

Abstract
Human mature IL-33 is a member of the IL-1 family and a potent regulator of immunity through its pro-T helper cell 2 activity. Its precursor form, full-length interleukin-33 (FLIL33), is an intranuclear protein in many cell types, including fibroblasts, and its intracellular levels can change in response to stimuli. However, the mechanisms controlling the nuclear localization of FLIL33 or its stability in cells are not understood. Here, we identified importin-5 (IPO5), a member of the importin family of nuclear transport proteins, as an intracellular binding partner of FLIL33. By overexpressing various FLIL33 protein segments and variants in primary human lung fibroblasts and HEK293T cells, we show that FLIL33, but not mature interleukin-33, physically interacts with IPO5 and that this interaction localizes to a cluster of charged amino acids (positions 46–56) but not to an adjacent segment (positions 61–67) in the FLIL33 N-terminal region. siRNA-mediated IPO5 knockdown in cell culture did not affect nuclear localization of FLIL33. However, the IPO5 knockdown significantly decreased the intracellular levels of overexpressed FLIL33, reversed by treatment with the 20S proteasome inhibitor bortezomib. Furthermore, FLIL33 variants deficient in IPO5 binding remained intranuclear and exhibited decreased levels, which were also restored by the bortezomib treatment. These results indicate that the interaction between FLIL33 and IPO5 is localized to a specific segment of the FLIL33 protein, is not required for nuclear localization of FLIL33, and protects FLIL33 from proteasome-dependent degradation.
Keywords: fibroblast, nuclear translocation, proteasome, protein degradation, protein-protein interaction, interleukin-33
Introduction
IL-33 is a member of the IL-1 family of cytokines that has been implicated as a major regulator of immune homeostasis and a contributor to pathologic inflammatory and fibrotic processes (1, 2). Increased levels of IL-33 have been shown in asthma (3–5), chronic obstructive pulmonary disease (6, 7), and idiopathic pulmonary fibrosis (8, 9). Receptor-mediated cytokine activity of IL-33 is driven by its C-terminal region, termed mature IL-33 (MIL33),2 which is released as a proteolytic product of the precursor, or full-length IL-33 (FLIL33). Both FLIL33 and MIL33 can bind to the T1/ST2 receptor (3, 10, 11), but MIL33 binds with much higher affinity and is a far more potent inducer of Th2 responses (12). The majority of IL-33 research has focused on MIL33, but many of the diseases in which IL-33 has been implicated are not mediated by Th2-type inflammation. Intracellular FLIL33 is quantitatively predominant in tissues and is constitutively expressed in a wide variety of cell types, with increasing evidence suggesting a role in Th2-independent processes (1, 2, 8, 9, 12–14).
At the molecular level, nuclear FLIL33 can bind to histones and is associated with euchromatin and heterochromatin (13, 15–17). FLIL33 can also bind to NF-κB, interfering with pro-inflammatory intracellular signaling (13). At the cellular level, studies report that FLIL33 mediates the mechanoresponsiveness of fibroblasts (4) and contributes to non-Th2-mediated fibrosis in vivo (8, 12). Additionally, the N-terminal domain of the protein targets FLIL33 to the nucleus and contains the target sites for proteolytic enzymes, cleaving FLIL33 into MIL33 (18). Genetically modified mice that express IL-33 lacking the N-terminal domain have markedly elevated systemic levels of IL-33 and die of massive sterile inflammation (19). More recently, alternatively spliced variants of IL-33 lacking segments of the N terminus were primarily localized to the cytoplasm, secreted extracellularly, and retained the capacity for receptor binding and immunologic effects. Increased levels of these transcripts were found in the airways of asthma patients with type 2 inflammation (20). We have recently reported that intracellular levels of FLIL33 protein are regulated by Th1 and Th2 cytokines post-translationally through proteasomal degradation (21). Given the complexities of subcellular distribution, proteolytic processing, and the differential biological effects of FLIL33 and MIL33, in-depth investigations into the intracellular regulation of FLIL33 through its N-terminal domain are warranted.
Importins are chaperone proteins primarily involved in the delivery of macromolecules across the nuclear pore complex through recognition of a nuclear localization sequence on their cargoes (22). Importins are also known to perform other functions, such as regulation of intracellular stability of proteins (23) or protein secretion (24). Importin-5 (IPO5, also referred to as karyopherin β3 (KPNB3) and Ras-related nuclear protein-binding protein 5 (RANBP5)) is one of these carriers and is a member of the importin-β superfamily (24–30). Here, we demonstrate that IPO5 is a novel intracellular binding partner of FLIL33 but is not required for its nuclear localization and does not control its secretion. Rather, we show that the interaction between IPO5 and FLIL33 results in stabilization of the intracellular FLIL33 protein by protecting it from proteasome-dependent degradation.
Results
FLIL33 interacts with IPO5
To identify intracellular partners of IL-33, the human FLIL33 gene was delivered to primary adult human pulmonary fibroblasts using infection with FLIL33-encoding recombinant replication-deficient adenovirus (AdV) or electroporations with FLIL33-encoding recombinant plasmids. Similar constructs with the same AdV backbone or plasmid backbone but encoding full-length precursor form of human IL-37 (FLIL37, a member of the IL-1 family) were used as controls. In all of these constructs, the encoded proteins were fused to an HA tag through a flexible peptide linker on the C terminus of the molecule. The resulting fusion proteins are referred to as FLIL33HA and FLIL37HA. Following overexpression of these HA-tagged proteins in deidentified adult primary normal human lung fibroblast (NHLF) cell cultures, cell lysates were incubated with anti-HA antibody covalently immobilized to magnetic beads. In a set of six preliminary experiments, eluates from the beads used to capture FLIL33HA or FLIL37HA were analyzed by LC-MS/MS. The results suggested that FLIL33HA but not FLIL37HA co-immunoprecipitated with IPO5. To validate these observations, Western blotting assays of the co-immunoprecipitates were performed, confirming that FLIL33HA, but not FLIL37HA, bound IPO5 (Fig. 1, A and B). Identical experiments were performed with FLIL33HA- and FLIL37HA-overexpressing HEK293T cells with similar results (Fig. 1C). As an additional control, Western blots of the same immunoprecipitates for selected other importins did not show an association with FLIL33HA. These included blots probed with an antibody against importin β1 and an antibody reactive to importins α1, α3, α5, and α7.
Figure 1.

IPO5 co-immunoprecipitates with FLIL33. Overexpression of FLIL33HA, FLIL37HA, MIL33HA (aa 112–270), or HA-tagged N-terminal segment of FLIL33 (aa 1–111, N-term) was achieved in cultured NHLF (A and B) or HEK293T cells (C–E) using electroporations with plasmids (A, D, and E) or infections with recombinant replication-deficient AdV (B and C), as indicated. Non-coding plasmid and AdV vehicles were used as controls (NULL). Cell lysates were loaded whole (W) or after IP, and Western blots were developed with antibodies against IPO5 or HA, as indicated (A–D). In E, IPO5-Myc was immunoprecipitated with anti-Myc antibody and incubated with anti-HA beads loaded with MIL33HA, FLIL33HA, or control beads (NULL) for 1 h at room temperature. The beads were then washed, and the eluates were analyzed by Western blotting using antibodies against Myc or HA, as indicated. The experiments in NHLF were performed on 10 separate occasions with each plasmid-based and AdV gene delivery utilizing primary cell cultured from four separate adult healthy lungs, with consistent results. Experiments in HEK293T cells were repeated on multiple independent occasions (at least three and for some experiments up to nine times) with selected results shown in C–E and subsequent figures.
Separate plasmid constructs were prepared that encoded N-terminal (aa 1–111) and C-terminal (aa 112–270) segments of FLIL33; the encoded proteins were tagged with HA similarly to FLIL33HA and FLIL37HA. The aa 112–270 construct encoding mature IL-33 was synonymically designated as MIL33HA. Co-immunoprecipitation experiments revealed that the N-terminal segment interacts with IPO5 similarly to FLIL33HA, whereas MIL33HA showed no detectable binding of IPO5 in HEK293T cells (Fig. 1D). In all of these experiments, intracellular interaction with IPO5 was assessed. An additional experiment was performed to determine whether IL-33 can bind IPO5 in vitro. HEK293T cells were electroporated with a non-coding control NULL plasmid or MIL33HA- or FLIL33-encoding plasmids, and the proteins were immunoprecipitated from cells lysates utilizing anti-HA antibody immobilized on magnetic beads. Separately, HEK293T cells were electroporated with a plasmid encoding an IPO5-Myc fusion construct, which was then immunoprecipitated from cell lysates using anti-Myc antibody immobilized on agarose beads. Following elution of IPO5-Myc from the beads, the eluent was applied to magnetic beads coated with MIL33HA or FLIL33HA prepared as described above. The resulting protein complexes were eluted from the magnetic beads and analyzed by Western blotting for Myc and HA. It was found that, similar to their intracellular interactions, FLIL33HA but not MIL33HA binds IPO5 (Fig. 1E). A conclusion was made that FLIL33 interacts with IPO5 through its N-terminal segment in living cells and in vitro.
Interaction with IPO5 localizes to the N terminus of FLIL33
Having observed that FLIL33 and its N-terminal segment, but not MIL33, binds IPO5, we sought to narrow down the IPO5-binding region within the N-terminal segment of FLIL33. To localize the site(s) of interaction with IPO5 to a specific area of FLIL33, recombinant plasmids encoding truncated segments of FLIL33 were constructed. All of the segments were HA-tagged on the C terminus through the peptide linker identical to that in the constructs described above. Electroporations of HEK293T cells with the constructs were performed followed by immunoprecipitation for HA and Western blotting for IPO5 and HA. Electroporation of unmanipulated HEK293T cells with the constructs encoding the truncated segments of FLIL33 were not detectable by Western blotting, suggesting their rapid proteasomal degradation. Indeed, HEK293T cells treated with the proteasome inhibitor bortezomib readily expressed all of the FLIL33 segments (Fig. 2). It was observed that aa 1–90 and 45–135 fragments co-immunoprecipitated IPO5 (Fig. 2A), confirming its binding to the N-terminal segment of FLIL33 and suggesting that the aa 45–90 region of IL-33 may be sufficient for IPO5 binding. To more precisely localize IPO5 binding within the N-terminal segment of FLIL33, shorter peptide fragments were similarly overexpressed and co-immunoprecipitated with IPO5, revealing that fragment aa 45–90, but not fragment aa 67–113, bound IPO5 (Fig. 2B). These findings indicated that IPO5 is likely to bind within the aa 45–67 region (Fig. 2C). Overexpression of the aa 1–90 fragment containing a mutated nuclear localization sequence (15) was still able to co-immunoprecipitate IPO5 (Fig. 2D).
Figure 2.

IPO5 co-immunoprecipitates with HA-tagged N-terminal segments of FLIL33 prior to aa 67. 90-amino acid-long (A) and 45-amino acid-long (B) segments of FLIL33 show differential binding of IPO5. The findings suggest that IPO5 binds in the region between amino acids 45 and 65 (C). In C, the peptide segments are drawn to scale; the IPO5-binding segments are indicated as shaded bars, whereas non-binding segments are shown as open bars. The aa 1–90 segment with double-mutated nuclear localization sequence (R67A and K71A) co-immunoprecipitated with IPO5 (D). Each of the peptides was overexpressed in HEK293 cells and tested for IPO5 co-immunoprecipitation on at least two occasions, with consistent results.
FLIL33 nuclear localization or secretion is not affected by IPO5 knockdown
Based on the well known predominant nuclear localization of FLIL33 and the main function of IPO5 as an importin, it was hypothesized that the interaction of FLIL33 with IPO5 facilitates nuclear import of FLIL33. To investigate this possibility, IPO5 levels were attenuated using RNA interference in HEK293T cells and NHLFs. Additional attempts to fully abrogate IPO5 using commercially available shRNA or clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 constructs were unsuccessful in that the cells became non-viable in the absence of IPO5. This observation is not surprising considering that IPO5 is a highly conserved protein that participates in indispensable cell function.
Cultured cells were electroporated with either non-targeting siRNA control or with IPO5-targeting siRNA. After 48 h, recombinant FLIL33HA plasmid was additionally introduced by electroporation. Nuclear and cytoplasmic extracts were prepared, and Western blots were performed. Despite robust depletion of IPO5 in HEK293T cells (Fig. 3A), no difference was seen in the nuclear fraction of FLIL33HA between cells treated with non-targeting siRNA and IPO5-specific siRNA as visualized by Western blot probed with anti-HA antibody (Fig. 3B). These findings were confirmed by performing similar experiments in NHLFs. IPO5 levels were attenuated using IPO5-specific siRNA with subsequent introduction of FLIL33HA (Fig. 3C). Western blot analysis of nuclear and cytoplasmic extracts probed with anti-HA antibody revealed unchanged nuclear predominance in the setting of attenuated IPO5 (Fig. 3D). Retained nuclear localization of FLIL33HA was confirmed using immunofluorescence in HEK293T cells transfected with either non-targeting or IPO5-specific siRNA and probed with anti-HA antibody (Fig. 3E). Thus, substantial attenuation of IPO5 levels did not affect nuclear localization of FLIL33.
Figure 3.
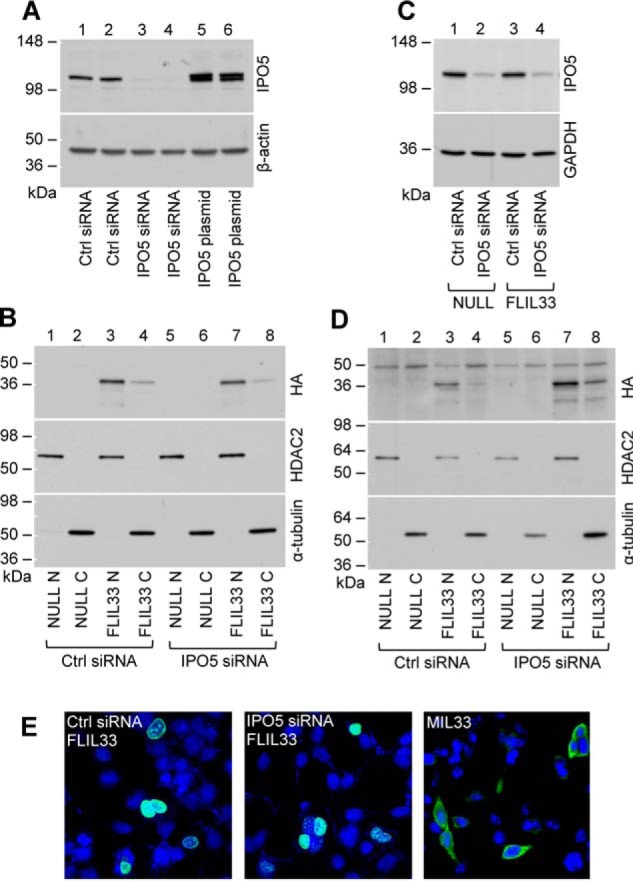
Attenuation of IPO5 levels with siRNA does not prevent nuclear localization of FLIL33. A, transfection of HEK293T cells with non-targeting siRNA (Ctrl) does not, whereas IPO5-specific siRNA does attenuate the basal levels of IPO5. For comparison, overexpression IPO5 using electroporation of an IPO5-encoding plasmid is also shown. The membranes were stripped and redeveloped for β-actin. B, overexpressed FLIL33HA detected with anti-HA antibody is predominantly intranuclear in both non-targeting siRNA-transfected (Ctrl) and IPO5-targeting siRNA-transfected HEK293T cells. Electroporations with a non-coding plasmid (NULL) were used as controls. Nuclear fractions are labeled with N, and cytoplasmic fractions are labeled with C. The membranes were stripped of antibodies and redeveloped for nuclear and cytoplasmic markers, HDAC2 and α-tubulin, respectively, as indicated. C, transfection of primary NHLF with non-targeting siRNA (Ctrl) does not, whereas IPO5-specific siRNA does, attenuate the basal levels of IPO5. Such attenuation is not affected by the subsequent FLIL33HA overexpression compared with electroporation with a non-coding plasmid (NULL). D, overexpressed FLIL33HA detected with anti-HA antibody is predominantly intranuclear in both non-targeting siRNA-transfected (Ctrl) and IPO5-targeting siRNA-transfected NHLF. Electroporations with a non-coding plasmid (NULL) were used as controls. Nuclear fractions are labeled with N, and cytoplasmic fractions are labeled with C. The membranes were stripped of anti-HA antibodies and redeveloped for nuclear and cytoplasmic markers, HDAC2 and α-tubulin, respectively, as indicated. E, immunofluorescent staining of cultured HEK293T cells for the HA tag (green) after electroporation with indicated siRNAs and then FLIL33HA demonstrates that FLIL33 remains intranuclear without (left panel) or with (middle panel) IPO5 attenuation. For comparison, cells similarly electroporated with MIL33HA express mature IL-33 in the cytoplasm (right panel). Nuclei were stained with DAPI (blue). The experiments in HEK293T cells were performed twice, and in NHLF, the experiments were performed in cultures from two different donors on two separate occasions, with consistent results.
Importins, including IPO5, perform other functions in addition to facilitating nuclear import of their cargo. One report found that IPO5 is responsible for the secretion of apolipoprotein A1 from cells (24). A possibility was evaluated that IPO5 may control FLIL33 secretion from cells, which is normally minimal (12). ELISA of cell lysates and supernatants of HEK293T cells and NHLFs transfected with the FLIL33HA-encoding plasmid revealed no difference in the levels of secreted IL-33 with or without IPO5-specific siRNA delivery.
IPO5 protects FLIL33 from degradation
The observations that attenuation of IPO5 levels did not alter subcellular localization or secretion of FLIL33 prompted an additional consideration that IPO5 binding may control intracellular degradation of FLIL33. This possibility was based on previous reports that karyopherins regulate intracellular degradation of target proteins (23, 31). To assess this possibility, NHLFs were transfected with the FLIL33HA-encoding plasmid following attenuation of IPO5 levels with siRNA as described above, and IL-33 protein levels in cell lysates were assessed using Western blotting and ELISA (Fig. 4). By Western blotting, attenuation of IPO5 resulted in decreased levels of FLIL33HA but not MIL33HA (Fig. 4A). The proteasome has been reported to be a regulator of intracellular IL-33 levels (21), suggesting that IPO5 may specifically protect IL-33 from proteasome-dependent degradation. To assess the extent to which proteasome-dependent degradation was responsible for the decreased IL-33 levels in the setting of IPO5 attenuation, cells were treated with bortezomib to suppress the 20S proteasome. ELISA tests of cell lysates revealed that IL-33 levels were partially rescued by proteasome inhibition in the setting of IPO5 attenuation (Fig. 4B). RT-quantitative PCR tests revealed that the basal levels of endogenous IL-33 mRNA were low and not affected by bortezomib treatment in these experiments. These findings indicate that the interaction between FLIL33 and IPO5 results in stabilization of the FLIL33 protein and that this stabilization is a result of protection from proteasome-dependent degradation. The IPO5-dependent FLIL33 protein stabilization was observed in NHLFs, primary cell cultures, but not HEK293T cells, a transformed cell line. This observation in NHLFs indicates that IPO5-controlled FLIL33 stabilization is applicable to FLIL33 stability in cells known to contribute to the IL-33 pool in vivo. At the same time, this observation allows for the use of HEK293T cells as a tool to study intracellular interactions and subcellular localization of FLIL33 even in the absence of IPO5-mediated protection.
Figure 4.

Attenuation of IPO5 levels with siRNA results in reduced levels of IL-33 when overexpressed in NHLFs. A, NHLFs treated with IPO5-targeting siRNA then electroporated with the FLIL33HA-encoding plasmid show a reduction in FLIL33HA protein as detected by anti-HA antibody, with two separate experiments shown (left panel). By contrast, MIL33HA levels were not affected by IPO5 attenuation (right panel). This experiment was repeated in two different primary NHLF cultures, with similar results. B, ELISA for IL-33 was performed with NHLF lysates following siRNA-mediated depletion of IPO5 and electroporation with the FLIL33HA-encoding (left panel) or vehicle control (NULL, right panel). Mean ± S.D. values pooled from three separate experiments with each condition tested in duplicates are shown. Significantly decreased (p < 0.05) IL-33 levels were observed with IPO5 attenuation in FLIL33HA-overexpressing cells as indicated with asterisks; there was a tendency to lower intracellular endogenous IL-33 in NULL-transfected cells, but it did not reach statistical significance. Treatment with the 20S proteasome inhibitor, bortezomib, was used as indicated and resulted in partial restoration of IL-33 levels.
IPO5 binding–deficient FLIL33 mutants retain nuclear localization
To further investigate the consequence of FLIL33 binding to IPO5, alanine substitution mutants within the IPO5-binding domain in the aa 45–67 region of FLIL33HA (see Fig. 2C and related text) were created. Given the high density of charged amino acids within this domain that could participate in protein-protein interactions, two clusters of charged amino acids were targeted for substitution with alanines or deletion. The resulting HA-tagged constructs were designated FLIL33(46/56A11), FLIL33(61/67A7), FLIL33(46/56DEL), and FLIL33(61/67DEL) (Fig. 5A). Electroporation of HEK293T cells with the constructs was performed followed by immunoprecipitation for HA and Western blotting for IPO5 and HA. FLIL33(46/56A11) was observed to have no detectable binding to IPO5 by co-immunoprecipitation, whereas FLIL33(61/67A7) retained IPO5 binding (Fig. 5B). The deletion mutants revealed similar expression levels and IPO5-binding patterns as the corresponding substitution mutants. Following similar electroporations with deletion mutant-encoding plasmid constructs, FLIL33(61/67DEL) protein was abundantly present, whereas FLIL33(46/56DEL) was minimally expressed in the absence of bortezomib (Fig. 5C). Immunoprecipitation for HA revealed that IPO5 co-immunoprecipitated with FLIL33(61/67DEL) and non-mutated FLIL33, whereas FLIL33(46/56DEL) bound IPO5 minimally (Fig. 5C).
Figure 5.

Mutations within the IPO5 binding region of FLIL33 do not affect nuclear localization. A, charged cluster-to-alanine mutagenesis of the IPO5 binding domain of FLIL33 resulted in two FLIL33HA mutants, termed FLIL33(46/56A11) and FLIL33(61/67A7); the corresponding deletion mutants were termed FLIL33(46/56DEL) and FLIL33(61/67DEL). B, Western blot of co-immunoprecipitates from HEK293T cells transfected with plasmids encoding FLIL33(46/56A11) and FLIL33(61/67A7) was performed and probed with anti-IPO5 and anti-HA antibodies, as indicated. C, Western blot of co-IPs and whole cell lysates (W) from HEK293T cells transfected with plasmids encoding FLIL33(46/56DEL) and FLIL33(61/67DEL) was performed and probed with anti-IPO5 and anti-HA antibodies, as indicated. D, nuclear and cytoplasmic extraction was performed on HEK293T cells transfected with plasmids encoding FLIL33(46/56A11), FLIL33(61/67A7), or WT FLIL33HA. The membranes were stripped of anti-HA antibodies and redeveloped for nuclear and cytoplasmic markers, HDAC2 and α-tubulin, respectively, as indicated. E, immunofluorescent staining of cultured HEK293T cells for the HA tag (green) after electroporation with plasmids encoding FLIL33(46/56A11), FLIL33(46/56DEL), FLIL33(61/67A7), and FLIL33(61/67DEL). Nuclei were stained with DAPI (blue). Experiments were performed on at least two occasions for each mutant with consistent results.
To assess the subcellular localization of the alanine substitution mutants, nuclear and cytoplasmic extracts of HEK293T cells transfected with FLIL33(46/56A11) or FLIL33(61/67A7) were analyzed by Western blot for HA. Both mutants retained nuclear localization, similarly to native FLIL33 (Fig. 5D). Nuclear localization was confirmed using immunofluorescence in HEK293T cells transfected with FLIL33(46/56A11) or FLIL33(61/67A7), as well as FLIL33(46/56DEL) or FLIL33(61/67DEL), by staining with anti-HA antibody (Fig. 5E). Although both mutants were predominantly nuclear, FLIL33(46/56A11) and FLIL33(46/56DEL) consistently showed a granular appearance within the nucleus, whereas the distribution of FLIL33(61/67A7) and FLIL33(61/67DEL) was homogenous; the mechanistic reason for this difference remains to be investigated.
Thus, nuclear localization does not appear to be dependent on the charged clusters of amino acids 46–56 or 61–67. The reduction in IPO5 binding by FLIL33(46/56A11) and FLIL33(46/56DEL) does not affect their nuclear localization, further supporting the notion that the interaction with IPO5 is not involved in FLIL33 nuclear localization.
IPO5 binding–deficient FLIL33 mutants are degraded by the proteasome
In NHLFs, FLIL33(61/67A7) was easily expressed following electroporation, and IPO5 attenuation with IPO5-targeting siRNA led to decreased levels of FLIL33(61/67A7), similarly to FLIL33 (Fig. 6A). Conversely, FLIL33(46/56A11) was expressed at low levels in NHLF lysates analyzed by Western blotting for HA (Fig. 6A, right panels). The levels of FLIL33(46/56A11) in NHLFs were rescued by proteasome inhibition with bortezomib (Fig. 6B). Rescue of FLIL33(46/56A11) in NHLFs occurred similarly in NHLF cultures with and without IPO5 attenuation with IPO5-targeting siRNA, consistent with the notion that IPO5 does not bind or protect this mutant (Fig. 6B).
Figure 6.

Mutations within the IPO5 binding region of FLIL33 result in protein degradation. A, whole cell lysates from NHLFs transfected with plasmids encoding WT FLIL33HA, FLIL33(46/56A11), or FLIL33(61/67A7), with or without IPO5 attenuation by IPO5-targeting siRNA, were analyzed by Western blot. B, Whole cell lysates of NHLFs transfected with plasmid encoding FLIL33(46/56A11) and IPO5-targeting siRNA, with or without bortezomib were analyzed by Western blot. C, NHLFs transfected with plasmids encoding FLIL33(46/56A11) or FLIL33(61/67A7) were treated with cycloheximide, and lysates were collected at the time points indicated. Western blot was performed and probed with anti-HA antibody at indicated times (h). The experiments were performed in NHLFs from at least two different donors with consistent results.
To further assess the reduction of FLIL33(46/56A11) protein levels, chase experiments using a protein synthesis inhibitor, cycloheximide, were performed. Following electroporation of NHLFs with FLIL33(46/56A11) and FLIL33(61/67A7), cells were treated with bortezomib to boost basal levels, and then bortezomib-containing medium was exchanged for media containing cycloheximide. The cells were lysed at sequential time points, and protein levels were measured by Western blotting for HA. Protein levels of FLIL33(46/56A11) decreased more rapidly than FLIL33(61/67A7) (Fig. 6C), consistent with increased rate of degradation in the absence of IPO5 binding and protection. These results show that amino acids 46–56 are necessary for IPO5 binding and IPO5-mediated protection of FLIL33 from proteasomal degradation, whereas amino acids 61–67 are not.
Discussion
In this study, initial observation by LC-MS/MS suggested, and Western blotting experiments confirmed, that FLIL33, but not MIL33 or FLIL37, binds IPO5 intracellularly and in vitro (Fig. 1). This binding activity is localized to the N-terminal portion of FLIL33 and requires the aa 45–67 region of the protein (Fig. 2). Attenuation of IPO5 levels through RNA interference does not alter the predominant nuclear localization of FLIL33 (Fig. 3), but it induces its proteasomal degradation in primary lung fibroblasts (Fig. 4). Mutations in FLIL33, abrogating its ability to bind IPO5, result in proteasome-dependent degradation of the mutants in primary lung fibroblasts, but their nuclear localization is preserved (Figs. 5 and 6).
The intracellular activity and regulation of FLIL33 has been studied since its first description as a nuclear factor over a decade ago, but these investigations have lagged in quantity and breadth when compared with the studies of MIL33 and T1/ST2 receptor-mediated biology. The unique intracellular behaviors of FLIL33 are of substantial biologic importance for at least three specific reasons. First, IL-33, like its IL-1 family member IL-1α, is a potent cytokine that is basally expressed in a variety of cell types throughout the human body and is implicated in many disease processes (1, 5–8, 12, 32). Mechanisms that maintain the intracellular pool of FLIL33 will impact the amount of mature cytokine available for inflammatory processes. Second, nuclear FLIL33 levels are elevated in some non-Th2 diseases and, in these cases, has been implicated in cellular processes contributing to pathology, although the exact mechanisms of such contributions remain to be fully elucidated (5–7, 13, 33–36). Third, the intracellular trafficking of FLIL33 is essential to maintain homeostasis, and failure of this process can lead to untoward or exuberant inflammation through unintentional release or secretion (19, 20).
The interaction between IPO5 and FLIL33 described in this report brings us a step closer to understanding intracellular FLIL33 biology but also raises additional questions. Our results confirm that FLIL33 is primarily nuclear but indicate that IPO5, a binding partner of FLIL33, is unlikely responsible for its nuclear translocation. Inability to completely knock out IPO5 protein expression, instead relying on knockdown through RNA interference, is a limitation of this study. The observed lethality of IPO5 knock-out in our experiments with CRISPR/Cas9 and shRNA supports the notion that IPO5 is indispensable for cell function, making FLIL33 binding all the more intriguing. RNA interference of IPO5 was well tolerated by the cells, suggesting that reduced levels of IPO5 are still sufficient to sustain cellular processes. A corollary of this notion is a possibility that even low levels of IPO5 may be sufficient for nuclear translocation of FLIL33. To investigate this possibility in greater depth, we devised additional experiments based on the previously reported binding of a yeast homolog of IPO5, Kap121p, to its cargo at a consensus sequence K(V/I)XKX1–2(K/H/R), with the (V/I)XK sequence being essential for such binding (26). This sequence is only present once in the FLIL33 peptide sequence in the area of amino acids 46–56. The alanine substitution FLIL33(46/56A11) and deletion FLIL33(46/56DEL) mutants did not bind IPO5 or minimally associated with it. Still, FLIL33(46/56A11) localized to the nucleus, further supporting non-involvement of IPO5 in nuclear translocation of FLIL33, yet not fully excluding a possible contribution of residual IPO5-FLIL33 interaction. Overall, the conclusion on IPO5 non-involvement in FLIL33 nuclear translocation is rather well supported but should still be made with caution.
Other members of the importin family have been implicated in the regulation of the intracellular degradation of their targets (23, 31). We have shown that IPO5 protects FLIL33 from proteasome-dependent degradation, but the specific mechanism remains unclear. A key step in degradation by the proteasome involves ubiquitination of the target protein on a designated lysine by a specific E3 ubiquitin ligase. It is possible that IPO5 shields FLIL33 from its corresponding E3 ubiquitin ligase. Another possibility is an interaction with deubiquitinases, which have been shown to stabilize IL-33 (37, 38), leading to deubiquitination of FLIL33. A third possibility is for IPO5 to prevent delivery to the proteasome itself, regardless of ubiquitination status. These postulations will require further investigation.
In summary, FLIL33 and IPO5 associate intracellularly, leading to protection of FLIL33 from proteasome-dependent degradation, but not affecting its nuclear translocation. Further investigations are necessary to better understand the nature and consequences of this interaction. With increasing evidence of IL-33 being a key mediator of human disease, further elucidation of its complex intracellular biology could lead to improved therapies.
Experimental procedures
Cell culture
Deidentified NHLFs derived from healthy adult volunteers were purchased from Lonza (Walkersville, MD). Each experiment was performed in primary fibroblast cultures from at least two different donors. Overall, NHLFs from seven different donors were used throughout the course of this study. The cultures were maintained in T75 culture flasks (NEST Biotechnology, Rahway, NJ) in a humidified atmosphere of 5% CO2 at a temperature of 37 °C in DMEM supplemented with 4.5 g/liter glucose, l-glutamine, and sodium pyruvate (Corning, Corning, NY), 10% bovine calf serum, minimal essential medium non-essential amino acids (Gibco/Thermo Fisher Scientific), and antibiotic-antimycotic mixture (Gibco) to a final concentration of 100 units/ml of penicillin, 100 μg/ml of streptomycin, and 0.25 μg/ml of amphotericin B. For experiments, the cells were harvested by trypsinization at passages 4–6, washed, counted, and seeded on 6-well culture plates (NEST Biotechnology) at a density of 5 × 105 cells/well. HEK293T cells (American Type Culture Collection, Manassas, VA) were cultured and maintained in a similar fashion. Each experiment in HEK293T cells was performed on multiple independent occasions as stated in the corresponding figure legends for each experiment. Gene delivery was achieved using electroporation of cells with recombinant plasmids utilizing the Amaxa Nucleofector (Lonza). In each reaction, 5 × 105–1 × 106 cells and 0.5–2.0 μg of plasmid were used, based on preliminary experiments to optimize expression of each delivered recombinant protein. Alternatively, infections of cultured cells with replication-deficient recombinant AdV constructs were used to overexpress proteins or peptides of interest. For these infections, 1 × 105–5 × 106 plaque-forming units/ml of AdVs were used per 2.5 × 105 cells in culture. Overexpression of the proteins and peptides of interest was confirmed by Western blotting. The proteasome inhibitor bortezomib (Cell Signaling Technology, Danvers, MA) was used in cell culture at a concentration of 500 nm; bortezomib was added 24 h after plasmid delivery for an additional 24 h of culture. Cycloheximide (Sigma–Aldrich) was used in cell culture at a concentration of 100 μg/ml.
Recombinant plasmid and adenoviral constructs
All recombinant constructs in this study were cloned into the VQAd5CMVK-NpA plasmid (ViraQuest, North Liberty, IA) downstream of the cytomegalovirus promoter. This plasmid can be used for gene expression in mammalian cells as well as a shuttle plasmid for constructing AdV in the RAPAd system (39). The mRNA sequences of IL-33 and IL-37 from National Center for Biotechnology Information GenBankTM (accession numbers NM_033439 and NM_014439, respectively) were used to design the recombinant constructs. The stop codon was removed, and a linker peptide (GGGGSGGGGSGGGGS)-encoding sequence was added on the 3′-end, followed by the HA tag (YPYDVPDYA)-encoding sequence and a stop codon. The DNA sequence was codon-optimized for mammalian expression, synthesized (Genscript, Piscataway, NJ), and transferred into the VQAd5CMVK-NpA plasmid. Additionally, recombinant constructs similarly encoding FLIL33 segments and mutants (described under “Results”) were all prepared identically. To produce FLIL33-encoding and FLIL37-encoding replication-deficient AdV vectors, the corresponding plasmid constructs were utilized using RAPAd technology (ViraQuest); the viruses were purified and concentrated as described elsewhere (21). Human IPO5-Myc-encoding plasmid was purchased from Origene (Rockville, MD).
Liquid chromatography tandem mass spectrometry (LC-MS/MS)
Proteins were extracted from cell pellets in 50 mm Tris, pH 8.0, 150 mm NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% Igepal CA630. After sonication, disulfide bonds were reduced with dithiothreitol, and the resulting sulfhydryl groups were alkylated with chloroacetamide. Proteins were digested with trypsin overnight at 37 °C. The reaction mixture was then acidified with formic acid, insoluble material was removed by centrifugation, and the peptides were recovered free of detergent by hydrophilic interaction solid phase extraction on PolyHYDROXYETHYL A TopTips (PolyLC, Columbia, MD) according to the manufacturer's recommendations. The purified peptides were analyzed by LC-MS/MS on a Thermo LTQ-Orbitrap. Peptides were separated using a 2-h chromatographic gradient online with a data-dependent MS/MS duty cycle of the top 10 most abundant ions. Database search and peptide quantification were performed using MaxQuant (40) version 1.5.5.1.
Reagents and molecular biology techniques
Concentrations of IL-33 protein in cell lysates and cell culture supernates were tested in ELISAs (R&D Systems). Immunoprecipitation of HA-tagged proteins was performed using a Pierce HA tag magnetic IP/co-IP kit (ThermoFisher) according to the manufacturer's recommendations. To assure nuclear membrane disruption, lysates were passed 10 times through a 26-gauge hypodermic needle prior to proceeding with immunoprecipitation. Samples were then incubated with the provided magnetic beads bound to a high-affinity mouse IgG1 monoclonal antibody that recognizes the HA-epitope tag. Bound proteins were eluted using a low-pH elution buffer, and the beads were removed using a magnetic stand. Non-reducing loading buffer was added with DTT to a final concentration of 50 mm and then boiled for 10 min to prepare for reducing gel analysis.
IPO5-Myc immunoprecipitation was performed using Pierce Myc-tag IP/co-IP kit (ThermoFisher) according to the manufacturer's recommendations to purify recombinant IPO5. For in vitro FLIL33-IPO5 binding assays, the immunoprecipitated and eluted IPO5 preparation was diluted 1:10 in TBS containing 0.05% Tween 20 and 2.5% bovine serum albumin. 300 μl of this mixture was added to HA magnetic beads that were preincubated with HA-tagged protein. Following 2 h of incubation at room temperature, beads were washed, and elution procedures were completed, followed by Western blotting analyses of the eluate.
Nuclear and cytoplasmic extracts were prepared using an active motif (Carlsbad, CA) nuclear extract kit per the manufacturer's recommendations. The cells were first collected in cold PBS with phosphatase inhibitors, collected by centrifugation at 800 × g, and then resuspended in a hypotonic solution with detergent to release proteins from the cytoplasmic compartment while keeping the nuclei intact. Cytoplasmic disruption was confirmed using light microscopy, and the lysate was centrifuged at 14,000 rpm for 10 min to form the nuclear pellet. The supernatant was collected as the cytoplasmic fraction. The remaining nuclear pellet was lysed in the presence of protease inhibitors. Protein concentration was determined using a Pierce BCA protein assay kit. Each fraction was diluted 1:1 with Laemmli sample buffer and boiled for 10 min to prepare for reducing gel analysis.
IPO5 RNA interference was performed using FlexiTube siRNA (Qiagen). The cells were transfected with siRNA or AllStars negative control siRNA (Qiagen) at a concentration of 300 nm using electroporation as described above and incubated for 48 h in a 75-cm2 flask. A second transfection was performed with the recombinant plasmid, and the cells were incubated an additional 48 h in 6-well plates. IPO-5 knockdown was confirmed by Western blotting.
Western blotting was performed using the Novex (ThermoFisher) system. All samples were run on Tris-glycine gels per the manufacturer's recommendations. Wet transfer was performed using a XCell II blot module (ThermoFisher) at 22 V for 2 h to a PVDF membrane (Bio-Rad). All membranes were blocked and incubated with primary and secondary antibody using Tris-buffered saline with 0.1% Tween 20 and 5% bovine serum albumin. The antibodies used include anti-HA antibody from Abcam (Cambridge, UK; catalog no. ab18181), anti-IPO5 antibody from Sigma–Aldrich (catalog no. SAB4200178), anti-Myc antibody from Cell Signaling Technology (catalog no. 2278), β-actin antibody from Cell Signaling Technology (catalog no. 4967S), HDAC2 from Abcam (catalog no. 32117), α-tubulin from Cell Signaling Technology (catalog no. 2144), and GAPDH from Cell Signaling Technology (catalog no. 5174). All listed antibodies were developed in rabbit, except the anti-HA antibody, which was mouse. Secondary antibodies used were goat anti-rabbit from EMD Millipore (Billerica, MA) (catalog no. 12–348) and goat anti-mouse from Santa Cruz Biotechnology (Dallas, TX) (catalog no. sc-2005). Following incubation with the corresponding HRP-bound secondary antibody, membranes were developed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and autoradiography film. Stripping was performed using ReBlot Plus strong antibody stripping solution (EMD Millipore).
For immunocytochemistry, cultured HEK293T cells were transfected as described above and seeded at a density of 15–20 × 103 cells/well in two-well chamber slides (Falcon; Becton Dickinson Labware). Staining was performed 48 h after transfection. The cells were fixed by incubating with methanol at 20 °C for 20 min, washed with PBS, and blocked for 1 h at room temperature with 5% BSA in 0.1% Tween 20 in PBS. The cells were incubated overnight with primary antibody to HA (Abcam) at 1:100 dilution, washed, incubated with a secondary FITC-labeled goat anti-mouse antibody from Jackson ImmunoResearch (West Grove, PA) at 1:1,000 dilution for 1 h, and visualized with a Keyence (Itasca, IL) BZ-X700 fluorescent microscope.
Author contributions
S. P. A. and I. G. L. conceived the idea of the study. A. C., S. P. A., and I. G. L. designed the experiments. A. C., Z. N., R. F., V. L., B. S. H., M. V. S., and N. W. T. performed the experiments. A. C., N. G. S., S. P. A., and I. G. L. analyzed and interpreted the data. A. C., S. P. A., and I. G. L. composed the figures and wrote the manuscript. All authors reviewed the results, contributed to editing the manuscript, and approved its submission.
Acknowledgment
We thank Jean-Paul Courneya for assistance with database searches and sequence alignments.
This work was supported, in whole or in part, by NHLBI, National Institutes of Health Grant R01HL126897 (to S. P. A.). This work was also supported by Veterans Affairs Merit Awards I01CX000101 (to I. G. L.) and I01BX002499 (to S. P. A.) and a Scleroderma Foundation award (to S. P. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MIL33
- mature IL-33
- Th2
- T helper cell 2
- FLIL33
- full-length interleukin-33
- IPO5
- importin-5
- AdV
- adenovirus
- NHLF
- normal human lung fibroblast
- aa
- amino acid(s)
- CRISPR
- clustered regularly interspaced short palindromic repeats
- IP
- immunoprecipitation
- HDAC2
- histone deacetylase 2.
References
- 1. De la Fuente M., MacDonald T. T., and Hermoso M. A. (2015) The IL-33/ST2 axis: role in health and disease. Cytokine Growth Factor Rev. 26, 615–623 [DOI] [PubMed] [Google Scholar]
- 2. Molofsky A. B., Savage A. K., and Locksley R. M. (2015) Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity 42, 1005–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cayrol C., and Girard J.-P. (2009) The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. U.S.A. 106, 9021–9026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kakkar R., Hei H., Dobner S., and Lee R. T. (2012) Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J. Biol. Chem. 287, 6941–6948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liew F. Y., Pitman N. I., and McInnes I. B. (2010) Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat. Rev. Immunol. 10, 103–110 [DOI] [PubMed] [Google Scholar]
- 6. Holtzman M. J., Byers D. E., Brett J. A., Patel A. C., Agapov E., Jin X., and Wu K. (2014) Linking acute infection to chronic lung disease: the role of IL-33-expressing epithelial progenitor cells. Ann. Am. Thorac. Soc. 11 (suppl. 5), S287–S291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Byers D. E., Alexander-Brett J., Patel A. C., Agapov E., Dang-Vu G., Jin X., Wu K., You Y., Alevy Y., Girard J. P., Stappenbeck T. S., Patterson G. A., Pierce R. A., Brody S. L., and Holtzman M. J. (2013) Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J. Clin. Invest. 123, 3967–3982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luzina I. G., Kopach P., Lockatell V., Kang P. H., Nagarsekar A., Burke A. P., Hasday J. D., Todd N. W., and Atamas S. P. (2013) Interleukin-33 potentiates bleomycin-induced lung injury. Am. J. Respir. Cell Mol. Biol. 49, 999–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luzina I. G., Todd N. W., Sundararajan S., and Atamas S. P. (2015) The cytokines of pulmonary fibrosis: much learned, much more to learn. Cytokine 74, 88–100 [DOI] [PubMed] [Google Scholar]
- 10. Lüthi A. U., Cullen S. P., McNeela E. A., Duriez P. J., Afonina I. S., Sheridan C., Brumatti G., Taylor R. C., Kersse K., Vandenabeele P., Lavelle E. C., and Martin S. J. (2009) Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31, 84–98 [DOI] [PubMed] [Google Scholar]
- 11. Talabot-Ayer D., Lamacchia C., Gabay C., and Palmer G. (2009) Interleukin-33 is biologically active independently of caspase-1 cleavage. J. Biol. Chem. 284, 19420–19426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luzina I. G., Pickering E. M., Kopach P., Kang P. H., Lockatell V., Todd N. W., Papadimitriou J. C., McKenzie A. N., and Atamas S. P. (2012) Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion. J. Immunol. 189, 403–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ali S., Mohs A., Thomas M., Klare J., Ross R., Schmitz M. L., and Martin M. U. (2011) The dual function cytokine IL-33 interacts with the transcription factor NF-κB to dampen NF-κB-stimulated gene transcription. J. Immunol. 187, 1609–1616 [DOI] [PubMed] [Google Scholar]
- 14. Pei C., Barbour M., Fairlie-Clarke K. J., Allan D., Mu R., and Jiang H. R. (2014) Emerging role of interleukin-33 in autoimmune diseases. Immunology 141, 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carriere V., Roussel L., Ortega N., Lacorre D.-A., Americh L., Aguilar L., Bouche G., and Girard J.-P. (2007) IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. 104, 282–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baekkevold E. S., Roussigné M., Yamanaka T., Johansen F.-E., Jahnsen F. L., Amalric F., Brandtzaeg P., Erard M., Haraldsen G., and Girard J.-P. (2003) Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am. J. Pathol. 163, 69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roussel L., Erard M., Cayrol C., and Girard J.-P. P. (2008) Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Reports 9, 1006–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lefrançais E., Duval A., Mirey E., Roga S., Espinosa E., Cayrol C., and Girard J. P. (2014) Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. U.S.A. 111, 15502–15507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bessa J., Meyer C. A., de Vera Mudry M. C., Schlicht S., Smith S. H., Iglesias A., and Cote-Sierra J. (2014) Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J. Autoimmun. 55, 33–41 [DOI] [PubMed] [Google Scholar]
- 20. Gordon E. D., Simpson L. J., Rios C. L., Ringel L., Lachowicz-Scroggins M. E., Peters M. C., Wesolowska-Andersen A., Gonzalez J. R., MacLeod H. J., Christian L. S., Yuan S., Barry L., Woodruff P. G., Ansel K. M., Nocka K., et al. (2016) Alternative splicing of interleukin-33 and type 2 inflammation in asthma. Proc. Natl. Acad. Sci. U.S.A. 113, 8765–8770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kopach P., Lockatell V., Pickering E. M., Haskell R. E., Anderson R. D., Hasday J. D., Todd N. W., Luzina I. G., and Atamas S. P. (0500) IFN-γ directly controls IL-33 protein level through a STAT1- and LMP2-dependent mechanism. J. Biol. Chem. 289, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chook Y. M., and Süel K. E. (2011) Nuclear import by karyopherin-βs: recognition and inhibition. Biochim. Biophys. Acta 1813, 1593–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhong Y., Wang Y., Yang H., Ballar P., Lee J. G., Ye Y., Monteiro M. J., and Fang S. (2011) Importin β interacts with the endoplasmic reticulum-associated degradation machinery and promotes ubiquitination and degradation of mutant alpha1-antitrypsin. J. Biol. Chem. 286, 33921–33930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chung K. M., Cha S. S., and Jang S. K. (2008) A novel function of karyopherin β3 associated with apolipoprotein A-I secretion. Mol. Cells 26, 291–298 [PubMed] [Google Scholar]
- 25. Baas R., Sijm A., van Teeffelen H. A., van Es R., Vos H. R., and Marc Timmers H. T. (2016) Quantitative proteomics of the SMAD (suppressor of mothers against decapentaplegic) transcription factor family identifies importin 5 as a bone morphogenic protein receptor SMAD-specific importin. J. Biol. Chem. 291, 24121–24132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi J., and Matsuura Y. (2013) Structural basis for cell-cycle-dependent nuclear import mediated by the karyopherin Kap121p. J. Mol. Biol. 425, 1852–1868 [DOI] [PubMed] [Google Scholar]
- 27. Wischnewski J., Sölter M., Chen Y., Hollemann T., and Pieler T. (2000) Structure and expression of Xenopus karyopherin-β3: definition of a novel synexpression group related to ribosome biogenesis. Mech. Dev. 95, 245–248 [DOI] [PubMed] [Google Scholar]
- 28. Jäkel S., and Görlich D. (1998) Importin β, transportin, RanBP5 and RanBP7 mediate nuclear import of ribosomal proteins in mammalian cells. EMBO J. 17, 4491–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deane R., Schäfer W., Zimmermann H. P., Mueller L., Görlich D., Prehn S., Ponstingl H., and Bischoff F. R. (1997) Ran-binding protein 5 (RanBP5) is related to the nuclear transport factor importin-β but interacts differently with RanBP1. Mol. Cell Biol. 17, 5087–5096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ross A. E., Vuica M., and Desiderio S. (2003) Overlapping signals for protein degradation and nuclear localization define a role for intrinsic RAG-2 nuclear uptake in dividing cells. Mol. Cell Biol. 23, 5308–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huber F. M., and Hoelz A. (2017) Molecular basis for protection of ribosomal protein L4 from cellular degradation. Nat. Commun. 8, 14354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murphy G. E. J., Xu D., Liew F. Y., and McInnes I. B. (2010) Role of interleukin 33 in human immunopathology. Ann. Rheum. Dis. 69 (Suppl. 1), i43–i47 [DOI] [PubMed] [Google Scholar]
- 33. Luheshi N. M., Rothwell N. J., and Brough D. (2009) Dual functionality of interleukin-1 family cytokines: implications for anti-interleukin-1 therapy. Br. J. Pharmacol. 157, 1318–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohta S., Tago K., Funakoshi-Tago M., Matsugi J., and Yanagisawa K. (2016) Intracellular NF-HEV/IL-33 harbors essential roles in Ras-induced cellular transformation by contributing to cyclin D1 protein synthesis. Cell Signal. 28, 1025–1036 [DOI] [PubMed] [Google Scholar]
- 35. Oshio T., Komine M., Tsuda H., Tominaga S. I., Saito H., Nakae S., and Ohtsuki M. (2017) Nuclear expression of IL-33 in epidermal keratinocytes promotes wound healing in mice. J. Dermatol. Sci. 85, 106–114 [DOI] [PubMed] [Google Scholar]
- 36. Shan J., Oshima T., Wu L., Fukui H., Watari J., and Miwa H. (2016) Interferon γ-induced nuclear interleukin-33 potentiates the release of esophageal epithelial derived cytokines. PLoS One 11, e0151701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tao L., Chen C., Song H., Piccioni M., Shi G., and Li B. (2014) Deubiquitination and stabilization of IL-33 by USP21. Int. J. Clin. Exp. Pathol. 7, 4930–4937 [PMC free article] [PubMed] [Google Scholar]
- 38. Ni Y., Tao L., Chen C., Song H., Li Z., Gao Y., Nie J., Piccioni M., Shi G., and Li B. (2015) The deubiquitinase USP17 regulates the stability and nuclear function of IL-33. Int. j. Mol. Sci. 16, 27956–27966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anderson R. D., Haskell R. E., Xia H., Roessler B. J., and Davidson B. L. (2000) A simple method for the rapid generation of recombinant adenovirus vectors. Gene Ther. 7, 1034–1038 [DOI] [PubMed] [Google Scholar]
- 40. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]