

Abstract
K+/Cl− cotransporter 2 (KCC2) is selectively expressed in the adult nervous system and allows neurons to maintain low intracellular Cl− levels. Thus, KCC2 activity is an essential prerequisite for fast hyperpolarizing synaptic inhibition mediated by type A γ-aminobutyric acid (GABAA) receptors, which are Cl−-permeable, ligand-gated ion channels. Consistent with this, deficits in the activity of KCC2 lead to epilepsy and are also implicated in neurodevelopmental disorders, neuropathic pain, and schizophrenia. Accordingly, there is significant interest in developing activators of KCC2 as therapeutic agents. To provide insights into the cellular processes that determine KCC2 activity, we have investigated the mechanism by which N-ethylmaleimide (NEM) enhances transporter activity using a combination of biochemical and electrophysiological approaches. Our results revealed that, within 15 min, NEM increased cell surface levels of KCC2 and modulated the phosphorylation of key regulatory residues within the large cytoplasmic domain of KCC2 in neurons. More specifically, NEM increased the phosphorylation of serine 940 (Ser-940), whereas it decreased phosphorylation of threonine 1007 (Thr-1007). NEM also reduced with no lysine (WNK) kinase phosphorylation of Ste20-related proline/alanine-rich kinase (SPAK), a kinase that directly phosphorylates KCC2 at residue Thr-1007. Mutational analysis revealed that Thr-1007 dephosphorylation mediated the effects of NEM on KCC2 activity. Collectively, our results suggest that compounds that either increase the surface stability of KCC2 or reduce Thr-1007 phosphorylation may be of use as enhancers of KCC2 activity.
Keywords: chloride transport, epilepsy, GABA receptor, membrane protein, phosphorylation, KCC2, N-ethylmaleimide
Introduction
K+-Cl− cotransporter 2 (KCC2)2 is a membrane protein that lowers intracellular Cl− concentrations by a secondary active transport mechanism (1). This process allows Cl− to passively reenter the cell upon opening of Cl− channels such as GABAA and glycine receptors, resulting in membrane hyperpolarization (2, 3). Due to the lack of ligand-gated K+ channels, fast synaptic inhibition in the mammalian central nervous system is mediated exclusively by GABAA and glycine receptors. KCC2 is expressed in most adult neurons (4), and expression levels correlate well with the maturation state of neurons. Specifically, KCC2 expression levels are low in immature neurons and high in mature neurons, which underlies the developmental shift in reversal potential of GABAA-mediated currents (EGABA) (5).
Dysregulation of KCC2 is associated with a number of neurological disorders, including epilepsy (6–11) and neuropathic pain (12–17). Thus, there is great interest in understanding the mechanisms that regulate the activity of this transporter.
KCC2 activity can be enhanced by modulation of its phosphorylation state (18–21). Specifically, three key phosphorylation sites in the C-terminal domain of KCC2 are associated with the regulation of its transporter activity. Dephosphorylation of residues Thr-906 and Thr-1007 correlates with increased KCC2 activity (19), whereas increased levels of Ser-940 phosphorylation correlate with both up-regulated KCC2 surface levels and increased function (18). The importance of KCC2 Ser-940 phosphorylation in the regulation of transporter activity is highlighted by studies that demonstrated an enhanced onset and severity of status epilepticus upon kainate treatment in a KCC2-S940A knock-in mouse (22).
N-Ethylmaleimide (NEM) has been used as a tool compound in the field for numerous years to activate KCC transport under isotonic conditions (1, 23, 24). NEM contains a Michael acceptor functionality that modifies the sulfhydryl group of cysteine residues through the formation of a covalent (possibly reversible) thioether bond. Although the chemical properties of NEM are known, it is unclear which cysteine moieties are modified in a cellular context. Prior work has demonstrated that NEM does not likely act directly on these transporters but rather modulates a kinase or phosphatase involved in the activation of KCCs (25–27). However, the specific mechanism by which NEM functions to activate KCCs has yet to be established.
Here we investigated the precise mechanism by which NEM affects KCC2 to rapidly increase its function in both HEK293 cells and neurons. We also developed a new phosphospecific antibody to study the modulation of a known phosphorylation site of KCC2. We show that although NEM does not affect total KCC2 levels it does modulate its surface levels and phosphorylation state in a cell type–dependent manner. We demonstrate that NEM both increased KCC2 Ser-940 phosphorylation and decreased KCC2 Thr-1007 phosphorylation. Further analysis with single-point mutant constructs indicated that the dephosphorylation of the Thr-1007 residue alone mediates the effects of NEM. These studies provide a novel mechanistic understanding of the activation of KCC2 by NEM and demonstrate that post-translational modifications of KCC2 lead to a rapid enhancement of Cl− extrusion.
Results
NEM potentiates KCC2 function in HEK293 cells as measured using thallium flux
To recapitulate historical findings using unidirectional tracer experiments, we performed thallium influx fluorescence assays in KCC2-transfected HEK293 cells to demonstrate the activation of KCC2 with NEM treatment (Fig. 1A) (28). We observed a 1.4-fold increase in the rate of thallium influx in cells treated with NEM (100 μm; 15 min) compared with cells treated with DMSO vehicle control (0.1%; 15 min) (DMSO, 3.1 ± 0.1 counts/s (n = 32); NEM, 4.5 ± 0.2 counts/s (n = 16); unpaired t test, p < 0.0001). These results demonstrate that the NEM treatment conditions used in our study result in increased KCC2 transporter activity.
Figure 1.

NEM acutely potentiates KCC2-mediated thallium influx and Cl− extrusion in HEK293 cells. A, thallium influx assay showing increased KCC2 activity upon 15-min exposure to NEM (red trace) compared with DMSO control (green trace). Each trace is an average of multiple experiments (DMSO, n = 32; NEM, n = 16) with error bars representing S.E. (unpaired t test, p < 0.0001). B, example leak-subtracted traces show EGly shifts in response to NEM (baseline in blue; 15-min time point in red) as compared with DMSO (baseline in black; 15-min time point in green). C, summary of data displaying changes in EGly over 15-min exposure to DMSO or NEM. D, summary data displaying change in [Cl−]i in response to 15-min exposure to DMSO or NEM. Error bars represent S.E. in all panels. Correlation graphs relating baseline EGly (E) and [Cl−]i (F) to NEM-induced shifts for KCC2-WT reveal greater NEM-induced shifts in cells with higher initial [Cl−]i. Asterisks indicate that the best fit of the linear regression to the NEM data set deviates significantly from a slope of zero (see text). **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
NEM potentiates KCC2 activity in HEK293 cells as measured using patch clamp recording
To confirm our results using thallium, we investigated the ability of NEM to acutely potentiate KCC2 function using the gramicidin perforated-patch technique in HEK293 cells (Fig. 1, B–F). In HEK293 cells transiently transfected with WT KCC2 and the α1 glycine receptor, the basal reversal potential of glycine-activated currents (EGly) was −69 ± 5 mV prior to treatment with DMSO in our control set and −67 ± 6 mV prior to NEM exposure in our treatment group. We then applied DMSO or NEM (100 μm) for 15 min while continuously monitoring changes in EGly every 3 min. NEM significantly decreased EGly values after 15 min (Fig. 1C; DMSO, 0.6 ± 1.3 mV, n = 12; NEM, −8.1 ± 1.8 mV, n = 9; unpaired t test, p = 0.0009). We then calculated the intracellular concentration of Cl− from the observed EGly values using the Nernst equation. The EGly shifts were reflected in decreases in the calculated [Cl−]i values (Fig. 1D; DMSO, 0.44 ± 0.5 mm; NEM, −4.37 ± 1.4 mm; unpaired t test, p = 0.0022). Further analysis revealed that cells with higher basal EGly values exhibited larger NEM-induced reductions of EGly over 15 min (Fig. 1E; DMSO, R2 < 0.001, F = 0.003, p = 0.9589; NEM, R2 = 0.407, F = 4.801, p = 0.0646). An even tighter relationship was found after converting EGly to values of [Cl−]i (Fig. 1F; DMSO, R2 = 0.005, F = 0.051, p = 0.8251; NEM, R2 = 0.899, F = 62.4, p < 0.0001). Importantly, these data support our thallium flux data and indicate that NEM rapidly increases KCC2-mediated Cl− extrusion in a self-limiting manner with cells having the least amount of basal KCC2 activity exhibiting the highest degree of potentiation and vice versa.
NEM potentiates KCC2 activity in neurons
To understand whether NEM can potentiate KCC2 activity in neurons, we measured EGABA using the selective agonist muscimol (1 μm) in DIV 10 rat cortical neurons. To isolate the influence of KCC2, we applied the NKCC1 inhibitor bumetanide (10 μm) to all of our solutions and added tetrodotoxin, (2R)-amino-5-phosphonovaleric acid, and 6,7-dinitroquinoxaline-2,3-dione to suppress activity-dependent shifts in EGABA (29).
In the vehicle DMSO group of neurons, baseline EGABA values were −74 ± 4 mV with calculated [Cl−]i values of 9.5 ± 1.9 mm (n = 10 neurons). After continuous exposure to DMSO for 15 min, the measured EGABA and [Cl−]i values remained statistically similar (EGABA = −75 ± 4 mV; ΔEGABA = −0.9 ± 0.5 mV, p = 0.0878; Cl− = 9.2 ± 1.8 mm, ΔCl− = −0.30 ± 0.14 mm, p = 0.0712, paired t tests; Fig. 2, A–C). In sharp contrast, NEM (100 μm) exposure significantly reduced baseline EGABA values from −72 ± 4 to −81 ± 3 mV (n = 10 neurons; ΔEGABA = −8.2 ± 1.2 mV, p < 0.0001, paired t test; Fig. 2B). This was equivalent to a significant reduction of [Cl−]i from 9.4 ± 1.2 to 6.7 ± 0.8 mm (ΔCl− = −2.7 ± 0.51 mm, p = 0.0004, paired t test; Fig. 2C). Interestingly, we noticed that the sensitivity of neurons to NEM was proportional to their baseline EGABA/[Cl−]i values with neurons having the highest values exhibiting the greatest reductions (EGABA, R2 = 0.4374, F = 6.22, p = 0.0373, linear regression; Cl−, R2 = 0.8112, F = 34.37, p = 0.0004, linear regression; Fig. 2, D and E). Correlations for the DMSO group were not significantly different from zero (EGABA, R2 = 0.0011, F = 0.01, p = 0.9274; Cl−, R2 = 0.0576, F = 0.49, p = 0.5042, linear regression). This indicated that the modification of KCC2 by NEM exhibited a floor effect.
Figure 2.

NEM acutely enhances Cl− extrusion in immature cortical neurons. A, muscimol-activated currents recorded during voltage ramps in cells treated with DMSO or NEM. The time (in minutes) indicates when the recordings were taken, either at 0 or 15 min after each treatment. B and C, summary of data displaying changes in the EGABA values (B) and the [Cl−]i values (C) during the 15-min exposure to DMSO or NEM. Error bars represent S.E. in both panels. D and E, correlation graphs relating the basal EGABA values (D) and the basal [Cl−]i values (E) to the NEM-induced shifts (red) or DMSO-induced shifts (black) for each value. Each data point represents an individual neuron. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001.
NEM treatment modifies the cell surface stability of KCC2 in neurons
It is well established that transporter activity can be potentiated by increased total or surface protein levels (30). We therefore first tested whether NEM increased the total protein level of KCC2. We treated KCC2-transfected HEK293 cells and immature cortical neurons with 100 μm NEM or DMSO as a vehicle control for 15 min. Lysate from treated cells was probed for KCC2, and levels were quantified relative to the DMSO control (Fig. 3). NEM did not significantly alter total KCC2 levels in HEK293 cells (1.0 ± 0.2 relative to DMSO control, n = 5, unpaired t test, p = 0.9139). Although we observed a trend for increased total KCC2 levels in immature cortical neurons, this increase was not significant (1.8 ± 0.5 relative to DMSO control, n = 3, unpaired t test, p = 0.1736). Thus, NEM-induced activation of KCC2 is unlikely to be attributed to an overall increase in total KCC2 protein levels.
Figure 3.

NEM treatment does not alter total KCC2 levels. A, cell lysates from DMSO-treated and NEM-treated HEK293 cells were probed for total KCC2. Total KCC2 levels were normalized to GAPDH and quantified relative to the DMSO control (n = 5; unpaired t test, p = 0.9139). Error bars represent S.E. B, cell lysates from DMSO-treated and NEM-treated immature cortical neurons were probed for total KCC2. Total KCC2 levels were normalized to GAPDH and quantified relative to the DMSO control (n = 3; unpaired t test, p = 0.1736). Error bars represent S.E. U, untransfected.
We next examined whether NEM treatment alters KCC2 surface levels using a surface biotinylation assay (18, 22, 31). We compared the ratio of surface KCC2 to total KCC2 for DMSO- versus NEM-treated cells. Cytosolic markers were examined to demonstrate the proper isolation of surface proteins without contamination of cytosolic proteins. There was no significant increase in surface KCC2 levels in HEK293 cells treated with NEM (Fig. 4A; 1.2 ± 0.1 relative to DMSO control, n = 3, unpaired t test, p = 0.0983). However, we observed a significant NEM-induced increase of surface KCC2 levels in immature cortical neurons (Fig. 4B; 2.5 ± 0.4 relative to DMSO control, n = 3, unpaired t test, p = 0.0275). Therefore, increased surface KCC2 levels likely contribute to the potentiation of KCC2 activity observed in neurons but not in HEK293 cells.
Figure 4.
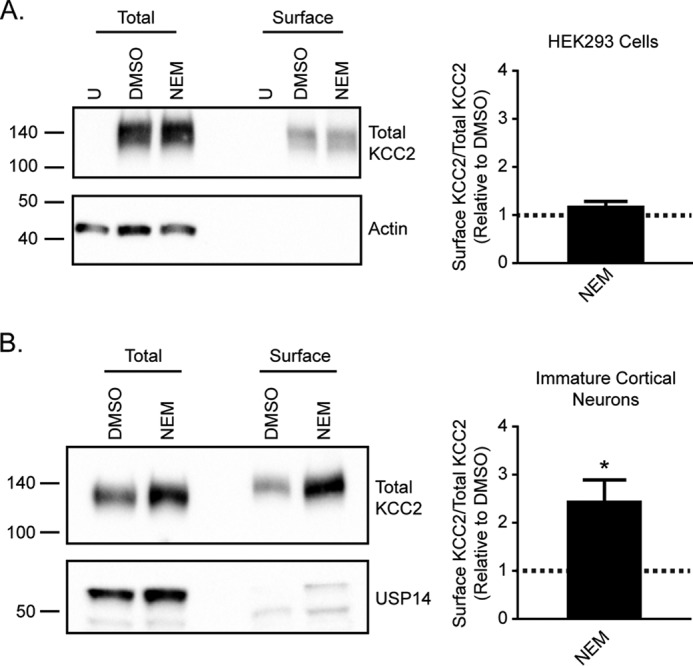
NEM treatment increases KCC2 surface levels in immature cortical neurons. A, surface biotinylation assays to compare surface KCC2 levels between DMSO- and NEM-treated KCC2-transfected HEK293 cells. The ratio of surface to total KCC2 is shown relative to the DMSO control (n = 3; unpaired t test, p = 0.0983). B, surface biotinylation assays to compare surface KCC2 levels between DMSO- and NEM-treated immature cortical neurons. The ratio of surface to total KCC2 is shown relative to the DMSO control (n = 3; unpaired t test, p = 0.0275). *, p < 0.05. U, untransfected.
NEM treatment regulates KCC2 Ser-940 phosphorylation
To further examine the mechanism underlying the effects of NEM, we assessed its effects on the phosphorylation of Ser-940, a modification that increases KCC2 activity and surface stability (Fig. 5) (18). To measure changes in the phosphorylation state of KCC2 Ser-940, we used a previously characterized phosphospecific antibody directed against Ser-940 (32). KCC2 Ser-940 phosphorylation was significantly increased in lysates of HEK293 cells treated with NEM compared with DMSO (Fig. 5A; 3.6 ± 0.9 relative to DMSO control, n = 3, unpaired t test, p = 0.0484). Likewise, KCC2 Ser-940 phosphorylation was also significantly increased in immature cortical neurons treated with NEM (Fig. 5B; 2.4 ± 0.3 relative to DMSO control, n = 3, unpaired t test, p = 0.0098). These results demonstrated that NEM increases KCC2 Ser-940 phosphorylation in both cell types, which could contribute to the mechanism that increases KCC2 activity with NEM treatment.
Figure 5.

NEM modulates KCC2 Ser-940 phosphorylation. A, KCC2 was immunoprecipitated from untransfected (U) or DMSO-treated or NEM-treated KCC2-transfected HEK293 cell lysate. Immunoprecipitated KCC2 was probed for both total KCC2 and KCC2 pSer-940. KCC2 pSer-940 levels were quantified relative to the DMSO control (n = 3; unpaired t test, p = 0.0484). Error bars represent S.E. B, cell lysates from DMSO-treated and NEM-treated immature cortical neurons were probed for both total KCC2 and KCC2 pSer-940. KCC2 pSer-940 levels were quantified relative to the DMSO control (n = 3; unpaired t test, p = 0.0098). Error bars represent S.E. *, p < 0.05; **, p < 0.01.
NEM treatment decreases KCC2 Thr-1007 phosphorylation
We next examined the effect of NEM treatment on the phosphorylation state of KCC2 Thr-1007. This site is conserved across all human KCC transporters, and decreased phosphorylation of this residue is associated with increased KCC2 activity (19, 33). To understand whether NEM modulates Thr-1007 phosphorylation, we developed a phosphospecific antibody directed against KCC2 Thr-1007. To validate the specificity of this antibody, we generated a KCC2-T1007A mutant to mimic the non-phosphorylated state of this residue. KCC2-WT or -T1007A was overexpressed in HEK293 cells. As the pThr-1007 site is not specific to KCC2 but conserved in all KCC proteins (19), we first immunoprecipitated total KCC2 from cell lysates prior to probing for KCC2 Thr-1007 phosphorylation. We showed a clear signal with the pThr-1007 antibody in KCC2-WT lysate that was lost in KCC2-T1007A cell lysate, which demonstrated the specificity of this antibody for the Thr-1007–phosphorylated form of KCC2 (Fig. 6A).
Figure 6.

NEM decreases KCC2 Thr-1007 phosphorylation levels. A, validation of KCC2 pThr-1007 antibody in KCC2-transfected HEK293 cells. Both KCC2-WT and KCC2-T1007A constructs were expressed in HEK293 cells. Immunoprecipitated KCC2 was probed for both pThr-1007 and total KCC2. The loss of pThr-1007 signal with expression of the KCC2-T1007A construct demonstrates the specificity of this antibody for the phosphorylated state of Thr-1007. B, KCC2 was immunoprecipitated from untransfected (U) or DMSO-treated or NEM-treated KCC2-transfected HEK293 cell lysate. Immunoprecipitated KCC2 was probed for both total KCC2 and KCC2 pThr-1007. KCC2 pThr-1007 levels were quantified relative to the DMSO control (n = 3; unpaired t test, p = 0.0007). Error bars represent S.E. C, KCC2 was immunoprecipitated from DMSO-treated or NEM-treated immature cortical neuron lysate. Immunoprecipitated KCC2 was probed for both total KCC2 and KCC2 pThr-1007. KCC2 pThr-1007 levels were quantified relative to the DMSO control (n = 3; unpaired t test, p < 0.0001). Error bars represent S.E. ***, p < 0.001; ****, p < 0.0001.
We then used this phosphospecific antibody to understand whether NEM modulated the phosphorylation state of this site. We immunoprecipitated KCC2 from both KCC2-transfected HEK293 cells and immature cortical neurons treated with either DMSO or NEM and probed for Thr-1007 phosphorylation (Fig. 6, B and C). KCC2 Thr-1007 phosphorylation was significantly decreased in NEM-treated HEK293 cells compared with control DMSO-treated cells (Fig. 6B; 0.3 ± 0.1 relative to DMSO control, n = 3, unpaired t test, p = 0.0007). Similarly, phosphorylation of KCC2 Thr-1007 was significantly reduced in NEM-treated immature cortical neurons (Fig. 6C; 0.08 ± 0.003 relative to DMSO control, n = 3, unpaired t test, p < 0.0001). This demonstrated that NEM also affected the phosphorylation state of KCC2 Thr-1007 in both HEK293 cells and neurons.
NEM regulates KCC2 activity through Thr-1007 phosphorylation
To determine the roles that Ser-940 and Thr-1007 play in mediating the effects of NEM, we used mutant KCC2 molecules in which Ser-940 was mutated to alanine (S940A), preventing phosphorylation at this site, whereas Thr-1007 was mutated to glutamate (T1007E), mimicking phosphorylation at this site. Similar to cells expressing KCC2-WT, KCC2-S940A was also sensitive to NEM exposure. Basal EGly values shifted from −65 ± 4 to −75 ± 5 mV (n = 10 cells, p = 0.0012, paired t test; Fig. 7A), and the calculated [Cl−]i values were reduced from 13.0 ± 2.1 to 9.1 ± 2.2 mm (p = 0.0028, paired t test; Fig. 7B). In parallel experiments, DMSO-treated cells did not exhibit significant shifts in EGly values (baseline, −63 ± 3 mV; 15-min DMSO, −60 ± 3 mV, n = 7 cells, p = 0.0954, paired t test). Based on our findings that the NEM potentiation of KCC2-WT exhibited a floor effect (Fig. 1), we chose to examine the T1007E mutant for further analysis to avoid the possibility of encountering a baseline floor with the gain-of-function T1007A mutant (33). Such an effect would occlude NEM reactivity and result in a false negative. In contrast to both KCC2-WT and -S940A, HEK293 cells expressing KCC2-T1007E were insensitive to NEM treatment with a 15-min application not resulting in a negative shift in EGly values (Fig. 7C; n = 10; baseline, −77.1 ± 4.3 mV; NEM, −78.3 ± 4.6 mV, paired t test, p = 0.1175) or the calculated [Cl−]i values (Fig. 7D; baseline, 8.4 ± 1.6 mm; NEM, 8.2 ± 1.8 mm, paired t test, p = 0.3914). Combined with our biochemical analyses, these data indicated that the NEM-induced activation of KCC2 function can be mediated exclusively through the dephosphorylation of the Thr-1007 site.
Figure 7.
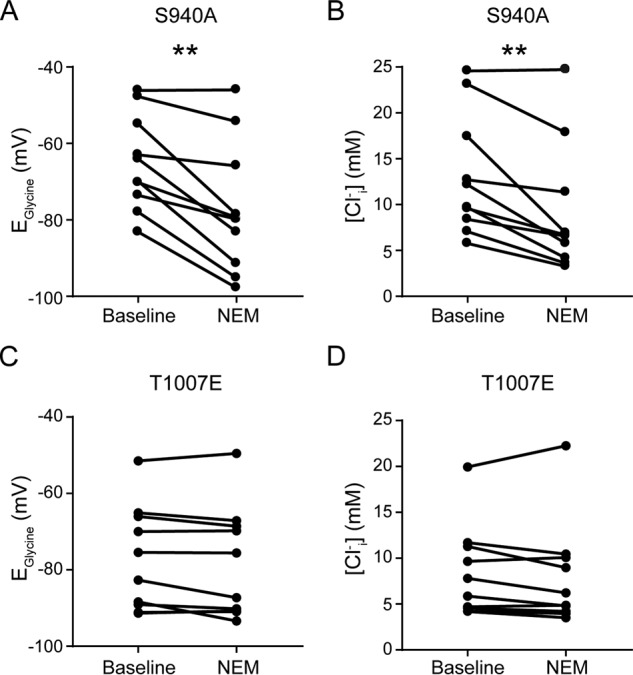
KCC2-T1007E point mutant is insensitive to NEM. A and B, summary data demonstrating a shift in EGly and [Cl−]i values exhibited by the KCC2-S940A mutant following a 15-min exposure to NEM. C and D, summary data showing lack of shift in EGly and [Cl−]i values exhibited by the KCC2-T1007E mutant after a 15-min NEM application. **, p < 0.01.
NEM targets the WNK/SPAK kinase pathway to modulate Thr-1007 phosphorylation
To gain insight into the mechanism by which NEM modulates changes in KCC2 Thr-1007 phosphorylation, we investigated whether NEM targets the WNK/SPAK kinase pathway known to be involved in the phosphorylation of this site in multiple KCCs (34). Although SPAK is known to directly phosphorylate KCC2 Thr-1007 (35), SPAK activity itself is activated via WNK phosphorylation (36). We examined the phosphorylation state of SPAK Ser-373, an established WNK phosphorylation site (36) (Fig. 8). We demonstrated a significant reduction in SPAK Ser-373 phosphorylation upon NEM treatment in HEK293 cells (Fig. 8A; 0.2 ± 0.01 relative to DMSO control, n = 3, unpaired t test, p < 0.0001) and immature cortical neurons (Fig. 8B; 0.1 ± 0.03 relative to DMSO control, n = 3, unpaired t test, p < 0.0001). In addition, we observed a significant increase in total SPAK levels in cell lysate of HEK293 cells treated with NEM (Fig. 8A; 1.2 ± 0.1 relative to DMSO control, n = 3, unpaired t test, p = 0.0367). However, no significant change in total SPAK levels was observed with NEM treatment in immature cortical neurons (Fig. 8B; 1.5 ± 0.4 relative to DMSO control, n = 3, unpaired t test, p = 0.2352). The effect of NEM treatment on SPAK Ser-373 phosphorylation observed in both HEK293 cells and neurons suggests that NEM targets the WNK/SPAK kinase pathway as a mechanism for KCC2 activation.
Figure 8.

NEM reduces the phosphorylation of SPAK by WNK kinases. A, cell lysates from DMSO-treated and NEM-treated HEK293 cells were probed for both total SPAK and SPAK pSer-373. SPAK pSer-373 levels were quantified relative to the DMSO control (n = 3; unpaired t test, p < 0.0001). Total SPAK levels were quantified relative to the DMSO control (n = 3; unpaired t test, p = 0.0367). Error bars represent S.E. B, cell lysates from DMSO-treated and NEM-treated immature cortical neurons were probed for both total SPAK and SPAK pSer-373. SPAK pSer-373 levels were quantified relative to the DMSO control (n = 3; unpaired t test, p < 0.0001). Total SPAK levels were quantified relative to the DMSO control (n = 3; unpaired t test, p = 0.2352). Error bars represent S.E. *, p < 0.05; ****, p < 0.0001.
Discussion
NEM has been used as a KCC activator for many years; however, the precise mechanism by which it modulates KCC2 activity remains unknown (1, 23, 24, 37). Here, we have carried out studies to determine the mechanism by which NEM rapidly increases KCC2 activity. Although we did not observe any effect of NEM on total KCC2 protein levels, we showed that NEM increased surface KCC2 levels in neurons but not in HEK293 cells. Using phosphospecific antibodies directed against key regulatory phosphorylation sites of KCC2, we also demonstrated that NEM modulated the phosphorylation profile in a specific pattern that was consistent in both neurons and HEK293 cells. Our results demonstrated that NEM quickly alters the biochemical profile of KCC2 to increase its Cl− extrusion capacity.
We showed that KCC2 surface levels were increased in immature neurons with NEM treatment but not in HEK293 cells. This discrepancy could be due to differences in KCC2 trafficking between the two cell types. NEM is known to inhibit the activity of N-ethylmaleimide–sensitive factor, a AAA+ ATPase involved in the disassembly of the SNARE complex required for SNARE-mediated membrane fusion (38, 39). Thus, it is expected that trafficking of KCC2 to the plasma membrane would be affected by NEM treatment. However, the extent to which trafficking is altered with NEM treatment likely differs between cell types as SNARE machinery composition and its role in membrane fusion vary between cell types and are dependent on cell type–specific requirements (40).
We showed that KCC2 Ser-940 phosphorylation is increased with NEM treatment in both HEK293 cells and neurons. This change suggested a correlation with the potentiation of KCC2 activity as increased Ser-940 phosphorylation is associated with increased surface stability of KCC2 and increased transporter activity (18). Interestingly, our electrophysiological analysis of the KCC2-S940A mutant indicated that modulation of this residue was not required for potentiation by NEM, which supports our previously published Rb+ influx measurements (18). Our data are also consistent with NEM-dependent increases of KCC1, KCC3, and KCC4 activity (41–43). Among mammalian KCC proteins, the Ser-940 residue is unique to KCC2; thus, only residues conserved across the KCCs are likely to mediate their shared sensitivity to NEM.
Another key regulatory phosphorylation site is KCC2 Thr-1007, and mutation of this site increases KCC1, KCC2, and KCC3 activity (19, 33, 44). We developed and characterized an antibody to specifically recognize the phosphorylated state of KCC2 Thr-1007. Using this antibody, we showed for the first time that Thr-1007 phosphorylation is significantly decreased following NEM treatment. To test the relative contribution of the modulation of Thr-1007 phosphorylation by NEM treatment, we generated a KCC2-T1007E construct. The KCC2-T1007E mutant was insensitive to NEM treatment, consistent with the idea that dephosphorylation of Thr-1007 alone is able to modulate the rapid NEM-induced potentiation of KCC2 activity.
The changes in the phosphorylation state of KCC2 that we observed align with previous studies that suggest NEM acts via modulation of a kinase or phosphatase involved in the regulation of KCCs rather than acting directly on the transporters (25–27). Dephosphorylation of the Thr-1007 residue suggests that NEM causes either a decrease in the kinase activity or an increase in the phosphatase activity associated with this site. It is known that SPAK phosphorylates KCC2 at Thr-1007 (35), and SPAK itself is activated by WNK phosphorylation at sites Thr-233 and Ser-373 (36). To gain insight into whether WNK activity is disrupted by NEM treatment, we studied SPAK Ser-373 phosphorylation. SPAK Ser-373 is positioned within the C-terminal autoinhibitory domain that is relieved upon WNK phosphorylation (45). We demonstrated decreased phosphorylation of SPAK Ser-373 upon NEM treatment, suggesting that NEM may be inhibiting WNK kinases.
Our study clearly suggests that measuring total KCC2 levels is an insufficient gauge of its function and that a more complete investigation of its phosphorylation profile is necessary. Importantly, the mechanism by which the phosphorylation state of KCC2 could alter its intrinsic activity remains an open question. One possibility is that phosphorylation of the transporter could induce a conformational change that alters the transport velocity or substrate affinity of the transporter. Indeed, phosphoregulation of the plant nitrate transporter NRT1.1 modulates its affinity for the substrate, thereby controlling transporter-mediated nitrate uptake (46, 47).
Our correlation analysis of KCC2-WT data indicated that cells with higher intracellular Cl− concentrations were more sensitive to NEM, suggesting that these cells had higher Thr-1007 phosphorylation levels or lower Ser-940 phosphorylation and surface levels in the case of neurons. This implies that in the central nervous system any drugs that act by modulating phosphorylation or surface levels would only exert their effects in tissue areas or cells where pThr-1007 levels were high or pSer-940 and surface levels were low, perhaps after neuronal injury. This mode of KCC2 potentiation is therefore self-limiting and spatially restricted.
Intriguingly, KCC2 and NKCC1 are both substrates for SPAK phosphorylation with SPAK decreasing KCC2 activity and increasing NKCC1 activity (25, 35, 48–51). By contrast, NEM has the exact opposite effects on KCC2 and NKCC1 activity (1, 52). Therefore, the ability of NEM to target the WNK/SPAK pathway may explain its reciprocal effects on KCC2 versus NKCC1. It is therefore possible that our conclusions can also apply to NKCC1 activity. Changes in the phosphorylation state of NKCC1 should parallel those of KCC2 Thr-1007, and its activity should also respond in a self-limiting and spatially restricted manner.
These studies provide the first detailed mechanistic understanding of how NEM functions to rapidly potentiate KCC2 activity. Although we demonstrate that NEM acts through two distinct mechanisms in neurons, through modulation of both surface levels and the phosphorylation state of the transporter, we show that NEM is able to potentiate KCC2 activity through modulation of the Thr-1007 site alone to result in rapid activation of this transporter. This work provides evidence suggesting that manipulation of pathways involved in the regulation of the phosphorylation state of Thr-1007 could prove therapeutically relevant.
Experimental procedures
Constructs
The KCC2b construct used in these studies was obtained from Origene (SC304801) and contains human SLC12A5 cDNA cloned into the pCMV6-XL5 vector. This construct was used to generate the KCC2-T1007A, -T1007E, and -S940A mutants using site-directed mutagenesis.
Antibodies
Anti-KCC2 antibody was used for KCC2 detection in Western blotting and was purchased from Millipore and used at a 1:2,000 dilution. Anti-KCC2 transporter NeuroMab clone N1/12 was used for immunoprecipitation of KCC2 and was purchased from Antibodies Inc. Anti-β-actin antibody, mouse monoclonal clone AC-15, was purchased from Sigma-Aldrich and used at a 1:10,000 dilution. Monoclonal anti-USP14 antibody was purchased from Sigma-Aldrich and used at a 1:1,000 dilution. GAPDH antibody (6C5) was purchased from Santa Cruz Biotechnology and used at a 1:3,000 dilution. Transferrin receptor/CD71 antibody (H68.4) was purchased from Thermo Scientific and used at a 1:1,000 dilution. KCC2 pSer-940 and pThr-1007 antibodies were both generated by PhosphoSolutions and used at 1:1,000 dilutions. KCC2 pSer-940 antibody has been characterized previously (32). Anti-serine/threonine-protein kinase 39 antibody was purchased from Millipore and used at a 1:500 dilution to detect total SPAK. Anti-phospho-SPAK antibody (Ser-373)/phospho-OSR1 antibody (Ser-325) was purchased from Millipore and used at a 1:500 dilution.
KCC2 pThr-1007 antibody generation
To produce antibodies specific for phosphorylated Thr-1007, rabbits were injected with a synthetic peptide corresponding to residues 996–1116 of murine KCC2 in which the threonine residue corresponding to Thr-1007 (residue Thr-1006 in mouse sequence only) was chemically phosphorylated: ERETDPEVHLTWTKDKSVAEK. The resulting antiserum was subjected to affinity purification. The specificity of this antibody was confirmed by immunoblotting extracts of HEK293 cells expressing wild-type and mutant KCC2 constructs. The respective antibody did not recognize forms of KCC2 in which Thr-1007 was mutated to an alanine residue.
Immature rat cortical neuron preparation
Neurons were prepared as described previously (53). Briefly, cultured cortical neurons were obtained from E18 embryos from Sprague-Dawley rats (Charles River). Cortical neurons were maintained in Neurobasal medium containing B27 (2%), glucose (0.6%), GlutaMAX (1%), and penicillin/streptomycin (1%). Neurons were grown in culture for 10–11 days prior to experimental use. All animal procedures were performed in accordance with the National Institutes of Health and approved by the Institutional Animal Care and Use Committee of Tufts University.
NEM treatment
Immature rat cortical neurons (DIV 10–11) or HEK293 cells transfected with KCC2 using Lipofectamine 2000 reagent for 72 h were treated with either DMSO or 100 μm NEM in DMSO (Sigma) in saline solution (140 mm NaCl, 2.5 mm KCl, 2.5 mm CaCl2, 2.5 mm MgCl2, 10 mm HEPES, 11 mm glucose, pH 7.4). Immature rat cortical neurons and HEK293 cells were treated for 15 min at 37 °C and room temperature, respectively. Cells were then cooled on ice and washed three times with PBS. Cells were then lysed in lysis buffer (10 mm sodium phosphate, 100 mm NaCl, 5 mm EGTA, 5 mm EDTA, 2% Triton X-100, 0.5% deoxycholate, 10 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 25 mm NaF, 250 μg/ml 4-(2-aminoethyl)benzenesulfonyl fluoride, 10 μg/ml antipain, 1 μg/ml pepstatin, 10 μg/ml leupeptin, pH 7.4) for 30 min at 4 °C with rotation. In the case of KCC2 pSer-940 experiments in HEK293 cells, cells were lysed in radioimmune precipitation assay buffer (50 mm Tris-HCl, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 5 mm EDTA, 0.1% SDS, pH 7.4) containing protease inhibitors (cOmplete tablets, mini, EDTA-free, Roche Applied Science) and phosphatase inhibitors (PhosSTOP, Roche Applied Science). Following lysis, insoluble material was removed by centrifugation at 15,700 × g for 10 min.
Immunoprecipitation
Total KCC2 was immunoprecipitated from immature rat cortical neuron cell lysate or HEK293 cell lysate prior to detection of KCC2 pThr-1007. Immunoprecipitation was also carried out for detection of pSer-940 from HEK293 cell lysate. A total of 20 μl of mouse IP matrix (Santa Cruz Biotechnology) was washed in IP buffer (10 mm Tris-HCl, pH 8.0, 7.5 mm NaCl, 0.5% Triton X-100). Beads were then mixed with 350 μg of lysate and either 4 μg/ml anti-KCC2 antibody or 4 μg/ml mouse IgG in lysis buffer overnight at 4 °C with rotation. Beads were then washed three times in IP buffer. After the final wash, beads were mixed with NuPAGE lithium dodecyl sulfate sample buffer and incubated for 20 min at room temperature. Beads were then centrifuged, and the supernatant was analyzed by Western blotting.
Western blotting
Cell lysate or immunoprecipitated KCC2 samples were separated by SDS-PAGE and transferred to nitrocellulose membrane. Following the transfer, membranes were blocked at room temperature for 1 h in 5% milk, 1% BSA in PBS-Tween. In the case of total and phospho-SPAK antibodies, blocking and antibody dilutions were carried out in 5% BSA in PBS-Tween. Membranes were incubated with primary antibody diluted in blocking solution overnight at 4 °C. Membranes were then washed three times in PBS-Tween and incubated with the respective HRP-conjugated secondary antibody. Secondary antibody incubations were carried out in blocking solution for 1 h at room temperature. Membranes were then washed three times in PBS-Tween followed by one wash in PBS. Chemiluminescence signal was detected using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific). Quantification of chemiluminescence signal was carried out using Image Lab 5.0 (Bio-Rad).
Biotinylation
Immature rat cortical neurons (DIV 10–11) or KCC2-transfected HEK293 cells plated on poly-l-lysine–coated plates were treated with either DMSO or 100 μm NEM in DMSO for 15 min in saline solution. Immature rat cortical neurons and HEK293 cells were treated for 15 min at 37 °C and room temperature, respectively. Following NEM treatment, all steps were carried out on ice using ice-cold solutions. Cells were cooled on ice and washed three times with wash solution (1 mm MgCl2, 0.1 mm CaCl2 in PBS) with the final wash carried out for 15 min at 4 °C with gentle shaking. Cells were then incubated with biotin solution (0.5 mg/ml EZ-Link Sulfo-NHS-LC-Biotin (Thermo Scientific) in wash solution) for 30 min at 4 °C with gentle shaking. Following incubation with biotin, cells were washed three times with glycine (100 mm glycine in wash solution) with the final wash carried out for 20 min at 4 °C with gentle shaking. Cells were then washed twice with wash solution and lysed in radioimmune precipitation assay buffer containing protease inhibitors (cOmplete tablets, mini, EDTA-free) and phosphatase inhibitors (PhosSTOP). Insoluble material was removed by centrifugation at 15,700 × g. Lysate (350 μg) was added to Pierce streptavidin-agarose beads (Thermo Scientific) in binding buffer (1% Nonidet P-40 in PBS) containing protease inhibitors and incubated with beads overnight at 4 °C with rotation. Beads were then washed with binding buffer four times. After the final wash, beads were mixed with NuPAGE lithium dodecyl sulfate sample buffer and incubated for 20 min at room temperature. Beads were then centrifuged for 5 min at 15,700 × g, and the supernatant was saved as the biotinylated fraction. KCC2 levels were then measured by Western blotting.
Electrophysiology
HEK293 cells were transiently transfected with cDNA for GFP, GlyR1, and either full-length human KCC2b (WT KCC2) or KCC2-T1007E and incubated for 48 h prior to recording. Recordings were conducted at room temperature (22 °C) or 34° C for HEK293 cells and neurons, respectively in bath saline containing 140 mm NaCl, 2.5 mm KCl 2.5 mm, MgCl2, 2.5 mm CaCl2, 10 mm HEPES, 11 mm glucose, pH 7.4 with NaOH. All chemicals were purchased from Sigma-Aldrich. Isolated GFP+ cells were visualized by epifluorescence and perforated with gramicidin (50 μg/ml) inside a patch pipette containing 140 mm KCl, 10 mm HEPES, pH 7.4 with KOH. All solutions were applied through a three-barrel microperfusion system (700 μm; Warner Instruments, Hamden, CT) closely positioned above the cell. After establishing series resistance <100 megaohms, bumetanide (10 μm) was added, and initial EGly or EGABA values were obtained by application of glycine (50 μm) or muscimol (1 μm) during positive-going voltage ramps (10 or 20 mV; 1-s duration). EGly or EGABA was calculated from the reversal potential of leak-subtracted glycine or muscimol currents. NEM (100 μm) in DMSO (0.1%) or DMSO alone was applied for 15 min. Test pulses of agonists (100-ms duration) at a holding potential of −40 mV every 20 s were used to monitor changes in between the voltage-ramp protocols conducted every 3 min. Data were acquired at 10 kHz with an Axopatch 200B amplifier and Clampex 10 software (Molecular Devices, Sunnyvale, CA). Differences between treatments were assessed by paired or unpaired Student's t tests as appropriate with data expressed as mean ± S.E. For correlations, linear regressions were fit to the data and tested for divergence of the best fit slope from zero.
Thallium FLIPR assay
The effect of NEM on human KCC2 activity was tested in a cellular thallium assay. Briefly, cDNA encoding human KCC2 (NM_001134771.1) was transiently transfected in HEK293 cells. The cells were plated in 384-well, black-walled, clear-bottom plates. The Tl+ FLIPR (Fluorometric Imaging Plate Reader) assay was performed using the FluxORTM Potassium Ion Channel Assay (F10017). First, cells were washed with HBSS buffer using the BioTek Elx 405. FluxOR dye was added in assay buffer (HBSS supplemented with 20 mm HEPES, pH 7.3, BackDrop® background suppressor (B10512, Life Technologies), 20 μm ouabain, 10 μm bumetanide) using a Multidrop Combi. Compound plates were prepared with DMSO (0.1%; 15 min) or NEM (100 μm; 15 min) in HBSS assay buffer. KCC2 activity was calculated from the rate of thallium influx over time. Each trace was fit with a linear regression between 20 and 220 s with the slope calculated as the rate.
Author contributions
L. C. C., R. A. C., Y. E. M., K. J., L. J. M., D. J. B., and T. Z. D. carried out experiments. L. C. C., R. A. C., D. J. B., and T. Z. D. analyzed the data. L. C. C., R. A. C., K. J., M. P. B., R. W. B., Q. W., N. J. B., S. J. M., and T. Z. D. designed the experiments. L. C. C., R. A. C., N. J. B., S. J. M., and T. Z. D. wrote the paper.
Acknowledgments
We thank Paul M. Sharpe, Graham G. Sproat, Thomas A. Ollerhead, and Jayanta Mukherjee for preparation of reagents and constructs and Christopher Lucaj and Peter Andrew for the preparation of rat cortical neurons.
This work was supported in part by funding from AstraZeneca. K. J., L. J. M., D. J. B., M. P. B., R. W. B., Q. W., and N. J. B. are full-time employees and shareholders of AstraZeneca. S. J. M. serves as a consultant for SAGE Therapeutics and AstraZeneca, relationships that are regulated by Tufts University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- KCC
- K+-Cl− cotransporter
- EGABA
- reversal potential of GABAA-mediated currents
- NEM
- N-ethylmaleimide
- EGly
- reversal potential of glycine-activated currents
- WNK
- with no lysine kinase
- SPAK
- Ste20-related proline/alanine-rich kinase
- DIV
- day(s) in vitro
- IP
- immunoprecipitation
- HBSS
- Hanks' balanced salt solution.
References
- 1. Payne J. A. (1997) Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am. J. Physiol. Cell Physiol. 273, C1516–C1525 [DOI] [PubMed] [Google Scholar]
- 2. Kaila K. (1994) Ionic basis of GABAA receptor channel function in the nervous system. Prog. Neurobiol. 42, 489–537 [DOI] [PubMed] [Google Scholar]
- 3. Moore Y. E., Kelley M. R., Brandon N. J., Deeb T. Z., and Moss S. J. (2017) Seizing control of KCC2: a new therapeutic target for epilepsy. Trends Neurosci. 40, 555–571 [DOI] [PubMed] [Google Scholar]
- 4. Payne J. A., Stevenson T. J., and Donaldson L. F. (1996) Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J. Biol. Chem. 271, 16245–16252 [DOI] [PubMed] [Google Scholar]
- 5. Rivera C., Voipio J., Payne J. A., Ruusuvuori E., Lahtinen H., Lamsa K., Pirvola U., Saarma M., and Kaila K. (1999) The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255 [DOI] [PubMed] [Google Scholar]
- 6. Miles R., Blaesse P., Huberfeld G., Wittner L., and Kaila K. (2012) Chloride homeostasis and GABA signaling in temporal lobe epilepsy, in Jasper's Basic Mechanisms of the Epilepsies (Noebels J. L., Avoli M., Rogawski M. A., Olsen R. W., and Delgado-Escueta A. V., eds) 4th Ed., National Center for Biotechnology Information, Bethesda, MD: [PubMed] [Google Scholar]
- 7. Löscher W., Puskarjov M., and Kaila K. (2013) Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 69, 62–74 [DOI] [PubMed] [Google Scholar]
- 8. Kahle K. T., Merner N. D., Friedel P., Silayeva L., Liang B., Khanna A., Shang Y., Lachance-Touchette P., Bourassa C., Levert A., Dion P. A., Walcott B., Spiegelman D., Dionne-Laporte A., Hodgkinson A., et al. (2014) Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 15, 766–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stödberg T., McTague A., Ruiz A. J., Hirata H., Zhen J., Long P., Farabella I., Meyer E., Kawahara A., Vassallo G., Stivaros S. M., Bjursell M. K., Stranneheim H., Tigerschiöld S., Persson B., et al. (2015) Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun. 6, 8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saitsu H., Watanabe M., Akita T., Ohba C., Sugai K., Ong W. P., Shiraishi H., Yuasa S., Matsumoto H., Beng K. T., Saitoh S., Miyatake S., Nakashima M., Miyake N., Kato M., et al. (2016) Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci. Rep. 6, 30072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kahle K. T., Staley K. J., Nahed B. V., Gamba G., Hebert S. C., Lifton R. P., and Mount D. B. (2008) Roles of the cation-chloride cotransporters in neurological disease. Nat. Clin. Pract. Neurol. 4, 490–503 [DOI] [PubMed] [Google Scholar]
- 12. Coull J. A., Boudreau D., Bachand K., Prescott S. A., Nault F., Sík A., De Koninck P., and De Koninck Y. (2003) Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942 [DOI] [PubMed] [Google Scholar]
- 13. Price T. J., Cervero F., Gold M. S., Hammond D. L., and Prescott S. A. (2009) Chloride regulation in the pain pathway. Brain Res. Rev. 60, 149–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou H. Y., Chen S. R., Byun H. S., Chen H., Li L., Han H. D., Lopez-Berestein G., Sood A. K., and Pan H. L. (2012) N-Methyl-d-aspartate receptor- and calpain-mediated proteolytic cleavage of K+-Cl− cotransporter-2 impairs spinal chloride homeostasis in neuropathic pain. J. Biol. Chem. 287, 33853–33864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doyon N., Ferrini F., Gagnon M., and De Koninck Y. (2013) Treating pathological pain: is KCC2 the key to the gate? Expert Rev. Neurother. 13, 469–471 [DOI] [PubMed] [Google Scholar]
- 16. Zhang Z., Wang X., Wang W., Lu Y. G., and Pan Z. Z. (2013) Brain-derived neurotrophic factor-mediated downregulation of brainstem K+-Cl− cotransporter and cell-type-specific GABA impairment for activation of descending pain facilitation. Mol. Pharmacol. 84, 511–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lavertu G., Côté S. L., and De Koninck Y. (2014) Enhancing K-Cl co-transport restores normal spinothalamic sensory coding in a neuropathic pain model. Brain 137, 724–738 [DOI] [PubMed] [Google Scholar]
- 18. Lee H. H., Walker J. A., Williams J. R., Goodier R. J., Payne J. A., and Moss S. J. (2007) Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 282, 29777–29784 [DOI] [PubMed] [Google Scholar]
- 19. Rinehart J., Maksimova Y. D., Tanis J. E., Stone K. L., Hodson C. A., Zhang J., Risinger M., Pan W., Wu D., Colangelo C. M., Forbush B., Joiner C. H., Gulcicek E. E., Gallagher P. G., and Lifton R. P. (2009) Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138, 525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kahle K. T., Deeb T. Z., Puskarjov M., Silayeva L., Liang B., Kaila K., and Moss S. J. (2013) Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 36, 726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Medina I., Friedel P., Rivera C., Kahle K. T., Kourdougli N., Uvarov P., and Pellegrino C. (2014) Current view on the functional regulation of the neuronal K+-Cl− cotransporter KCC2. Front. Cell Neurosci. 8, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Silayeva L., Deeb T. Z., Hines R. M., Kelley M. R., Munoz M. B., Lee H. H., Brandon N. J., Dunlop J., Maguire J., Davies P. A., and Moss S. J. (2015) KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. U.S.A. 112, 3523–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lauf P. K., and Theg B. E. (1980) A chloride dependent K+ flux induced by N-ethylmaleimide in genetically low K+ sheep and goat erythrocytes. Biochem. Biophys. Res. Commun. 92, 1422–1428 [DOI] [PubMed] [Google Scholar]
- 24. Lauf P. K. (1985) K+:Cl− cotransport: sulfhydryls, divalent cations, and the mechanism of volume activation in a red cell. J. Membr. Biol. 88, 1–13 [DOI] [PubMed] [Google Scholar]
- 25. Jennings M. L., and Schulz R. K. (1991) Okadaic acid inhibition of KCl cotransport. Evidence that protein dephosphorylation is necessary for activation of transport by either cell swelling or N-ethylmaleimide. J. Gen. Physiol. 97, 799–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaji D. M., and Tsukitani Y. (1991) Role of protein phosphatase in activation of KCl cotransport in human erythrocytes. Am. J. Physiol. Cell Physiol. 260, C176–C180 [DOI] [PubMed] [Google Scholar]
- 27. Bize I., Güvenç B., Buchbinder G., and Brugnara C. (2000) Stimulation of human erythrocyte K-Cl cotransport and protein phosphatase type 2A by N-ethylmaleimide: role of intracellular Mg++. J. Membr. Biol. 177, 159–168 [DOI] [PubMed] [Google Scholar]
- 28. Delpire E., Days E., Lewis L. M., Mi D., Kim K., Lindsley C. W., and Weaver C. D. (2009) Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. U.S.A. 106, 5383–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thompson S. M., and Gähwiler B. H. (1989) Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl− in hippocampal CA3 neurons. J. Neurophysiol. 61, 512–523 [DOI] [PubMed] [Google Scholar]
- 30. Stein W. D. (1986) Intrinsic, apparent, and effective affinities of co- and countertransport systems. Am. J. Physiol. Cell Physiol. 250, C523–C533 [DOI] [PubMed] [Google Scholar]
- 31. Lee H. H., Jurd R., and Moss S. J. (2010) Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci. 45, 173–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee H. H., Deeb T. Z., Walker J. A., Davies P. A., and Moss S. J. (2011) NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 14, 736–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Titz S., Sammler E. M., and Hormuzdi S. G. (2015) Could tuning of the inhibitory tone involve graded changes in neuronal chloride transport? Neuropharmacology 95, 321–331 [DOI] [PubMed] [Google Scholar]
- 34. Richardson C., and Alessi D. R. (2008) The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J. Cell Sci. 121, 3293–3304 [DOI] [PubMed] [Google Scholar]
- 35. de Los Heros P., Alessi D. R., Gourlay R., Campbell D. G., Deak M., Macartney T. J., Kahle K. T., and Zhang J. (2014) The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl− co-transporters. Biochem. J. 458, 559–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vitari A. C., Deak M., Morrice N. A., and Alessi D. R. (2005) The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J. 391, 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weber M., Hartmann A. M., Beyer T., Ripperger A., and Nothwang H. G. (2014) A novel regulatory locus of phosphorylation in the C terminus of the potassium chloride cotransporter KCC2 that interferes with N-ethylmaleimide or staurosporine-mediated activation. J. Biol. Chem. 289, 18668–18679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Glick B. S., and Rothman J. E. (1987) Possible role for fatty acyl-coenzyme A in intracellular protein transport. Nature 326, 309–312 [DOI] [PubMed] [Google Scholar]
- 39. Söllner T., Bennett M. K., Whiteheart S. W., Scheller R. H., and Rothman J. E. (1993) A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75, 409–418 [DOI] [PubMed] [Google Scholar]
- 40. Jung J. J., Inamdar S. M., Tiwari A., and Choudhury A. (2012) Regulation of intracellular membrane trafficking and cell dynamics by syntaxin-6. Biosci. Rep. 32, 383–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gillen C. M., and Forbush B. 3rd (1999) Functional interaction of the K-Cl cotransporter (KCC1) with the Na-K-Cl cotransporter in HEK-293 cells. Am. J. Physiol. Cell Physiol. 276, C328–C336 [DOI] [PubMed] [Google Scholar]
- 42. Mercado A., Song L., Vazquez N., Mount D. B., and Gamba G. (2000) Functional comparison of the K+-Cl− cotransporters KCC1 and KCC4. J. Biol. Chem. 275, 30326–30334 [DOI] [PubMed] [Google Scholar]
- 43. Shen M. R., Chou C. Y., Hsu K. F., Liu H. S., Dunham P. B., Holtzman E. J., and Ellory J. C. (2001) The KCl cotransporter isoform KCC3 can play an important role in cell growth regulation. Proc. Natl. Acad. Sci. U.S.A. 98, 14714–14719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brown F. C., Conway A. J., Cerruti L., Collinge J. E., McLean C., Wiley J. S., Kile B. T., Jane S. M., and Curtis D. J. (2015) Activation of the erythroid K-Cl cotransporter Kcc1 enhances sickle cell disease pathology in a humanized mouse model. Blood 126, 2863–2870 [DOI] [PubMed] [Google Scholar]
- 45. Gagnon K. B., and Delpire E. (2010) On the substrate recognition and negative regulation of SPAK, a kinase modulating Na+-K+-2Cl− cotransport activity. Am. J. Physiol. Cell Physiol. 299, C614–C620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Parker J. L., and Newstead S. (2014) Molecular basis of nitrate uptake by the plant nitrate transporter NRT1.1. Nature 507, 68–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun J., and Zheng N. (2015) Molecular mechanism underlying the plant NRT1.1 dual-affinity nitrate transporter. Front. Physiol. 6, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dunham P. B., Stewart G. W., and Ellory J. C. (1980) Chloride-activated passive potassium transport in human erythrocytes. Proc. Natl. Acad. Sci. U.S.A. 77, 1711–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lytle C., and Forbush B. 3rd (1992) The Na-K-Cl cotransport protein of shark rectal gland. II. Regulation by direct phosphorylation. J. Biol. Chem. 267, 25438–25443 [PubMed] [Google Scholar]
- 50. Adragna N. C., Di Fulvio M., and Lauf P. K. (2004) Regulation of K-Cl cotransport: from function to genes. J. Membr. Biol. 201, 109–137 [DOI] [PubMed] [Google Scholar]
- 51. Thastrup J. O., Rafiqi F. H., Vitari A. C., Pozo-Guisado E., Deak M., Mehellou Y., and Alessi D. R. (2012) SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem. J. 441, 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Muzyamba M. C., Cossins A. R., and Gibson J. S. (1999) Regulation of Na+-K+-2Cl− cotransport in turkey red cells: the role of oxygen tension and protein phosphorylation. J. Physiol. 517, 421–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sivakumaran S., Cardarelli R. A., Maguire J., Kelley M. R., Silayeva L., Morrow D. H., Mukherjee J., Moore Y. E., Mather R. J., Duggan M. E., Brandon N. J., Dunlop J., Zicha S., Moss S. J., and Deeb T. Z. (2015) Selective inhibition of KCC2 leads to hyperexcitability and epileptiform discharges in hippocampal slices and in vivo. J. Neurosci. 35, 8291–8296 [DOI] [PMC free article] [PubMed] [Google Scholar]