

Abstract
Thrombolites are unlaminated carbonate deposits formed by the metabolic activities of microbial mats and can serve as potential models for understanding the molecular mechanisms underlying the formation of lithifying communities. To assess the metabolic complexity of these ecosystems, high throughput DNA sequencing of a thrombolitic mat metagenome was coupled with phenotypic microarray analysis. Functional protein analysis of the thrombolite community metagenome delineated several of the major metabolic pathways that influence carbonate mineralization including cyanobacterial photosynthesis, sulfate reduction, sulfide oxidation, and aerobic heterotrophy. Spatial profiling of metabolite utilization within the thrombolite-forming microbial mats suggested that the top 5 mm contained a more metabolically diverse and active community than the deeper within the mat. This study provides evidence that despite the lack of mineral layering within the clotted thrombolite structure there is a vertical gradient of metabolic activity within the thrombolitic mat community. This metagenomic profiling also serves as a foundation for examining the active role individual functional groups of microbes play in coordinating metabolisms that lead to mineralization.
Keywords: Thrombolites, Microbial mats, Metagenome, Photosynthesis, Carbonate mineralization, Microbialites
Introduction
Microbialites are carbonate-depositing ecosystems that are the products of multiple microbial metabolisms and complex biogeochemical cycling. Microbialites have a long fossil record dating back billions of years and are generally thought to be one of the oldest known ecosystems on Earth (Grotzinger and Knoll 1999). Microbialites are distinguished by their carbonate macrostructure. Those microbialites with a laminated macrostructure are referred to as stromatolites, whereas microbialites with unlaminated clotted fabrics are known as thrombolites. The laminated stromatolites are the result of iterative microbial growth that accrete through the precipitation of calcium carbonate as well as the trapping and binding of inorganic sediment (Reid et al. 2000; Macintyre et al. 2000; Paerl et al. 2001; Andres et al. 2006). In contrast to the stromatolites, the thrombolites show a high degree of heterogeneity and variability in carbonate precipitation; however, the underlying microbial processes that form these thrombolitic clotted structures are not well understood.
To explore this issue further, the thrombolites of High-borne Cay, an island located in the Exuma Sound, The Bahamas were targeted. At Highborne Cay, both stromatolites and thrombolites actively grow in the subtidal and intertidal zones, respectively. The thrombolites of High-borne Cay form large, unlaminated carbonate structures ranging in size from a few centimeters to several meters wide (Fig. 1a). Overlaying these thrombolitic carbonate structures is a thick (~1 cm) microbial mat (Fig. 1b) comprised of a diverse microbial consortium, which is thought to drive the precipitation of extracellular carbonate through their various metabolic activities (Planavsky et al. 2009; Myshrall et al. 2010; Mobberley et al. 2012). There have been several recent studies that have characterized the taphonomy of the thrombolite carbonate structures (Planavsky and Ginsburg 2009), the surrounding oolitic sand grains (Edgcomb et al. 2013), as well as the biogeochemistry and microbial diversity of the thrombolite-forming microbial mats, from here on referred to as thrombolitic mats (Desnues et al. 2008; Myshrall et al. 2010; Mobberley et al. 2012). These previous analyses have revealed several key functional groups of organisms associated with the mats and include: phototrophs, aerobic heterotrophic bacteria, sulfate-reducing bacteria, sulfide-oxidizing bacteria, and fermentative bacteria. All these guilds have been shown to be critical for carbonate precipitation and/or dissolution in other lithifying microbial mat ecosystems, such as stromatolites (e.g., Dupraz and Visscher 2005; Visscher and Stolz 2005; Baumgartner et al. 2009). Together, these groups of organisms generate steep vertical chemical gradients within the thrombolites with large diel fluctuations. For example, O2 levels throughout the diel cycle can go from undetectable to several-fold supersaturated in the top 1–5 mm of the lithifying mat surface (Myshrall et al. 2010). The dominant organisms responsible for this O2-rich layer are photosynthetic cyanobacteria, which have been shown to be a driving force in the biogeochemical cycling in thrombolites (Myshrall et al. 2010).
Fig. 1.

Modern thrombolites of Highborne Cay, Bahamas. a Thrombolitic build-ups along the intertidal zone. Bar = 30 cm. b Cross section of thrombolitic microbial mat demarking three zones within the mat. Zone 1 comprised the upper 3 mm of the mat, whereas Zone 2 consisted of 3–5 mm beneath the surface of the mat, and Zone 3 contained the lower portion of the mat between 5 and 9 mm. Bar = 1 cm. c Within the upper Zone 1 there is an increase in the relative abundance of the filamentous cyanobacterium Dichothix spp. (arrows) which appear as hot spots of carbonate deposition within the thrombolitic mats. Bar = 0.5 mm
In the thrombolitic mats of Highborne Cay, one of the prominent cyanobacteria that comprises the upper few millimeters of the thrombolitic mats are calcified filamentous Dichothrix spp. (Fig. 1c; Planavsky et al. 2009; Mobberley et al. 2012), which are characterized by their basal heterocysts and tapered apical ends (Planavsky et al. 2009). These bundles of vertically orientated filaments produce copious amounts of exopolymeric substances (EPS) and appear to be “hot spots” for carbonate precipitation in the thrombolitic mats. The carbonate deposition is thought to be due to the elevated rates of photosynthesis, which increases the pH and the carbonate saturation state within the mats and facilitates carbonate precipitation (Dupraz and Visscher 2005).
The production of EPS material has been shown to be a critical factor associate with carbonate precipitation in modern microbialites. The EPS matrix within microbial mats not only serves as a structural support for the growing community (Decho 1990), but it can also chelate cations, such as Ca2+ and function as nucleation sites for calcium carbonate nucleation (Decho 2000; Dupraz and Visscher 2005). The EPS associated with the adjacent stromatolites of Highborne Cay are predominantly generated by cyanobacteria and sulfate-reducing bacteria (Decho 2000; Braissant et al. 2007). The cyanobacterial EPS material contains approximately 50 % carbohydrates, such as mannose, xylose and fucose, whereas the remaining material is a mixture of amino acids, uronic acids, and glycans (Kawaguchi and Decho 2000). Through the heterotrophic degradation of the EPS material in the stromatolites, the Ca2+ binding capacity of the EPS can be altered resulting in the release of Ca2+ ions, which can increase the saturation index of the microenvironment and promote localized areas of carbonate precipitation within the mat community (Dupraz and Visscher 2005).
Despite recent studies regarding the viral and microbial diversity, sedimentology, and biogeochemistry of the Bahamian thrombolitic mats (Desnues et al. 2008; Planavsky and Ginsburg 2009; Planavsky et al. 2009; Myshrall et al. 2010; and Mobberley et al. 2012; Edgcomb et al. 2013) there remains an overall lack of understanding of the molecular mechanisms that underlie and regulate the metabolisms associated with these communities. In this study, we survey the metabolic potential of thrombolitic mats by sequencing the metagenome using massively parallel DNA sequencing. The metagenomic sequencing was coupled with phenotypic microarray analysis, which assessed vertical metabolite utilization patterns throughout the thrombolitic mats, thereby generating a spatial profile of metabolic activities. These two approaches help to increase our understanding of the underlying molecular pathways and overall genomic complexity associated with
Materials and methods
Thrombolitic mat sample collection
Thrombolitic mats were collected from the island of Highborne Cay, The Bahamas (76°49′ W, 24°43′N) in February 2010 from an intertidal thrombolite platform located at Site 5 (Fig. 1a; Andres and Reid 2006). Based on previous studies there are four known types of thrombolitic mats (Myshrall et al. 2010; Mobberley et al. 2012). For this study, we have chosen the ‘button’ thrombolitic mats as they have been shown to be the most abundant and most productive at Highborne Cay (Myshrall et al. 2010). The ‘button’ mats were cut into three 9-mm vertical sections and immediately placed in the RNAlater (Life Technologies, Inc, Grand Island, NY) to preserve the nucleic acids for later metagenomic analysis. Corresponding thrombolitic mat sections for metabolic phenotypic microarray studies were collected and placed into a sterile Nalgene container and transported to the Space Life Sciences Lab at the Kennedy Space Center, FL. The mats were incubated in a walk-in growth chamber maintained at Highborne Cay ambient conditions (24 °C; 2,000 μE/m2/s; 12-h light/dark cycle) for 2 weeks until they were processed for the phenotypic microarray analysis as described below.
DNA extraction and Illumina sequencing
Total community DNA was extracted from each of the three 9-mm RNAlater sections in triplicate using a previously described modified xanthogenate method (Mobberley et al. 2012). The DNA was then pooled and concentrated for sequencing with the Wizard Genomic DNA Purification kit (Promega, Madison, WI) following manufacturers protocol. The pooled thrombolite community DNA (i.e., a total of nine replicate DNA extractions) was independently sequenced in triplicate using an Illumina GAIIx platform generating three distinct libraries of non-overlapping paired-end reads that each represented the entire 9-mm region of the thrombolite mat sections (Table 1). All raw sequencing data was deposited into GenBank NCBI short read archive SRP021141.
Table 1.
Summary of metagenomic sequences and MG-RAST analysis
| Sample | Sequence readsa | rRNA gene readsb (%) | Protein encoding readsc (%) | Annotated proteinsd (%) | Proteins assigned functione (%) |
|---|---|---|---|---|---|
| Thr-A | 34234300 | 38674 (0.1) | 25055197 (73.2) | 7581161 (22.1) | 4638130 (13.6) |
| Thr-B | 34958742 | 39324 (0.1) | 25742370 (73.6) | 7824431 (22.4) | 4791538 (13.7) |
| Thr-C | 34704789 | 39283 (0.1) | 25792847 (74.3) | 7741612 (22.3) | 4782100 (13.8) |
Total number of high quality sequences following preprocessing and de-replication
Number of reads annotated against the SILVA SSU and LSU databases (ver. 104) at 97 % identity with bit scores above 50
Number of reads predicted by FragGeneScan to encode protein
Annotation against the MD5NR database, 60 % identity, 60 bp alignment, E value >10−5
Annotated proteins assigned to either a SEED Subsystem or KEGG ontology functional category
Analysis of thrombolitic mat metagenomes
Each of the three metagenomic libraries (Thr-A, Thr-B, and Thr-C) was analyzed and annotated with the Me-taGenome Rapid Annotation using Subsystem Technology (MG-RAST) version 3.3 pipeline (Meyer et al. 2008) and are publically available under project 3438. Within the MG-RAST pipeline, the sequences in each library were quality screened using the preprocessing program DynamicTrim (Cox et al. 2010) in which the phred score was set to 20 and only five bases or less below this threshold was allowed. Sequences of low quality were trimmed from each database resulting in 11.5, 10.7, and 10.1 % of the raw sequences being removed from the Thr-A, Thr-B, and Thr-C libraries, respectively. In addition, the libraries were screened for artificially replicated sequences as previously described (Gomez-Alvarez et al. 2009) with 1.9, 2.0, and 1.9 % of the sequences from the Thr-A, Thr-B, and Thr-C libraries removed, respectively.
The MG-RAST analysis of the replicate metagenomic libraries consisted of BLAT comparisons of the sequences against the SILVA SSU and LSU databases (version 104) for ribosomal gene reads and the multiple sourced non-redundant M5NR database (version 1) for the protein encoding reads (Wilke et al. 2012). Ribosomal reads were annotated to the two SILVA databases at 97 % identity with bit scores above 50. Protein encoding reads were assigned to SEED subsystems and KEGG pathways using only matches of >60 % similarity and >60 base pairs that had an E value of ≤10−5. To acquire the broadest representation, taxonomic assignment of reads was performed using the MG-RAST Lowest Common Ancestor (LCA) approach with phylogenetic trees using the NCBI taxonomy created in MEGAN (version 4.70.4; Huson et al. 2007). Deeper taxonomic exploration of metabolic genes in SEED subsystems and KEGG pathways were carried out in MEGAN through LCA assignment using default parameters of the ten best NCBI nr database BLASTX hits for reads of interest.
Comparison of thrombolite metagenome to other microbial mat habitats
To explore functional differences between the Bahamian thrombolites and other lithifying and non-lithifying mat ecosystems, comparisons of whole metagenome datasets were carried out in MG-RAST. Comparisons were made using SEED subsystems with the following parameters: minimum E value of 10−5 and minimum identity of 60 %. The following publically available datasets were used in this analysis (citation and MG-RAST IDs given): thrombolitic mat replicates (this study); non-lithifying (Type 1) and lithifying (Type 3) Highborne Cay stromatolitic mats (Khodadad and Foster 2012; 4449590.3, 4449591.3), Cuatro Ciénegas lithifying oncolites (layered spherical microbialites) and thrombolites (Breitbart et al. 2009; 44440060.4, 4440067.3), Cuatro Ciénegas non-lithifying mats (Peimbert et al. 2012; 4442466.3, 4441363.3), Guerrero Negro hypersaline mats (Kunin et al. 2008; 4440964.3–4440972.3), Octopus Springs mat, Yellowstone National Park (Bhaya et al. 2007; 4443749.3), and a Global Ocean Sampling Sargasso Sea water column sample served as an outgroup (Rusch et al. 2007; 4441570.3). Multivariate analysis of the metagenomes functional subsystems was performed within the R statistical program (R Development Core Team 2010) using the random forest (version 4.6-2; Liaw and Weiner 2002) and bpca (version 1.0–10; Faria and Demetrio 2008) as previously described (Dinsdale et al. 2013). To account for differences in sampling depth, the subsystem abundance counts were normalized to the total number of reads that were able to be assigned a subsystem for each metagenome. To predict the most important subsystems (i.e., response variables) for separating out the datasets, a supervised random forest was carried out the normalized subsystem counts for each environmental dataset using 5,000 bootstrap iterations. Principle Component Analysis (PCA) for all metagenomes was performed and the directionality of the six most important subsystems, based on the highest variance, was calculated and overlain as a biplot.
Metabolic profiling with phenotypic microarrays
Following acclimation in the growth chamber, the living thrombolitic mat samples transported from Highborne Cay were further sectioned into three discrete zones that corresponded to known in situ oxygen depth profiles that occur at the peak of photosynthetic activity (Myshrall et al. 2010). The three zones included the upper 0–3 mm (Zone 1), the middle 3–5 mm (Zone 2), and the lower 5–9 mm (Zone 3) region of the mats (Fig. 1b). The metabolic capacity of each of the thrombolitic mat zones was characterized by diluting 250 mg of the sections into 1.7 mL of filter-sterilized artificial seawater. The samples were then homogenized by vortexing for 15 min and centrifuged to remove the larger sand particles. The cell density within the homogenates was examined spectrophotometrically (A590nm; Genesys 20, Thermo Fisher Scientific, Waltham, MA) and normalized to 1 × 106 cells per mL of seawater. Aliquots (100 μl) of the mat slurries were then placed into Phenotypic Microarray (PM) plates that contained either carbon (PM1), nitrogen (PM3B), phosphorus (PM4A) or sulfur (PM4A) substrates (Biolog Inc., Hayward, CA). With the exception of the carbon plates, all other plates were supplemented with 20-mM sodium succinate and 2-μM ferric citrate as described in the manufacturer’s protocol. The inoculated plates were incubated for 24 h at 30°C and then examined every 15 min at A590nm using an Omnilog reader, which measures cellular respiration of each substrate (Biolog, Inc., Hayward, CA). Only those substrates with absorbance readings above the set threshold were considered utilized by the mat community. The utilization threshold was considered to be 20 % of the highest recorded absorbance on the PM plates. All samples were examined in triplicate and the differences between the different thrombolitic mat zones were statistically compared using ANOVA with a significance of P ≤ 0.05. Heat map analyses were carried out Euclidean complete linkage clustering using the pheatmap package (version 0.7.4; Kolde 2013) for the statistical program (R Development Core Team 2010).
Results
Three replicate metagenome libraries representing the entire 9-mm thrombolitic mat profile were sequenced independently resulting in a combined 103,897,831 high quality sequencing reads derived from the thrombolitic mats and were highly reproducible (Table 1; Supplemental Fig. S1). An ANOVA analysis of the three independently sequenced libraries showed no statistical differences between replicates (P ≥ 0.986), and from here on will be described as a single data set. The average guanine–cytosine (GC) content of the thrombolitic mat metagenome was 46 % and most of the recovered reads were protein encoding genes representing between 73 and 74 % of the recovered sequences with less than 0.1 % of the recovered metagenome sequences corresponding to rRNA genes (Table 1). Although most of the reads recovered were protein encoding only 22 % of the reads could be specifically annotated and only 13 % were assigned to a SEED subsystem or KEGG functional category.
Community diversity associated with thrombolitic mat metagenome
Analysis of the annotated protein encoding and ribosomal features revealed that 91.7 % were assigned to the domain Bacteria, whereas 7.4 % were assigned to Eukaryota and only 0.8 % to Archaea (Fig. 2). Within the domain Bacteria a total of 16 phyla were represented in the metagenomic libraries with extensive differences in the distribution of each of phyla (Fig. 2a). The dominant phyla represented in the metagenomic library were the Cyanobacteria, which comprised 32.8 % of the total annotated sequences. These results differed from previous clone and barcoded 16S rRNA gene libraries where the Proteobacteria were the dominant recovered taxa (Myshrall et al. 2010; Mobberley et al. 2012). In the metagenomic libraries, Proteobacteria comprised 21.7 % of the total number of bacterial sequences, while Bacteroidetes (10.4 %) and Planctomycetes (1.1 %) were also abundant. Five additional phyla were detected in the metagenomic libraries not previously identified through a barcoded 16S rRNA gene-pyrosequencing approach (Mobberley et al. 2012), although they comprised less than 1 % of the total sequencing reads. These phyla included the Chlamydiae, Chlorobi, Syngeristetes, Tenericutes, and Nitrospirae.
Fig. 2.

Taxonomic distribution of the thrombolite metagenome based on MG-RAST Lowest Common Ancestor analysis. a Overview of reads assigned to Bacterial phyla (>100 reads) with Proteobacteria assigned to order level. b Higher resolution of Archaea reads. c Phyla level (>10 reads) resolution of Eukaryota reads. All trees were created in MEGAN using MG-RAST annotation server read abundance data for each respective taxonomic level is given in parentheses
Although the Cyanobacteria were the dominant bacterial taxa, 70 % of the reads assigned to the Cyanobacteria were unable to be designated to a taxon beyond the phylum level (Fig. 3). Those sequences that could be further classified 47.6 % were assigned to the order Chroococcales, with 39.4 % of the classifiable Chroococcales sequences associating with the genus Cyanothece. The Nostocales were also prevalent, comprising 43.8 % of the cyanobacterial sequences, with most sharing similarity to the genes typically associated with the genera Nostoc and Anabena (Fig. 3). The Oscillatoriales were also present (7.7 %) although 95 % of the sequences associated with this order could not be assigned beyond the order level.
Fig. 3.
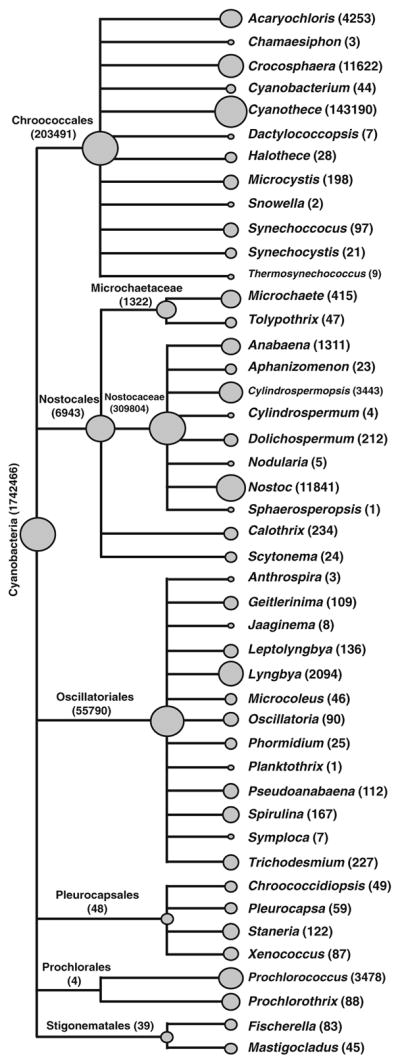
Genus level distribution of cyanobacteria reads from the thrombolite metagenome based on MG-RAST Lowest Common Ancestor analysis. Tree created in MEGAN using MG-RAST annotation server read abundance data for each respective taxonomic level is given in parentheses
In the archaeal population, most of the recovered sequences were assigned to the phyla Thaumarchaeota (92.1 %) with most assigned to the ammonia-oxidizing genus Nitrosopumilus (63.5 %; Fig. 2b). Metagenomic sequencing also revealed sequences associated with methanogenic archaea, which had not been previously detected with 16S rRNA gene analysis (Mobberley et al. 2012). Specifically, sequences similar to the Euryarchaeota families Methanosarcinaceae and Methanobacteriaceae were recovered (Fig. 2b). Other archaea phyla, such as Crenarchaeota, comprised <0.2 % of the archaeal taxa associated with the thrombolitic mat metagenome.
The recovered reads assigned to Eukaryota taxa were diverse containing more than ten superphyla and phyla within the thrombolitic mat metagenome (Fig. 2c). Of those Eukaryota sequences that could be classified, Metazoans accounted for 9.6 % of the total eukaryotic reads with the majority of the reads associated with Arthropoda (6.8 %), Chordata (1.2 %), Annelida (0.8 %) and Nematoda (0.4 %). Bacillariophyta, or diatoms, were also prevalent within eukaryotic population comprising 8.6 % of the protein encoding and ribosomal genes. Other prevalent eukaryotic taxa included the Viridiplantae, specifically the Streptophyta (4.7 %), Ascomycota (3.4 %), Alveolata (3.0 %), Rhodophyta (2.1 %), and the Foraminifera (0.5 %).
Overview of the functional genes of the thrombolitic mat metagenome
The protein encoding genes within the thrombolitic mat metagenome were compared to the M5NR database using the MG-RAST platform and the KEGG database using BLAT. Approximately 73.7 % of the recovered sequences were identified as protein encoding genes, with 26.2 % of the sequences unable to be characterized (Table 1). Subsystem-level analysis of the thrombolitic mat metagenome indicated that most of the protein encoding genes were of unknown function (75.5 %); however, those sequences that were annotated were assigned to 28 different SEED subsystems (Fig. 4a). The two subsystems with the highest relative abundance of annotated sequencing reads were Protein Metabolism and RNA Metabolism subsystems representing a wide range of housekeeping genes involved in the thrombolitic mats (Fig. 4a). Another subsystem with a high relative abundance of sequencing reads was the Carbohydrate Metabolism subsystem, which contained numerous genes associated with central carbon metabolism and carbon dioxide uptake. There was also a high representation of genes associated with mono-, di- and oligosaccharide metabolism and biosynthesis, specifically xylose, mannose, fructose, and trehalose (data not shown). Screening the metagenome for genes associated with various energy metabolisms showed a diverse metabolic potential within the thrombolitic mats (Fig. 4b). Genes associated with oxidative phosphorylation and photosynthesis were abundant in annotated metagenome as were hydrogenases and genes associated with fermentation, methane metabolism, sulfur oxidation, and reduction (Fig. 4b).
Fig. 4.

MG-RAST Functional assignment of thrombolite metagenome protein features. a Overview of SEED subsystems (Level 1). b SEED subsystems (Level 3) associated with energy metabolism. Protein feature abundances are given in parentheses
A similar trend was observed when the mat metagenome was compared to the KEGG database. There was an increase in the relative abundance of genes associated with the functional categories Carbohydrate and Energy Metabolism (Table 2). Specifically, in the Carbohydrate Metabolism genes associated with the core carbon metabolisms, such as glycolysis and gluconeogenesis, the citric acid cycle, and pentose phosphate pathway were enriched. The dominant taxa associated with these pathways were primarily Cyanobacteria, with most reads sharing similarity to the orders Chroococcales and Nostocales with bacterial orders Rhodobacterales, Rhizobiales, and Planctomycetales also represented (Table 2). Interestingly, there were also several high frequency matches to order Nitrosopumilales of the phylum Thaumarchaeota in the functional categories associated with core carbon metabolisms and in fructose and mannose metabolisms. The majority of the gene reads associated with glyoxylate and dicarboxylate metabolism were similar to the Ribulose-1,5-bisphosphate carboxylase oxygenase (RuBisCO, Form III) genes from the methanogenic Methanosarcinales.
Table 2.
Relative abundance of dominant metabolic pathways in thrombolitic mat metagenome
| Functional categorya | KEGG ID | Protein or gene name with high relative abundancebc | Total Reads | Dominant taxa (No. Reads)d |
|---|---|---|---|---|
| Carbohydrate metabolism | 1,110 | |||
| Glycolysis, gluconeogenesis | 10 | ALDO, fbaAB, GAPDHS, gap2, cel, porAB (aceE, pfkA, pckA) | 19,212 |
Chroococcales (503) Rhodobacterales (320) Nitrosopumilales (288) Nostocales (175) Planctomycetales (136) Bacillariophyta (73) |
| Citric acid (TCA cycle) | 20 | acnB, fumAB, sdhA, SDH1, frdA (gltA, sucC, korA) | 14,449 |
Chroococcales (533) Nostocales (166) Rhodobacterales (163) Planctomycetales (149) Nitrosopumilales (83) |
| Pentose phosphate pathway | 30 | Phosphoketolase, pgl, kguK (UGDH, rbsK) | 644 |
Nostocales (602) Rhizobiales (11) |
| Glyoxylate, dicarboxylate metabolism | 630 | Formamidase, purU, rbcSL, (phbB, glcD, ttdB) | 1,871 |
Methanosarcinales (180) Chroococcales (178) Rhizobiales (57) |
| Fructose, mannose metabolism | 51 | manB, rhaB, GMPP, fucK, gmd, fcl (rhaA, gutB) | 3,939 |
Chroococcales (553) Nostocales (324) Chloroflexales (30) Nitrosopumilales (18) |
| Energy metabolism | 1,120 | |||
| Oxidative phosphorylation | 190 | ATPF1, ATPeF1, ATPV, ccoN, coxC, ndhACEJL (MQCRB, hoxF) | 23,872 |
Chroococcales (3134) Nostocales (1276) Planctomycetales (339) Rhodobacterales (266) Archaea (101) Viridiplante (12) |
| Photosynthesis | 195 | psaABEX, psbABCDF28, petACEF | 20,942 |
Chroococcales (2411) Nostocales (908) Oscillatoriales (541) Chlorophyta (97) Bacillariophyta (74) |
| Methane metabolism | 680 | fbaA, frhA, fwbB/fmdB, mtd, mtrF, hdr (coxSL, fwdDCFH) | 5,410 |
Chroococcales (235) Nostocales (75) Bacillariophyta (46) Rhodobacterales (6) Archaea (6) |
| Nitrogen metabolism | 910 | nifHDNV (napACE, narVW, nosZ, norDF, nirAB, narB, amoC) | 1,743 |
Chloroflexales (60) Chroococcales (46) Nostocales (31) Chlorobiales (18) |
| Sulfur metabolism | 920 | met3, aprAB, cysC, cysD (cysQHJ, dsrB, soxBYZ) | 6212 |
Chroococcales (411) Rhodobacterales (260) Nitrosopumilales (61) Desulfobacterales (37) Desulfovibrionales (11) |
Categories based on KEGG classification system
Genes that were highly represented within the thrombolitic mat metagenomes
Genes similarities found from 10−3 to 10−5 in parentheses
Reads assigned based on LCA algorithm in MEGAN
Within the Energy Metabolism category there were five dominant pathways including oxidative phosphorylation, photosynthesis, methane, nitrogen, and sulfur metabolism (Table 2). The dominant taxa associated with each metabolism varied; however, the Cyanobacteria, specifically the order Chroococcales, had the highest number of matches in each category. These included the majority of the reads for genes encoding ATP synthase complexes, cytochromes, and Photosystems I and II, although there were smaller contributions from other bacterial phyla as well as diatoms, plants, and cyanophage (psbA). In methane metabolism, genes associated with the oxidation of methane (e.g., fbaA, metF) had a high frequency of matches to the Chroococcales, specifically there was a similarity to the genus Cyanothece sp. Surprisingly, most of the hits for coenzyme F420 hydrogenase (frhA) were more similar to [NiFe]-hydrogenase homologs found in cyanobacteria instead of methanogens. No methyl-coenzyme reductase genes were detected at the cut-offs of this analysis; however, there was a low abundance of archaeal genes associated with methanogenesis, such as tetrahydromethanopterin S-methyltransferase (mtr) and formylmethanofuran dehydrogenase (fwd) found in the metagenome (Table 2). Soluble methane monooxygenase genes were also recovered suggesting that methane oxidation could be occurring, although it should be noted that these genes shared a high sequence similarity to ammonia monooxygenase genes.
In nitrogen metabolism, the vast majority of nitrogen fixation genes were associated with Cyanobacteria (i.e., Chroococcales and Nostocales) and Chloroflexales. With regard to other nitrogen metabolisms, genes involved in assimilatory nitrate reduction were predominately associated with Cyanobacteria, while dissimilatory reduction genes were found to be similar to those in Alphaproteo-bacteria, Gammaproteobacteria, and Planctomycetes. Denitrification genes (e.g., nor) were associated with Flavobacteriales, Rhodobacterales, and Nitrosopumilales. There were very few nitrification genes detected within the thrombolite metagenomes, those that were recovered appeared similar to ammonia monooxygenase genes from the archaea Nitrosopumilus maritimus.
In sulfur metabolism, most of the recovered genes were associated with assimilatory sulfate reduction, such as sulfate adenylyltransferases, and shared similarity to Chroococcales (met3, cysC), Rhodobacterales (met3, cysD), and Nitrosopumilales (cysD). Although pathways associated with assimilatory sulfate reduction had the highest relative abundance in the thrombolitic mat metagenome, genes associated dissimilatory sulfate reduction pathways (e.g., dsrB, aprAB) were also detected and were associated with the Deltaproteobacteria, specifically the orders Desulfobacterales and Desulfovibrionales. Thrombolitic mat metagenome reads associated with sulfur oxidation were most similar to the sox genes found in Rhodobacterales and Rhizobiales. In addition, there were reads similar to genes from Rhodobacterales, Cytophagales, and Chroococcales that were involved in the transport and utilization of organic sulfur compounds including sulfonates and DMSP.
Comparison of thrombolite metagenome to other functional metagenomes
The three replicate thrombolite metagenomes of this study were compared to nine publically available metagenomes derived from both lithifying and non-lithifying microbial mat habitats using MG-RAST (Fig. 5). These environments included: the non-lithifying hypersaline mats of Guerrero Negro, Mexico; nonlithifying thermophilic mats of Octopus Spring, Yellowstone National Park; non-lithifying and lithifying freshwater mats of Cuatros Ciénegas, Mexico; and the marine stromatolites of Highborne Cay, The Bahamas. A PCA plot of the functional metagenomes is visualized in Fig. 5a. Overlaying the PCA plot of the different metagenomes were the six SEED subsystems that appeared to drive several of the differences between the communities. The results indicated that the Bahamian thrombolitic mats were distinct from the other nine microbial mat metagenomes with the analysis explaining 92.85 % of the variance. Protein encoding genes associated with the subsystem Respiration appeared to be the most important subsystem driving the differences between Highborne Cay thrombolites and the other metagenomes. Additionally, an independent multivariate analysis of the 12 metagenomes was conducted using the random forest package in R (Fig. 5b; Liaw and Weiner 2002; Dinsdale et al. 2013). These results also indicated that genes associated with Respiration were the most important variable that distinguished the thrombolite metagenomes from other lithifying and non-lithifying mat communities. Genes associated with subsystems such as Photosynthesis, Nitrogen Metabolism, Cofactors and Pigments all appear to be conserved between the 12 metagenomes (Fig. 5b).
Fig. 5.

Comparison of thrombolite metagenome with metagenomes of previously sequenced lithifying and non-lithifying microbial mat ecosystems. a Principle Component Analysis of SEED subsystems (Level 1) derived from several distinct habitats. These habitats include the hypersaline non-lithifying mats of Guerrero Negro, Mexico; thermophilic microbial mats of Octopus Spring, Yellow Stone National Park (YNP); freshwater microbial mats of Los Venados and Pozas Azules found in Cuatros Ciénegas Basin, Mexico (CCB); lithifying freshwater oncolites and thrombolites of Rio Mesquites and Pozas Azules in CCB; non-lithifying (Type 1) and lithifying (Type 3) stromatolites of Highborne Cay, The Bahamas and the unlaminated thrombolites of this study. A metagenome from the Sargasso Sea was used as an outgroup. The first two principle components represent 92.85 % of the variation between samples. The red biplot lines represent the directionality of the six most important subsystems driving differences between the metagenomes included in the analysis. b SEED subsystem (i.e., variable) importance determined by random forest analysis of lithifying and non-lithifying microbial mat metagenomes. Importance is reported as mean decrease in accuracy
Substrate utilization patterns within thrombolitic microbial mats
A spatial profile of the metabolic activity within thrombolitic mats was generated using phenotypic microarray analysis, which assesses the ability of the mat communities to grow on a wide range of carbon (C), nitrogen (N), phosphorus (P), and sulfur (S) substrates. Live mat samples were sectioned into three discrete zones ranging from 0–3 mm (Zone 1), l 3–5 mm (Zone 2), and 5–9 mm (Zone 3; Fig. 1b) and homogenized into slurries that were normalized for cell density. An overview of the substrate utilization patterns for each of the zones within the thrombolitic mat is visualized in Fig. 6. A full list of the substrates, their usage in the three mat zones and significance values are listed in supplemental Tables S1, S2, S3, and S4. The upper 3 mm of the thrombolitic mat utilized the greatest number of tested C, N, P, S substrates (n = 149) compared to the 3–5 mm (n = 131) and 5–9 mm (n = 60) zones under our experimental conditions. However, when each substrate category was examined individually there was a pronounced difference in the extent of the substrate utilization within the different zones.
Fig. 6.

Clustered heat map visualizing the substrate utilization patterns throughout the spatial profile of the thrombolitic mats using phenotypic microarrays at 24 h. Dendograms generated based on Euclidean complete linkage clustering. Color scale reflects the intensity of usage with Biolog absorbance units ranging from 0 to 250. White boxes indicate that absorbance readings were below the threshold level. a Carbon (C) and nitrogen (N) substrate utilization within the three zones of the thrombolitic mats. b Phosphate (P) and Sulfur (S) substrate utilization within the three zones of the thrombolitic mat
Although only 31 % of the carbon substrates were metabolized by the thrombolite mat slurries most were utilized in Zones 1 (n = 21) and 2 (n = 24; Fig. 6a). Zone 2 had the highest number of exclusive carbon sources (n = 9) with most containing an amine group, while Zone 1 (n = 4) exclusively used D-fructose-6-phosphate and a subset of carboxylic acids. Several of the substrates were utilized at significantly different levels between the zones. For example, the substrates adenosine and D-mannose were utilized at a significantly higher level in Zones 1 and 2, whereas Zone 3 utilized alpha-ketoglutaric acid at a higher level than the other two zones (Fig. 6a; Table S1). The C sources metabolized by all three zones were mostly substrates and intermediates in energy generating processes (e.g., TCA cycle), such as pyruvic acid and citric acid (Fig. 6a; Table S1). Of the nitrogen substrates, more than half (53 %) of them were utilized as sole nitrogen sources by the thrombolitic mat zones (Fig. 6a; Table S2). Zone 1 metabolized the most N substrates (51 %), and had high levels of utilization of cytidine, alanine and glycine dipeptides, and most amino acids compared to Zones 2 and 3. Although Zone 2 utilized the least number of substrates, it used adenosine at a higher rate than any of the other zones. Complete linkage cluster analysis of total C and N substrate usage shows that the overall mat metabolism for Zone 1 is distinct from those found in Zones 2 and 3 (Fig. 6a).
The thrombolitic mat slurries metabolized P (94 %) and S (71 %) substrates at an overall higher rate compared to the C (31 %) and N (53 %; Fig. 6b). For P substrate utilization, Zones 1 and 2 had the highest utilization rate (94 %), while Zone 3 slurries were only able to metabolize nine P substrates by 24 h (Table S3). The P usage pattern for Zones 1 and 2 were similar and exhibited high rates of utilization for adenosine-containing nucleotides and thiophosphates. All three thrombolitic mat zones metabolized cyclic nucleotides, pyrophosphate, and glycerol phosphate. As with P substrate utilization, S metabolism patterns in Zones 1 and 2 were similar and included high rates of growth on L-djenkolic acid and L-cysteine (Fig. 6b; Table S4). In addition, these two thrombolitic mat zones were able to use inorganic sulfate, thiosulfate, and thiophosphates. Interestingly, the organisms in Zone 3 were able to metabolize two sulfone-containing substrates as their sole sulfur source with L-methionine sulfone being exclusively used. Since Zone 3 was not metabolizing most P and S substrates at 24 h, we examined utilization at 48 h and found that more of the P and S substrates (all on PM4 plates) were used suggesting delayed utilization under the experimental testing conditions (data not shown). Cluster analysis revealed that overall P and S utilization was most similar between Zones 1 and 2 with little or slower utilization rates by the organisms in Zone 3.
Discussion
This study represents the first metagenomic survey of the functional gene complexity associated with Bahamian thrombolites and provides a spatial profile of the substrate utilization patterns within the thrombolitic mat community. The results of this study suggest that (1) cyanobacterial molecular pathways dominate the thrombolite metagenome; (2) all microbial energy metabolisms that have been proposed to promote carbonate precipitation are represented within the metagenome; (3) archaea, although low in diversity and relative abundance, contributes to metabolic cycling within the mats; and (4) discrete gradients of metabolite utilization occur within the depth profile of the thrombolitic mat.
Cyanobacterial molecular pathways dominate the thrombolite metagenome
Cyanobacteria have long been known to dominate microbialite forming communities (e.g., Reid et al. 2000; Breitbart et al. 2009; Myshrall et al. 2010; Santos et al. 2010; Foster and Green 2011; Khodadad and Foster 2012). The metagenomic sequencing of the thrombolitic mats reinforces this concept and shows that the majority of the recovered reads from the Bahamian thrombolitic mats were derived from cyanobacteria. The thrombolite metagenome was enriched in genes with sequence similarity to nitrogen fixing organisms such as Cyanothece, Nostoc, and Anabaena, whereas common microbialite mat builders such as Schizothrix sp. and Dicho-thrix spp. were not well represented. The metagenome results differed from previous 16S rRNA gene amplicon studies where Oscillatoriales and Pleurocapsales were highly prevalent in the thrombolites (Myshrall et al. 2010; Mobberley et al. 2012). This discrepancy between gene-centered surveys and whole metagenomic sequencing may result from biases in ribosomal primer design, variation in copies of the 16S rRNA genes between organisms, as well as database differences and limitations (Liu et al. 2007; Steven et al. 2012). In general, most complete cyanobacterial genomes found in genomic databases, including those used by MG-RAST, are from ubiquitous aquatic ecotypes such as Nostoc spp. and Anabaena spp., whereas trapping and binding filamentous Oscillatoriales and endolithic Pleurocapsales typically associated with microbialite communities are significantly under-represented. Despite these caveats regarding the assignment of specific genes and pathways to individual taxa the metagenome does provide important insight into the specific metabolisms associated with the cyanobacterial community.
It has been previously established, based on biogeochemical profiling and stable isotope analyses, that cyanobacteria play a major role in energy generation and carbonate mineralization in Highborne Cay thrombolitic mats (Planavsky et al. 2009; Myshrall et al. 2010). These findings are supported by the recovered metagenome results with most of the cyanobacterial genes being similar to the well-characterized Photosystems I and II predominately from the orders Chroococcales and Nostocales. Besides converting light energy into usable biological energy, the thrombolitic mat cyanobacteria are involved in processing of fixed organic carbon through a variety of anabolic (e.g., gluconeogenesis, sugar biosynthesis) and catabolic (e.g., glycolysis, oxidative pentose phosphate, fermentation) pathways to create and consume complex carbohydrates. For example, the presence of many genes involved in the conversion of mannose and fructose to molecules such as rhamnose, alginate, xylose, and trehalose indicates that a portion of thrombolitic mat organic carbon is utilized in the formation of the EPS matrix of the thrombolitic mats. The EPS matrix is integral to biologically influence mineralization via the binding of calcium cations (Kawaguchi and Decho 2000; Dupraz et al. 2009).
In addition to energy metabolism and carbohydrate synthesis, cyanobacterial genes associated with nitrogen fixation were abundant in the metagenome. Nitrogen is often a limiting nutrient in marine microbialite forming systems (Pinckney et al. 1995) and biological nitrogen fixation may represent an important metabolism for the growth and maintenance of the thrombolitic mats. Numerous cyanobacterial nitrogenases (e.g., nifDH) and associated genes were recovered from the thrombolitic mats, as were other diazotrophic organisms (Table 2). Nitrogenase activity has been measured in the adjacent stromatolites of Highborne Cay and has been shown to peak at night, with the activity strongly influenced by photosynthesis and the availability of reduced organic carbon (Steppe et al. 2001). The thrombolites, however, are dominated by heterocystous-forming cyanobacteria (Mobberley et al. 2012), which are not readily found in the adjacent stromatolites. These population differences in the thrombolitic mats may result in different temporal and spatial patterns of nitrogen fixation compared to the stromatolites and needs to be explored further. Nitrogen fixation is also of interest as it is closely associated with the evolution of H2. Genes encoding other H2-producing enzymes, such as [NiFe]-hydrogenases were prevalent in the thrombolite metagenome. Overall genes encoding hydrogenases, including both uptake (e.g., hya, hyb) and bidirectional hydrogenases (hox operon) comprised 5 % of the Energy Metabolism subsystem. Although in most mat ecosystems, the microbes are quite effective in sequestering the evolved H2 (Peters et al. 2013), the rates of H2 production in these lithifying thrombolitic mats are unknown and needs to be examined. The novelty of many of the cyanobacteria within the thrombolitic mats and their various hydrogenases may help to improve the understanding of H2 evolution in lithifying ecosystems, and this system has the potential to be exploited for renewable energy carrier production (Bothe et al. 2010; Peters et al. 2013).
Energy metabolisms that influence carbonate precipitation in lithifying microbial mats
The thrombolitic mat metagenome contained genes and molecular pathways associated with all the proposed functional guilds associated with the precipitation and dissolution of carbonate including phototrophs, sulfate-reducing bacteria, sulfide-oxidizing bacteria, aerobic heterotrophs, and fermentative bacteria (Visscher and Stolz 2005; Dupraz et al. 2009). As discussed above, cyanobacteria were the major oxygenic phototrophs detected within the thrombolitic mats; however, eukaryotic algae (i.e., Streptophyta; Chlorophyta) and diatoms (i.e., Bacillariophyta) were also present at low levels. Genes associated with anoxygenic photosynthetic photopigments, such as bacteriochlorophylls and proteorhodopsins were also recovered from the metagenome suggesting a diverse phototrophic community involved in thrombolitic mat photosynthesis. However, comparison to other microbial mat metagenomes indicated that genes associated with photosynthesis and pigment synthesis are highly conserved between both lithifying and non-lithifying metagenomes, whereas genes associated with the subsystem respiration was an important variable in distinguishing the Bahamian thrombolites from other known microbial mat metagenomes (Fig. 5).
In microbialite forming mats the role of anaerobic respiration, specifically sulfate reduction, in promoting carbonate precipitation through remineralization has been well documented (Visscher et al. 1998, 2000; Paerl et al. 2001; Baumgartner et al. 2006; Nitti et al. 2012; Gallagher et al. 2013). Amplicon libraries of the 16S rRNA gene amplicon studies have previously shown the presence of several sulfate-reducing bacteria (SRB) mostly associated with the Desulfovibrionales in the thrombolitic mats (Mobberley et al. 2012; Myshrall et al. 2010, Planavsky et al. 2009). The metagenomic survey of the top 9 mm of the thrombolitic mats, however, showed that the relative abundance of dissimilatory sulfate reduction genes was low compared to metagenomic studies of other microbialite forming communities (Breitbart et al. 2009; Khodadad and Foster 2012). Additionally, most of the recovered genes in thrombolitic mats were similar to Desulfobacterales, although a few genes associated with Desulfovibrionales were detected (Table 2). The low relative abundance may suggest that the sulfate-reducing metabolisms are located deeper in the thrombolitic mat communities compared to the adjacent stromatolites. Future assessment of SRB activity through biogeochemical profiling (e.g., HS−) and dissimilatory sulfate reduction gene expression will be required to fully assess this important metabolism within the thrombolitic mat communities.
The thrombolitic metagenome also contained many bacterial organic substrate degradation pathways that have also been recovered from other microbialite forming systems (Breitbart et al. 2009; Khodadad and Foster 2012) and were primarily derived from the phyla Proteobacteria, Bacteroidetes, and Planctomycetes. Additionally, genes were recovered from lactate and mixed acid fermentation pathways from a variety of bacteria with most reads sharing similarity to cyanobacteria. Although not well documented in microbialite forming mats, cyanobacterial fermentation under anoxic conditions in non-lithifying mats leads to the production of organic acids, H2, and CO2 (van der Meer et al. 2007; Burow et al. 2013). Together these heterotrophic metabolisms represented in the metagenome may play an important role in organic matter cycling within the thrombolitic mat. For example, the degradation and alteration of cyanobacterial EPS by heterotrophic metabolisms can lead to the release of calcium cations from the polymers, which can subsequently be used for carbonate precipitation (Decho 2000).
Contribution of archaea in thrombolitic mat metabolic cycling
Microbial diversity studies have detected the presence of archaea in several lithifying mats ecosystems (Burns et al. 2004; Goh et al. 2009; Couradeau et al. 2011; Arp et al. 2012; Khodadad and Foster 2012; Mobberley et al. 2012), but little work has been done investigating the contribution of these organisms to microbialite physiology. Although archaea comprised only a small portion of the thrombolitic mat metagenome, the recovery of reads in major carbohydrate and energy KEGG functional categories (Table 2) suggests that these organisms are not insignificant contributors to thrombolitic mat metabolic cycling. The overabundance of thrombolitic metagenomic reads similar to the ammonia-oxidizing chemolithoautotroph Nitrosopumilus maritimus is different from other metagenomic analyses of microbialite forming ecosystems, such as the adjacent stromatolites of Highborne Cay (Khodadad and Foster 2012) and freshwater microbialites of Cuatros Ciénegas, Mexico (Breitbart et al. 2009). In these other microbialite forming systems most of the recovered archaeal protein encoding genes were assigned to the Euryarchaeota. Genomic sequencing of cultured isolates of Nitrosopumilus maritimus has identified several metabolic pathways for ammonia oxidation and carbon fixation through a modified hydroxypropionate–hydroxybutylate cycle and an incomplete TCA cycle (Walker et al. 2010), both of which are detected in the thrombolitic mat metagenome. Additionally, the presence of Nitrosopumilales-like genes in glycolysis/gluconeogenesis and sugar metabolisms indicates that archaeal carbohydrate processing may occur in the thrombolitic mats. These results suggest that Nitrosopumilus maritimus-like archaea may be significant contributors to nitrification and carbon cycling in the thrombolitic mats and may potentially influence carbonate precipitation.
The metagenomic results also suggest that archaea may also be contributing the thrombolitic mat carbon cycling through methanogenesis. To date, the role of methanogenesis in modern marine microbialite formation is unclear as methanogenic ecotypes have been recovered from only a few hypersaline stromatolites, such as Shark Bay and the Kiritimati Atoll (Burns et al. 2004; Leuko et al. 2007; Goh et al. 2009; Arp et al. 2012). Methanogenic ecotypes have not been previously identified in the Bahamian thrombolitic mats based on 16S rRNA gene analyses (Myshrall et al. 2010; Mobberley et al. 2012), nor have they been identified in the metagenome of the adjacent stromatolites (Khodadad and Foster 2012). Analyses of the thrombolitic mat metagenome in this study did not recover any methyl-coenzyme reductase genes; however, genes encoding accessory enzymes that are typically associated with methanogenesis, such as tetrahydromethanopterin S-methyltransferase (mtr) were present and appear to be derived from the Methanosarcinaceae. Members of this family undergo methanogenesis with CO2, acetate, and methylated one-carbon compounds (Feist et al. 2006), suggesting that methanogenesis may be occurring in Bahamian thrombolitic mats; however, its role in carbonate mineralization, if any, needs to be explored further.
Gradients of metabolic potential occur within the depth profile of the thrombolitic mat
The phenotypic microarrays measured the respiration of carbon, nitrogen, phosphorous, and sulfate substrates within the thrombolitic mats thereby serving as proxies of heterotrophic metabolism. The overall substrate utilization patterns in the upper 5 mm of the thrombolitic mat, as represented by Zones 1 and 2, contained the most active communities. This result combined with the wide range of heterotrophic respiratory and degradation pathways found in the metagenome (Fig. 4; Table 2), suggests that the top two zones of the thrombolitic mat support a more robust and adaptable community of heterotrophic metabolisms. Although it is likely that the full metabolic capability of the lower zone of the mat was not captured, as it typically remains anoxic throughout the day (Myshrall et al. 2010), this approach did detect differential patterns of substrate utilization that could be linked to the metagenome.
The differential usage of carboxylic acids and saccharides by the three discrete zones was correlated to the diverse array of carbohydrate metabolisms observed in the metagenome, such as decarboxylases, and sugar transporters and processing pathways. For example, Zone 1 of the thrombolite mats used a broader range of carboxylic acids than other zones of the thrombolitic mats suggesting that the microbes in this upper portion of the mat more readily processed organic acids. Preferential degradation of acidic residues within the thrombolitic mat EPS could liberate Ca2+ cations that could serve as nucleation sites for carbonate precipitation (Decho 2000). The Zone 2 microarray results indicated that these microbial communities utilized a wide range of sugars particularly those with amine groups, which also suggests a diverse, but different range of EPS degradation capabilities in this zone of the thrombolitic mat. Together, these examples indicate discreet spatial differences in carbohydrate metabolism by the microbial communities within the thrombolitic mats, suggesting differences in the potential for carbonate precipitation throughout the vertical profile of the thrombolitic mat.
With regard to N substrate utilization, distinct differences were also observed throughout the vertical profile of the mats. The varied uptake and assimilation of different nitrogen sources between the thrombolitic mat zones suggested the potential partitioning of nitrogen metabolisms. As with carbohydrate metabolisms, the greater utilization of dipeptides and amino acids by the heterotrophic community in Zone 1 could also promote carbonate precipitation through alteration of the EPS chemistry during degradation (Dupraz et al. 2009). In contrast to the C and N patterns, the S and P utilization were comparable in all three zones of the thrombolitic mats, although in Zone 3 the microbial communities required an additional 48 h for utilization of many of the various substrates (data not shown). This delay likely reflects the difficulty these anaerobic communities may have had growing under the aerobic experimental conditions.
Taken together, the results of the phenotypic micro-arrays suggest the presence of distinct vertical gradients of metabolic activity within the clotted, unlaminated thrombolitic mats. Similar trends have also been reported in the freshwater microbialites of Cuatros Ciénegas, Mexico (Nitti et al. 2012). Results from these studies, which used a combined genomic, lipid and stable isotope analyses, also showed distinct spatial differences within the lithifying communities although no lithified layers were present (Nitti et al. 2012) reinforcing the idea that the microbial communities can form discrete metabolic zones within unlaminated microbialite forming communities. Although the metagenomic survey correlated many of the specific pathways associated with the use of these different substrates, future work is required to delineate the specific expression of these molecular pathways to assess how the various metabolic activities are coordinated and regulated throughout the vertical profile of these lithifying microbial communities.
Supplementary Material
Acknowledgments
The authors would like to thank Louis Sherman and Pramod Wangikar for organizing the first Indo-US workshop on Cyanobacteria: Molecular Networks to Biofuels, which led to this manuscript. The research was supported by the NASA: Exobiology and Evolutionary Biology Program Element (NNX12AD64G) awarded to Jamie S. Foster. Jennifer M. Mobberley was supported by a NASA Graduate Student Research Program fellowship (NNX10AO18H).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11120-013-9890-6) contains supplementary material, which is available to authorized users.
References
- Andres MS, Reid RP. Growth morphologies of modern marine stromatolites: a case study from Highborne Cay, Bahamas. Sediment Geol. 2006;185(3–4):319–328. [Google Scholar]
- Andres M, Sumner D, Reid RP, Swart PK. Isotopic fingerprints of microbial respiration in aragonite from Bahamian stromatolites. Geology. 2006;34(11):973–976. [Google Scholar]
- Arp G, Helms G, Kalinska K, Schumann G, Reimer A, Reitner J, Trichet J. Photosynthesis versus exopolymer degradation in the formation of microbialites on the atoll of Kiritimati, Republic of Kiribati, Central Pacific. Geomicrobiol J. 2012;29(1):29–65. [Google Scholar]
- Baumgartner LK, Reid RP, Dupraz C, Decho AW, Buckley DH, Spear JR, Przekop KM, Visscher PT. Sulfate reducing bacteria in microbial mats: changing paradigms, new discoveries. Sediment Geol. 2006;185:131–145. [Google Scholar]
- Baumgartner LK, Spear JR, Buckley DH, Pace NR, Reid RP, Visscher PT. Microbial diversity in modern marine stromatolites, Highborne Cay, Bahamas. Environ Microbiol. 2009;11(10):2710–2719. doi: 10.1111/j.1462-2920.2009.01998.x. [DOI] [PubMed] [Google Scholar]
- Bhaya D, Grossman AR, Steunou AS, Khuri N, Cohan FM, Hamamura N, Melendrez MC, Bateson MM, Ward DM, Heidelberg JF. Population level functional diversity in a microbial community revealed by comparative genomic and metagenomic analyses. ISME J. 2007;1(8):703–713. doi: 10.1038/ismej.2007.46. [DOI] [PubMed] [Google Scholar]
- Bothe H, Schmitz O, Yates MG, Newton WE. Nitrogen fixation and hydrogen metabolism in cyanobacteria. Microbiol Mol Biol Rev. 2010;74(4):529–551. doi: 10.1128/MMBR.00033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braissant O, Decho AW, Dupraz C, Glunk C, Przekop KM, Visscher PT. Exopolymeric substances of sulfate-reducing bacteria: interactions with calcium at alkaline pH and implication for formation of carbonate minerals. Geobiology. 2007;5(4):401–411. [Google Scholar]
- Breitbart M, Hoare A, Nitti A, Siefert J, Haynes M, Dinsdale E, Edwards R, Souza V, Rohwer F, Hollander D. Metagenomic and stable isotopic analyses of modern freshwater microbialites in Cuatro Ciénegas, Mexico. Environ Microbiol. 2009;11(1):16–34. doi: 10.1111/j.1462-2920.2008.01725.x. [DOI] [PubMed] [Google Scholar]
- Burns BP, Goh F, Allen M, Neilan BA. Microbial diversity of extant stromatolites in the hypersaline marine environment of Shark Bay, Australia. Environ Microbiol. 2004;6(10):1096–1101. doi: 10.1111/j.1462-2920.2004.00651.x. [DOI] [PubMed] [Google Scholar]
- Burow LC, Woebken D, Marshall IP, Lindquist EA, Bebout BM, Prufert-Bebout L, Hoehler TM, Tringe SG, Pett-Ridge J, Weber PK, Spormann AM, Singer SW. Anoxic carbon flux in photosynthetic microbial mats as revealed by metatranscriptomics. ISME J. 2013;7(4):817–829. doi: 10.1038/ismej.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couradeau E, Benzerara K, Moreira D, Gerard E, Kazmierczak J, Tavera R, Lopez-Garcia P. Prokaryotic and eukaryotic community structure in field and cultured microbialites from the alkaline Lake Alchichica (Mexico) PLoS One. 2011;6(12):e28767. doi: 10.1371/journal.pone.0028767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MP, Peterson DA, Biggs PJ. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decho AW. Microbial exopolymer secretions in ocean environments - their role(s) in food webs and marine processes. Oceanogr Mar Biol. 1990;28:73–153. [Google Scholar]
- Decho AW. Microbial biofilms in intertidal systems: an overview. Cont Shelf Res. 2000;20:1257–1273. [Google Scholar]
- Desnues CG, Rodriguez-Brito B, Rayhawk S, Kelley S, Tran T, Haynes M, Lui H, Hall D, Angly FE, Edwards RA, Thurber RV, Reid RP, Siefert J, Souza V, Valentine D, Swan B, Breitbart M, Rohwer F. Biodiversity and biogeography of phages in modern stromatolites and thrombolites. Nature. 2008;452:340–345. doi: 10.1038/nature06735. [DOI] [PubMed] [Google Scholar]
- Dinsdale EA, Edwards RA, Bailey BA, Tuba I, Akhter S, McNair K, Schmieder R, Apkarian N, Creek M, Guan E, Hernandez M, Isaacs K, Peterson C, Regh T, Ponomarenko V. Multivariate analysis of functional metagenomes. FGENE. 2013;4:41. doi: 10.3389/fgene.2013.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupraz C, Visscher PT. Microbial lithification in marine stromatolites and hypersaline mats. Trends in Microbiol. 2005;13(9):429–438. doi: 10.1016/j.tim.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Dupraz C, Reid RP, Braissant O, Decho AW, Norman RS, Visscher PT. Processes of carbonate precipitation in modern microbial mats. Earth Sci Rev. 2009;96(3):141–162. [Google Scholar]
- Edgcomb VP, Bernhard JM, Beaudoin D, Pruss S, Welander PV, Schubotz F, Mehay S, Gillespie AL, Summons RE. Molecular indicators of microbial diversity in oolitic sands of Highborne Cay, Bahamas. Geobiology. 2013;11(3):234–251. doi: 10.1111/gbi.12029. [DOI] [PubMed] [Google Scholar]
- Faria JC, Demetrio CGB. R package version 1.02 edn. UESC and ESALQ; Ilheus, Bahia, Brasil and Piracicaba, Sao Paulo, Brasil: 2008. BPCA: bioplot of multivariate data based on principal components analysis. [Google Scholar]
- Feist AM, Scholten JCM, Palsson BO, Brockman FJ, Ideker T. Modeling methanogenesis with a genome-scale metabolic reconstruction of Methanosarcina barkeri. Mol Syst Biol. 2006;2:20060004. doi: 10.1038/msb4100046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JS, Green SJ. Microbial diversity in modern stromatolites. In: Seckbach J, Tewari V, editors. Cellular origin, life in extreme habitats and astrobiology: interactions with sediments. Springer; Berlin: 2011. pp. 385–405. [Google Scholar]
- Gallagher KL, Braissant O, Kading TJ, Dupraz C, Visscher PT. Phosphate-related artifacts in carbonate mineralization experiments. J Sediment Res. 2013;83(1):37–49. [Google Scholar]
- Goh F, Allen MA, Leuko S, Kawaguchi T, Decho AW, Burns BP, Neilan BA. Determining the specific microbial populations and their spatial distribution within the stromatolite ecosystem of Shark Bay. ISME J. 2009;3:383–396. doi: 10.1038/ismej.2008.114. [DOI] [PubMed] [Google Scholar]
- Gomez-Alvarez V, Teal TK, Schmidt TM. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 2009;3(11):1314–1317. doi: 10.1038/ismej.2009.72. [DOI] [PubMed] [Google Scholar]
- Grotzinger JP, Knoll AH. Stromatolites in Precambrian carbonates: evolutionary mileposts or environmental dipsticks? Annu Rev Earth Planet Sci. 1999;27:313–358. doi: 10.1146/annurev.earth.27.1.313. [DOI] [PubMed] [Google Scholar]
- Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17(3):377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi T, Decho AW. Biochemical characterization of cyanobacterial extracellular polymers (EPS) from modern marine stromatolites (Bahamas) Prep Biochem Biotech. 2000;30(4):321–330. doi: 10.1080/10826060008544971. [DOI] [PubMed] [Google Scholar]
- Khodadad CL, Foster JS. Metagenomic and metabolic profiling of nonlithifying and lithifying stromatolitic mats of Highborne Cay, The Bahamas. PLOS One. 2012;7(5):e38229. doi: 10.1371/journal.pone.0038229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R. pheatmap: Pretty Heatmaps. version 0.7.4 edn. R package. 2013. [Google Scholar]
- Kunin V, Raes J, Harris JK, Spear JR, Walker JJ, Ivanova N, von Mering C, Bebout BM, Pace NR, Bork P, Hugenholtz P. Millimeter-scale genetic gradients and community-level molecular convergence in a hypersaline microbial mat. Mol Syst Biol. 2008;4:198. doi: 10.1038/msb.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuko S, Goh F, Allen MA, Burns BP, Walter MR, Neilan BA. Analysis of intergenic spacer region length polymorphisms to investigate the halophilic archaeal diversity of stromatolites and microbial mats. Extremophiles. 2007;11(1):203–210. doi: 10.1007/s00792-006-0028-z. [DOI] [PubMed] [Google Scholar]
- Liaw A, Weiner M. Classification and regression by random-forest. R News. 2002;2(3):18–22. [Google Scholar]
- Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007;35(18):e120. doi: 10.1093/nar/gkm541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre IG, Prufert-Bebout L, Reid RP. The role of endolithic cyanobacteria in the formation of lithified lamiae in Bahamian stromatolites. Sedimentology. 2000;47:915–921. [Google Scholar]
- Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobberley JM, Ortega MC, Foster JS. Comparative microbial diversity analyses of modern marine thrombolitic mats by barcoded pyrosequencing. Environ Microbiol. 2012;14:82–100. doi: 10.1111/j.1462-2920.2011.02509.x. [DOI] [PubMed] [Google Scholar]
- Myshrall K, Mobberley JM, Green SJ, Visscher PT, Havemann SA, Reid RP, Foster JS. Biogeochemical cycling and microbial diversity in the modern marine thrombolites of Highborne Cay, Bahamas. Geobiology. 2010;8:337–354. doi: 10.1111/j.1472-4669.2010.00245.x. [DOI] [PubMed] [Google Scholar]
- Nitti A, Daniels CA, Siefert J, Souza V, Hollander D, Breitbart M. Spatially resolved genomic, stable isotopic, and lipid analyses of a modern freshwater microbialite from Cuatro Cienegas, Mexico. Astrobiology. 2012;12(7):685–698. doi: 10.1089/ast.2011.0812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paerl HW, Steppe TF, Reid RP. Bacterially mediated precipitation in marine stromatolites. Environ Microbiol. 2001;3(2):123–130. doi: 10.1046/j.1462-2920.2001.00168.x. [DOI] [PubMed] [Google Scholar]
- Peimbert M, Alcaraz LD, Bonilla-Rosso G, Olmedo-Alvarez G, Garcia-Oliva F, Segovia L, Eguiarte LE, Souza V. Comparative metagenomics of two microbial mats at Cuatro Ciénegas Basin I: ancient lessons on how to cope with an environment under severe nutrient stress. Astrobiology. 2012;12(7):648–658. doi: 10.1089/ast.2011.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JW, Boyd ES, D’Adamo S, Mulder DW, Posewitz MC. Hydrogenases, nitrogenases, anoxia, and H2 production in water-oxidizing phototrophs. In: Borowitzka MA, Moheimani NR, editors. Algae for biofuels and energy developments in applied phycology. Vol. 5. Springer; Dordrecht: 2013. pp. 37–75. [Google Scholar]
- Pinckney J, Paerl HW, Reid RP, Bebout BM. Ecophysiology of stromatolitic microbial mats, Lee Stocking Island, Exuma Cays, Bahamas. Microb Ecol. 1995;29:19–37. doi: 10.1007/BF00217420. [DOI] [PubMed] [Google Scholar]
- Planavsky N, Ginsburg RN. Taphonomy of modern marine Bahamian microbialites. Palaios. 2009;24:5–17. [Google Scholar]
- Planavsky N, Reid RP, Andres M, Visscher PT, Myshrall KL, Lyons TW. Formation and diagenesis of modern marine calcified cyanobacteria. Geobiology. 2009;7:566–576. doi: 10.1111/j.1472-4669.2009.00216.x. [DOI] [PubMed] [Google Scholar]
- Reid RP, Visscher PT, Decho AW, Stolz JF, Bebout BM, Dupraz C, Macintyre IG, Paerl HW, Pinckney JL, Prufert-Bebout L, Steppe TF, DesMarais DJ. The role of microbes in accretion, lamination and early lithification of modern marine stromatolites. Nature. 2000;406(6799):989–992. doi: 10.1038/35023158. [DOI] [PubMed] [Google Scholar]
- Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, Wu D, Eisen JA, Hoffman JM, Remington K, Beeson K, Tran B, Smith H, Baden-Tillson H, Stewart C, Thorpe J, Freeman J, Andrews-Pfannkoch C, Venter JE, Li K, Kravitz S, Heidelberg JF, Utterback T, Rogers YH, Falcon LI, Souza V, Bonilla-Rosso G, Eguiarte LE, Karl DM, Sathyendranath S, Platt T, Bermingham E, Gallardo V, Tamayo-Castillo G, Ferrari MR, Strausberg RL, Nealson K, Friedman R, Frazier M, Venter JC. The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5(3):e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos F, Pena A, Nogales B, Soria–Soria E, Del Cura MA, Gonzalez-Martin JA, Anton J. Bacterial diversity in dry modern freshwater stromatolites from Ruidera Pools Natural Park, Spain. Syst Appl Microbiol. 2010;33(4):209–221. doi: 10.1016/j.syapm.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Steppe TF, Pinckney JL, Dyble J, Paerl HW. Diazotrophy in modern marine Bahamian stromatolites. Microb Ecol. 2001;41(1):36–44. doi: 10.1007/s002480000066. [DOI] [PubMed] [Google Scholar]
- Steven B, Gellegos-Graves L, Starkenburg SR, Chain PS, Kuske CR. Targeted and shotgun metagenomic approaches provide different descriptions of dryland soil microbial communities in a manipulated field study. Environ Microbiol Rep. 2012;4(2):249–256. doi: 10.1111/j.1758-2229.2012.00328.x. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. A language and environment for statistical computing. 3-900051-07-0 edn. R Foundation for Statistical Computing; Vienna, Austria: 2010. [Google Scholar]
- van der Meer MT, Schouten S, Damste JS, Ward DM. Impact of carbon metabolism on 13C signatures of cyanobacteria and green non-sulfur-like bacteria inhabiting a microbial mat from an alkaline siliceous hot spring in Yellowstone National Park (USA) Environ Microbiol. 2007;9(2):482–491. doi: 10.1111/j.1462-2920.2006.01165.x. [DOI] [PubMed] [Google Scholar]
- Visscher PT, Stolz JF. Microbial mats as bioreactors: populations, processes and products. Palaeogeogr Palaeoclimatol Palaeoecol. 2005;219:87–100. [Google Scholar]
- Visscher PT, Reid RP, Bebout BM, Hoeft SE, Macintyre IG, Thompson JA. Formation of lithified micritic laminae in modern marine stromatolites (Bahamas): the role of sulfur cycling. Amer Mineral. 1998;83:1482–1493. [Google Scholar]
- Visscher PT, Reid RP, Bebout BM. Microscale observations of sulfate reduction: correlation of microbial activity with lithified micritic laminae in modern marine stromatolites. Geology. 2000;28(10):919–922. [Google Scholar]
- Walker CB, de la Torre JR, Klotz MG, Urakawa H, Pinel N, Arp DJ, Brochier-Armanet C, Chain PS, Chan PP, Gollabgir A, Hemp J, Hugler M, Karr EA, Konneke M, Shin M, Lawton TJ, Lowe T, Martens-Habbena W, Sayavedra-Soto LA, Lang D, Sievert SM, Rosenzweig AC, Manning G, Stahl DA. Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc Natl Acad Sci USA. 2010;107(19):8818–8823. doi: 10.1073/pnas.0913533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke A, Harrison T, Wilkening J, Field D, Glass EM, Kyrpides N, Mavrommatis K, Meyer F. The M5nr: a novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinform. 2012;13:141. doi: 10.1186/1471-2105-13-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.