

Abstract
During influenza A virus (IAV) infection, cytokine storms play a vital and critical role in clinical outcomes. We have previously reported that microRNA (miR)-302c regulates IAV-induced IFN expression by targeting the 3′-UTR of nuclear factor κB (NF-κB)–inducing kinase. In the current study, we found that miR-302a, another member of the miR-302 cluster, controls the IAV-induced cytokine storm. According to results from cell-based and knockout mouse models, IAV induces a cytokine storm via interferon regulatory factor-5 (IRF-5). We also found that IAV infection up-regulates IRF-5 expression and that IRF-5 in turn promotes IAV replication. Furthermore, we observed that IRF-5 is a direct target of miR-302a, which down-regulated IRF-5 expression by binding its 3′-UTR. Moreover, IAV increased IRF-5 expression by down-regulating miR-302a expression. Interestingly, miR-302a inhibited IAV replication. In IAV-infected patients, miR-302a expression was down-regulated, whereas IRF-5 expression was up-regulated. Taken together, our work uncovers and defines a signaling pathway implicated in an IAV-induced cytokine storm.
Keywords: cytokine induction, cytokine storm, influenza virus, interferon regulatory factor (IRF), microRNA (miRNA), signaling
Introduction
Influenza A viruses (IAVs)2 are respiratory pathogens responsible for major pandemics and annual epidemics that have serious economic and health consequences (1). IAVs are negative-sense, single-stranded RNA viruses encoding 10 or 11 proteins (2). In the 20th century, three novel influenza virus strains emerged that caused the 1918, 1957, and 1968 pandemics and resulted in high mortality rates (3). The cytokine storm refers to the massive release of inflammatory cytokines and chemokines caused by uncontrolled activation of the host's innate immune system (4). The development of a cytokine storm coupled with pulmonary edema, pneumonia, and alveolar hemorrhage has been consistently observed in severe cases of influenza infection in humans (5). Accumulating evidence supports the hypothesis that exaggerated production of inflammatory cytokines and chemokines plays an essential and commanding role in the clinical outcome and pathogenesis of influenza virus infection (6). However, the mechanisms by which IAV induces a cytokine storm and an understanding of why some individuals exhibit an excessive response to the virus to an extent that leads to hospitalization or death remain relatively unexplored.
MicroRNAs (miRNAs) are small and short RNA molecules that are usually composed of 18–23 nucleotides and regulate mRNA transcription in various cellular processes (7). The mechanism of miRNA silencing involves complementary binding of the miRNA and 3′-UTR of the target mRNA and subsequent association with the Argonaute (Ago) and Dicer1 proteins (7). Recently, miRNAs have been shown to be involved in the immune response during IAV infection. For example, miR-323, miR-491, and miR-654 inhibit the replication of the H1N1 IAV by binding to the PB1 gene (8). According to a previous study, miR-155 is remarkably up-regulated in macrophages during IAV infection and positively regulates the host antiviral innate immune response through a feedback mechanism by promoting type I IFN signaling through the targeting of SOCS1 (suppressor of cytokine signaling 1) (9). Through a posttranscriptional mechanism, miR-146 targets and down-regulates the expression of TRAF6 (tumor necrosis factor receptor-associated factor 6) and IRAK1 (interleukin 1 receptor–associated kinase 1) genes and affects viral production by functioning in the antiviral response (10).
IRF-5 (interferon-regulatory factor 5) is a member of the IRF family that participates in the virus-mediated activation of interferon and other cytokines (11). Among the members of the IRF family, IRF-3 is constitutively expressed in all cell types, whereas constitutive IRF-7 expression is predominantly detected in cells of lymphoid origin (12). Compared with IRF-3 and IRF-7, IRF-5 is transcribed into different splice variants that exhibit distinct cell type–specific expression patterns, cellular localization, differential regulation, and dissimilar functions in virus-mediated IFN induction (13). IRF-5 is not a target of the Toll-like receptor-3 (TLR-3) signaling pathway but is activated by TLR-7 or TLR-8 signaling (14). IRF-5 interacts with and is activated by myeloid differentiation primary response gene 88 (MyD88) and TRAF6, and TLR activation results in the nuclear translocation of IRF-5 to activate cytokine gene transcription (15). Moreover, IKKα kinase and alkaline phosphatase negatively regulate of IRF-5 activity in the MyD88 pathway, indicating that these proteins control the inflammatory response by attenuating IRF-5 activity (16). TLR-activated IRF-5 mediates cooperativity of the enhancer with the promoter that contains IFN-stimulated response elements (17). IRF-5 cooperates with CREB-binding protein/p300 to further transactivate target promoter-reporter constructs and binds to the endogenous IL-6 and TNFα promoters in response to virus infection (18). Recent studies from Irf-5 knockout mice have confirmed a critical role for IRF-5 in virus-induced expression of type I IFN and the proinflammatory cytokines IL-6, IL-12, and TNF-α (19). Regarding IRF-5, the virus induces the formation of IRF-5/IRF-7 or IRF-5/IRF-3 heterodimers that act as both activators and repressors by binding to IFNα promoters and modulating the expression profile of IFNα subtypes (20).
As shown in our previous studies, miR-302c mediates IAV-induced IFNβ expression by targeting NF-κB–inducing kinase (21). In this study, the expression of miR-302a, another member of the miR-302 cluster, was inhibited during IAV infection. Furthermore, miR-302a inhibited IRF-5 expression by directly targeting the IRF-5 3′-UTR, resulting in the overexpression of inflammatory cytokines and chemokines.
Results
IAV induces cytokine expression through miR-302a
Based on the results from our previous studies, IAV infection induces IFNβ mRNA expression by down-regulating miR-302c expression (21). In this study, we investigated whether miR-302a affected IAV-induced cytokine expression. IAV infection slightly elevated the production of IFN-β and CCL5 and significantly increased the levels of the TNF-α, IL-6, IL-8, and CCL2 mRNAs (Fig. 1A). Moreover, transient transfection of PBMCs with miR-302a mimics led to a significant decrease in the production of those proinflammatory cytokines and chemokines (Fig. 1A). miR-28-5p mimics were included as a negative control for comparison. Decreased levels of those proinflammatory cytokines and chemokines in culture supernatants were also observed using ELISAs (Fig. 1B). The effect of miR-302a inhibition on an IAV-induced cytokine expression was studied to confirm the results obtained with the miR-302a mimics. Consistent with these results, the miR-302a inhibitor increased the levels of the IFN-β, TNF-α, IL-6, IL-8, CCL2, and CCL5 mRNAs and proteins (Fig. 1, C and D). The efficiency of either overexpression or suppression of miR-302a by oligonucleotide mimics or inhibitors was also explored. Real-time RT-PCR results showed that transfection with miR-302a mimics resulted in a remarkable increase of miR-302a levels in A549 cells (supplemental Fig. 1A), whereas the miR-302a inhibitor could lead to a reduction of miR-302a levels compared with controls (supplemental Fig. 1B). Similarly, transfection of A549 cells with miR-28-5p mimic up-regulated the miR-28-5p level, and conversely, the level of endogenous miR-28-5p declined after transfection of miR-28-5p inhibitor (supplemental Fig. 1, C and D). Based on these data, miR-302a inhibits IAV-induced cytokine expression.
Figure 1.

Involvement of miR-302a in IAV-induced inflammatory cytokine expression. A and B, PBMCs were transfected with the indicated miRNA mimics or mimic controls for 24 h, infected with the WSN virus (MOI = 1) or mock-infected for 24 h, and subjected to real-time RT-PCR (A) and ELISA (B). C and D, experiments were performed similar to those described in A and B, except that the indicated miRNA inhibitor or inhibitor controls were used. In the real-time RT-PCR experiments, the control was designated as 1. Bar graphs present means ± S.D. (error bars); n = 3 (**, p < 0.01; *, p < 0.05; n.s., not significant).
miR-302a down-regulates IRF-5 expression by directly targeting its 3′-UTR
Because miR-302a inhibited the IAV-induced production of proinflammatory cytokines and chemokines, we reasoned that the specific genes suppressed by miR-302a should predominantly function in this process. Using computational tools, we predicted that the 3′-UTR of the IRF-5 mRNA contains two miR-302a–binding sites. The IRF-5 3′-UTR was cloned into a firefly luciferase reporter plasmid, and two distinct mutations were generated in the 3′-UTR of IRF-5 at the predicted seed-matching sites to test the interaction between miR-302a and the IRF-5 3′-UTR and to further confirm the effect of miR-302a on IRF-5 expression (Fig. 2A). According to the results of the luciferase activity assay, the miR-302a mimic reduced the activity of the plasmid controlled by the IRF-5 3′-UTR, but the attenuation of luciferase activity was impaired by the IRF-5 3′-UTR–binding site mutation (Fig. 2B). Conversely, the miR-302a inhibitor significantly stimulated the luciferase activity of the WT IRF-5 3′-UTR, without any effect on the Mut IRF-5 3′-UTR (Fig. 2C). In addition, miR-302a overexpression significantly reduced the levels of the IRF-5 mRNA and protein (Fig. 2D). In contrast, miR-302a inhibitor–mediated knockdown of endogenous miR-302a expression increased the levels of the IRF-5 mRNA and protein (Fig. 2D). Thus, IRF-5 is a direct functional target gene of miR-302a.
Figure 2.

IRF-5 is a target of miR-302a. A, predicted miR-302a–binding sites in the 3′-UTR of the IRF-5 mRNA. Perfect matches in seed regions are indicated by lines. Mutations (underlined) were generated in the binding sites in the 3′-UTR for the reporter gene assay. B, A549 cells were transfected with the WT or the indicated mutants of the IRF-5 3′-UTR reporter plasmid and miR-302a for 48 h before luciferase assays. C, experiments were performed as described in B, except that cells were transfected with the miR-302a inhibitor or inhibitor controls. D, THP-1 cells were transfected with the miR-302a mimic or mimic controls for 24 h before real-time RT-PCR (top) and Western blot (bottom) analyses. E, experiments were performed as described in D, except that a nonspecific inhibitor control or miR-302a inhibitor was used. In the real-time RT-PCR experiments, the control was designated as 1. Bar graphs present means ± S.D. (error bars), n = 3 (**, p < 0.01; *, p < 0.05; n.s., not significant).
IRF-5 plays an important role in an IAV-induced cytokine storm
Because IRF-5 is a target of miR-302a, we hypothesized that IRF-5 may affect an IAV-induced cytokine storm. PBMCs were infected with IAV and transfected with an IRF-5 expression vector (or empty vector) to test this hypothesis. Based on the real-time RT-PCR and ELISA results, IRF-5 overexpression promotes IAV-induced production of the IFN-β, TNF-α, IL-6, IL-8, CCL2, and CCL5 mRNAs and proteins (Fig. 3, A and B). The expression of IRF-5 and IAV viral proteins were also detected (supplemental Fig. 2A). We designed three specific siRNAs for IRF-5 (siRNA-IRF-5 1, 2, and 3) and tested their efficiency to confirm the effects of IRF-5 on an IAV-induced cytokine storm (Fig. 3C); siRNA-IRF-5 2 and 3 were selected for the experiments described below. In real-time RT-PCRs and ELISAs, IRF-5 knockdown inhibited IAV-induced production of the IFN-β, TNF-α, IL-6, IL-8, CCL2, and CCL5 mRNAs and proteins (Fig. 3, C and D). Moreover, the efficiency of siRNA transduction and the expression of IAV viral proteins were detected (supplemental Fig. 2B).
Figure 3.

Effects of IRF-5 on IAV-induced inflammatory cytokine expression. A and B, PBMCs were transfected with the indicated expression constructs or empty vector for 24 h, infected with the WSN virus (MOI = 1) or mock-infected for 24 h, and subjected to real-time RT-PCR (A) and ELISA (B). C, THP-1 cells were transfected with the indicated IRF-5 siRNAs or siRNA control for 48 h before real-time RT-PCR (left) and Western blot (right) analyses. D and E, experiments were performed similar to those described in A and B, except that IRF-5 siRNAs or siRNA control were used. All experiments were repeated at least three times with consistent results. Bar graphs present means ± S.D., n = 3 (**, p < 0.01; *, p < 0.05; n.s., not significant).
We generated mice deficient in the Irf-5 gene (Irf-5−/− mice) using standard CRISPR/Cas9 technology (Fig. 4A) and confirmed the IRF-5 deficiency using semiquantitative RT-PCR and immunoblot analyses (Fig. 4B) to further confirm the involvement of IRF-5 in an IAV-induced cytokine storm in vivo. We next infected WT or Irf-5−/− mice with the WSN strain of the influenza virus and monitored the body weights. The Irf-5−/− mice exhibited higher body weights than WT mice during the IAV infection (Fig. 4C, left). Moreover, in response to the IAV infection, IRF-5−/− mice displayed a prolonged survival time and higher survival rate than WT mice (Fig. 4C, right). Similar results were also obtained using the lethal mouse-adapted influenza virus A/FM/1/47 (H1N1) (Fig. 4D). We next investigated whether the production of proinflammatory cytokines and chemokines was altered in Irf-5−/− mice during the IAV infection. As expected, the levels of the IFN-β, TNF-α, IL-6, IL-8, CCL2, and CCL5 mRNAs and proteins were significantly reduced in the Irf-5−/− mice compared with WT mice during the IAV infection (Fig. 4, E and F). Based on these results, IAV induces a cytokine storm through IRF-5 in vitro and in vivo.
Figure 4.

IRF-5 regulates IAV-induced inflammatory cytokine expression in vivo. A, schematic showing the single guide RNA (sgRNA)–binding site in the mouse Irf-5 gene (top). Sequence of the genomic single guide RNA–binding site (bottom). B, Irf-5 expression in the lung was detected using RT-PCR (top) and Western blot (bottom) analyses. C, WT (black square, n = 15) and Irf-5−/− (red circle, n = 15) mice were intranasally infected with 1 × 104 pfu of WSN IAV, and body weights were recorded daily (left). Survival curves show data collected until day 14 postinfection (right). The statistical analysis was performed using a log-rank test. D, experiments were performed similar to those described in C, except that mice were intranasally infected with the FM IAV. E and F, WT (black square; n = 5) and Irf-5−/− (red circle; n = 5) mice were infected with 1 × 104 pfu of WSN IAV (E) or 1 × 104 PFU FM IAV (F). Levels of proinflammatory cytokines and chemokines in the bronchoalveolar lavage fluid (BALF) were measured 48 h postinfection using ELISAs. Bar graphs present means ± S.D. (error bars) or means ± S.E. (error bars) (**, p < 0.01; *, p < 0.05; n.s., not significant).
IAV induces IRF-5 expression through miR-302a
Because both miR-302a and IRF-5 were involved in an IAV-induced cytokine storm and IRF-5 is a direct target gene of miR-302a, we next examined the role of miR-302a in IAV-induced IRF-5 expression. As shown in Fig. 5 (A and B), miR-302a overexpression decreased IAV-induced IRF-5 expression, and conversely, the miR-302a inhibitor increased IAV-induced IRF-5 expression in A549 cells. Similar experiments were performed in PBMCs and MLE-12 cells to determine whether miR-302a–mediated IAV-induced IRF-5 expression is a common feature in cells (Fig. 5 (C and D) and supplemental Fig. 3). Thus, miR-302a is required for IAV-induced IRF-5 expression.
Figure 5.

IAV induces IRF-5 expression via miR-302a. A, A549 cells were transfected with the miR-302a mimic or mimic controls for 24 h, infected with the WSN virus (MOI = 1) or mock-infected for 24 h, and subjected to real-time RT-PCR (top) and Western blot (bottom) analyses. B, experiments were performed similar to those described in A, except that the miR-302a inhibitor or inhibitor controls were used. C and D, experiments were performed as described in A and B, except that PBMCs were used. All experiments were repeated at least three times with consistent results. In the real-time RT-PCR experiments, the control was designated as 1. Bar graphs present means ± S.D., n = 3 (**, p < 0.01; *, p < 0.05, n.s., not significant).
The relationship between IAV replication and miR-302a and IRF-5 expression
Freshly isolated PBMCs from healthy donors were infected with 1 MOI of IAV at different time points to verify whether IAV infection regulates miR-302a expression. Levels of miR-302a were detected by real-time RT-PCR, and the miR-302a levels decreased over time (Fig. 6A). Consistently, freshly isolated PBMCs from healthy donors were infected with different concentrations of IAV for 12 h. Based on the results from real-time RT-PCR analyses, miR-302a levels were down-regulated by the IAV infection in a dose-dependent manner (Fig. 6B). We next investigated whether miR-302a plays a role in viral transcription and replication by measuring the production of three different forms of the IAV RNA (mRNA, cRNA, and vRNA) using an approach described previously (22). According to the real-time RT-PCR analyses, the levels of the nucleoprotein (NP)-specific mRNA, cRNA, and vRNA were reduced by miR-302a mimics (Fig. 6C, left). Similarly, in Western blot assays, miR-302a overexpression also significantly inhibited the expression of the IAV viral proteins (NP and M1) (Fig. 6C, right). In contrast, high levels of IAV viral proteins and NP-specific mRNA, cRNA, and vRNA are present in PBMCs in which miR-302a expression has been inhibited by the miR-302a inhibitor (Fig. 6D).
Figure 6.
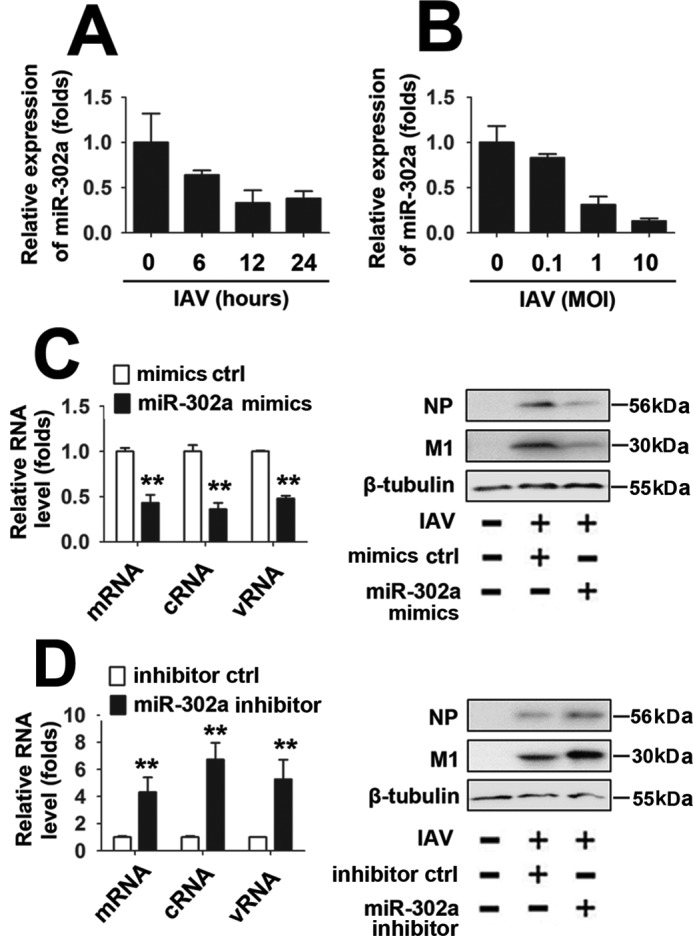
The relationship between miR-302a and IAV replication. A, PBMCs were infected with the WSN virus (MOI = 1) for the indicated times. Levels of miR-302a were quantified using real-time RT-PCR. B, PBMCs were infected with the indicated concentrations of the WSN virus for 12 h. Levels of miR-302a were quantified using real-time RT-PCR. The expression levels were normalized to the U6 snRNA. C, PBMCs were transfected with the miR-302a mimic or mimic controls for 24 h, infected with the WSN virus (MOI = 1) or mock-infected for 24 h, and subjected to real-time RT-PCR (left) and Western blot (right) analyses. D, experiments were performed as described in C, except that a nonspecific inhibitor control or miR-302a inhibitor was used. All experiments were repeated at least three times with consistent results. Bar graphs present means ± S.D. (error bars), n = 3 (**, p < 0.01; *, p < 0.05; n.s., not significant).
We also validated the relationship between IAV replication and IRF-5 expression. As shown in Fig. 7 (A and B), IAV infection induced IRF-5 mRNA and protein expression in a time- and dose-dependent manner. In addition, IRF-5 overexpression significantly increased the expression of the three different forms of IAV RNA and viral protein (NP and M1) (Fig. 7C). Conversely, low levels of IAV RNA and protein were present in PBMCs in which IRF-5 was knocked down with siRNA-IRF-5 2 (Fig. 7D). Interestingly, Irf-5−/− mice exhibited a significantly decreased viral load in the lungs compared with WT mice (Fig. 7E). Because it has been reported that IRF-5 does not participate in IFNα induction (11), we investigated whether miR-302a or IRF-5 plays a role in IAV-induced IFNα production. Real-time RT-PCRs and ELISAs confirmed that overexpression or knockdown of miR-302a had no effect on IAV-induced IFNα production (supplemental Fig. 4, A and B). Similar results were observed by overexpression or knockdown of IRF-5 (supplemental Fig. 4, C and D). These findings support a critical role for the miR-302a/IRF-5 axis in regulating IAV replication.
Figure 7.

The relationship between IRF-5 and IAV replication. A, PBMCs were infected with the WSN virus (MOI = 1) for the indicated times. IRF-5 RNA levels were quantified by real-time RT-PCR (top) and protein levels were detected by Western blotting (bottom). B, PBMCs were infected with the indicated concentrations of the WSN virus for 12 h. IRF-5 RNA levels were quantified by real-time RT-PCR (top), and protein levels were detected by Western blotting (bottom). C, PBMCs were transfected with the indicated constructs or empty vector for 24 h, infected with the WSN virus (MOI = 1) or mock-infected for 24 h, and subjected to real-time RT-PCR (left) and Western blot (right) analyses. D, experiments were performed as described in C, except that IRF-5 siRNAs or siRNA control were used. E, WT (n = 10) and Irf-5−/− (n = 10) mice were infected with 1 × 104 pfu of WSN IAV for 3 days. The viral titers in the lungs were determined using a plaque assay. In the real-time RT-PCR experiments, the lowest value was designated as 1. All experiments were repeated at least three times with consistent results. Bar graphs present means ± S.D. (error bars) or means ± S.E. (error bars) (**, p < 0.01; *, p < 0.05).
Expression of miR-302a and IRF-5 in patients infected with IAV
To further elucidate the roles of the miR-302a/IRF-5 axis in IAV infection, we analyzed their expression levels in throat swabs from IAV-infected patients (n = 36) and healthy individuals (controls) (n = 36). As shown in Fig. 8A, miR-302a expression was down-regulated in IAV-infected patients compared with healthy individuals. Because IRF-5 is the direct target gene of miR-302a, we sought to determine whether IRF-5 expression was increased in IAV patients. As determined by a real-time RT-PCR assay, IRF-5 was expressed at significantly higher levels in IAV-infected patients than in the healthy individuals (Fig. 8B). Interestingly, elevated IRF-5 expression in IAV-infected patients was tightly correlated with low levels of miR-302a expression (Fig. 8C). Furthermore, miR-302a and IRF-5 expression was examined in PBMCs from IAV-infected patients (n = 40) and healthy individuals (n = 40) using a real-time RT-PCR assay. IAV-infected patients showed higher levels of IRF-5 and low levels of miR-302a expression compared with healthy individuals (Fig. 8, D and E). Elevated IRF-5 expression in PBMCs also negatively correlated with low levels of miR-302a expression (Fig. 8F). These findings provide further evidence that alterations in miR-302a and IRF-5 expression are involved in the same pathway and are potentially involved in an IAV-regulated cytokine storm.
Figure 8.

Correlation between IRF-5 expression and suppressed miR-302a levels in IAV-infected patients. A and B, real-time RT-PCR assays of miR-302a (A) and IRF-5 (B) expression levels in throat swabs from healthy individuals (n = 36) or IAV-infected patients (n = 36). C, the relative IRF-5 mRNA and miR-302a levels in the throat swabs from IAV-infected patients were subjected to Pearson's correlation analysis. D and E, real-time RT-PCR assays of miR-302a (D) and IRF-5 (E) expression levels in PBMCs from healthy individuals (n = 40) or IAV-infected patients (n = 40). F, the relative IRF-5 mRNA and miR-302a levels in the PBMCs from IAV-infected patients were subjected to Pearson's correlation analysis. Box plots illustrate medians with 25 and 75% values, and error bars represent the 5th and 95th percentiles. For A–D, the lowest value was designated as 1. IRF-5 or miR-302a expression data are expressed as -fold induction (-fold change) relative to the lowest value (**, p < 0.01; *, p < 0.05).
Discussion
In this study, we defined a novel signaling pathway implicated in controlling an IAV-induced cytokine storm. First, IAV infection down-regulated miR-302a expression and up-regulated IRF-5 expression. In turn, miR-302a decreased and IRF-5 increased IAV replication. Furthermore, miR-302a down-regulated IRF-5 expression by directly targeting its 3′-UTR, and IAV regulated IRF-5 expression via miR-302a. Finally, we provided in vivo evidence showing that IAV regulated miR-302a and IRF-5 expression in IAV-infected patients.
Influenza virus infection is occasionally associated with dysregulated and overabundant cytokine/chemokine production and innate inflammatory infiltrates, known as a cytokine storm (23, 24). Based on the results from numerous studies, a cytokine storm accompanying an influenza virus infection is an essential component of the morbidity and mortality of the infection (23, 24). However, little is known about the molecular mechanism underlying this phenomenon. To our knowledge, only a few investigations have identified some molecules that play a vital role in an IAV-induced cytokine storm. For example, MyD88 is required for the early amplification of cytokine production in the lung following influenza virus infection (25). SOCS4 inhibits an IAV-induced cytokine storm and enhances viral clearance (26). c-Jun, which lies downstream of JNK, regulates H5N1 influenza virus replication and host inflammation (27). NF-κB, a famous transcription factor in the inflammatory network, also plays an important role in an IAV-induced cytokine storm (28). In this study, IAV induced a cytokine storm via the miR-302a/IRF-5 axis. Our study reveals a previously undescribed mechanism by which IAV regulates a cytokine storm and may provide a new therapeutic target for IAV infection.
The miR-302/367 cluster comprises five members, including miR-302b, miR-302c, miR-302a, miR-302d, and miR-367 (29). The miR-302–367 cluster was shown to play an important role in self-renewal and stemness maintenance in pluripotent cells, as well as differentiation, cell cycle progression, and apoptosis regulation in embryonic stem cells or primed pluripotent stem cells (29). Recently, the miR-302–367 cluster has been shown to be involved in cell proliferation and tumor formation, although the results are limited and inconsistent (30). However, to date, few studies have investigated the possible role of the miR-302–367 cluster in an IAV-induced cytokine storm. As shown in our previous study, IAV infection down-regulates miR-302c expression, leading to the activation of IFNβ expression and the inhibition of viral replication (21). Here, IAV induced the production of inflammatory cytokines and chemokines (IFN-β, TNF-α, IL-6, IL-8, CCL2, and CCL5) through miR-302a. To the best of our knowledge, miR-302a is the first microRNA reported to be involved in an IAV-induced cytokine storm. Interestingly, the miR-302 cluster targets IL-8 and inhibits the expression of its mRNA in gastric cancer. Our results are consistent with this study and provide another mechanism by which the miR-302 cluster induces IL-8 expression (31). Because the miR-302/367 cluster is located in the first intron of the LARP7 gene on human chromosome 4 (32), we tested whether IAV infection had an effect on LARP7 expression. As shown in supplemental Fig. 5A, there was no association between IAV infection and LARP7 expression. It has been reported that Oct4 and Sox2 bind to the promoter region of the miR-302 cluster of miRNAs in human embryonic stem cells (hESCs) (32), so we also investigated the role of Oct4 and Sox2 in miR-302a expression. In ChIP assays, Sox2 and Oct4 were not bound to the putative promoter region of miR-302 during IAV infection, and hESCs were included as a positive control for comparison (supplemental Fig. 5, B and C). It seems that IAV did not regulate miR-302 expression at a transcriptional level. As mentioned above, the miR-302 cluster comprises five members. Do other members of the miR-302 cluster also participate in an IAV-induced cytokine storm? Further studies are needed to verify these putative functions.
Following the identification of the function of miR-302a in an IAV-induced cytokine storm, we further explored the underlying mechanism. In this study, we identified IRF-5 as a novel target of miR-302a. IRF-5, a member of the IRF family, is involved in both the type I interferon and Toll-like receptor signaling pathways. IRF-5 is associated with systemic lupus erythematosus, rheumatoid arthritis, and inflammatory bowel diseases (11). In addition, IRF-5 also functions in M1 macrophage polarization and IgG class switching in B cells (13). However, little is currently known about the role of IRF-5 in an IAV-induced cytokine storm. In this report, we present evidence that IRF-5 mediated an IAV-induced cytokine storm in vitro and in vivo. Further studies confirm that overexpression of IRF-5 significantly increased the expression of IAV RNA and protein, and conversely, knockdown of IRF-5 decreased the expression of IAV RNA and protein (Fig. 7). Intriguingly, IAV infection induces IRF-5 mRNA and protein expression at a very early stage of the viral replication (as early as 6 h) (Fig. 7A). The IAV replication cycle does not contain a DNA stage, and transcription factors did not act directly on the IAV promoter (33), so we suspected that IRF-5 was involved in the establishment of IAV replication. However, whether IRF-5 induction is needed for viral entry is yet to be determined. Previously, two reports have described the mechanism by which IRF-5 is activated. According to one study, MyD88 activates IRF-5 via Lys-63–linked polyubiquitination (34). As shown in the other study, IKKβ is an IRF-5 kinase that instigates inflammation (35). In addition, IRF-5 is also a target of miR-146b (36). Are the MyD88–IRF-5 and IKKβ–IRF-5 signaling pathways required for an IAV-induced cytokine storm? Does miR-146b participate in an IAV-induced cytokine storm? The answers to these questions may provide valuable information regarding the mechanisms and lead to the development of new tools to control the dysregulated pro-inflammatory cytokine and chemokine production in the lungs during IAV infection. Although the present study is designed to provide insights into these questions, the dissection of each mechanism is beyond the scope of this paper.
We propose a working model of the role of the miR-302a/IRF-5 axis in an IAV-induced cytokine storm (Fig. 9). In this model, IAV infection strongly induces IRF-5 expression through inhibiting miR-302a expression. Enhanced IRF-5 expression results in an overabundant cytokine production, leading to promotion of IAV replication. In summary, although more research is needed to understand the delicate regulatory mechanisms by which IAV induces the inflammatory response, our current findings highlight how the miR-302a/IRF-5 axis functions as a critical regulator of an IAV-induced cytokine storm and provide a potential target for the treatment of IAV infection in the future.
Figure 9.
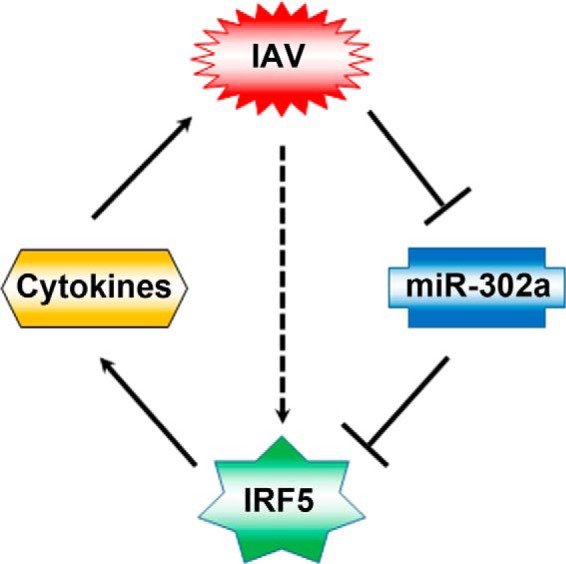
A hypothetical model for the role of miR-302a/IRF-5 axis in IAV-induced inflammatory cytokine expression. Solid arrows, signaling pathways identified in this study; broken arrows, potential signaling pathways.
Materials and methods
Ethics statement
The collection of human PBMCs from blood samples and throat swabs was conducted according to the principles of the Declaration of Helsinki and approved by the institutional review board of the College of Life Sciences, Wuhan University, in accordance with guidelines for the protection of human subjects. All study participants provided written informed consent for the collection of samples and subsequent analyses.
All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol was approved by the institutional animal care and use committee of Wuhan University (project license WDSKY0201302).
Cell culture and virus
A549 cells were used for all experiments and cultured in minimum Eagle's medium (Gibco) containing 10% fetal bovine serum (Invitrogen). THP-1 cells were cultured in RPMI 1640 medium (Gibco) containing 10% FBS. 293T cells were cultured in DMEM (Gibco) containing 10% FBS. MLE-12 cells were cultured in a 50:50 mix of DMEM/Ham's F-12 supplemented with 2% FCS, 5 mg/ml insulin, 10 mg/ml transferrin, 30 mm sodium selenite, 10 nm hydrocortisone, 10 nm β-estradiol, 2 mm glutamine, and 2 mg/ml gentamicin. hESC lines H9 (Wicell, Madison, WI) were grown on mitomycin C–inactivated mouse embryonic fibroblasts in medium containing Dulbecco's modified Eagle's medium F-12 supplemented with 20% knockout serum replacement, 1 mm l-glutamine, 0.1 mm β-mercaptoethanol, 1% nonessential amino acids, and 4 ng/ml human basic fibroblast growth factor. One to two passages before beginning our experiments, hESCs were transferred to Matrigel (BD Biosciences)-coated plates with mTeSR (Stem Cell, Vancouver, Canada). Differentiation through embryoid body formation was induced by collecting hESCs and culturing them in suspension with hESC medium without basic fibroblast growth factor. All cell lines were grown at 37 °C in an aerobic incubator with 5% CO2.
Recombinant human IAV A/WSN/33 (H1N1) was generated by transfecting MDCK cells with an eight-plasmid transfection system, as described previously (37). The eight-plasmid transfection system was a gift from Dr. Webster RG (Department of Infectious Diseases, St. Jude Children's Research Hospital, Memphis, Tennessee). The IAV A/FM/1/47 (H1N1) was a gift from Wei Hou (School of Basic Medical Sciences, Wuhan University). The stock virus was propagated in 10-day-old embryonated chicken eggs (Shijun Li laboratory at Central China Agricultural University) for 36–48 h at 37 °C. The allantoic fluid was then harvested, and aliquots were stored at −80 °C before use.
Plasmids, miRNA, siRNA, antibodies, and biochemical reagents
The complete luciferase reporter gene was PCR-amplified from pGL3-basic vector (Invitrogen), digested with BamHI and EcoRI, and inserted into the pCMV-FLAG2B (Invitrogen) vector to form a subclone vector, pCMV-FLAG2B-Luc. The full-length 3′-UTR of the human IRF-5 gene (GI: 629266059) was amplified using the following primers: sense, 5′-CCCAAGCTTGGTGCGGACTGATGTGGAGA-3′; antisense, 5′-CCGCTCGAGGGCAGAAAGTGGGCTGGAGT-3′. The PCR product was digested with HindIII/XhoI and cloned into the pCMV-FLAG2B-Luc vector. The mutated 3′-UTR of IRF-5 was constructed using PCR-based mutagenesis with the following primers: Mut-I sense, 5′-CTCTGAGATAATATGAGTGGGTATCATATCAGATG-3′; Mut-I antisense, 5′-CATCTGATATGATACCCACTCATATTATCTCAGAG-3′; Mut-II sense, 5′-CCAACTGGCCCACTCCCCACCCCCAGCTTGCGACC-3′; Mut-II antisense, 5′-GGTCGCAAGCTGGGGGTGGGGAGTGGGCCAGTTGG-3′. All plasmids were sequenced and confirmed.
The miR-302a mimics, inhibitors, and negative controls were obtained from GenePharma (Shanghai, China): miR-302a mimics (sense, 5′-UAAGUGCUUCCAUGUUUUGGUGA-3′; antisense, 5′-UCACCAAAACAUGGAAGCACUUA-3′); miR-302a inhibitors, 5′-UCACCAAAACAUGGAAGCACUUA-3′. The small interfering RNAs for the IRF-5 gene were designed and synthesized by Ribobio: siIRF-5-1 (5′-GACGGAGAUAACACCAUCUTT-3′), siIRF-5-2 (5′-ATACACCGAAGGCGTGGAT-3′), and siIRF-5-3 (5′-GCAGTTTAAATAGCTTCATTT-3′). Antibodies against human IRF-5 (ab33478), human β-tubulin (ab6046), human GAPDH (ab8245), IAV NP (ab128193), and M1 (ab25919) were purchased from Abcam. Antibodies against mouse β-actin (A5441) and mouse IRF-5 (SAB1403991) were purchased from Sigma-Aldrich. Unless specified otherwise, all biochemical reagents were purchased from Sigma-Aldrich. Goat anti-mouse IgG-HRP (sc-2055) and goat anti-rabbit IgG-HRP (sc-2030) were purchased from Santa Cruz Biotechnology, Inc.
Mice
Wild-type C57BL/6 mice were purchased from the Animal Facility at Wuhan University ZhongNan Hospital. We designed a pair of synthesized oligonucleotides for gRNA targeting exon 2 of IRF-5, GGCTTCAGCCTCACACGGCGGGG and CCCCGCCGTGAGGCTGAAGCC, which were annealed and cloned into the pUC57-gRNA expression vector (plasmid 51132, Addgene, Cambridge, MA) to generate the IRF-deficient mouse (Irf-5−/−). The gRNA expression plasmids were linearized with DraI and used as templates for in vitro transcription using the MEGAshortscript kit (Ambion, AM1354). The Cas9 expression plasmid (plasmid 44758, Addgene, Cambridge, MA) was linearized with AgeI and used as the template for in vitro transcription using the T7 Ultra Kit (Ambion, AM1345). The transcribed Cas9 mRNA and gRNA were both purified using the MEGAclear kit (Ambion, AM1908), and then a mixture of transcribed Cas9 mRNA and gRNA was microinjected into C57/BL6 mouse zygotes to generate the Irf-5−/− mouse. Microinjections were performed in the cytoplasm of zygotes using a Nikon microinjection system under standard conditions. The mouse was genotyped by PCR with the primers 5′-AAAGCCCTTTGCCATGAACCA-3′ and 5′-GCGCCAGGGTATATAGAAGAGTT-3′. For genotyping, a 157-bp fragment of the WT allele and a 126-bp fragment of the Irf-5 knockout allele were amplified with 30 PCR cycles consisting of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s. The mutant Irf-5 allele 2 was used in future study. All mice were maintained under specific pathogen-free conditions. Irf-5−/− mice developed normally, and no overt difference was observed in hematopoietic cell population. The phenotypes of the Irf-5−/− mice are in line with previously reported findings (11). Mice were humanely euthanized when they met certain clinical criteria or at the end of the experiments.
Isolation and transfection of human PBMCs
The isolation and transfection of human PBMCs were described previously (22, 38). Briefly, human PBMCs were isolated from venous blood samples obtained from healthy volunteers by density centrifugation using Histopaque, according to the manufacturer's instructions (Sigma-Aldrich). PBMCs were transfected with plasmid DNA by electroporation with an Amaxa Nucleofector II device according to the manufacturer's protocol and then resuspended in RPMI 1640 supplemented with penicillin (100 units/ml) and streptomycin (100 mg/ml).
RNA extraction, reverse transcription, and quantitative real-time RT-PCR
Quantitative real-time RT-PCR was described previously (39, 40). Briefly, total RNA was extracted using TRIzol reagent (Invitrogen), according to the manufacturer's protocol. 2–4 μg of RNA were reverse transcribed with oligo(dT) primers or the specific stem–loop primers for mature miR-302a and the normalization control, U6 snRNA. The miR-302a–specific reverse primer was 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCACCAAA-3′, and the U6- specific reverse primer was 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAAATA-3′. Other primers used in this study are listed in supplemental Table 1. Amplifications were performed in the Prism 7200 RT-PCR system (Applied Biosystems) using standard settings: 50 °C for 2 min, 95 °C for 2 min, and 40 cycles of 15 s at 95 °C and 1 min at 60 °C. The data were collected and processed to calculate the gene expression levels using Step One software version 2.0 (Applied Biosystems). The detection of IAV replication was performed using quantitative RT-PCR and primers designed to amplify the NP viral RNA.
Cytokine and chemokine analyses
The tracheas of euthanized mice were exposed, transected, and intubated with a blunt 18-gauge needle. One milliliter of PBS supplemented with Complete Mini, EDTA-free protease inhibitor mixture (Roche Applied Science) was infused and collected four times. The recovered bronchoalveolar lavage fluid was centrifuged at 3,000 × g for 3 min at 4 °C and stored at −80 °C until use. A multiplex ELISA was performed on supernatants using ELISA kits (R&D Systems).
Transfection and luciferase reporter gene assays
Cells were seeded in 24-well dishes and transfected using Lipofectamine 2000 (Invitrogen) for 24 h, after which cells were serum-starved for an additional 24 h before harvest. Transfection complexes were prepared according to the manufacturer's instructions. A Renilla luciferase reporter vector, pRL-TK, was used as an internal control. Both the firefly and Renilla luciferase activities were measured using a GLOMAX 20/20 luminometer (Promega).
ChIP
Cells were incubated for 24 h after transfection and then were serum-starved for 24 h. Formaldehyde was added to the culture medium to a final concentration of 1%. The cells were then washed twice with PBS, scraped, and lysed in lysis buffer (1% SDS, 10 mm Tris-HCl, pH 8.0, 10% protease inhibitor mixture, 50 mg/ml both aprotinin and leupeptin) for 10 min at 4 °C. The lysates were sonicated on ice, and the debris was removed by centrifugation at 12,000 rpm for 15 min at 4 °C. One-fourth of the supernatant was used as DNA input control. The remaining sample was diluted 10-fold with dilution buffer (0.01% SDS, 1% Triton X-100, 1 mm EDTA, 10 mm Tris-HCl, pH 8.0, and 150 mm NaCl), followed by incubation with antibodies against Sox2 (Sigma) and Oct4 (Sigma) overnight at 4 °C. Immunoprecipitated complexes were collected using protein A/G-Sepharose. The pellets were washed for four times with dialysis buffer containing 2 mm EDTA and 50 mm Tris-HCl, pH 8.0. After washing, the precipitates were incubated in an elution buffer (1% SDS and 0.1 m NaHCO3) at room temperature. Supernatants were transferred to clean tubes, and RNase A was added to destroy bound RNA in the sample. Samples were incubated at 65 °C for 5 h to reverse formaldehyde cross-links, and DNA was precipitated with ethanol and extracted two times with phenol/chloroform. Finally, pellets were resuspended in TE buffer and subjected to PCR amplification using the miR-302 cluster promoter containing the Oct4-Sox2 binding sites. The following primers were used: sense, 5′-CCCGTGGAAGCAATCTATTTATTT-3′; antisense, 5′-TGTGTTTCTATCTGGAGGAACTCTGT-3′.
Western blot analysis
Western blot analyses were described in a previous study (41). Briefly, cells were harvested by low-speed centrifugation and washed with PBS. The whole-cell lysates were prepared in PBS buffer (pH 7.4) containing 0.01% Triton X-100, 0.01% EDTA, and a 10% protease inhibitor mixture (Roche Applied Science). The protein concentration was measured using a Bradford assay kit (Bio-Rad). Forty micrograms of each protein sample were separated by 12% SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). The membrane was blocked with PBS buffer containing 0.05% Tween 20 and 5% (w/v) nonfat milk for 1 h at room temperature. Then the membrane was incubated with primary antibodies overnight at 4 °C. Following an incubation with a horseradish peroxidase-linked secondary antibody for an additional 1 h, proteins were detected with the chemiluminescence plus reagent (Millipore) and visualized using an LAS-4000 instrument (Fujifilm, Tokyo, Japan).
Statistical analysis
The data presented in this article were obtained from three independent reproducible experiments. The results are presented as means ± S.D. Student's t test was performed for statistical comparisons between two groups. A one-way analysis of variance was used to compare three or more groups. Kaplan–Meier analysis was used for the survival analysis. A p value < 0.05 was considered significant and is indicated with an asterisk.
Author contributions
X. C. participated in design of the study, carried out all of the experiments, analyzed the results, and drafted the manuscript. L. Z., N. P., and H. Y. participated in gene cloning. M. L., Z. C., J. W., and C. Z. collected and analyzed clinical samples. Y. L., X. W., Q. L., and Y. S. conducted mouse experiments. M. L. and Y. Z. participated in design of the study. S. L. participated in design of the study and critical review of the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Adolfo García-Sastre (Global Health and Emerging Pathogens Institute, Icahn School of Medicine at Mount Sinai) for providing influenza A/PR/8/34 viruses as well as the Wei Hou laboratory (Medical School of Wuhan University) for providing stocks of mouse-adapted influenza virus A/FM/1/47.
This work was supported by National Natural Science Foundation of China Grants 31500149 (to S. L.), 31570870 (to Y. Z.), and 81301428 (to L. Z.); Fundamental Research Funds for the Central Universities Grants 2042017kf0221 (to S. L.) and 2042015kf0188 (to L. Z.); National Basic Research Program of China (973 Program) Grant 2014CB542603 (to S. L.); and Deutsche Forschungsgemeinschaft Grants TRR60 and RTG1949 (to M. L.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Table 1 and Figs. 1–5.
- IAV
- influenza A virus
- miRNA or miR
- microRNA
- IRF
- interferon regulatory factor
- CREB
- cAMP-response element–binding protein
- PBMC
- peripheral blood mononuclear cell
- MOI
- multiplicity of infection
- NP
- nucleoprotein.
References
- 1. Si L., Xu H., Zhou X., Zhang Z., Tian Z., Wang Y., Wu Y., Zhang B., Niu Z., Zhang C., Fu G., Xiao S., Xia Q., Zhang L., and Zhou D. (2016) Generation of influenza A viruses as live but replication-incompetent virus vaccines. Science 354, 1170–1173 [DOI] [PubMed] [Google Scholar]
- 2. Banerjee I., Miyake Y., Nobs S. P., Schneider C., Horvath P., Kopf M., Matthias P., Helenius A., and Yamauchi Y. (2014) Influenza A virus uses the aggresome processing machinery for host cell entry. Science 346, 473–477 [DOI] [PubMed] [Google Scholar]
- 3. Morens D. M., Subbarao K., and Taubenberger J. K. (2012) Engineering H5N1 avian influenza viruses to study human adaptation. Nature 486, 335–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harrison C. (2010) Sepsis: calming the cytokine storm. Nat. Rev. Drug. Discov. 9, 360–361 [DOI] [PubMed] [Google Scholar]
- 5. Teijaro J. R., Walsh K. B., Cahalan S., Fremgen D. M., Roberts E., Scott F., Martinborough E., Peach R., Oldstone M. B., and Rosen H. (2011) Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146, 980–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iwasaki A., and Medzhitov R. (2011) A new shield for a cytokine storm. Cell 146, 861–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheloufi S., Dos Santos C. O., Chong M. M., and Hannon G. J. (2010) A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465, 584–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Song L., Liu H., Gao S., Jiang W., and Huang W. (2010) Cellular microRNAs inhibit replication of the H1N1 influenza A virus in infected cells. J. Virol. 84, 8849–8860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang H., Zhao Z., Pang X., Yang J., Yu H., Zhang Y., Zhou H., and Zhao J. (2017) Genistein protects against Ox-LDL-induced inflammation through MicroRNA-155/SOCS1-mediated repression of NF-κB signaling pathway in HUVECs. Inflammation 40, 1450–1459 [DOI] [PubMed] [Google Scholar]
- 10. Gao M., Wang X., Zhang X., Ha T., Ma H., Liu L., Kalbfleisch J. H., Gao X., Kao R. L., Williams D. L., and Li C. (2015) Attenuation of cardiac dysfunction in polymicrobial sepsis by microRNA-146a is mediated via targeting of IRAK1 and TRAF6 expression. J. Immunol. 195, 672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takaoka A., Yanai H., Kondo S., Duncan G., Negishi H., Mizutani T., Kano S., Honda K., Ohba Y., Mak T. W., and Taniguchi T. (2005) Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249 [DOI] [PubMed] [Google Scholar]
- 12. Honda K., Takaoka A., and Taniguchi T. (2006) Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360 [DOI] [PubMed] [Google Scholar]
- 13. Krausgruber T., Blazek K., Smallie T., Alzabin S., Lockstone H., Sahgal N., Hussell T., Feldmann M., and Udalova I. A. (2011) IRF-5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238 [DOI] [PubMed] [Google Scholar]
- 14. Schoenemeyer A., Barnes B. J., Mancl M. E., Latz E., Goutagny N., Pitha P. M., Fitzgerald K. A., and Golenbock D. T. (2005) The interferon regulatory factor, IRF-5, is a central mediator of Toll-like receptor 7 signaling. J. Biol. Chem. 280, 17005–17012 [DOI] [PubMed] [Google Scholar]
- 15. Yanai H., Chen H. M., Inuzuka T., Kondo S., Mak T. W., Takaoka A., Honda K., and Taniguchi T. (2007) Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc. Natl. Acad. Sci. U.S.A. 104, 3402–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ren J., Chen X., and Chen Z. J. (2014) IKKβ is an IRF-5 kinase that instigates inflammation. Proc. Natl. Acad. Sci. U.S.A. 111, 17438–17443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Negishi H., Yanai H., Nakajima A., Koshiba R., Atarashi K., Matsuda A., Matsuki K., Miki S., Doi T., Aderem A., Nishio J., Smale S. T., Honda K., and Taniguchi T. (2012) Cross-interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nat. Immunol. 13, 659–666 [DOI] [PubMed] [Google Scholar]
- 18. Feng D., Sangster-Guity N., Stone R., Korczeniewska J., Mancl M. E., Fitzgerald-Bocarsly P., and Barnes B. J. (2010) Differential requirement of histone acetylase and deacetylase activities for IRF-5-mediated proinflammatory cytokine expression. J. Immunol. 185, 6003–6012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Purtha W. E., Swiecki M., Colonna M., Diamond M. S., and Bhattacharya D. (2012) Spontaneous mutation of the Dock2 gene in IRF-5−/− mice complicates interpretation of type I interferon production and antibody responses. Proc. Natl. Acad. Sci. U.S.A. 109, E898–E904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bi X., Yang L., Mancl M. E., and Barnes B. J. (2011) Modulation of interferon regulatory factor 5 activities by the Kaposi sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3 contributes to immune evasion and lytic induction. J. Interferon Cytokine Res. 31, 373–382 [DOI] [PubMed] [Google Scholar]
- 21. Gui S., Chen X., Zhang M., Zhao F., Wan Y., Wang L., Xu G., Zhou L., Yue X., Zhu Y., and Liu S. (2015) Mir-302c mediates influenza A virus-induced IFNβ expression by targeting NF-κB inducing kinase. FEBS Lett. 589, 4112–4118 [DOI] [PubMed] [Google Scholar]
- 22. Liu S., Hao Q., Peng N., Yue X., Wang Y., Chen Y., Wu J., and Zhu Y. (2012) Major vault protein: a virus-induced host factor against viral replication through the induction of type-I interferon. Hepatology 56, 57–66 [DOI] [PubMed] [Google Scholar]
- 23. Clark I. A. (2007) The advent of the cytokine storm. Immunol. Cell Biol. 85, 271–273 [DOI] [PubMed] [Google Scholar]
- 24. Liu Q., Zhou Y. H., and Yang Z. Q. (2016) The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol. Immunol. 13, 3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Teijaro J. R., Walsh K. B., Rice S., Rosen H., and Oldstone M. B. (2014) Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc. Natl. Acad. Sci. U.S.A. 111, 3799–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kedzierski L., Linossi E. M., Kolesnik T. B., Day E. B., Bird N. L., Kile B. T., Belz G. T., Metcalf D., Nicola N. A., Kedzierska K., and Nicholson S. E. (2014) Suppressor of cytokine signaling 4 (SOCS4) protects against severe cytokine storm and enhances viral clearance during influenza infection. PLoS Pathog. 10, e1004134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xie J., Zhang S., Hu Y., Li D., Cui J., Xue J., Zhang G., Khachigian L. M., Wong J., Sun L., and Wang M. (2014) Regulatory roles of c-jun in H5N1 influenza virus replication and host inflammation. Biochim. Biophys. Acta 1842, 2479–2488 [DOI] [PubMed] [Google Scholar]
- 28. Schmolke M., Viemann D., Roth J., and Ludwig S. (2009) Essential impact of NF-κB signaling on the H5N1 influenza A virus-induced transcriptome. J. Immunol. 183, 5180–5189 [DOI] [PubMed] [Google Scholar]
- 29. Barroso-delJesus A., Romero-López C., Lucena-Aguilar G., Melen G. J., Sanchez L., Ligero G., Berzal-Herranz A., and Menendez P. (2008) Embryonic stem cell-specific miR302–367 cluster: human gene structure and functional characterization of its core promoter. Mol. Cell. Biol. 28, 6609–6619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cai N., Wang Y. D., and Zheng P. S. (2013) The microRNA-302–367 cluster suppresses the proliferation of cervical carcinoma cells through the novel target AKT1. RNA 19, 85–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen L., Min L., Wang X., Zhao J., Chen H., Qin J., Chen W., Shen Z., Tang Z., Gan Q., Ruan Y., Sun Y., Qin X., and Gu J. (2015) Loss of RACK1 promotes metastasis of gastric cancer by inducing a miR-302c/IL8 signaling loop. Cancer Res. 75, 3832–3841 [DOI] [PubMed] [Google Scholar]
- 32. Card D. A., Hebbar P. B., Li L., Trotter K. W., Komatsu Y., Mishina Y., and Archer T. K. (2008) Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol. Cell. Biol. 28, 6426–6438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kumar N., Xin Z. T., Liang Y., Ly H., and Liang Y. (2008) NF-κB signaling differentially regulates influenza virus RNA synthesis. J. Virol. 82, 9880–9889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Balkhi M. Y., Fitzgerald K. A., and Pitha P. M. (2008) Functional regulation of MyD88-activated interferon regulatory factor 5 by K63-linked polyubiquitination. Mol. Cell. Biol. 28, 7296–7308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bergstrøm B., Aune M. H., Awuh J. A., Kojen J. F., Blix K. J., Ryan L., Flo T. H., Mollnes T. E., Espevik T., and Stenvik J. (2015) TLR8 senses Staphylococcus aureus RNA in human primary monocytes and macrophages and induces IFN-β production via a TAK1-IKKβ-IRF-5 signaling pathway. J. Immunol. 195, 1100–1111 [DOI] [PubMed] [Google Scholar]
- 36. Peng L., Zhang H., Hao Y., Xu F., Yang J., Zhang R., Lu G., Zheng Z., Cui M., Qi C. F., Chen C., Wang J., Hu Y., Wang D., Pierce S., et al. (2016) Reprogramming macrophage orientation by microRNA 146b targeting transcription factor IRF-5. EBioMedicine 14, 83–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoffmann E., Neumann G., Kawaoka Y., Hobom G., and Webster R. G. (2000) A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. U.S.A. 97, 6108–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peng N., Liu S., Xia Z., Ren S., Feng J., Jing M., Gao X., Wiemer E. A., and Zhu Y. (2016) Inducible major vault protein plays a pivotal role in double-stranded RNA- or virus-induced proinflammatory response. J. Immunol. 196, 2753–2766 [DOI] [PubMed] [Google Scholar]
- 39. Jing M., Wang J., Zhu S., Ao F., Wang L., Han T., Yue X., Zhu Y., Ye L., and Liu S. (2016) Development of a more efficient hepatitis B virus vaccine by targeting hepatitis B virus preS to dendritic cells. Vaccine 34, 516–522 [DOI] [PubMed] [Google Scholar]
- 40. Zhu S. L., Wang L., Cao Z. Y., Wang J., Jing M. Z., Xia Z. C., Ao F., Ye L. B., Liu S., and Zhu Y. (2016) Inducible CYP4F12 enhances hepatitis C virus infection via association with viral nonstructural protein 5B. Biochem. Biophys. Res. Commun. 471, 95–102 [DOI] [PubMed] [Google Scholar]
- 41. Liu S., Peng N., Xie J., Hao Q., Zhang M., Zhang Y., Xia Z., Xu G., Zhao F., Wang Q., Han T., and Zhu Y. (2015) Human hepatitis B virus surface and e antigens inhibit major vault protein signaling in interferon induction pathways. J. Hepatol. 62, 1015–1023 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.