

Abstract
Phosphocholine (pCho) is a precursor for phosphatidylcholine and osmoprotectants in plants. In plants, de novo synthesis of pCho relies on the phosphobase methylation pathway. Phosphoethanolamine methyltransferase (PMT) catalyzes the triple methylation of phosphoethanolamine (pEA) to pCho. The plant PMTs are di-domain methyltransferases that divide the methylation of pEA in one domain from subsequent methylations in the second domain. To understand the molecular basis of this architecture, we examined the biochemical properties of three Arabidopsis thaliana PMTs (AtPMT1–3) and determined the X-ray crystal structures of AtPMT1 and AtPMT2. Although each isoform synthesizes pCho from pEA, their physiological roles differ with AtPMT1 essential for normal growth and salt tolerance, whereas AtPMT2 and AtPMT3 overlap functionally. The structures of AtPMT1 and AtPMT2 reveal unique features in each methyltransferase domain, including active sites that use different chemical mechanisms for phosphobase methylation. These structures also show how rearrangements in both the active sites and the di-domain linker form catalytically competent active sites and provide insight on the evolution of the PMTs in plants, nematodes, and apicomplexans. Connecting conformational changes with catalysis in modular enzymes, like the PMT, provides new insights on interdomain communication in biosynthetic systems.
Keywords: Arabidopsis thaliana, enzyme kinetics, X-ray crystallography, protein structure, S-adenosylmethionine (SAM)
Introduction
In plants, choline is an essential metabolite that serves as a building block for phosphocholine (pCho),2 the predominant membrane phospholipid phosphatidylcholine (PtdCho), and the osmoprotectants choline O-sulfate and glycine betaine (1–3). Synthesis of PtdCho occurs through three different pathways in various organisms (Fig. 1A). The major route from choline to PtdCho in mammals, fungi, and some bacteria is the de novo choline or Kennedy pathway, which involves the phosphorylation of choline and attachment of pCho to lipids (1). In yeast and mammals, the Bremer-Greenberg pathway provides a second route to the phospholipid and involves methylation of phosphatidylethanolamine to PtdCho (4). In plants, nematodes, and Plasmodium apicomplexan parasites, conversion of phosphoethanolamine (pEA) to pCho via the phosphobase methylation pathway is the primary route to pCho and its entry into the Kennedy pathway (5–12). Phosphoethanolamine methyltransferases (PMT; also known as PEAMT) catalyze the reactions of the phosphobase methylation pathway.
Figure 1.
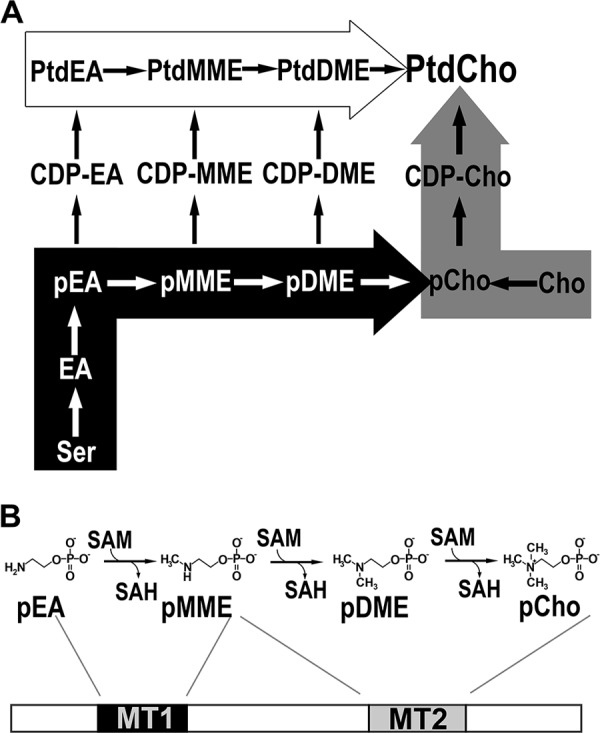
Plant phosphatidylcholine biosynthesis and the di-domain architecture of the type I PMT from plants. A, phosphatidylcholine biosynthesis. The de novo choline or Kennedy pathway (gray arrow), the Bremer-Greenberg pathway (white arrow), and the phosphobase methylation pathway (black arrow) are shown. Metabolite names consist of a prefix (p, phospho; CDP-, cytidine 5′-diphosphate; or Ptd, phosphatidyl) and a core name (EA, ethanolamine; MME, monomethylethanolamine; DME, dimethylethanolamine; Cho, choline). B, reactions and domain structure of the type I PMT from plants. Methylation of phosphoethanolamine (pEA) to phosphomethylethanolamine (pMME) is catalyzed by the first methyltransferase domain (MT1) and the methylation reactions converting pMME to phosphocholine (pCho) catalyzed by the second methyltransferase domain (MT2).
Although the PMT from plants, nematodes, and apicomplexans catalyze a sequential series of S-adenosylmethionine (SAM)-dependent methylation reactions that convert pEA to phosphomonomethylethanolamine (pMME), phosphodimethylethanolamine (pDME), and pCho, the organization of methyltransferase domains in the enzymes differs in each organism. The plant PMT (type I PMT) are bi-functional di-domain proteins with tandem methyltransferase domains, in which the N-terminal domain (MT1) catalyzes the conversion of pEA to pMME and the C-terminal domain (MT2) methylates pMME to pDME and pDME to pCho (Fig. 1B) (8, 13, 14). The type II PMT from Plasmodium falciparum and other apicomplexan parasites are half the length of the plant enzyme and use a single MT2-like active site to methylate all three phosphobases (9, 15–20). In nematodes, such as Caenorhabditis elegans and Haemonchus contortus, two genes encode the mono-functional di-domain type III enzymes, which contain either the N-terminal methyltransferase domain (MT1) or the C-terminal methyltransferase domain (MT2), along with vestigial versions of the other domain (10, 11, 17, 20).
The type I PMT from plants were first identified from Spinacia oleracea (spinach) by functional complementation of yeast mutants and shown to convert pEA to pCho (8). Likewise, cloning and biochemical analysis of two PMT from Triticum aestivum (wheat) indicated a similar domain organization (14, 21). Reports describe clones for Brassica napus (canola) and Zea mays (corn) PMT, but these have not been tested for biochemical function (22, 23). There are three PMT encoded in the genome of Arabidopsis thaliana (thale cress), AtPMT1 (At3g18000; xipotl1), AtPMT2 (At1g48600), and AtPMT3 (At1g73600). AtPMT1 is the best studied isoform in this model plant and, like the spinach PMT, was first identified by functional complementation of a yeast mutant (13). Subsequent work demonstrated that silencing of atpmt1 resulted in temperature-sensitive male sterility and salt hypersensitivity and that the Arabidopsis xipotl1 mutant, which disrupts atpmt1, is characterized by a short primary root, increased lateral root formation, and short epidermal cells (24, 25). Limited analysis of AtPMT2 suggests that this protein may not catalyze the methylation of pEA, but only the conversion of pMME to pCho (26). No phenotypic or functional information is available on AtPMT3, and no detailed biochemical examination of either AtPMT1 or AtPMT3 has been described.
The plant PMT are important for the synthesis of PtdCho; however, the biochemical roles of the three PMT isoforms in the phosphobase methylation pathway of Arabidopsis remain to be fully examined. In addition, the structural basis for the di-domain architecture of the type I PMT from plants that separate enzymatic steps of the pathway (i.e. pEA to pMME in MT1 and pMME to pDME and pDME to pCho in MT2) is unclear. Here, we present the biochemical analysis of AtPMT1–3, which indicates that although each isoform catalyzes all the steps in the phosphobase methylation pathway, atpmt1 is the only isoform with a growth phenotype and hypersensitivity to salinity in T-DNA knock-out plants. This appears linked to expression differences of atpmt1 versus atpmt2 and atpmt3. The X-ray crystal structures of AtPMT1 in complex with S-adenosylhomocysteine (SAH) and pCho and of AtPMT2 in complex with SAH at 1.34- and 1.50-Å resolution, respectively, provide details that differentiate the MT1 and MT2 domains both structurally and mechanistically. These structures also reveal the conformational flexibility in the di-domain architecture and provide insight on the molecular evolution of the different types of PMT in various organisms.
Results
Biochemical analysis of Arabidopsis PMT
To examine the biochemical activities of the three Arabidopsis PMT isoforms, the coding region of each enzyme was PCR-amplified from a cDNA library with the obtained sequences matching those reported in GenBankTM. AtPMT1 (491 amino acids; 56.1 kDa) shares 89 and 80% amino acid sequence identity with AtPMT2 (491 amino acids; 56.1 kDa) and AtPMT3 (504 amino acids; 57.8 kDa), respectively. AtPMT2 and AtPMT3 are 80% identical. The AtPMTs share 33–36% sequence identity with the nematode PMT and 42% identity with the Plasmodium enzyme, but this is localized entirely to the MT2 domain of the plant enzymes (supplemental Fig. S1). The three AtPMTs were expressed in Escherichia coli as N-terminal His-tagged proteins and purified using Ni2+-affinity and size-exclusion chromatographies. Each isoform migrated with a molecular mass of ∼55 kDa on SDS-PAGE (supplemental Fig. S2, A–C, insets) and eluted from the size-exclusion column as a monomer (∼50–60 kDa) (supplemental Fig. S2D), which was similar to the spinach and nematode enzymes (8, 10, 11, 17, 20).
Steady-state kinetic parameters for AtPMT1–3 were determined using pEA, pMME, or pDME and SAM as substrates (Table 1). The kcat and Km values for pEA and SAM were similar to those reported for the spinach, wheat, nematode, and Plasmodium enzymes (8–11, 20, 21). Comparison of kcat/Km values indicates that AtPMT1 displays a modest 3-fold preference for pEA versus either pMME or pDME. Both AtPMT2 and AtPMT3 also prefer pEA with a 1.5–2-fold higher catalytic efficiency compared with pMME and pDME.
Table 1.
Steady-state kinetic analysis of AtPMT isoforms
Enzyme assays were performed using purified protein as described under “Experimental procedures.” Kinetic parameters for each phosphobase substrate were determined at 2 mm SAM. Kinetic parameters for SAM were determined at 3 mm pEA and at 10 mm pDME. Values shown are the mean ± S.D. for n = 3.
| Substrate | Parameter | AtPMT1 | AtPMT2 | AtPMT3 |
|---|---|---|---|---|
| pEA | kcat (min−1) | 12.3 ± 0.6 | 16.1 ± 2.5 | 5.2 ± 1.0 |
| Km (μm) | 156 ± 24 | 408 ± 143 | 284 ± 96 | |
| kcat/Km (m−1 s−1) | 1,314 | 658 | 305 | |
| pMME | kcat (min−1) | 8.2 ± 0.4 | 12.7 ± 0.6 | 2.5 ± 0.3 |
| Km (μm) | 278 ± 38 | 577 ± 72 | 201 ± 59 | |
| kcat/Km (m−1 s−1) | 489 | 367 | 209 | |
| pDME | kcat (min−1) | 13.1 ± 1.4 | 16.0 ± 1.0 | 2.4 ± 0.2 |
| Km (μm) | 507 ± 115 | 653 ± 98 | 249 ± 69 | |
| kcat/Km (m−1 s−1) | 433 | 408 | 161 | |
| SAM (pEA) | kcat (min−1) | 7.8 ± 0.4 | 16.0 ± 0.7 | 3.7 ± 0.2 |
| Km (μm) | 89.7 ± 11.8 | 308 ± 33 | 69.1 ± 9.6 | |
| kcat/Km (m−1 s−1) | 1,453 | 866 | 897 | |
| SAM (pDME) | kcat (min−1) | 9.9 ± 0.9 | 12.8 ± 0.3 | 1.5 ± 0.1 |
| Km (μm) | 215 ± 41 | 212 ± 13 | 92.8 ± 7.5 | |
| kcat/Km (m−1 s−1) | 767 | 1,006 | 266 |
Multiple studies suggest that the plant PMT contain two methyltransferase domains defined by SAM-binding sequence motifs (8, 13, 14). Isothermal titration calorimetry (ITC) was used to evaluate the stoichiometry and energetics of ligand binding to AtPMT2. The other two isoforms were not stable enough during the time of experiment for analysis. Titration of AtPMT2 with SAM, pEA, and pCho did not yield sufficient heat to accurately determine binding constants. Addition of SAH to AtPMT2 (supplemental Fig. S3) showed a 2:1 ligand/protein stoichiometry consistent with the predicted di-domain structure. The Kd value for SAH (4.6 ± 0.4 μm) and the binding energy (ΔG = −7.19 ± 0.20 kcal mol−1), which was largely driven by the enthalpic contribution (ΔH = −10.8 ± 0.2 kcal mol−1; TΔS = 3.6 kcal mol−1), were similar to those reported for the nematode and Plasmodium PMT (15–17).
In planta analysis of Arabidopsis PMT
Examination of the role of each AtPMT isoform in Arabidopsis has been limited to the xipotl1 mutant phenotype of atpmt1, which suggests a key role for this enzyme in root development (24, 25), and a report of a change in one phospholipid form in atpmt2 knock-out lines (26). For a comparison of the role of each isoform in planta, T-DNA insertion mutants for each atpmt were obtained with the knock-out confirmed by reverse transcript PCR (Fig. 2, A and B). As described previously (24, 25), the atpmt1 mutant displayed a short root phenotype under normal growth conditions and hypersensitivity, as determined by root length, under salt-stress conditions. Comparison of all three lines showed the reported phenotype with atpmt1 and no phenotypic differences with either atpmt2 or atpmt3 under normal conditions (Fig. 2C). Under salt stress, atpmt1 also showed the hypersensitive short primary root phenotype with the atpmt2 and atpmt3 lines comparable with wild type (Fig. 2D). Earlier work suggested that NaCl treatment induces PMT expression (8). Comparison of isoform expression changes in Arabidopsis seedlings treated with either 0 or 50 mm NaCl indicates that expression of atpmt1 increased 2-fold relative to untreated seedlings with unchanged atpmt2 expression and a 2-fold reduction in atpmt3 transcript (Fig. 2E). At the metabolic level, previous studies describe a modest 10% decrease in PtdCho in roots and leaves of the atpmt1/xipotl1 mutant (25) and only a 2-fold decrease for one polar lipid (34:3) species in the atpmt2 knock-outs (26). We compared the polar lipid profiles of wild-type and atpmt3 knock-out plants, which indicate no significant differences (supplemental Fig. S4). As our results suggest a singular role for AtPMT1 versus functional redundancy of AtPMT2 and AtPMT3, the tissue-specific expression for each isoform was examined by qRT-PCR using gene-specific primers (Fig. 2F). In roots, atpmt1 and atpmt2 are more highly expressed than atpmt3, but in leaves the expression of atpmt2 and atpmt3 predominates. The differences in expression of atpmt1 in root tissue compared with the higher transcript levels of atpmt2 and atpmt3 in leaf tissues, along with induction of atpmt1 expression by NaCl, likely explain the effects of the atpmt1/xipotl1 mutant under salt stress versus the phenotype of the knock-out lines for the other two isoforms.
Figure 2.

Comparison of atpmt T-DNA knock-out mutants and tissue-specific expression of AtPMT isoforms. A, schematic of the genomic structures of atpmt1, atpmt2, and atpmt3 with T-DNA insertion sites indicated. Boxes represent exons, and the lines represent introns. B, RT-PCR analysis of homozygous mutant plants using AtPMT isoform-specific primers. β-Tubulin (TUB) was used as a control. C, effect of T-DNA knock-out on root length. Wild-type, atpmt1, atpmt2, and atpmt3 A. thaliana seedlings were grown for 10 days on half-strength MS agar plates and photographed. D, effect of T-DNA knock-out on salt tolerance. As in C, seedlings were grown for 10 days on half-strength MS agar plates ± 50 mm NaCl, and then primary root length was determined. Error bars represent the standard error of the mean (n = 25). E, effect of NaCl treatment on atpmt1–3 expression. Wild-type A. thaliana seedlings were grown as in D. Total RNA was extracted from 7-day-old seedlings for qRT-PCR amplifications. Relative expression values for atpmt1 (black), atpmt2 (white), and atpmt3 (gray) in NaCl-untreated and -treated seedlings were normalized against tub gene expression. Error bars are the standard error of the mean (n = 3). F, tissue-specific expression of AtPMT isoforms. Total RNA was extracted from 7-day-old seedlings and 1-month-old plant tissues. The qRT-PCR amplifications were performed in triplicate independently for each target gene and the data averaged. The relative expression values for atpmt1 (black), atpmt2 (white), and atpmt3 (gray) in different tissues were normalized against act7 gene expression. Error bars represent the standard error of the mean (n = 6).
X-ray crystal structure of AtPMT1: Overall di-domain structure
To understand the molecular basis for the distinct methyltransferase activities of the PMT from plants, the 1.34-Å resolution X-ray crystal structure of AtPMT1 in complex with SAH and pCho was determined by molecular replacement (Table 2). The overall structure of AtPMT1 reveals the N-terminal MT1 and C-terminal MT2 domains connected by a linker between β1g and α9 (Fig. 3A). The MT1 domain, which is responsible for conversion of pEA to pMME, contains a 7-stranded β-sheet (β1a–β1g) and multiple α-helices (α1–α8). The MT2 domain, which catalyzes the methylations of pMME to pDME and pDME to pCho, retains the core β-sheet structure (β2a–β2g) but with 11 α-helices (α9–α19) arranged differently compared with MT1 (Fig. 3, B and C, left). Clear electron density for SAH and pCho in the active site of each domain was observed (supplemental Fig. S5, A and B). In each domain, SAH binds along the central β-sheet; however, the regions around the phosphobase-binding site comprise structurally different arrangements of α-helices and loops that cap the active site of each domain. For example, the β1e–α7 loop caps the MT1 active site, whereas two α-helices (α18 and α19) serve a similar role in the MT2 active site. Thus, the MT1 and MT2 domains are structurally distinct in terms of both overall three-dimensional fold and local active-site architecture.
Table 2.
Summary of x-ray crystallographic data collection and refinement statistics
| Crystal | AtPMT1·SAH·pCho | AtPMT2·SAH |
|---|---|---|
| Space group | P21 | P21 |
| Cell dimensions | a = 49.26 Å, b = 86.03 Å, c = 68.24 Å; β = 106.2° | a = 48.06 Å, b = 90.22 Å, c = 122.9 Å; β = 100.1° |
| Data collection | ||
| Wavelength (Å) | 0.979 | 0.979 |
| Resolution (Å) (highest shell) | 26.7–1.34 (1.36–1.34) | 30.0–1.50 (1.53–1.50) |
| Reflections (total/unique) | 238,716/121,932 | 742,803/162,824 |
| Completeness (highest shell) | 99.0% (99.4%) | 99.1% (99.6%) |
| 〈I/σ〉 (highest shell) | 30.2 (2.0) | 37.4 (2.1) |
| Rsym (highest shell) | 6.2 (53.9)% | 16.2 (60.6)% |
| Refinement | ||
| Rcryst/Rfree | 15.2/16.6% | 16.0/17.9% |
| No. of protein atoms | 4,022 | 7,543 |
| No. of water molecules | 650 | 1,175 |
| No. of ligand atoms | 74 | 104 |
| r.m.s.d., bond lengths (Å) | 0.006 | 0.006 |
| r.m.s.d., bond angles (°) | 1.106 | 1.071 |
| Average B-factor (Å2), protein, water, ligand | 21.3, 35.8, 14.3 | 29.1, 42.2, 22.2 |
| Stereochemistry: favored, allowed, outlier | 97.6, 2.4, 0.0% | 97.4, 2.4, 0.2% |
Figure 3.

Overall three-dimensional structure of AtPMT1. A, di-domain structure of AtPMT1. The MT1 and MT2 domains are indicated in the ribbon diagram. The schematic represents α-helices as gold cylinders and β-strands as blue arrows with numbering as shown. SAH and pCho are represented as space-filling molecules. The β1e–α7 loop that caps the active site of the MT1 domain is indicated. B, comparison of the secondary structure features of the MT1 domain of AtPMT1 (left) and the non-catalytic vestigial N-terminal domain of HcPMT2 (right). Coloring of the MT1 domain is as in A with the α9-helix of the AtPMT1 MT2 domain shown for reference. The schematic of the vestigial domain shows α-helices as rose cylinders and β-strands as green arrows. C, comparison of the secondary structure features of the MT2 domain of AtPMT1 (left) and the non-catalytic vestigial C-terminal domain of HcPMT1 (right). Coloring is as in A and B. The AtPMT1 MT2 domain α9 is shown for reference. The position of the insertion loop in the vestigial domain of HcPMT1 that occludes the SAM/SAH-binding site is indicated.
The structure of AtPMT1 reveals the conservation of the overall fold of each MT domain with the respective catalytic domains from the type III PMT of the parasitic nematode H. contortus (17) but also key differences with the vestigial non-catalytic domains of the nematode enzymes (Fig. 3, B and C). The MT1 domain of AtPMT1 is similar to the N-terminal catalytic domain of HcPMT1 (1.2 Å2 root mean squared deviation (r.m.s.d.) for 224 Cα atoms; Z = 31.9; 34% identity) but differs from the vestigial non-catalytic N-terminal domain of HcPMT2 (2.4 Å2 r.m.s.d. for 192 Cα atoms; Z = 20.0; 14% identity). The loss of regions corresponding to α1, α2, α5, and β1c (Fig. 3B), along with sequence variations (supplemental Fig. S1), yields a vestigial MT1 domain in HcPMT2. The MT2 domain of AtPMT1 shares a common fold (1.0 Å2 r.m.s.d. for 255 of Cα atoms; Z = 40.5; 42% identity) with the type II PMT from P. falciparum (15, 16) and the C-terminal domain of HcPMT2 (1.1 Å2 r.m.s.d. for 257 Cα atoms; Z = 40.3; 41% identity). It should be noted that a DALI search using the MT2 domain of AtPMT1 does not identify any portion of the nematode PMT1 as similar in fold. Comparison of the AtPMT1 MT2 domain and the vestigial non-catalytic C-terminal domain of HcPMT1 highlights key structural differences, including shortening of the α18 and α19 helices compared with the plant enzyme and a longer β2a–α12 loop, which occludes the SAM/SAH-binding site (Fig. 3C). These changes lead to loss of the MT2 domain activity in HcPMT1.
X-ray crystal structure of AtPMT2: Changes in di-domain structure and formation of the MT2 active site
The reported structures of the nematode PMT (17) and the structure of AtPMT1 (Fig. 3A) show the N- and C-terminal domains oriented adjacent to each other; however, the linker region connecting the domains could be conformationally flexible. During initial crystallization efforts, AtPMT2 (89% amino acid identity to AtPMT1) was used to increase the odds of obtaining crystals of a plant PMT. Fortuitously, the X-ray crystal structure of AtPMT2 reveals how changes in di-domain structure are linked to MT2 active-site formation. The 1.50-Å resolution structure of AtPMT2 in complex with SAH was solved by molecular replacement (Table 2) with clear electron density for an SAH molecule observed in each active site (supplemental Fig. S5, C and D). In contrast to previously reported PMT structures, AtPMT2 crystallized in a domain-swapped form (Fig. 4A). In the asymmetric unit, two AtPMT2 molecules pack with the MT1 and MT2 domains in a crossed orientation, instead of a side-by-side arrangement. An overlay of the MT2 domains of AtPMT1 and AtPMT2 emphasizes the global movement of the MT1 domain and shows how a structural transition of an α-helix positions one domain relative to the other (Fig. 4B).
Figure 4.
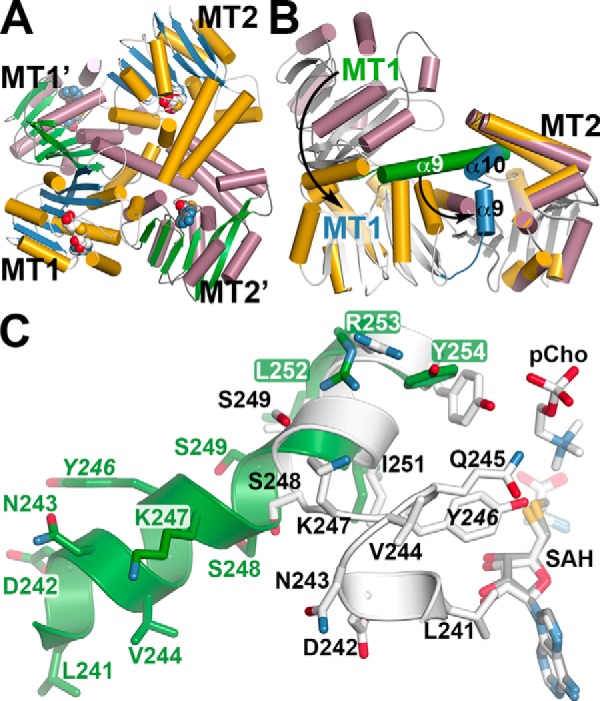
Crystal structure of AtPMT2 and conformational changes in di-domain architecture. A, domain-swapped organization of the MT1 and MT2 domains of AtPMT2. The ribbon diagram shows the arrangement of each domain in two distinct AtPMT2 molecules. For one AtPMT2 molecule, α-helices as gold cylinders and β-strands as blue arrows with each domain labeled (MT1 and MT2) are shown. The α-helices and β-strands of the second AtPMT2 molecule are shown as rose cylinders and green arrows, respectively, with each domain labeled (MT1′ and MT2′). B, comparison of MT1 and MT2 domain positions in AtPMT1 and AtPMT2. The MT2 domains of AtPMT1 (colored gold) and AtPMT2 (colored rose) were overlaid to highlight the movement of the MT1 domain between the two structures. The α9-helix of AtPMT1 is colored green with the corresponding α9 and α10 helices of AtPMT2 colored blue. Arrows emphasize the rearrangements between the two structures leading to active-site formation in the MT2 domain. C, details of the structural transition in the di-domain linker helix. A section of the extended structure of α9 (Leu-241–Tyr-254) in AtPMT2 is shown in green. The position of SAH in the AtPMT2 MT2 active site is shown as a stick model (gray). Portions of α9 (Leu-241–Asn-243), the active-site loop (Val-244–Ser-248), and α10 (Ser-249–Tyr-254), along with SAH and pCho, in AtPMT1 are shown in white stick models.
In the AtPMT2 structure, residues from Gly-236–Gly-259 form the α9-helix that extends between the two domains (Fig. 4B). The same residues in the AtPMT1 structure form α9 (Gly-236–Asn-243) and α10 (Ser-248–Phe-258) with a short loop between. The transition from one extended α-helix (AtPMT2) to two shorter α-helices (AtPMT1), which are packed closer to the core structure of the MT2 domain and in proximity to the active site, effectively shifts the MT1 domain by 30–40 Å and into a position adjacent to the MT2 domain. Structural comparison of the residues in this transition region highlights the repositioning of key residues into the MT2 active site (Fig. 4C). The N-terminal residues of α9 (Gly-236–Asn-243) retain an α-helical structure and swing toward the active site of the MT2 domain; however, Val-244–Ser-248 adopt a loop conformation that places Gln-245 and Tyr-246 into proximity with the bound SAH and pCho in the AtPMT1-active site. The remaining stretch of α9, including Ser-249–Tyr-254, undergoes minor side-chain movement between the two structures to become α10 in the AtPMT1 structure.
Structure of the MT2 domain active site
The changes in di-domain orientation and conformational changes in the linker helix affect access to the MT2 domain active site. In the AtPMT2·SAH complex structure, the extended conformation of α9 leaves the MT2 domain active site open for potential access of substrates (Fig. 5A). In comparison, the AtPMT1·SAH·pCho complex structure suggests that breaking the linker helix into two segments allows for movement of α9 and α10 into the MT2 active site for substrate binding (Fig. 5B). Residues from the core regions of the MT2 domain provide multiple interactions with SAH (Fig. 5C). The amino acid group of SAH binds in a pocket formed by Val-263, Ser-264, and Arg-355. A bidendate interaction between the ribose hydroxyl groups of SAH and Asp-312 anchors the ligand in the active site. Leu-241, Leu-313, Ser-314, Cys-339, and Ile-361 provide a largely apolar binding site for the adenosine ring of SAH with Asp-388 forming a hydrogen bond to the nitrogen of the adenine ring. These interactions position the sulfur group, which would be methylated in SAM, toward the phosphobase-binding site of the active site.
Figure 5.

Structure of the MT2 domain active site. A, open MT2 domain active site in the AtPMT2·SAH complex structure. The surface view shows the MT2 domain active site with bound SAH (stick model). The α9 linker helix, which is extended in this structure, is shown as a green schematic. B, closed MT2 domain active site in the AtPMT1·SAH·pCho complex structure. The surface view shows the MT2 domain active site with bound pCho and SAH (stick models). The α9- and α10-helices, along with Tyr-246 (stick model), are shown in rose. C, MT2 domain SAH/SAM-binding site in the AtPMT1·SAH·pCho complex structure. Secondary structure features are shown in white with side chains contacting SAH shown as stick models. D, stereo view of the MT2 domain phosphobase-binding site in the AtPMT1·SAH·pCho complex structure. The view shows secondary structure as white ribbon diagrams with side chains contacting pCho shown as stick models.
Within the MT2 domain active site of AtPMT1, extensive protein–ligand contacts are involved in phosphobase binding (Fig. 5D). Hydrogen bonds from Gln-245, Tyr-254, Tyr-386, Tyr-400, and Tyr-406 and charge–charge interactions from Arg-404 and Lys-472 lock the pCho phosphate group into the active site to orient the choline group toward SAH. Two residues from the α9–α10 loop form key contacts with pCho, and the amide side-chain nitrogen of Gln-245 forms a hydrogen bond with the bridging oxygen of pCho. As noted above, the structural transition from a single linker helix (i.e. α9) to two helices (α9 and α10) brings Tyr-246 into the active site (Fig. 4). In the functional active site, the hydroxyl group of Tyr-246 is positioned between the amine of pCho (3.9 Å) and the sulfur of SAH (3.8 Å). The Nϵ of His-360 interacts with the hydroxyl group of the tyrosine (2.8 Å), which would allow this residue to serve as a general base to activate Tyr-246. Previous studies of PfPMT and HcPMT2 suggest a critical role for these residues as a catalytic dyad during the methylations of pMME to pDME and pDME to pCho (15–17).
Structure of the MT1 domain active site
In addition to large-scale movements of the di-domain organization of AtPMT1 and AtPMT2, the three-dimensional structures of these enzymes reveal conformational changes in key features surrounding the MT1 active site (Fig. 6A). In the AtPMT2·SAH complex, Glu-7–Glu-18 form α1, but residues 19–41 are disordered. In addition, the β1e–α7 loop (Ser-159–Tyr-176) is oriented away from the active-site cleft where SAH binds. In comparison, the same features in the AtPMT1·SAH·pCho structure alter either their mobility (based on disorder) or position. With both ligands bound in the MT1 active site, α1 and α2 are ordered and contribute residues to pCho binding. There is also a corresponding shift of His-162–Ser-171 in the β1e–α7 loop, which now locks pCho into the active site. As with the MT2 domain active site, binding of SAH/SAM occurs along the core β-sheet of the MT1 domain (Fig. 6B). The amino acid group forms a charge–charge interaction via the carboxylate with Arg-66 and the hydrogen bond interactions with Asn-126. A bidendate interaction between the side chain of Asp-82 and the ribose hydroxyl groups of SAH helps anchor the methyl donor in the active site. The adenosine ring of SAH stacks between Phe-83 and Val-108 with N6 of the adenosine ring hydrogen bonding to Asp-107.
Figure 6.

Structure of the MT1 domain active site. A, conformational changes in the MT1 domain active site. The surface view shows the MT1 domain active site of AtPMT1 with bound pCho and SAH (stick models). The positions of the α1-helix and β1e–α7 loop of the AtPMT2·SAH complex are shown as a ribbon diagram (green). The positions of the α1- and α2-helices and β1e–α7 loop of the AtPMT1·SAH·pCho complex are shown in ribbon diagram (green). B, MT1 domain SAH/SAM-binding site in the AtPMT1·SAH·pCho complex structure. Secondary structure is shown in white with side chains contacting SAH as stick models. C, stereo view of the MT1 domain phosphobase-binding site in the AtPMT1·SAH·pCho complex structure. The view shows secondary structure as white ribbon diagrams with side chains contacting pCho as stick models.
In the phosphobase-binding site (Fig. 6C), the choline group of pCho is oriented into the binding site with the phosphate pointing toward the β1e–α7 loop. Ser-159, Gln-163, Asn-172, and Thr-174 from this loop form hydrogen bonds with the phosphate of pCho, and Arg-169 provides an ionic interaction to the ligand. Deeper into the pocket, residues from α1 (Tyr-15 and Trp-16) and α2 (Met-28, Met-29, and Leu-30), along with Trp-127 and Met-130, provide an apolar environment for phosphobase binding. In the region between pCho and SAH, there is one potential reactive group, Tyr-131, positioned to contribute to the methyl group transfer during conversion of pEA to pMME. The hydroxyl group of Tyr-131 is 3.6 Å from the amine of pCho and 5.5 Å from the sulfur of SAH, but it should be noted that binding of pCho likely alters the local structure to accommodate the methyl groups compared with binding of the unmethylated pEA substrate. In addition, there is a water bound between Tyr-19 (2.9 Å), Thr-174 (2.8 Å), the phosphate of pCho (2.8 Å), and Tyr-131 (2.7 Å). This arrangement of ligands, water, and key residues was also observed in the MT1 active site of HcPMT1 (17).
Mutagenesis of the catalytic tyrosines in the MT1 and MT2 domains of AtPMT2
To test the possible roles of Tyr-131 in MT1 and Tyr-246 in MT2 during the methylation reactions catalyzed by the plant PMT, each tyrosine was mutated to alanine and phenylalanine in AtPMT2 with the resulting mutant proteins expressed and purified as described for the wild-type enzyme. Enzymatic assays with either pEA or pDME as substrates indicates that these two residues are key for function (Fig. 7). The AtPMT2 Y131A and Y131F mutants exhibited decreased activity with pEA with less than 10% specific activity retained but were comparable with wild type and pDME. The AtPMT2 Y246A and Y246F mutants retained wild-type activity with pEA but were nearly inactive with pDME. This suggests that the plant PMT use similar residues as the nematode PMT for catalysis.
Figure 7.
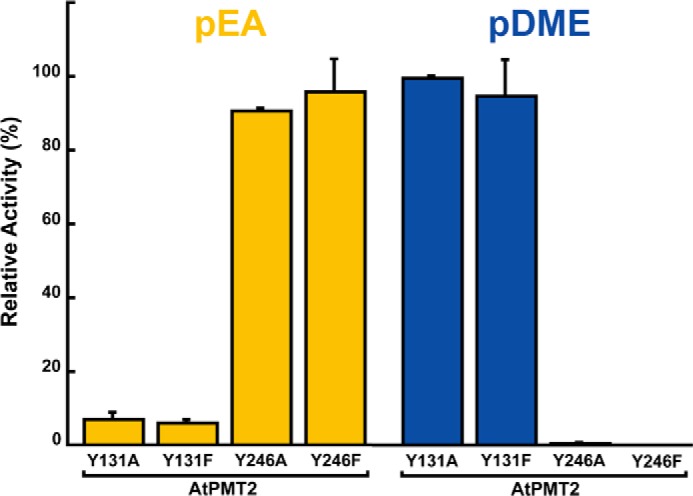
Comparison of MT1 and MT2 activities in AtPMT2 site-directed mutants. Activity of the AtPMT2 Y131A, Y131F, Y246A, and Y246F relative to wild-type enzyme is shown for either pEA (gold) or pDME (blue) as phosphobase substrate. All assays were performed using standard reaction conditions with values shown as the mean ± S.D. for n = 3.
Discussion
Synthesis of pCho in plants provides the headgroup for the predominant membrane phospholipid PtdCho and precursors for synthesis of the key osmoprotectants (1–3). Unlike animals, which can obtain choline from dietary sources, plants use the phosphobase methylation pathway to convert pEA into pCho (Fig. 1A) (5–8). The PMT that catalyze the triple methylation of pEA to pCho are the committed enzymes in this process (8). In plants, the PMT are structurally distinct from the nematode and Plasmodium enzymes (12). The di-domain organization of the bi-functional plant PMT divides the first methylation reaction from subsequent steps in the pathway (Fig. 1B). Here, our work reveals the molecular basis for distinct function and suggests how conformational transitions lead to catalytically competent active sites in each methyltransferase domain.
Initial cloning of the PMT from A. thaliana, S. oleracea (spinach), T. aestivum (wheat), B. napus (canola), and Z. mays (corn) identified homologs (supplemental Fig. S1), but only some were biochemically examined (8, 13, 14, 21–23). Of the three PMT isoforms in Arabidopsis, AtPMT1/XIOPTL is the best studied with in planta experiments showing that a loss of activity leads to temperature-sensitive male sterility, salt hypersensitivity, and altered root development (13, 24, 25). One report examining AtPMT2 suggests that this enzyme lacks the ability to convert pEA to pMME (i.e. the first step in the phosphobase methylation pathway) and that its gene knock-out alters levels of one phospholipid form (26). No information is available on AtPMT3, and the substrate preferences of the AtPMT have not been systematically examined.
Biochemical assays of the AtPMT demonstrate that each isoform catalyzes all three reactions in the phosphobase methylation pathway (Table 1). This result is consistent with the proposed di-domain structure of these enzymes (8). In contrast to previous work on AtPMT2, which reported that the enzyme does not methylate pEA (26), our data indicate that it accepts all three phosphobase substrates. In addition, the 2:1 stoichiometry of SAH binding to AtPMT2 (supplemental Fig. S3) and the 1.50-Å resolution X-ray crystal structure of the protein (Table 2; Fig. 4) reveal a structurally intact MT1 domain. The previous study (26) described a clone lacking the first 16 amino acids of full-length AtPMT2 (At1g48600). Deletion of the N-terminal residues would eliminate the α1-helix of the MT1 domain, which removes key structural features and protein–ligand contacts needed for substrate binding (Fig. 6). This explains the lack of activity with pEA for the N-terminally truncated protein.
Although the AtPMT isoforms share common biochemical properties, their physiological roles appear to be different and likely depend on specific expression patterns. The Arabidopsis xipotl1 phenotype associated with loss of atpmt1 expression suggests a key role for this enzyme in root development and pCho synthesis during salt stress (24, 25). No clear phenotype was reported for atpmt2 knock-out lines in Arabidopsis (26). Comparing the T-DNA knock-out lines of each isoform (Fig. 2, A and B) confirmed the previously described atpmt1/xipotl1 mutant effects when grown under either normal or salt-stress conditions (25), whereas the atpmt2 and atpmt3 knock-out lines displayed no unusual growth phenotypes under either condition (Fig. 2, C and D). In addition, only atpmt1 expression increased under salt stress (Fig. 2E). Earlier studies report that the Arabidopsis atpmt1 knock-out lines reduced PtdCho levels by 10% in roots and leaves (25) and that loss of atpmt2 decreased the level of one polar lipid (34:3) species by 2-fold (26). Likewise, comparison of the polar lipid profiles of wild-type Arabidopsis plants with the atpmt3 knock-out line shows no significant changes in phospholipid content of the T-DNA insertion line (supplemental Fig. S4). These results suggest functional redundancy for the AtPMT2 and AtPMT3 isoforms and that the phenotype associated with the atpmt1 knock-out may result from tissue-specific expression differences (Fig. 2F).
Elevated expression of atpmt1 in root compared with other tissues and the higher transcript levels of atpmt2 and atpmt3 in leaf may contribute to the differential effects of the atpmt1/xipotl1 mutant versus the other two isoforms. In the root, AtPMT1 is clearly key for normal growth and tolerance against salt stress (24, 25), whereas the other isoforms may have overlapping functions primarily in leaves. In Arabidopsis, the three PMT provide a route to pCho for production of PtdCho; however, production of downstream metabolites of the phosphobase methylation pathway may also contribute to phenotypic differences. For example, hydrolysis of PtdCho into choline releases phosphatidic acid, which is a lipid second messenger in stress signaling (21, 27). In addition, oxidation of choline yields the osmoprotectant glycine betaine (3). Thus, the roles of the PMT in plants may be broader than just pCho and PtdCho synthesis, and the resulting metabolic complexity in different tissues under normal growth and stress-response conditions requires additional effort to dissect their contributions.
At the molecular level, the three-dimensional structures of AtPMT1 and AtPMT2 provide new insights on the di-domain organization of these proteins (Fig. 3). The X-ray crystal structures of AtPMT1 and AtPMT2 show how the type I PMT retain an evolutionarily conserved core structure that forms the SAM/SAH-binding site in each methyltransferase domain, but they also show structural differences in the phosphobase-binding sites that lead to distinct MT1 and MT2 domain activities, which rely on two different sets of catalytic residues. As reported previously, key residues in the active sites of each domain are highly conserved across the PMT from a variety of organisms (12). Ultimately, the three types of PMT use different structural organizations for the same metabolic pathway (Fig. 8).
Figure 8.

Evolutionary relationships of PMT sequence and structure. The phylogenetic tree was generated with MEGA5 (49) with evolutionary relationships inferred using the Neighbor-Joining method (50). The bootstrap consensus tree was inferred from 100 replicates and taken to represent the evolutionary history of the taxa analyzed (51). Evolutionary distances were computed using the JTT matrix-based method (52). The analysis included 153 amino acid sequences (supplemental Table S1). Schematic images of the type I PMT from plants, type II PMT from Plasmodium, and the type III PMT from nematodes are shown to highlight changes in domain organization and function.
Based on sequence comparisons, the bi-functional di-domain architecture of the AtPMT is conserved across eudicots, monocots, conifers, mosses, and green algae, as the residues of each active site are highly conserved in their respective domain (Fig. 8). Interestingly, bioinformatic comparisons suggest that plant-like type I PMT may also be found in a variety of marine organisms, including oomycetes (water molds), tunicates, segmented seaworms, bivalves, gastropods, crustaceans, lancelets, bony fishes, frogs, toads, sea urchins, and anemones; however, the biochemical function and metabolic contribution of the PMT-like enzymes in these organisms remain to be determined. In the apicomplexans, which evolved from a plastid-containing ancestor, type II PMT perform all three methylation reactions in a single domain (Fig. 8). Structural and biochemical analysis of the P. falciparum PMT indicates that its active site contains substitutions to allow for methylation of pEA and that these residue changes are not found in the MT2 domains of the plant and nematode enzymes (15–18). It appears that the loss of the MT1 domain and alteration of the MT2 domain active site led to a multifunctional single domain PMT of the apicomplexans (12, 19). In contrast to the simplification of PMT architecture observed in the apicomplexans, the evolutionary path to the two mono-functional di-domain proteins found in nematodes appears to have involved gene duplication and structural alterations that yielded vestigial non-functional domains (Fig. 8) (10, 11, 20). Although the domain organization of each type of PMT varies, the overall structure within each domain is highly conserved across a range of evolutionarily distant and diverse organisms.
The published structures of the type II apicomplexan and type III nematode PMT show active sites that enclose reactants, suggesting that catalysis requires conformational changes (15–19). As described here, crystallization differences between AtPMT1 and AtPMT2 yielded structures that reveal the local and global conformational changes associated with each step in the phosphobase methylation pathway (Figs. 4–6). We propose an overall reaction sequence that takes into account the structural flexibility of the PMT.
The methylation of pEA to pMME occurs in the MT1 domain of the plant PMT. The open MT1 active site observed in the AtPMT2 structure allows sufficient contacts for SAM/SAH binding, which along with parts of a disordered N-terminal region and orientation of the β1e–α7 loop (Ser-159–Tyr-176) away from the active site provide access for phosphobase binding (Fig. 6A, green). Previous studies of C. elegans PMT1 and wheat PMT show that these enzymes use a random sequential kinetic mechanism in which either pEA or SAM binds first, followed by the other substrate (11, 21). Based on the structure of the AtPMT1·SAH·pCho complex (Fig. 6, A–C), we suggest that binding of both substrates leads to structural rearrangements that form a functional active site and that those movements are linked to local dynamic changes that stabilize residues of α1–α2 and reposition the β1e–α7 loop to enclose the site, as residues on this loop provide multiple contacts with pCho.
In the MT1 reaction complex, these structural transitions position the methyl group of SAM into proximity of pEA amine with Tyr-131 located between the two substrates. There is also a water molecule bound between the phosphate group of the pCho and the hydroxyl of Tyr-131. The active-site arrangement of AtPMT1 was also observed in the structure of the HcPMT1·SAH·pEA complex (17), which suggests a similar substrate-assisted reaction in the MT1 domain of the plant enzyme. The position of the substrate phosphate relative to the bound water molecule could provide a proton relay to activate the hydroxyl group of Tyr-131, which then promotes deprotonation of the substrate's primary amine for transfer of the methyl group from SAM (28). Site-directed mutagenesis of Tyr-131 confirms the importance of this residue, as substitutions resulted in a loss of pEA methylation (Fig. 7, left). Although pCho, the fully methylated product of the phosphobase pathway, can bind in the MT1 active site as an inhibitor, pMME and pDME are not substrates for this active site in either the nematode or plant PMT (8, 11, 17). The chemical mechanism of the MT1 domain likely takes advantage of the reactivity and/or pKa differences of the amine group of pEA versus the methylated phosphobases. Ultimately, the MT1 active site of the plant and nematode PMT is tuned for the conformational and chemical steps needed for the first methylation to pCho.
Following product release from the MT1 domain, pMME (and later pDME) is methylated in the MT2 domain of the plant and nematode PMT. In contrast to the localized structural changes observed in the MT1 domain, rearrangements leading to a functional MT2 active site require large-scale movements in the di-domain structure (Fig. 4). In the AtPMT2 structure, the open MT2 active site provides access for substrate binding (Fig. 5A). Kinetic studies of C. elegans PMT2 using pDME and SAM as substrates indicate a random sequential addition (10), which given the conserved domain structures likely holds true with the plant PMT. As with the MT1 domain, we suggest that binding of both substrates favors the conformational changes that lead to functional active-site formation, in particular the breaking of the extended α9-helix seen in AtPMT2 into the two shorter α-helices (α9 and α10) that enclose the active site to bring Tyr-246 into proximity of the substrates and within interaction distance of His-360 in the AtPMT1 structure (Figs. 4, B and C, and 5). Substitutions of Tyr-246 in AtPMT2 eliminate the catalytic activity of the MT2 domain and confirm the key role of this residue for methylation chemistry (Fig. 7, right). Extensive structural and mechanistic studies of the P. falciparum PMT identified a tyrosine–histidine dyad (corresponding to Tyr-246 and His-360 of AtPMT1 and AtPMT2) as essential for catalysis (15). Chemically, the histidine acts as a general base to abstract a proton from the tyrosine to polarize the amine group and facilitate methyl group transfer. The dyad was also proposed to act as a “catalytic latch” to lock substrates into the active site. Based on the structures presented here, the observed conformational changes appear linked to substrate binding and positioning of these key catalytic residues.
Multiple studies on a variety of methyltransferases imply a connection between conformational changes and catalysis (29–33); therefore, the structures of the plant PMT for the first time reveal the details of how those transitions occur and contribute to the growing appreciation of how dynamics and chemistry are linked (34, 35). How structural rearrangements occur both within modular domains and between distinct domains in biosynthetic “assembly line” systems is still a poorly understood topic. For example, how interdomain architecture links modules of polyketide synthases, non-ribosomal peptide synthetases, and other engineered modular proteins is critical for efficient production of a variety of natural products (36–41). In summary, the structures of the plant PMT, along with previous biochemical and structural studies of these enzymes from other organisms, indicate an important role for dynamic conformational processes that are linked to the reaction sequence in each methyltransferase domain and between those domains for the efficient synthesis of pCho.
Experimental procedures
Materials
E. coli Rosetta II (DE3) cells were purchased from Novagen. Ni2+-nitrilotriacetic acid-agarose beads were obtained from Qiagen. The HiLoad 26/60 Superdex-200 FPLC column was from Amersham Biosciences. Radiolabeled [methyl-14C]SAM (50.4 mCi/mmol) was bought from MP Biomedicals, and the pMME and pDME substrates were synthesized by Gateway Chemical. Kanamycin and isopropyl 1-thio-β-d-galactopyranoside (IPTG) were purchased from Research Products International. All other reagents were from Sigma. Seeds of A. thaliana ecotype Col-0 and T-DNA knock-out lines AtPMT1 (Salk_036291), AtPMT2 (Salk_204065), and AtPMT3 (Salk_016929C) were obtained from the Arabidopsis Biological Resource Center (Ohio State University, Columbus).
Cloning of AtPMT isoforms, generation of bacterial expression vectors, and site-directed mutagenesis
The coding regions of AtPMT1 (At3g18000), AtPMT2 (At1g48600), and AtPMT3 (At1g73600) were amplified by PCR from A. thaliana seedling cDNA with isoform-specific oligonucleotides containing NcoI (forward primer) and NotI (reverse primer) restriction sites. The primers used were as follows (restriction sites are underlined and start/stop codons are in bold): AtPMT1-F, 5′-dTTTCCATGGGGCTGCATCGTACGAAGAAG-3′, and AtPMT1-R, 5′-dTTTGCGGCCGCTTAATTCTGGTTGGCGATG-3′; AtPMT2-F, 5′-dTTTCCATGGGGCGACACCATACAAGGAAG-3′, and AtPMT2-R, 5′-dTTTGCGGCCGCTTACTTCTTGTCGGCTATG-3′; and AtPMT3-F, 5′-dTTTCCATGGGATGGCTCACAGTCACACT-3′, and AtPMT3-R, 5′-dTTTGCGGCCGCTCAGTTTCTCTTGGCG-3′. Each PCR product (∼1.5 kb) was cloned into the pCRII-TOPO vector (Invitrogen) for confirmation of sequence by automated nucleotide sequencing. Digestion of the TOPO vector with NcoI and NotI released the DNA fragment, which was then ligated into pHIS8 (42), a modified version of pET-28a, to generate bacterial protein expression constructs containing an N-terminal octahistidine tag followed by a thrombin cleavage site. Site-directed mutants of the putative catalytic tyrosine residues of the MT1 (Y131A and Y131F) and MT2 (Y246A and Y264F) domains of AtPMT2 were generated using the QuikChange PCR method (Agilent).
Protein expression and purification
The pHIS8-AtPMT1, -AtPMT2, and -AtPMT3 expression vectors were transformed into E. coli Rosetta II (DE3) cells. Isolated colonies were used to inoculate Terrific broth containing 50 μg ml−1 kanamycin and 35 μg ml−1 chloramphenicol and were grown (37 °C) until A600 nm ∼0.8–1.2. After addition of IPTG (1 mm final), cultures were grown at 20 °C overnight. Cells were harvested by centrifugation (10,000 × g; 10 min) and resuspended in lysis buffer (50 mm Tris (pH 8.0), 500 mm NaCl, 25 mm imidazole, 10% (v/v) glycerol, 1% (v/v) Tween 20). After sonication and centrifugation (45,000 × g; 1 h), the supernatant was passed over a 3–5-ml Ni2+-nitrilotriacetic acid column. Following washing with lysis buffer and wash buffer (lysis buffer without Tween 20), His-tagged PMT was isolated from the column using elution buffer (wash buffer with 300 mm imidazole). Eluted protein was next passed through a Superdex-200 26/60 size-exclusion FPLC column equilibrated in 25 mm Hepes (pH 7.5), 100 mm NaCl, 5 mm β-mercaptoethanol, and 10% (v/v) glycerol. Fractions containing protein were pooled, concentrated, and stored at −80 °C. Protein concentration was determined by the Bradford protein assay method (Bio-Rad) with bovine serum albumin as standard. Expression and purification of the AtPMT1 Y131A, Y131F, Y246A, and Y246F mutants used the same protocol as wild-type protein.
Enzyme assays and steady-state kinetic analysis
PMT activity was determined using a radiochemical assay, as described previously (10, 11). Standard assay conditions were 0.1 m Hepes·KOH (pH 8), 2 mm Na2EDTA, 10% (v/v) glycerol, 2 mm SAM (100 nCi of [methyl-14C]SAM), and either 3 mm pEA or 10 mm pDME in 100 μl. Protein amount and time of reaction provided a linear rate of product formation. Reactions were terminated, and the methylated product was isolated for scintillation counting. The kcat and Km values for SAM were determined under standard assay conditions at 3 mm pEA and 10 mm pMME and pDME. Kinetic parameters for pEA, pMME, and pDME were determined at 2 mm SAM. Initial velocity data were fitted to the Michaelis-Menten equation, v = (kcat[S])/(Km + [S]), using Kaleidagraph (Synergy Software).
Isothermal titration calorimetry of SAH binding
AtPMT2 was dialyzed overnight at a 1:500 volume ratio in 25 mm Hepes (pH 7.5), 100 mm NaCl, 5 mm β-mercaptoethanol, and 5% (v/v) glycerol at 4 °C. Ligands, including SAM, pEA, pCho, and SAH, were prepared in the same buffer. All ITC experiments were performed on a VP-ITC calorimeter (Microcal). Injections (10–12 μl) of ligand were added to a sample solution containing AtPMT2 using a computer-controlled 250-μl microsyringe at an interval of 5–6 min. Control experiments using buffer determined the heat of dilution for each injection, which was used as a correction in experimental titrations. Estimates of Kobs and ΔH were obtained by fitting the experimental data using the Microcal software to the following equation: Qitot = V0ΔHEtot((KobsL)/(1 + KobsL)), where Qitot is total heat after the ith injection; V0 is the volume of the calorimetric cell; Kobs is the equilibrium constant; ΔH is the enthalpy change; L is the ligand concentration; and Etot the protein concentration. Values for the change in free energy were calculated from ΔG = −RT ln(Kobs), where R is the gas constant (1.9872 cal K−1 mol−1), and T is absolute temperature. Changes in entropy were calculated using ΔG = ΔH − TΔS.
Plant growth and quantitative real-time PCR tissue-specific expression in Arabidopsis
For each T-DNA knock-out line, seeds were sterilized with 50% (v/v) bleach for 10 min and washed three times with sterilized water. Seeds were germinated on half-strength Murashige-Skoog agar medium containing 1% (w/v) sucrose and transferred to a growth chamber (16 h light at 22 °C and 8 h dark at 18 °C with 70% relative humidity) for 10 days. Seedlings were then transferred to soil and grown under the same conditions. For genotyping of T-DNA knock-out lines, genomic DNA was extracted from 3-week-old rosette leaves in 100 mm Tris-HCl (pH 9.5), 1 m KCl, and 10 mm EDTA. After incubation (70 °C; 30 min), the extract was centrifuged (30,000 × g; 10 min), and the supernatant was transferred into a new tube and diluted 2-fold with MilliQ water (Millipore). PCRs (melting: 95 °C, 60 s; annealing: 58 °C, 60 s; extension: 72 °C, 60 s; 35 cycles) were performed in a total volume of 50 μl with 1× PCR buffer, 1.5 mm MgCl2, 200 μm dNTPs, 50 pm forward and reverse primers, and 1 unit of TaqDNA polymerase (Invitrogen). The T-DNA-specific primer LBa1 (5′-dTGGTTCACGTAGTGGGCCATCG-3′) and AtPMT1–3-specific primers (AtPMT1-F, 5′-dACCGCTGAATCAAACACAATC-3′, and AtPMT1-R, 5′-dACATGATCTCTTTCGCATTGG-3′; AtPMT2-F, 5′-dTCCACCAATCGTAGATCAAGG-3′, and AtPMT2-R, 5′-dTTGACCCATCCAATCATTCTC-3′; and AtPMT3-F, 5′-dCTAACGGTCTAACAAACCCCC-3′, and AtPMT3-R, 5′-dATCAGCGCACAGAAATTTGAC-3′) were used for genotype analysis.
For qRT-PCR analysis, total RNA was isolated from different tissues of Arabidopsis using TRIzol reagent (Invitrogen). First-strand synthesis of cDNA from RNA (1 μg) was performed with the Superscript II reverse transcriptase kit (Invitrogen). Isoform-specific primers used for qRT-PCR were as follows: AtPMT1-F, 5′-dCGTGAATTAGACAGGGTGGAG-3′, and AtPMT1-R, 5′-dAGTCCCCATTTCTGCTCATC-3′; AtPMT2-F, 5′-dGAGTTTGAAGTCGCTGATTGC-3′, and AtPMT2-R, 5′-dAATGTCCTGAATAGAGCTGGC-3′; and AtPMT3-F, 5′-dAGTGCTAGAGTTTGGTGCTG-3′, and AtPMT3-R, 5′-dTTGACGTTCTTGTAGTGACCG-3′. The qRT-PCR analysis was performed according to the manufacturer's instructions on a Bio-Rad iCycler iQ system with platinum SYBR Green qPCR SuperMix-UDG (Invitrogen). The Arabidopsis actin7 (ACT7) gene was used as an internal control (43). The fold change of transcripts was calculated based on an efficiency-calibrated model and compared with the transcript level under normal conditions. Statistical differences between samples were evaluated by Student's t test. In each experiment, the mean of three biological replicates was used to generate means and statistical significance. Two experiments on independently grown plant material were performed to confirm that the results are reproducible.
Polar lipid analysis of wild-type and atpmt3 plants
Four-week-old wild-type and T-DNA knock-out plants were grown at the same time and in the same growth chamber. Dry weight of each leaf sample ranged from 13 to 35 mg. Total polar lipids were extracted using a published protocol recommended by the Kansas Lipidomics Research Center Analytical Laboratory (44). Briefly, leaves were quickly immersed in isopropyl alcohol containing 0.01% (v/v) butylated hydroxytoluene to prevent generation of phosphatidic acid by phospholipase D upon tissue damage. Polar lipid extracts were prepared using a series of chloroform/methanol and KCl extractions and then frozen for mass spectrometric analysis to provide a global profile of digalactosyldiacylglycerol, monogalactosyldiacylglycerol, phosphatidylglycerol, phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol, phosphatidylserine, and phosphatidic acid levels. Data analyzed were from three independent biological replicates.
Protein crystallography
Crystals of AtPMT1·SAH·pCho complex were grown by the vapor diffusion method at 4 °C in hanging drops of a 1:1 mixture of protein (10 mg ml−1) and crystallization buffer (16% (w/v) PEG-8000, 0.04 m potassium phosphate (monobasic), 20% (v/v) glycerol, 5 mm pCho, and 0.5 mm SAH). Crystals of AtPMT2·SAH complex were grown by the vapor diffusion method at 4 °C in hanging drops of a 1:1 mixture of protein (10 mg ml−1) and crystallization buffer (200 mm potassium formate, 20% (w/v) PEG-3350, and 0.5 mm SAH). Prior to data collection, crystals were frozen in cryoprotectant (mother liquor supplemented with 25% glycerol). All diffraction data were collected at beamline 19ID of the Argonne National Laboratory Advanced Photon Source. HKL3000 (45) was used to index, integrate, and scale the data sets. For solution of the AtPMT1 structure, molecular replacement using two independent search models was performed in PHASER (46). The structures of the H. contortus PMT1 N-terminal domain (PDB code 4KRG; residues 1–256; 17) and the H. contortus PMT2 C-terminal domain (PDB code 4KRH; residues 170–431; 17) were used as independent search models. COOT (47) and PHENIX (48) were used for iterative rounds of manual model building and refinement, respectively. Molecular replacement for the AtPMT2 dataset used a similar strategy. The final model of the AtPMT1·SAH·pCho complex includes residues Glu-7–Asn-491, two SAH molecules, two pCho molecules, and 650 waters. The final model of the AtPMT2·SAH complex includes residues Glu-7–Glu-18 and Arg-42–Lys-490 in chain A, residues Glu-7–Ser-20 and Glu-44–Lys-490 in chain B, two SAH molecules, and 1175 waters. Table 2 summarizes the data collection and refinement statistics for both of the determined structures. Coordinates and structure factors for the AtPMT1·SAH·pCho (PDB code 5WP4) and AtPMT2·SAH (PDB code 5WP5) complexes were deposited in the PDB.
Author contributions
S. G. L. and J. M. J. conceptualization; S. G. L. data curation; S. G. L. formal analysis; S. G. L. and J. M. J. validation; S. G. L. investigation; S. G. L. and J. M. J. writing original draft; S. G. L. and J. M. J. writing review and editing; J. M. J. supervision; J. M. J. funding acquisition; J. M. J. project administration.
Supplementary Material
Acknowledgments
Portions of this research were carried out at the Argonne National Laboratory Structural Biology Center of the Advanced Photon Source, a national user facility operated by the University of Chicago for the Department of Energy Office of Biological and Environmental Research under Contract No. DE-AC02–06CH11357. Lipid analysis was performed at the Kansas Lipidomics Research Center Analytical Laboratory, which is supported by National Science Foundation Grants EPS 0236913, MCB 045531, and DBI 0521587; Kansas Technology Enterprise Corp.; and K-IDeA Networks of Biomedical Research Excellence (INBRE) of National Institute of Health Grant P20RR16475, and Kansas State University.
This work was supported by National Institutes of Health Grant AI-097119. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains supplemental Figs. S1–S5 and Table S1.
The atomic coordinates and structure factors (codes 5WP4 and 5WP5) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- pCho
- phosphatidylcholine
- PMT
- phosphoethanolamine methyltransferase
- pEA
- phosphoethanolamine
- SAM
- S-adenosylmethionine
- PtdCho
- phosphatidylcholine
- pMME
- phosphomonomethylethanolamine
- pDME
- phosphodimethylethanolamine
- PDB
- Protein Data Bank
- SAH
- S-adenosylhomocysteine
- ITC
- isothermal titration calorimetry
- r.m.s.d.
- root mean squared deviation
- qRT
- quantitative RT
- F
- forward
- R
- reverse
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside.
References
- 1. Kent C. (1995) Eukaryotic phospholipid biosynthesis. Annu. Rev. Biochem. 64, 315–343 [DOI] [PubMed] [Google Scholar]
- 2. Hanson A. D., and Roje S. (2001) One-carbon metabolism in higher plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 119–137 [DOI] [PubMed] [Google Scholar]
- 3. McNeil S. D., Nuccio M. L., Ziemak M. J., and Hanson A. D. (2001) Enhanced synthesis of choline and glycine betaine in transgenic tobacco plants that overexpress phosphoethanolamine N-methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 98, 10001–10005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kanipes M. I., and Henry S. A. (1997) The phospholipid methyltransferases in yeast. Biochim. Biophys. Acta 1348, 138–141 [DOI] [PubMed] [Google Scholar]
- 5. Mudd S. H., and Datko A. H. (1986) Phosphoethanolamine bases as intermediates in phosphatidylcholine synthesis by Lemna. Plant Physiol. 82, 126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Datko A. H., and Mudd S. H. (1988) Phosphatidylcholine synthesis: differing patterns in soybean and carrot. Plant Physiol. 88, 854–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Datko A. H., and Mudd S. H. (1988) Enzymes of phosphatidylcholine synthesis in Lemna, soybean, and carrot. Plant Physiol. 88, 1338–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nuccio M. L., Ziemak M. J., Henry S. A., Weretilnyk E. A., and Hanson A. D. (2000) cDNA cloning of phosphoethanolamine N-methyltransferase from spinach by complementation in Schizosaccharomyces pombe and characterization of the recombinant enzyme. J. Biol. Chem. 275, 14095–14101 [DOI] [PubMed] [Google Scholar]
- 9. Pessi G., Kociubinski G., and Mamoun C. B. (2004) A pathway for phosphatidylcholine biosynthesis in Plasmodium falciparum involving phosphoethanolamine methylation. Proc. Natl. Acad. Sci. U.S.A. 101, 6206–6211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Palavalli L. H., Brendza K. M., Haakenson W., Cahoon R. E., McLaird M., Hicks L. M., McCarter J. P., Williams D. J., Hresko M. C., and Jez J. M. (2006) Defining the role of phosphomethylethanolamine N-methyltransferase from Caenorhabditis elegans in phosphocholine biosynthesis by biochemical and kinetic analysis. Biochemistry 45, 6056–6065 [DOI] [PubMed] [Google Scholar]
- 11. Brendza K. M., Haakenson W., Cahoon R. E., Hicks L. M., Palavalli L. H., Chiapelli B. J., McLaird M., McCarter J. P., Williams D. J., Hresko M. C., and Jez J. M. (2007) Phosphoethanolamine N-methyltransferase from Caenorhabditis elegans: a new nematicidal target. Biochem. J. 404, 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee S. G., and Jez J. M. (2014) Nematode phospholipid metabolism: an example of closing the genome-structure-function circle. Trends Parasitol. 30, 241–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bolognese C. P., and McGraw P. (2000) The isolation and characterization in yeast of a gene for Arabidopsis S-adenosylmethionine:phosphoethanolamine N-methyltransferase. Plant Physiol. 124, 1800–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Charron J. B., Breton G., Danyluk J., Muzac I., Ibrahim R. K., and Sarhan F. (2002) Molecular and biochemical characterization of a cold-regulated phosphoethanolamine N-methyltransferase from wheat. Plant Physiol. 129, 363–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee S. G., Kim Y., Alpert T. D., Nagata A., and Jez J. M. (2012) Structure and reaction mechanism of phosphoethanolamine methyltransferase from the malaria parasite Plasmodium falciparum–an anti-parasitic drug target. J. Biol. Chem. 287, 1426–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee S. G., Alpert T. D., and Jez J. M. (2012) Crystal structure of phosphoethanolamine methyltransferase from Plasmodium falciparum in complex with amodiaquine. Bioorg. Med. Chem. Lett. 22, 4990–4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee S. G., and Jez J. M. (2013) Evolution of structure and mechanistic divergence in di-domain methyltransferases from nematode phosphocholine biosynthesis. Structure 21, 1778–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saen-Oon S., Lee S. G., Jez J. M., and Guallar V. (2014) An alternate mechanism for the methylation of phosphoethanolamine catalyzed by Plasmodium falciparum phosphoethanolamine methyltransferase. J. Biol. Chem. 289, 33815–33825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garg A., Lukk T., Kumar V., Choi J. Y., Augagneur Y., Voelker D. R., Nair S., and Ben Mamoun C. (2015) Structure, function and inhibition of the phosphoethanolamine methyltransferases of the human malaria parasites Plasmodium vivax and Plasmodium knowlesi. Sci. Rep. 5, 9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee S. G., Haakenson W., McCarter J. P., Williams D. J., Hresko M. C., and Jez J. M. (2011) Thermodynamic evaluation of ligand binding in the plant-like phosphoethanolamine methyltransferases of the parasitic nematode Haemonchus contortus. J. Biol. Chem. 286, 38060–38068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jost R., Berkowitz O., Shaw J., and Masle J. (2009) Biochemical characterization of two wheat phosphoethanolamine N-methyltransferase isoforms with different sensitivities to inhibition by phosphatidic acid. J. Biol. Chem. 284, 31962–31971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ye C., Wu S., Yang Q., Ma C., Yang G., and Wang B. (2005) Cloning, sequencing and salt induced expression of PEAMT and BADH in oilseed rape (Brassica napus). DNA Seq. 16, 364–371 [DOI] [PubMed] [Google Scholar]
- 23. Wu S., Yu Z., Wang F., Li W., Ye C., Li J., Tang J., Ding J., Zhao J., and Wang B. (2007) Cloning, characterization, and transformation of the phosphoethanolamine N-methyltransferase gene (ZmPEAMT1) in maize (Zea mays L.). Mol. Biotechnol. 36, 102–112 [DOI] [PubMed] [Google Scholar]
- 24. Mou Z., Wang X., Fu Z., Dai Y., Han C., Ouyang J., Bao F., Hu Y., and Li J. (2002) Silencing of phosphoethanolamine N-methyltransferase results in temperature-sensitive male sterility and salt hypersensitivity in Arabidopsis. Plant Cell 14, 2031–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cruz-Ramírez A., López-Bucio J., Ramírez-Pimentel G., Zurita-Silva A., Sánchez-Calderon L., Ramírez-Chávez E., González-Ortega E., and Herrera-Estrella L. (2004) The xipotl mutant of Arabidopsis reveals a critical role for phospholipid metabolism in root system development and epidermal cell integrity. Plant Cell 16, 2020–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. BeGora M. D., Macleod M. J., McCarry B. E., Summers P. S., and Weretilnyk E. A. (2010) Identification of phosphomethylethanolamine N-methyltransferase from Arabidopsis and its role in choline and phospholipid metabolism. J. Biol. Chem. 285, 29147–29155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Munnik T. (2001) Phosphatidic acid: an emerging plant lipid second messenger. Trends Plant Sci. 6, 227–233 [DOI] [PubMed] [Google Scholar]
- 28. Dall'Acqua W., and Carter P. (2000) Substrate-assisted catalysis: molecular basis and biological significance. Protein Sci. 9, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fu Z., Hu Y., Konishi K., Takata Y., Ogawa H., Gomi T., Fujioka M., and Takusagawa F. (1996) Crystal structure of glycine N-methyltransferase from rat liver. Biochemistry 35, 11985–11993 [DOI] [PubMed] [Google Scholar]
- 30. Horton J. R., Sawada K., Nishibori M., Zhang X., and Cheng X. (2001) Two polymorphic forms of human histamine methyltransferase: structural, thermal, and kinetic comparisons. Structure 9, 837–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takata Y., Huang Y., Komoto J., Yamada T., Konishi K., Ogawa H., Gomi T., Fujioka M., and Takusagawa F. (2003) Catalytic mechanism of glycine N-methyltransferase. Biochemistry 42, 8394–8402 [DOI] [PubMed] [Google Scholar]
- 32. Komoto J., Yamada T., Takata Y., Konishi K., Ogawa H., Gomi T., Fujioka M., and Takusagawa F. (2004) Catalytic mechanism of guanidinoacetate methyltransferase: crystal structures of guanidinoacetate methyltransferase ternary complexes. Biochemistry 43, 14385–14394 [DOI] [PubMed] [Google Scholar]
- 33. Soriano A., Castillo R., Christov C., Andrés J., Moliner V., and Tuñón I. (2006) Catalysis in glycine N-methyltransferase: testing the electrostatic stabilization and compression hypothesis. Biochemistry 45, 14917–14925 [DOI] [PubMed] [Google Scholar]
- 34. Henzler-Wildman K., and Kern D. (2007) Dynamic personalities of proteins. Nature 450, 964–972 [DOI] [PubMed] [Google Scholar]
- 35. Henzler-Wildman K. A., Thai V., Lei M., Ott M., Wolf-Watz M., Fenn T., Pozharski E., Wilson M. A., Petsko G. A., Karplus M., Hübner C. G., and Kern D. (2007) Intrinsic motions along an enzymatic reaction trajectory. Nature 450, 838–844 [DOI] [PubMed] [Google Scholar]
- 36. Zhang Y., Li S. Z., Li J., Pan X., Cahoon R. E., Jaworski J. G., Wang X., Jez J. M., Chen F., and Yu O. (2006) Using unnatural protein fusions to engineer resveratrol biosynthesis in yeast and mammalian cells. J. Am. Chem. Soc. 128, 13030–13031 [DOI] [PubMed] [Google Scholar]
- 37. Khosla C., Tang Y., Chen A. Y., Schnarr N. A., and Cane D. E. (2007) Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu. Rev. Biochem. 76, 195–221 [DOI] [PubMed] [Google Scholar]
- 38. Frueh D. P., Arthanari H., Koglin A., Vosburg D. A., Bennett A. E., Walsh C. T., and Wagner G. (2008) Dynamic thiolation-thioesterase structure of a non-ribosomal peptide synthetase. Nature 454, 903–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y., Yi H., Wang M., Yu O., and Jez J. M. (2011) Structural and kinetic analysis of the unnatural fusion protein 4-coumaroyl-CoA ligase:stilbene synthase. J. Am. Chem. Soc. 133, 20684–20687 [DOI] [PubMed] [Google Scholar]
- 40. Miller B. R., Sundlov J. A., Drake E. J., Makin T. A., and Gulick A. M. (2014) Analysis of the linker region joining the adenylation and carrier protein domains of the modular nonribosomal peptide synthetases. Proteins 82, 2691–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reimer J. M., Aloise M. N., Harrison P. M., and Schmeing T. M. (2016) Synthetic cycle of the initiation module of a formylating nonribosomal peptide synthetase. Nature 529, 239–242 [DOI] [PubMed] [Google Scholar]
- 42. Jez J. M., Ferrer J. L., Bowman M. E., Dixon R. A., and Noel J. P. (2000) Dissection of malonyl-CoA decarboxylation from polyketide formation in the reaction mechanism of a plant polyketide synthase. Biochemistry 39, 890–902 [DOI] [PubMed] [Google Scholar]
- 43. Subramanian S., Hu X., Lu G., Odelland J. T., and Yu O. (2004) The promoters of the isoflavone synthase genes respond differentially to nodulation and defense signals in transgenic soybean roots. Plant Mol. Biol. 54, 623–639 [DOI] [PubMed] [Google Scholar]
- 44. Devaiah S. P., Roth M. R., Baughman E., Li M., Tamura P., Jeannotte R., Welti R., and Wang X. (2006) Quantitative profiling of polar glycerolipid species from organs of wild-type Arabidopsis and a phospholipase Dα1 knockout mutant. Phytochemistry 67, 1907–1924 [DOI] [PubMed] [Google Scholar]
- 45. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 46. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 48. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tamura K., Peterson D., Peterson N., Stecher G., Nei M., and Kumar S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saitou N., and Nei M. (1987) The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 [DOI] [PubMed] [Google Scholar]
- 51. Felsenstein J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791 [DOI] [PubMed] [Google Scholar]
- 52. Jones D. T., Taylor W. R., and Thornton J. M. (1992) The rapid generation of mutation data matrices from protein sequences. Comp. Appl. Biosci. 8, 275–282 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.