., Leal-Berumen I., Nielsen L., Glibetic M., and Jordana M. (1996) Interleukin (IL)-10 inhibits long-term IL-6 production but not preformed mediator release from rat peritoneal mast cells. J. Clin. Invest. 97, 1122–1128 10.1172/JCI118506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Beil W. J., Login G. R., Aoki M., Lunardi L. O., Morgan E. S., Galli S. J., and Dvorak A. M. (1996) Tumor necrosis factor α immunoreactivity of rat peritoneal mast cell granules decreases during early secretion induced by compound 48/80: an ultrastructural immunogold morphometric analysis. Int. Arch. Allergy Immunol. 109, 383–389 10.1159/000237267 [DOI] [PubMed] [Google Scholar]
- 72. Olszewski M. B., Trzaska D., Knol E. F., Adamczewska V., and Dastych J. (2006) Efficient sorting of TNF-α to rodent mast cell granules is dependent on N-linked glycosylation. Eur. J. Immunol. 36, 997–1008 10.1002/eji.200535323 [DOI] [PubMed] [Google Scholar]
- 73. Olszewski M. B., Groot A. J., Dastych J., and Knol E. F. (2007) TNF trafficking to human mast cell granules: mature chain-dependent endocytosis. J. Immunol. 178, 5701–5709 10.4049/jimmunol.178.9.5701 [DOI] [PubMed] [Google Scholar]
- 74. Dwyer D. F., Barrett N. A., Austen K. F., and Immunological Genome Project Consortium. (2016) Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat. Immunol. 17, 878–887 10.1038/ni.3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grimbaldeston M. A., Chen C. C., Piliponsky A. M., Tsai M., Tam S. Y., and Galli S. J. (2005) Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 167, 835–848 10.1016/S0002-9440(10)62055-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kanaoka Y., and Boyce J. A. (2004) Cysteinyl leukotrienes and their receptors: cellular distribution and function in immune and inflammatory responses. J. Immunol. 173, 1503–1510 10.4049/jimmunol.173.3.1503 [DOI] [PubMed] [Google Scholar]
- 77. Adachi R., Krilis S. A., Nigrovic P. A., Hamilton M. J., Chung K., Thakurdas S. M., Boyce J. A., Anderson P., and Stevens R. L. (2012) Ras guanine nucleotide-releasing protein-4 (RasGRP4) involvement in experimental arthritis and colitis. J. Biol. Chem. 287, 20047–20055 10.1074/jbc.M112.360388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kim K., Petrova Y. M., Scott B. L., Nigam R., Agrawal A., Evans C. M., Azzegagh Z., Gomez A., Rodarte E. M., Olkkonen V. M., Bagirzadeh R., Piccotti L., Ren B., Yoon J. H., McNew J. A., Adachi R., et al. (2012) Munc18b is an essential gene in mice whose expression is limiting for secretion by airway epithelial and mast cells. Biochem. J. 446, 383–394 10.1042/BJ20120057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Brock T. A., Dennis P. A., Griendling K. K., Diehl T. S., and Davies P. F. (1988) GTPγS loading of endothelial cells stimulates phospholipase C and uncouples ATP receptors. Am. J. Physiol. 255, C667–C673 [DOI] [PubMed] [Google Scholar]
- 80. Lindau M., and Neher E. (1988) Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflugers Arch. 411, 137–146 10.1007/BF00582306 [DOI] [PubMed] [Google Scholar]
- 81. Gillis K. D., Mossner R., and Neher E. (1996) Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron 16, 1209–1220 10.1016/S0896-6273(00)80147-6 [DOI] [PubMed] [Google Scholar]
- 82. Royet J. P. (1991) Stereology: a method for analyzing images. Prog. Neurobiol. 37, 433–474 10.1016/0301-0082(91)90009-P [DOI] [PubMed] [Google Scholar]
- 83. Tschanz S. A., Burri P. H., and Weibel E. R. (2011) A simple tool for stereological assessment of digital images: the STEPanizer. J. Microsc. 243, 47–59 10.1111/j.1365-2818.2010.03481.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.