

Abstract
RhoGC is a fusion protein from the aquatic fungus Blastocladiella emersonii, combining a type I rhodopsin domain with a guanylyl cyclase domain. It has generated excitement as an optogenetics tool for the manipulation of cyclic nucleotide signaling pathways. To investigate the regulation of the cyclase activity, we isolated the guanylyl cyclase domain from Escherichia coli with (GCwCCRho) and without (GCRho) the coiled-coil linker. Both constructs were constitutively active but were monomeric as determined by size-exclusion chromatography and analytical ultracentrifugation, whereas other class III nucleotidyl cyclases are functional dimers. We also observed that crystals of GCRho have only a monomer in an asymmetric unit. Dimers formed when crystals were grown in the presence of the non-cyclizable substrate analog 2′,3′-dideoxyguanosine-5′-triphosphate, MnCl2, and tartrate, but their quaternary structure did not conform to the canonical pairing expected for class III enzymes. Moreover, the structure contained a disulfide bond formed with an active-site Cys residue required for activity. We consider it unlikely that the disulfide would form under intracellular reducing conditions, raising the possibility that this unusual dimer might have a biologically relevant role in the regulation of full-length RhoGC. Although we did not observe it with direct methods, a functional dimer was identified as the active state by following the dependence of activity on total enzyme concentration. The low affinity observed for GCRho monomers is unusual for this enzyme class and suggests that dimer formation may contribute to light activation of the full-length protein.
Keywords: crystal structure, cyclic GMP (cGMP), disulfide, guanylate cyclase (guanylyl cyclase), optogenetics, rhodopsin, Blastocladiella emersonii, class III nucleotidyl cyclase
Introduction
The rhodopsin-guanylyl cyclase fusion protein RhoGC3 was first identified in the phototactic fungus Blastocladiella emersonii by Avelar et al. (1). It is a unique protein composed of a type I (microbial) rhodopsin (2, 3) domain fused to a guanylyl cyclase catalytic domain showing homology to the rod outer segment guanylyl cyclase from vertebrate retina. It was the first retinylidene protein described that is directly coupled to an enzyme. The protein is localized to the B. emersonii eyespot and is essential for zoospore phototaxis, a pathway known to signal through the second messenger cGMP. RhoGC contains four domains (Fig. 1): an intracellular N-terminal domain of unknown function (N-term); a type I rhodopsin domain (Rho); a CC domain; and a guanylyl cyclase domain (GC).
Figure 1.
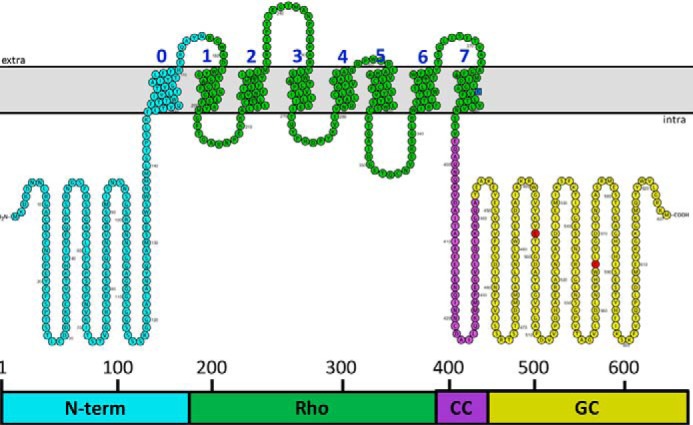
Model for the transmembrane topology and orientation of RhoGC. Domains are: N-term, N-terminal domain; Rho, microbial type I rhodopsin domain; CC, coiled-coil domain; and GC, guanylyl cyclase domain. Transmembrane helices were predicted and drawn by Protter (wlab.ethz.ch/protter/start (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site)) (34). Cyan, N-term; green, Rho; purple, CC; yellow, GC; blue square, Lys-384, presumed site of covalent attachment to the chromophore; and red circles, Glu-497 and Cys-566, residues controlling specificity for the GTP substrate.
RhoGC is of considerable interest as an optogenetic tool for control of cyclic nucleotide-signaling pathways (4, 5), and it has now been joined by another type I rhodopsin fusion protein, RhoPDE (6, 7), with potential as an optogenetic tool. The gene for RhoPDE was found in the genome of the choanoflagellate Salpingoeca rosetta and encodes a fusion of the rhodopsin domain with a cGMP-specific phosphodiesterase catalytic domain.
We have developed an expression/purification system and performed preliminary characterization for RhoGC in preparation of studies to elucidate the mechanism by which light controls guanylyl cyclase activity in the enzyme (8). We now turn our attention to the individual domains to more fully identify their functional contribution to the full-length RhoGC. We begin here with the guanylyl cyclase domain, GCRho.
Crystal structures of GC catalytic domains have been determined for bacterial (9), algal (10), and human (11) proteins (see Fig. S1 for sequence alignment). GC domains from the cyanobacterium Synechocystis PCC6803 (GCCya2) (9) and the unicellular algae Chlamydomonas reinhardtii (GCCYG12) (10) were crystallized as homodimers, whereas the GC domain (GCHum) from the human soluble guanylyl cyclase, sGC (11), was crystallized as both homodimer and heterodimer, although the enzyme is active only in the heterodimeric form. The active sites form at the dimer interface where catalytic residues are contributed by both monomers. Hence, dimerization is necessary for catalysis in this class of enzymes (12, 13). All three GC structures solved so far are the substrate-free form and are reported to be an inactive “open” conformation (11). Comparing the open GC structures with the active “closed” conformation of the adenylyl cyclase (AC) catalytic domain (14) revealed that the protomers in the inactive GC dimers are rotated by 7–8° for GCCya2 and GCCYG12 and by 26° for GCHum relative to the monomers in the active AC structure.
We have expressed the catalytic domain of RhoGC (GCRho) in Escherichia coli, purified the protein by nickel-affinity chromatography, and show here that the isolated protein is constitutively active in catalyzing the formation of cGMP. It is a monomer in solution and crystallizes with a monomer in the asymmetric unit, but it has a non-linear dependence of activity on enzyme concentration that is consistent with transient formation of an active-state dimer. Crystals of a dimeric form of GCRho were obtained from crystallization trials in the presence of the non-cyclizable substrate analog 2′,3′-dideoxyguanosine-5′-triphosphate (ddGTP), Mn2+, and added potassium sodium tartrate, but the two protomers are arranged in a head-to-head conformation distinctly different from the canonical head-to-tail wreath-like structures first described for adenylyl cyclases (13, 15) and then later for the guanylyl cyclases (9–11). We anticipate that both monomer and dimer structures reported here for GCRho will help guide mutagenesis experiments focused on the mechanism by which light controls activity of the cyclase domain in full-length RhoGC.
Results
Expression and purification of GCRho and GCwCCRho
Conventional PCR was used to construct genes for the isolated guanylyl cyclase domain, with (GCwCCRho, residues 397–627) and without (GCRho, residues 443–627) the connecting linker (CC, residues 397–442), from full-length RhoGC (Fig. 1). Both constructs contained a His6 tag on the C terminus for purification of the protein by nickel-affinity chromatography. Initial tests in E. coli showed that the proteins were expressed to a high level in T7-express cells under standard induction conditions (1 mm IPTG at A600 0.4–0.7 followed by incubation for 3 h at 37 °C), but neither protein was found in the soluble fraction after cell lysis and centrifugation. Subsequent attempts varying induction conditions, growth and expression temperature, and a change of cell lines met with limited success. In the end, coexpression with the molecular chaperone proteins GroEL and GroES (16) dramatically increased the amount of both GCwCCRho and GCRho in the soluble fraction such that we could purify (Fig. 2A) on average about 50 mg of protein (determined spectrophotometrically using an extinction coefficient ϵ280 nm = 31,065 m−1 cm−1) per liter of cell culture, a quantity sufficient to undertake biochemical and structural characterization of the proteins.
Figure 2.

Purification and initial characterization of the isolated GCwCCRho and GCRho domains. A, purification of GCwCCRho and GCRho. Left panel, Coomassie-stained SDS-polyacrylamide gel showing fractions for purification of the isolated GCwCCRho domain from transformed E. coli BL21(DE3)-pGro7 cells (co-expressing GroEL/ES) using nickel-affinity chromatography. Right panel, Coomassie-stained SDS-polyacrylamide gel showing fractions from purification of GCRho. The last lane contains purified GCwCCRho for size comparison. B, initial rate data for guanylyl cyclase activity from GCRho and GCwCCRho in the presence of MnCl2 (left panel) or MgCl2 (right panel). Reactions were performed as described under “Experimental procedures” and contained 10 μm enzyme, 5 mm GTP, and 10 mm divalent cation. C, Äkta FPLC profiles for size-exclusion chromatography of GCRho (solid line) and GCwCCRho (dashed line) on a Superdex-200 10/300 GL column in 25 mm HEPES buffer, pH 7.0, containing 100 mm NaCl. Both proteins were loaded onto the column at a concentration of 200 μm. Molecular mass standards: 1, blue dextran, 2000 kDa, 8.5 ml (void volume); 2, aldolase, 158 kDa, 13.15 ml; 3, albumin, 67 kDa, 14.57 ml; 4, ovalbumin, 43 kDa, 15.41 ml; 5, chymotrypsinogen-A, 25 kDa, 16.8 ml; and 6, ribonuclease-A, 14 kDa, 18 ml. Both GCRho and GCwCCRho elute as monomers under these conditions.
GCwCCRho and GCRho both displayed constitutive guanylyl cyclase activity that varied depending on whether Mn2+ or Mg2+ was used for the metal ion. As is shown in Fig. 2B, GCRho displayed much higher activity in the presence of Mn2+ than in Mg2+, consistent with observations from other isolated guanylyl (9–11, 16) and adenylyl cyclase (12) domains. However, this is not the case when the CC linker was included. GCwCCRho displays about the same activity in Mn2+ as in Mg2+, and both are significantly less active than the GC domain alone in Mn2+. These data seem to suggest that the role of CC might be to attenuate the constitutive activity of the GCRho domain, as has been suggested for other systems, but this is clearly not the case if GCwCCRho is compared with GC in the presence of Mg2+, where GCwCCRho displays the higher activity. At 10 μm enzyme concentration, the activities were as follows: GCRho with Mn2+ = 21 ± 1 μm/s; GCwCCRho with Mn2+ = 1.0 ± 0.1 μm/s; GCwCCRho with Mg2+ = 0.71 ± 0.06 μm/s; and GCRho with Mg2+ = 0.064 ± 0.002 μm/s.
The dependence of activity on GTP concentration for GCRho in the presence of MnCl2 is presented in the plot of Fig. S2, where the data are fit with a Hill-type equation using the parameters S0.5 = 0.9 ± 0.1 mm, Vmax = 16 ± 1 μm/s, and n = 1.2 ± 0.3.
All known class III cyclases function as dimers (12, 17, 18). They can be either hetero- or homodimers, with active sites formed at the dimeric interface. Given the robust constitutive activity of both GCwCCRho and GCRho, we anticipated that the proteins were assembled as homodimers in solution. Contrary to expectations, both proteins ran as monomers by size-exclusion chromatography (SEC) on a Superdex-200 column (Fig. 2C), with molecular mass estimates for the peak elution fractions from standards within 5% error of the calculated molecular mass of monomeric GCRho (21.5 kDa) and GCwCCRho (26.5 kDa).
Structure of GC
Initial crystallization trials were conducted with GCwCCRho. After 3 months, a small rectangular-shaped crystal appeared in a PEG3350 solution. The structure of the enzyme was determined to 1.6 Å resolution by molecular replacement using human soluble guanylyl cyclase (PDB entry 2WZ1) as a search model. The protein had crystallized in space group P22121 with one molecule in the asymmetric unit. Although the full-length GCwCCRho construct was purified and used in the crystallization trials, electron density was not observed for the CC linker, likely as a result of proteolysis or disorder in the crystal. Repeated trials in the presence of sodium azide to prevent bacterial growth did not yield crystals, and further attempts to optimize crystallization conditions were performed with GCRho alone. Crystals of GCRho formed in the same crystallization conditions after about 3 weeks, still in space group P22121 with one molecule in the asymmetric unit, and the structure was determined to 1.13 Å resolution and refined to an Rwork and Rfree of 0.188 and 0.195, respectively. Importantly, crystal packing did not reveal dimer formation with symmetry-related molecules.
The overall structure of the GCRho catalytic domain has five α-helices and eight β-strands (Fig. 3), as has been reported previously for other GC (9–11) and AC structures (13, 15). Clear electron density was observed for residues Thr-443 through Lys-626, with the exception of two missing loop regions not modeled in the final structure: Gly-559–Asn-562 in the β4–β5 loop, which has a major role in dimer formation, and Val-608–Lys-616 in the β7–β8 loop, which carries one of the eight conserved catalytic residues, Lys-612.
Figure 3.
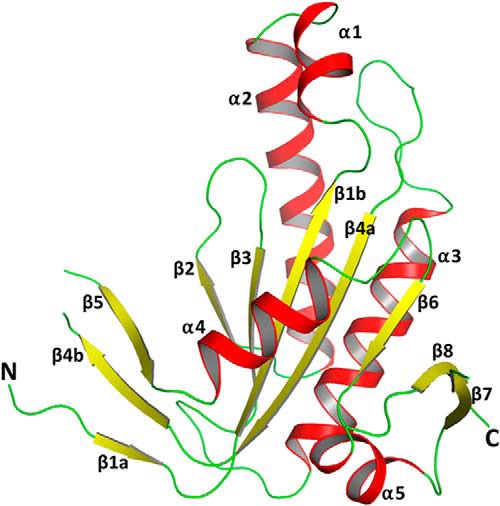
Overall structure of the GCRho monomer. Secondary structure elements are labeled according to conventional nomenclature and are color-coded: red, α-helix; yellow, β-strand; and green, loop.
Comparison with other guanylyl cyclases
Synechocystis guanylyl cyclase Cya2. The catalytic domain (GCCya2) of the Cya2 guanylyl cyclase from the cyanobacterium Synechocystis PCC6803 crystallized as a homodimer, and its structure was determined at 2.3 Å resolution (9). A superpose of the GCRho monomer and molecule A of GCCya2 shows that the two proteins are very similar in overall fold with r.m.s.d. for Cα atoms of 0.9 Å. The α3–β4 loop in GCRho is four residues shorter than the corresponding loop in GCCya2, and the α3 helix is two turns longer in GCCya2. This structural feature appears to be unique to GCCya2 because the respective regions in Chlamydomonas (10) and human sGCs (11) were similar to that of the GCRho monomer. The GCRho β4–β5 and β7–β8 loops are each one residue shorter than in GCCya2, whereas GCCya2 has an extended C terminus.
C. reinhardtii guanylyl cyclase CYG12
The catalytic domain (GCCYG12) of the soluble guanylate cyclase from the green algae C. reinhardtii crystallized as a homodimer, and its structure was solved at 2.6 Å resolution (10). A superpose of the GCRho monomer and molecule A of GCCYG12 shows that the two proteins are very similar in overall fold with r.m.s.d. for Cα atoms of 1.2 Å. The α2–β2 and α5–β7 loops in GCRho are one and two residues shorter, respectively, than the corresponding regions in GCCYG12. The C. reinhardtii protein also has an additional short α6 helix, which is not observed in other GC structures.
Human soluble guanylyl cyclase
The catalytic domain (GCHum) of the human soluble guanylate cyclase crystallized as a heterodimer, and its structure was solved at 1.6 Å resolution (11). A superpose of the GCRho monomer with the α-subunit of the human enzyme shows that the two proteins are very similar in overall fold with r.m.s.d. for Cα atoms of 1.2 Å but that they differ in conformation of the α3–β4 loop. In addition, the α1–α2, α2–β2, and α6–β7 loops in GCRho are five, three, and eight residues shorter than respective regions in GCHum.
Dimeric GCRho
Given that the active state of GCRho was expected to be a dimeric enzyme, we explored other crystallization conditions that might trap a dimer in the asymmetric unit. Because the enzyme displayed significant guanylyl cyclase activity when assayed in solution (Fig. 2B), we reasoned that substrate and metal ion might facilitate formation of the dimer. We used the substrate analog 2′,3′-dideoxyguanosine 5′-triphosphate (ddGTP), which lacks the ability to be cyclized due to the absence of a hydroxyl group at the 3′-position of the sugar ring. GCRho was mixed with ddGTP and Mn2+ and then monitored for formation of dimer by analytical ultracentrifugation (AUC). Sedimentation velocity profiles showed the existence of two separate sedimenting species consistent with the molecular mass expected for monomeric and dimeric GCRho (Fig. 4A). Notably, GCRho sediments exclusively as monomer in the absence of ddGTP and Mn2+. When a similar sample was applied to the Superdex-200 column, three major species were observed, corresponding to dimer, monomer, and free nucleotide (Fig. 4B). The percentage of dimer in the SEC experiment was less than that observed in AUC likely due to slight differences in sample preparation.
Figure 4.

Formation of GCRho dimers. A, normalized c(s) distribution plot for AUC of GCRho in the absence (black) and presence (red) of 1 mm MnCl2 and 60 μm ddGTP (ddGTP equimolar with GCRho). B, SEC profiles for GCRho in the absence (solid line) and presence (dotted line) of MnCl2 and ddGTP. Solid line, data for GCRho in the absence of MnCl2 and ddGTP are from Fig. 2C. Dotted line, data from protein that was subjected to the same conditions as for the AUC sample in A containing MnCl2 and ddGTP. Superdex-200 column was run in 25 mm HEPES, pH 7.0, containing 100 mm NaCl at 4 °C with a flow rate of 0.5 ml/min.
Crystal structure of the dimer
GCRho crystallized in space group C2221 from PEG3350 in the presence of 1 mm ddGTP, 10 mm MnCl2, and 200 mm potassium sodium tartrate additive. The structure was solved to 1.7 Å resolution by molecular replacement using the GCRho monomer (Fig. 3) as search model and yielded two molecules in the asymmetric unit (Fig. 5A). The final model was refined to Rwork and Rfree of 0.164 and 0.196, respectively. Clear electron density was observed for residues Met-442 to Met-627 in molecule A and Ala-445 to Lys-626 in molecule B. The β7–β8 loop region residues Glu-610–Gly-615 in molecule A and Lys-612–Lys-614 in molecule B were highly disordered and not modeled in the final structure.
Figure 5.

Structure of the GCRho homodimer. A, overall structure of the GCRho homodimer. Schematic representations of molecule A and B are colored green and gold, respectively. Selected secondary structure elements are labeled according to convention. Tartrate is modeled as sticks and shown in yellow. Manganese ions are shown as green spheres. Cysteine residues are shown as sticks and colored according to the respective main chain coloring. B, structural superposition of the GC homodimer with the “active conformation” of the human AC heterodimer (PDB entry 1CJU). Schematic representations are color-coded as follows: GCRho molecule A, green; GCRho molecule B, gold; and human AC molecules A and B, blue. N- and C-terminal amino acids are marked and color-coded according to the coloring of the respective molecule. C, disulfide cross-link at the dimer interface. Fo − Fc omit map is contoured at 3 σ cutoff. Parts of the protein backbone have been omitted for clarity. D, tartrate-binding site at the dimer interface. Fo − Fc omit map is contoured at 4 σ cutoff. Metal ion coordination and hydrogen bonding are indicated with dotted lines. Parts of the protein backbone and additional solvent molecules have been omitted for clarity.
Superposition of the GCRho monomer with both monomers of the dimer showed that there are no significant structural changes in the monomer units, with r.m.s.d. for Cα atoms of 0.4 and 0.3 Å for monomer A and monomer B, respectively, although minor changes were observed in part of the dimer interface around the α1 helix (Asn-460–Ser-469) and β4–β5 loop (Leu-558–His-564). Nonetheless, the monomer units do not form the signature head-to-tail wreath-like conformation observed in other GC (9–11) and AC (13–15) dimeric structures. Instead, the two monomers are in a head-to-head arrangement. Superposition of the GCRho dimer with the active conformation of the mammalian AC heterodimer (PDB entry 1CJU (14)), using monomer A as a reference, shows that monomer B of GCRho is rotated by 101° and displaced by 6.3 Å with respect to AC monomer B (Fig. 5B). The large rotation of monomer B compared with the AC structure suggests that the GCRho dimer is non-functional.
The GCRho dimer has a buried surface area of 1273 Å2 and geometric surface complementarity (Sc) (19) of 0.63. The buried surface area is comparable in magnitude to the buried surface area for the C. reinhardtii cyclase GCCYG12 (1370 Å2; 3ET6), for the cyclase GCCya2 from Synechocystis (1548.9 Å2; 2W01), and for the human sGC heterodimer GCHum (1230 Å2; 3UVJ). The shape complementarity statistic, Sc, is also similar in magnitude to those of other adenylyl and guanylyl cyclases (AC, 0.59; GCCYG12, 0.64; GCCya2, 0.67; GCHum, and 0.74).
Part of the dimer interface is formed by an unusual interaction of β-strands β4b and β5 of monomer A with those of monomer B. In other GC dimeric structures (9–11), this β-sheet interacts with strands β2 and β3 to form the dimer interface. Significantly, monomers A and B of GCRho are connected by a disulfide bridge involving the catalytically important residue Cys-566 (8, 20, 21) in β5 from both monomers (Fig. 5C), reinforcing the conclusion that this is an inactive form of the protein.
After initial refinement, difference Fourier density appeared at the cavity formed by the catalytically important residues Asp-457, Asp-501, and Arg-545 (12–14) from both monomers A and B (Fig. 5D). The density was further resolved into two 22 σ Fo − Fc peaks and an overlapping, elongated ∼8 σ Fo − Fc peak. Both 22 σ peaks were modeled with a Mn2+ ion. The 8 σ peak electron density was not fit well by ddGTP, cGMP, or PPi but was modeled well by the tartrate additive. Mn2+A is hexa-coordinate with Asp-457 Oδ2 of molecule B, Asp-501 Oδ1 of molecule A, O2 and O6 atoms of the tartrate molecule, and two water molecules as ligands. Mn2+B is also hexa-coordinate with Asp-457 Oδ1 and Asp-501 Oδ1 and Oδ2 of molecule B, O1 and O3 atom of the tartrate molecule, and a single water molecule as ligands. Tartrate is perfectly sandwiched between the side chains of the catalytically important residue R545 of both monomer A and monomer B. Apart from metal ion coordination, tartrate also forms hydrogen bonds with amino acids D457, T462, and D501 of molecule A, D457, I458, T462, and D501 of molecule of B, and with several water molecules.
Dependence on Mn2+
Discovery of the disulfide cross-link in the GCRho dimer caused us to revisit the AUC experiments where we could show that no dimer species was formed in the presence of a reducing agent (5 mm DTT). Likewise, subsequent crystal trials with ddGTP and Mn2+ in the presence of DTT produced only crystals with monomer in the asymmetric unit and cell dimensions and space group identical to those observed in the absence of nucleotide. Therefore, we conducted a series of experiments to determine what factors were important for formation of the disulfide cross-linked GCRho dimer as assayed by SDS-PAGE. As is shown in Fig. 6, the single factor most important was the presence of Mn2+. The presence of GTP, cGMP, and inorganic pyrophosphate in the absence of metal ion were without effect. Mg2+ could substitute for Mn2+ in this reaction but with reduced efficiency. Notably, addition of the CC domain dramatically decreased the amount of dimer formed under all conditions.
Figure 6.

Analysis of disulfide cross-linking by SDS-PAGE. Protein samples (GCRho or GCwCCRho) were incubated for 20 h under the conditions indicated in the figure before being loaded onto 10% polyacrylamide gels so as to simulate the AUC conditions in Fig. 4A. All samples were prepared in 25 mm HEPES, pH 7.0, 100 mm NaCl using either 1 μm GCRho or GCwCCRho. Top panels, GCRho; bottom panels, GCwCCRho; left panels, without DTT; and right panels, with 5 mm DTT. Other conditions are as indicated in the figure: Mn, 2 mm MnCl2; Hi Mn, 20 mm MnCl2; Mg, 10 mm MgCl2; cGMP, 1 mm cGMP; GTP, 1 mm GTP; and PPi, 1 mm pyrophosphate.
It should be noted that disulfide bond formation required a lengthy incubation period. The reactions shown in Fig. 6 included a 20-h incubation to mimic the original AUC and SEC conditions (Fig. 4). No cross-linked dimer band was observed when the incubation period was omitted.
Structure of the GCAC domain
Previously, we constructed a mutant of RhoGC in which the activity of the enzymatic domain was changed from a guanylyl cyclase to an adenylyl cyclase (8). This required the introduction of two mutations in the GC domain of the protein, E497K and C566D. Given that the reactive Cys-566 is replaced by Asp, it was of interest to determine a crystal structure for the isolated “adenylyl cyclase” domain, GCAC, of the mutant. GCAC displays the same substrate specificity (Fig. 7A) as was observed for the full-length mutant (8) and was used for crystal trials under a host of different conditions. Although GCAC did not form crystals under the same conditions as for the GCRho dimer, it did crystallize from PEG20000 in the presence of 10 mm potassium hydrogen tartrate with the same space group and unit cell dimensions as found with the GCRho monomer. The structure was solved by molecular replacement using the GCRho monomer as a search model. The final structure was determined to a resolution of 1.4 Å with Rwork/Rfree of 0.190/0.208. A single molecule was in the asymmetric unit with overall structure identical to that of the GCRho monomer. The r.m.s.d. for Cα atoms from superposition of the two structures is only 0.1 Å suggesting that the mutations had minimal effect on secondary and tertiary structure of the protein. Amino acids Gly-559 and Asp-560 in the β4–β5 loop and Glu-610–Gly-615 in the β7–β8 loop were highly disordered and not modeled in the final structure. The calculated omit map clearly revealed the two mutations in the protein (Fig. 7B), and Lys-497 and Asp-566 were modeled in the structure where their side chains were found to form a salt bridge.
Figure 7.

GCAC activity data and omit map. A, initial rate data for catalytic activity of GCAC when provided ATP (black circles) or GTP (red circles) as a substrate. The reactions contained 5 mm substrate (GTP or ATP) and 10 mm MnCl2. B, Fo − Fc omit map contoured at 3σ cutoff shows electron density for the two mutated active site residues Lys-497 and Asp-566 in GCAC. Secondary structure elements are colored green and labeled according to convention. Amino acid side chains are shown as sticks. Hydrogen bonding is indicated by the dotted line. Part of the protein backbone has been removed for clarity.
Indirect evidence for functional dimerization
Indirect evidence for formation of an active dimer was obtained by following the dependence of enzyme activity on the concentration of protein in the assays. As shown in Fig. 8, guanylyl cyclase activity showed a distinctly nonlinear dependence on total enzyme concentration that was well fit by Equation 1 under “Experimental procedures” for a monomer/dimer equilibrium in which only the dimer is active. The parameters used to fit Equation 1 to the data are presented in Table 1. The inclusion of DTT in the reaction had no effect on activity demonstrating that the disulfide cross-linked dimer observed in the crystal structure does not form during the short incubation times used here in the activity assays. It is evident from the data in Fig. 8 and Table 1 that the effect of the CC domain on quaternary structure of the protein is a rather modest 3–4-fold decrease in KD for formation of the dimer.
Figure 8.

Guanylyl cyclase activity as a function of enzyme concentration. A, rate of cGMP formation as determined by the HPLC assay is plotted as a function of total enzyme concentration showing a non-linear dependence that is well fit by Equation 1 under “Experimental procedures” relating initial rate to a monomer/dimer equilibrium model in which only the dimer is active. The construct and condition for each set of data are as indicated in the figure. B, data for GCRho with MgCl2 are re-plotted from A on a more sensitive scale to show clearly the non-linear dependence on enzyme concentration. All reactions contained 10 mm GTP and 20 mm metal ion (Mn2+ or Mg2+). The dotted curves are fits of Equation 1 to the data using the parameters of KD and kcat listed in Table 1. The enzyme concentration is total enzyme added to the reaction, expressed in terms of the micromolar concentration of monomer. An expanded view of the data for GCwCCRho-Mn is shown in Fig. S3A. A control experiment showing that the non-linear behavior is not a result of non-specific protein interactions at low concentrations is shown in Fig. S3B.
Table 1.
KD and kcat values for the fits of Equation 1 to the data of Fig. 8
| Construct and condition | KD | kcat |
|---|---|---|
| μm | s−1 | |
| GCRho–MnCl2 | 78 ± 12 | 10 ± 1 |
| GCRho–MnCl2 and DTT | 83 ± 30 | 9.6 ± 1.6 |
| GCRho–MgCl2 | 76 ± 25 | 0.16 ± 0.02 |
| GCwCCRho–MnCl2 | 23 ± 15 | 2.5 ± 0.5 |
To eliminate concern that the C-terminal His6 tag might interfere with GCRho dimerization, a new construct was generated with an N-terminal His6 tag and TEV-proteolysis site inserted between the His6 tag and the remainder of the GCRho catalytic domain, and the dependence of activity on enzyme concentration repeated with the TEV-proteolyzed enzyme. The data were again well fit by Equation 1 with an obviously non-linear dependence of activity on enzyme concentration giving a KD value of 64 ± 9 μm, well within experimental error of the KD values for GCRho reported in Table 1.
Active-state dimer
Because we could not crystallize an active-state dimer of GCRho, we modeled what the dimer might look like based upon superposition with the published structure of the mammalian adenylyl cyclase VC1IIC2 catalytic domain in complex with Mn2+ and ddATP (Fig. S4) (14). The active-site residues at the dimer interface align nicely between the two structures, although side chain conformations show some differences, as is expected given that GCRho crystallized as a monomer.
Discussion
The purpose of this study was to provide an initial purification and characterization of the isolated guanylyl cyclase domain of RhoGC, with (GCwCCRho) and without (GCRho) the CC domain, in preparation for a more in-depth mechanistic exploration of the full-length protein. Genes for His6-tagged versions of both proteins were co-expressed in E. coli with GroEL and GroES (16) to improve the yield of properly folded protein in the soluble fraction of cellular extracts. The proteins were purified by nickel-affinity chromatography in a yield of about 50 mg/liter cell culture.
Both proteins displayed robust guanylyl cyclase activity in a reaction that was dependent on added metal ion, Mg2+ or Mn2+. As has been observed with other cyclases, GCRho was significantly more active with Mn2+ than with Mg2+ (9–12, 16). Interestingly, including the CC domain in the GCwCCRho construct eliminated this difference in activity with the two metal ions. GCwCCRho displays a kcat (2.5 s−1) that is about 4-fold less than the GCRho domain with Mn2+ (10 s−1) but significantly more than GCRho with Mg2+ (0.16 s−1) and, perhaps most importantly, about the same as the apparent kcat for light-stimulated guanylyl cyclase activity for the full-length RhoGC (2.2 s−1) (8). The latter comparison suggests that a function of the remaining domains in RhoGC (i.e. N-term and Rho) is to suppress constitutive activity of GCwCCRho, such that the full-length protein expresses guanylyl cyclase activity only after activation with light. This appears to be the case also for ligand-activated guanylyl cyclases, where regulatory domains suppress constitutive activity of the catalytic domains (9–11, 16).
Although there were clear similarities of GCRho and GCwCCRho in comparison with other guanylyl cyclases, a striking difference was encountered in crystal structures of the GCRho domain where the protein crystallized under standard conditions with only a single subunit in the asymmetric unit. This was unexpected because both GCwCCRho and GCRho display significant guanylyl cyclase activity. All other guanylyl cyclases crystallize as dimers with two monomers in the asymmetric unit (9–11), and the active sites of adenylyl and guanylyl cyclases are known to be formed at the interface of the two protomers in a dimeric complex (17, 18). The unusual crystallization results for GCRho were supported by SEC and AUC experiments showing that the protein was 100% monomer in solution. To be sure, monomers of other guanylyl cyclases have been observed in solution studies (9, 16), but the monomer/dimer equilibria are more heavily shifted toward the dimer, and, as noted above, the other guanylyl cyclases all crystallized with a dimer in the asymmetric unit. There are two reports in which adenylyl cyclases crystallized as a monomer: one for a Phe to Arg mutant involving a residue in the dimer interface of the Mycobacterium tuberculosis guanylyl cyclase Rv1625c catalytic domain (22), and the other for the catalytic domains of the adenylyl cyclases, GRESAG4.1 and GRESAG4.3, from Trypanosoma brucei (23). Rv1625c forms an inactive dimer in solution (22) whereas the T. brucei enzymes display weak subunit affinity and transient formation of catalytically active dimers in solution (23).
We were able to crystallize a GCRho dimer in the presence of ddGTP, Mn2+, and added potassium sodium tartrate. The two subunits of the dimer are nearly identical in overall fold to the isolated GCRho monomer and to the individual subunits found in dimer structures of other guanylyl cyclases, but the arrangement of the two subunits is very different from the canonical wreathlike structure first described for adenylyl cyclase (13–15) and later found in the guanylyl cyclases (9–11). The subunits of GCRho dimer are displaced by 6 Å and rotated by 101° with respect to the arrangement of subunits in the adenylyl cyclase dimer (14) such that the conserved active-site residues are dispersed in the protein and are unlikely to support binding of nucleotide. In addition, the GCRho dimer contains a disulfide bond involving Cys-566, an active-site residue known to be critical for activity of the protein (8). Thus, it is clear that this dimer cannot be part of the catalytic cycle of the enzyme.
Unusual dimer structures have been described for both the Rv1264 (24) and Rv1625 (22) adenylyl cyclases from M. tuberculosis, and in this context the domain-swapped head-to-head dimer observed for WT Rv1625 (22) shares some similarity with the head-to-head dimer we see here for GCRho. It is unlikely that the disulfide in GCRho would form under intracellular reducing conditions, and it seems most probable that the disulfide forms subsequent to dimerization as a consequence of a high local concentration of the two Cys residues in an oxidizing environment. Thus, the disulfide could function as a kinetic trap for the dimer. The dimer structure does not appear to result from two monomers that were brought together in a random and haphazard association as would be expected if initial formation of a disulfide cross-link were followed by formation of the dimer. To begin, the GCRho dimer has a buried surface area and geometric surface complementarity (Sc) comparable in magnitude to those of other guanylyl and adenylyl cyclases. The C2 axis of the GCRho dimer is rotated by 90° with respect to the canonical AC dimer such that six of the conserved active-site residues, three identical residues from each subunit (Asp-457, Asp-501, and Arg-545), come together to form a ligand-binding pocket for tartrate and two Mn2+ ions. The Asp residues bind the Mn2+ ions much as do the corresponding residues in adenylyl cyclase, and the conserved Arg residues bind to the tartrate carboxylates much as the corresponding residue in adenylyl cyclase binds the terminal phosphate in ddATP (14). Interestingly, the disulfide bond and Mn2+/tartrate ligand-binding site are located on the C2 axis, at opposite ends of the GCRho dimer interface. Nonetheless, the disulfide-linked dimer is likely a crystallization artifact, and it remains to be determined experimentally whether this particular oligomeric state has any relevance for regulation of guanylyl cyclase activity.
Irrespective of whether or not the observed dimer is involved in GCRho function, it seems clear that the catalytically active state must be a dimer, similar in arrangement of subunits to that of adenylyl cyclase (13, 14). We have not been able to provide evidence for such a dimer by direct methods such as SEC, AUC, and crystallography, but we could provide evidence through the non-linear dependence of guanylyl cyclase activity on enzyme concentration. It is likely that Mn2+ and substrate assist in formation of the dimer under these conditions, but even so, the KD value for formation (20–80 μm) is much higher than for other guanylyl cyclases (e.g. the KD value for the catalytic domain of the mammalian soluble guanylyl cyclase is 0.45 μm) (16). The extremely high KD value suggests that formation of the active-state dimer might be part of the light-activation mechanism. Future work in the laboratory will explore the role of domain dimerization in the light-dependent activation of full-length RhoGC.
Experimental procedures
Materials and methods
Unless specified otherwise, all materials were as described previously (8).
Protein expression and purification
Two constructs containing the GC domain of RhoGC were used in these studies: one composed of the GC domain alone (“GCRho”; amino acids 443–627), and the other that included the CC domain as well (“GCwCCRho”; amino acids 397–627). Both were cloned into a pET15b vector for expression in E. coli BL21(DE3) cells and included a C-terminal His6 tag for purification on a nickel-affinity column. Both genes also included a Val codon after the first methionine to improve bacterial expression. The BL21(DE3) cell line harbored an additional pGro7 plasmid (Takara Bio Inc.) containing the genes for the GroEL and GroES molecular chaperone proteins under the control of the AraB promoter. Competent cells were transformed with either pET15b-GCRho or pET15b-GCwCCRho according to standard procedures and incubated overnight at 37 °C on LB-agar plates containing 100 μg/ml ampicillin and 20 μg/ml chloramphenicol. Single colonies were used to inoculate small growth cultures (10 ml of LB containing 100 μg/ml ampicillin and 20 μg/ml chloramphenicol), which were grown for 14–18 h with shaking at 220 rpm and 37 °C. One ml of the overnight culture was used to inoculate a liter of LB containing both antibiotics. 500 mg of l-(+)-arabinose (Acros Organics) was added upon inoculation to begin induction of GroEL/ES. Cells were grown at 37 °C until an A600 0.4–0.7 was reached, at which point expression of the protein was induced by addition of IPTG to a final concentration of 250 μm. Cells were then incubated with shaking at 20 °C for 20 h and harvested by centrifugation at 4000 rpm in a Beckman Coulter JLA-8.1000 rotor for 15 min at 4 °C. Cell pellets were stored at −80 °C until needed.
The frozen cell pellets were thawed on ice and resuspended to a final volume of 50 ml with lysis buffer (25 mm HEPES, pH 7.0, 100 mm NaCl, and 20 mm imidazole) containing 1 mm PMSF. The cell suspension was sonicated on ice with a Misonix Sonicator 3000 at 70-watt power with a cycle of 20 s on and 20 s off for a total of 4 min. Sonicated cells were centrifuged in a Beckman Coulter JA-20 rotor at 16,000 rpm for 30 min at 4 °C to pellet cell debris, and the supernatant fraction was passed through a 0.22-μm filter before loading onto a pre-equilibrated 5-ml prepacked HiTrapFF nickel-Sepharose column (GE Healthcare) at 1 ml/min. The column was washed with 10 column volumes of lysis buffer followed by 20 volumes of lysis buffer containing 40 mm imidazole, and the protein was eluted with an 80-ml linear gradient of 40–500 mm imidazole in lysis buffer. Protein was monitored by absorbance at 280 nm, and fractions containing GCRho (or GCwCCRho) were identified by SDS-PAGE and pooled before concentration with an exchange of buffer to remove imidazole (10-kDa molecular mass cutoff Amicon Ultra Centrifuge Filter from EMD Millipore). Purified GCRho and GCwCCRho were frozen in small concentrated aliquots at −80 °C until needed.
Size-exclusion chromatography (SEC) on an Äkta FPLC system (Amersham Biosciences, Uppsala, Sweden) equipped with a Superdex-200 10/300 GL gel filtration column (GE Healthcare) was used to determine the oligomeric state of the proteins. 300 μl of 200 μm purified GCRho (or GCwCCRho) was loaded onto the Superdex column that had been pre-equilibrated with 25 mm HEPES, pH 7.0, and 100 mm NaCl at 4 °C, and the column was run at a constant rate of 0.5 ml/min. Protein was monitored by absorbance at 280 nm, and elution volume was compared with a standard curve using soluble protein standards to estimate the molecular mass of the peak fraction.
A separate GCRho construct, GCNHisTEV, was designed with a TEV-protease site (ENLYFQG) inserted immediately after the N-terminal His6 tag and followed by a Met and the remainder of GCRho sequence to determine whether the affinity tag interfered with dimerization of the protein in solution. The protein was expressed and purified as presented above. The purified protein was proteolyzed with a 30:1 ratio of GCNHisTEV to TEV-protease for 9 h at 4 °C in 25 mm HEPES buffer, pH 7.0, containing 100 mm NaCl, 1 mm DTT, and 0.5 mm EDTA in a total volume of 1 ml. The cleaved protein was separated from undigested GCNHisTEV and TEV-protease by nickel-affinity chromatography, as confirmed by SDS-PAGE and anti-His6 Western blot analysis. Both TEV-cleaved and -uncleaved GCNHisTEV were shown to be monomeric in solution by SEC on a Superdex-200 column equilibrated in 25 mm HEPES, pH 7.0, 100 mm NaCl running at 0.5 ml/min at 4 °C.
Analytical ultracentrifugation
Sedimentation velocity experiments were designed to identify factors (e.g. salt, enzyme concentration, divalent cation, etc.) that could impact the oligomeric state of isolated GCRho protein (25). All centrifugation steps were carried out using a ProteomeLab Optima XL-A ultracentrifuge (Beckman Coulter). In all cases, 395 μl of protein was loaded into double sector centerpieces balanced by 400 μl of reference solution containing all components of the sample solution except the protein. All cell housings were loaded into a four-hole An-60 Ti rotor and spun at 35,000 rpm at 22 °C for 15 h. Absorbance at 280 nm was measured to track protein concentration during sedimentation. Data were processed and analyzed using SEDFIT software (National Institutes of Health, Version 14.4fb). Data were fit using a continuous c(s) distribution model based on theory for a soluble and globular (spherical) protein with self-association. Parameters used to fit the raw absorbance data to a confidence level of 95% included assumptions for average protein partial specific volume (0.73) as well as for buffer density (1.00 g/ml) and buffer viscosity (0.01002 units) being identical to pure water as no viscous agents such as glycerol were used to stabilize the protein in solution.
Enzymatic activity assays
Guanylyl cyclase activity was measured using reversed-phase (RP)-HPLC to follow formation of cGMP and disappearance of GTP. 100-μl reactions were prepared in 50 mm Tris buffer, pH 7.6, containing 50 mm NaCl and 0.5 mm EDTA. Reactions contained GTP and metal ion (MgCl2 or MnCl2) at the concentrations indicated in the figures and were initiated by addition of enzyme. 20-μl aliquots were quenched at specified time points (30 s to 5 h) by combining with an equal volume of 1 n HCl. Precipitated protein was removed with 0.22-μm Spin-X centrifugal filters (Corning Costar) by centrifugation at 5000 rpm for 5 min in a tabletop centrifuge. The samples were then neutralized with 20 μl of 1 m potassium phosphate, pH 8, and applied to a 250 × 2.1-mm ACE 5 C18-AR reversed-phase 5 μm column connected to an Agilent 1260 Infinity HPLC system with a G136D 1260 multiwavelength detector. Nucleotides were separated by isocratic elution with a 100 mm potassium phosphate buffer, pH 6.2, at a flow rate of 0.4 ml/min and monitored by absorbance at 254 nm. Peaks were integrated with OpenLab CDS ChemStation software and compared with peaks from standards for GTP and cGMP of known concentration.
Assays for adenylyl cyclase activity were performed identically except that the reaction contained ATP instead of GTP, and the HPLC column running buffer was 100 mm potassium phosphate, pH 6.2, containing 10% (v/v) methanol.
The concentration of stock solutions was determined spectrophotometrically using extinction coefficients of 13,700 m−1 cm−1 at 252 nm for GTP, 12,320 m−1 cm−1 at 260 nm for cGMP, 15,400 m−1 cm−1 at 259 nm for ATP, and 15,000 m−1 cm−1 at 260 nm for cAMP.
The dependence of activity on enzyme concentration was evaluated through a non-linear least-squares fit (MATLAB) of the rate data to Equation 1 for a reaction in which the only active species is a dimer of the enzyme in a monomer/dimer equilibrium,
| (Eq. 1) |
where ν = reaction rate (μm/s); kcat = turnover number (s−1); E = total enzyme concentration (μm); and KD is the equilibrium dissociation constant for dimerization of the enzyme, defined as KD = [monomer]2 / [dimer] with units of μm. KD and kcat are parameters determined through the fitting process. These reactions contained 10 mm GTP and 20 mm metal ion (Mn2+ or Mg2+), which was saturating for all enzyme concentrations tested.
SDS-PAGE analysis of disulfide cross-links
Samples of GCRho and GCwCCRho were incubated for 20 h under various conditions, as indicated in Fig. 6, to test for the formation of intermolecular disulfide bonds. Each sample contained 1 μm GCRho or GCwCCRho in 25 mm HEPES, pH 7.0, and 100 mm NaCl. Metal ion (Mn2+ or Mg2+), GTP substrate, and cGMP and inorganic pyrophosphate products were included as indicated in the figure. Each sample was diluted 1:2 with gel load buffer, with or without 5 mm DTT, and then loaded onto a 10% Mini-PROTEAN TGX polyacrylamide gel (Bio-Rad) for analysis. Protein bands were visualized with Coomassie Brilliant Blue stain.
Crystallization
Crystallization trials were performed by sitting drop vapor diffusion at room temperature using Hampton (Hampton Research, CA) and Jena Bioscience (Jena Bioscience, Jena, Germany) sparse matrix crystallization screens. Drops were set with a Phoenix robot (Art Robbins Instruments, CA) by mixing protein (GCRho, GCwCCRho, or GCAC; 20 mg/ml) in 25 mm HEPES, pH 7.0, containing 100 mm NaCl with crystallization mother liquor in a 1:1 ratio. Crystals of GCRho appeared after about 3 weeks in 25% w/v PEG3350, 100 mm BisTris, pH 5.5, and 200 mm NaCl, whereas GCAC crystals formed in 15% w/v PEG20000 and 10 mm potassium hydrogen tartrate. These crystals all contained a single subunit in the asymmetric unit. In some trials, the protein solution also contained 10 mm MgCl2 (or MnCl2) and 1 mm ddGTP. In this case (MnCl2), crystals appeared in 20% (w/v) PEG3350 and 200 mm potassium sodium tartrate and contained a disulfide cross-linked dimer in the asymmetric unit.
Data collection, processing, and refinement
Crystals were soaked in reservoir solution containing 15% glycerol as cryoprotectant before flash-freezing in liquid nitrogen. Diffraction data were collected at 100 K with beamline 8.2.1 at the Advanced Light Source (Lawrence Berkeley National Laboratory, Berkeley, CA) using an ADSC Q315R CCD detector (Area Detector Systems Corp.). The best crystals diffracted up to 1.1 and 1.4 Å resolution for the GCRho and GCAC monomers, respectively, and 1.7 Å resolution for the GCRho dimer. Data sets were integrated using iMosflm version 7.2 (26) and scaled using SCALA version 3.3 (27) from the CCP4 software suite version 7.0 (28, 29). Diffraction data were processed in space group P221214 for the GCRho and GCAC monomers and C2221 for the GCRho dimer. Complete data collection statistics are listed in Table 2.
Table 2.
Crystallographic data collection and refinement statistics
| GCRho-monomer | GCRho-dimer | GCAC-monomer | |
|---|---|---|---|
| PDB ID | 6AO9 | 6AOB | 6AOA |
| Data collection statistics | |||
| Space group | P22121 | C2221 | P22121 |
| Resolution range (Å) | 20 − 1.13 | 20 − 1.70 | 20 − 1.40 |
| Highest resolution shell (Å) | 1.19 − 1.13 | 1.79 − 1.70 | 1.48 − 1.40 |
| Unit cell parameters (Å) | a = 34.5, b = 65.6, c = 90.6 | a = 91.8, b = 95.9, c = 100.1 | a = 34.4, b = 65.4, c = 88.4 |
| Total reflections | 980,795 | 617,510 | 280,921 |
| Unique reflections | 77,710 | 48,364 | 40,049 |
| Completeness %a | 99.9 (99.7) | 99.3 (97.7) | 99.8 (100) |
| Rmerge %a | 7.7 (100) | 10.6 (130) | 11.1 (42.3) |
| I/σ (I) a | 15.2 (2.2) | 16.1 (2.0) | 8.7 (2.9) |
| Redundancy | 12.6 (10.2) | 12.8 (12.3) | 7.0 (7.0) |
| Refinement statistics | |||
| Resolution range (Å) | 20–1.13 | 20–1.70 | 18–1.40 |
| No. of reflections used | 77,532 | 48,333 | 39,937 |
| Rwork % | 18.8 | 16.4 | 19.0 |
| Rfree % | 19.5 | 19.6 | 20.8 |
| Protein atoms | 1384 | 2842 | 1388 |
| Ligand atoms | 0 | 10 | 0 |
| Metal atoms | 0 | 2 | 0 |
| Water molecules | 338 | 507 | 320 |
| r.m.s.d. in bond lengths (Å) | 0.005 | 0.006 | 0.005 |
| r.m.s.d. in bond angles (°) | 0.8 | 0.8 | 0.8 |
a Highest-resolution shell values are given in parentheses.
The GCRho monomer structure was solved by molecular replacement using PHASER version 2.6 (30) with the structure of the catalytic domain of homodimeric human soluble GC (PDB entry 2WZ1) molecule A as a search model. The GCAC monomer and GCRho dimer structures were solved using the final refined model of the GCRho monomer as a search model. Rigid body refinement followed by positional and B-factor refinement was carried out using phenix.refine (31) from the PHENIX software suite version 1.11 (32). Simulated annealing was included in earlier refinements to minimize the initial model bias. Manual model building was done using COOT version 0.8 (33). Water molecules were included in the final refinement after satisfying the criteria of 3σ Fo − Fc and 1σ 2Fo − Fc. Several iterative cycles of refinement were carried out before final submission of data. Data collection and final refinement statistics are given in Table 2. Data sets for the GCRho monomer (PDB entry 6AO9), GCAC (PDB entry 6AOA), and GCRho dimer (PDB entry 6AOB) have been submitted to the Protein Data Bank. All crystal structure figures in this paper were prepared using PyMOL version 1.8 (Schrödinger LLC, Portland, OR).
Author contributions
D. D. O. conceived and coordinated the study. R. P. K. and B. R. M. were responsible for all aspects of the experimental program. J. F. was involved in the initial development of the expression and purification system for the guanylyl cyclase domain. M. M. T. was involved in the development of the HPLC method for enzymatic assays and in the construction of the GCAC mutant. D. H. Z. performed assays for dependence of activity on enzyme concentration (Fig. 8). M. O. L. developed and performed disulfide cross-linking assays (Fig. 6). D. D. O., R. P. K., and B. R. M. wrote the paper. All authors discussed and commented on the manuscript.
Supplementary Material
Acknowledgments
This research used resources of the Advanced Light Source, which is a Department of Energy Office of Science User Facility under Contract No. DE-AC02-05CH11231. We are grateful to the staff at the Advanced Light Source–Berkeley Center for Structural Biology for their assistance in X-ray data collection. We thank the Adar family and friends for continued support throughout this work. We also thank Prof. Timothy Street and Jackson Halpin for sharing their HPLC and providing technical advice. We thank Prof. Bruce Foxman for helpful discussions on crystallography.
This work was supported by National Institutes of Health Grants T32GM007596 (to B. R. M. and D. H. Z.) and EY007965 (to D. D. O.) and the Brandeis University Provost's research fund. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
The atomic coordinates and structure factors (codes 6AO9, 6AOA, and 6AOB) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The equivalent positions for this non-standard setting of P21212 are (x, y, z); (x, −y, −z); (−x, ½ + y, ½ − z); (−x, ½ − y, ½ + z).
- RhoGC
- rhodopsin-guanylate cyclase
- GCRho
- isolated RhoGC guanylyl cyclase domain
- CC
- RhoGC coiled-coil domain
- GCwCCRho
- GCRho with the CC domain
- GCAC
- GCRho double mutant with adenylyl cyclase activity
- GCCya2
- catalytic domain of the guanylyl cyclase Cya2 from Synechocystis PCC6803
- GCCYG12
- catalytic domain of the guanylyl cyclase CYG12 from C. reinhardtii
- GCHum
- catalytic domain of the human soluble guanylyl cyclase sGC
- ddGTP
- 2′,3′-dideoxyguanosine 5′-triphosphate
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- SEC
- size-exclusion chromatography
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- TEV
- tobacco etch virus
- AUC
- analytical ultracentrifugation
- sGC
- soluble guanylyl cyclase
- AC
- adenylyl cyclase.
References
- 1. Avelar G. M., Schumacher R. I., Zaini P. A., Leonard G., Richards T. A., and Gomes S. L. (2014) A rhodopsin-guanylyl cyclase gene fusion functions in visual perception in a fungus. Curr. Biol. 24, 1234–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ernst O. P., Lodowski D. T., Elstner M., Hegemann P., Brown L. S., and Kandori H. (2014) Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 114, 126–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spudich J. L., Yang C. S., Jung K. H., and Spudich E. N. (2000) Retinylidene proteins: structures and functions from archaea to humans. Annu. Rev. Cell Dev. Biol. 16, 365–392 [DOI] [PubMed] [Google Scholar]
- 4. Gao S., Nagpal J., Schneider M. W., Kozjak-Pavlovic V., Nagel G., and Gottschalk A. (2015) Optogenetic manipulation of cGMP in cells and animals by the tightly light-regulated guanylyl-cyclase opsin CyclOp. Nat. Commun. 6, 8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scheib U., Stehfest K., Gee C. E., Körschen H. G., Fudim R., Oertner T. G., and Hegemann P. (2015) The rhodopsin-guanylyl cyclase of the aquatic fingus Blastocladiella emersonii enables fast optical control of cGMP signaling. Sci. Signal. 8, rs8. [DOI] [PubMed] [Google Scholar]
- 6. Lamarche L. B., Kumar R. P., Trieu M. M., Devine E. L., Cohen-Abeles L. E., Theobald D. L., and Oprian D. D. (2017) Purification and characterization of RhoPDE, a retinylidene/phosphodiesterase fusion protein and potential optogenetic tool from the choanoflagellate Salpingoeca rosetta. Biochemistry 56, 5812–5822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoshida K., Tsunoda S. P., Brown L. S., and Kandori H. (2017) A unique choanoflagellate enzyme rhodopsin with cyclic nucleotide phosphodiesterase activity. J. Biol. Chem. 292, 7531–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Trieu M. M., Devine E. L., Lamarche L. B., Ammerman A. E., Greco J. A., Birge R. R., Theobald D. L., and Oprian D. D. (2017) Expression, purification, and spectral tuning of RhoGC, a retinylidene/guanylyl cyclase fusion protein and optogenetics tool from the aquatic fungus Blastocladiella emersonii. J. Biol. Chem. 292, 10379–10389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rauch A., Leipelt M., Russwurm M., and Steegborn C. (2008) Crystal structure of the guanylyl cyclase Cya2. Proc. Natl. Acad. Sci. U.S.A. 105, 15720–15725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winger J. A., Derbyshire E. R., Lamers M. H., Marletta M. A., and Kuriyan J. (2008) The crystal structure of the catalytic domain of a eukaryotic guanylate cyclase. BMC Struct. Biol. 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allerston C. K., von Delft F., and Gileadi O. (2013) Crystal structures of the catalytic domain of human soluble guanylate cyclase. PLoS one 8, e57644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hurley J. H. (1999) Structure, mechanism, and regulation of mammalian adenylyl cyclase. J. Biol. Chem. 274, 7599–7602 [DOI] [PubMed] [Google Scholar]
- 13. Tesmer J. J., Sunahara R. K., Gilman A. G., and Sprang S. R. (1997) Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα.GTPγS. Science 278, 1907–1916 [DOI] [PubMed] [Google Scholar]
- 14. Tesmer J. J., Sunahara R. K., Johnson R. A., Gosselin G., Gilman A. G., and Sprang S. R. (1999) Two-metal-ion catalysis in adenylyl cyclase. Science 285, 756–760 [DOI] [PubMed] [Google Scholar]
- 15. Zhang G., Liu Y., Ruoho A. E., and Hurley J. H. (1997) Structure of the adenylyl cyclase catalytic core. Nature 386, 247–253 [DOI] [PubMed] [Google Scholar]
- 16. Winger J. A., and Marletta M. A. (2005) Expression and characterization of the catalytic domains of soluble guanylate cyclase: interaction with the heme domain. Biochemistry 44, 4083–4090 [DOI] [PubMed] [Google Scholar]
- 17. Lucas K. A., Pitari G. M., Kazerounian S., Ruiz-Stewart I., Park J., Schulz S., Chepenik K. P., and Waldman S. A. (2000) Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev. 52, 375–414 [PubMed] [Google Scholar]
- 18. Sinha S. C., and Sprang S. R. (2006) Structures, mechanism, regulation and evolution of class III nucleotidyl cyclases. Rev. Physiol. Biochem. Pharmacol. 157, 105–140 [DOI] [PubMed] [Google Scholar]
- 19. Lawrence M. C., and Colman P. M. (1993) Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 [DOI] [PubMed] [Google Scholar]
- 20. Sunahara R. K., Beuve A., Tesmer J. J., Sprang S. R., Garbers D. L., and Gilman A. G. (1998) Exchange of substrate and inhibitor specificities between adenylyl and guanylyl cyclases. J. Biol. Chem. 273, 16332–16338 [DOI] [PubMed] [Google Scholar]
- 21. Tucker C. L., Hurley J. H., Miller T. R., and Hurley J. B. (1998) Two amino acid substitutions convert a guanylyl cyclase, RetGC-1, into an adenylyl cyclase. Proc. Natl. Acad. Sci. U.S.A. 95, 5993–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barathy D., Mattoo R., Visweswariah S., and Suguna K. (2014) New structural forms of a mycobacterial adenylyl cyclase Rv1625c. IUCrJ. 1, 338–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bieger B., and Essen L. O. (2001) Structural analysis of adenylate cyclases from Trypanosoma brucei in their monomeric state. EMBO J. 20, 433–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tews I., Findeisen F., Sinning I., Schultz A., Schultz J. E., and Linder J. U. (2005) The structure of a pH-sensing mycobacterial adenylyl cyclase holoenzyme. Science 308, 1020–1023 [DOI] [PubMed] [Google Scholar]
- 25. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 28. Potterton E., Briggs P., Turkenburg M., and Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 29. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Omasits U., Ahrens C. H., Müller S., and Wollscheid B. (2014) Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.