

Abstract
The potential for adoptive cell immunotherapy as a treatment against cancers has been demonstrated by the remarkable response in some patients with hematological malignancies using autologous T cells endowed with chimeric antigen receptors (CARs) specific for CD19. Clinical efficacy of CAR-T cell therapy for the treatment of solid tumors, however, is rare due to physical and biochemical factors. This review focuses on different aspects of multiple mechanisms of immunosuppression in solid tumors. We characterize the current state of CAR-modified T cell therapy and summarize the various strategies to combat the immunosuppressive microenvironment of solid tumors, with the aim of promoting T cell cytotoxicity and enhancing tumor cell eradication.
Keywords: Chimeric antigen receptor T cell, Adoptive cell therapy, Solid tumor, Immunosuppressive microenvironment, Cancer immunotherapy
Background
With advances in tumor biology and immunology, cancer immunotherapy has become a new approach for cancer treatment in recent years [1–3]. The adoptive transfer of genetically engineered T cells expressing a chimeric antigen receptor (CAR) specific for tumor antigens, a novel form of cancer immunotherapy, has been remarkably successful in the treatment of some human hematological malignancies, including leukemia and lymphoma [4–8]. The integration of a single chain variable fragment (scFv) and the signaling domain can endow CAR with specificity as well as cytotoxicity in a human leukocyte antigen (HLA)-independent manner [9, 10]. The initial CAR mode comprising the scFv and the CD3ζ signaling domain gives T cells transient activation and cytotoxicity [11]. To improve the cytotoxicity and persistence of CAR-T cells, a costimulatory signaling domain, such as CD28 or CD137 (4/1BB), has been integrated into the intracellular signaling domain in some studies and clinic trials [12, 13].
Despite remarkable success in treating hematological malignancies, particularly in acute lymphoblastic leukemia (ALL) where the T cell therapy achieves high clinical response rates in some trials (e.g., NCT02588456, NCT02535364, and NCT01475058), the adoptive transfer of CAR-T cells has faced a number of challenges for solid tumors [14–18]. Theoretically, modified-T cells have poor homing ability to tumor sites, and a hostile tumor-microenvironment (TME) containing many immunosuppressive cells and other inhibitory factors impairs migrated CAR-T cell cytotoxicity. Although CAR-T cell treatment of solid tumors has not shown promising response, a comprehensive understanding of the multiple barriers seen in the TME is necessary to advance CAR engineering in cancer immunology. In this review, we analyze the factors that limit the application of CAR-T cell therapy in the treatment of solid tumors. We then characterize some new approaches that are being considered to overcome these hurdles, providing guidance for researchers and physicians to effectively fight solid tumors.
Barriers in the solid tumor microenvironment
Physical barriers
The extracellular matrix (ECM) in the TME, including proteoglycans and glycopeptidases, has multiple effects on the biological behaviors of tumors and the remodeling of the immune system. Some studies have shown that proteins in ECM that are nonstructural matrix proteins, such as heparan sulfate proteoglycans (HSPGs), have a major role in the maintenance of tumor cell proliferation and migration. [19, 20]. T cells attacking stroma-rich solid tumors have little ability to penetrate and aggregate in tumor sites, resulting in lower antitumor activity [21]. Therefore, improving the capacity of modified-T cells to specifically degrade the ECM in stroma-rich solid tumors, yet without compromising their cytotoxicity, would enhance their antitumor activity.
Immunosuppressive cells and secreted cytokines
In the TME, immune suppression is mainly mediated by immunosuppressive cells, including regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and M2 macrophages. It has become clear that these cells and released cytokines, such as transforming growth factor-β (TGF-β) and interleukin (IL) 10, inside solid tumors seriously dampen the efficacy of infused CAR-T cells.
Tregs
Tregs play an important role in the regulation of immune responses, including CD4+CD25+ Tregs and type 1 Tregs. TGF-β is essential for CD4+CD25+ Treg function while Foxp3, a regulator of its transcription factor, is highly expressed on Tregs [22]. Type 1 Tregs exert their suppressive activity through the secretion of the cytokine IL-10. In general, Tregs are enriched at the site of inflammation and tumors where they modulate the immune reaction via various mechanisms [23, 24]. Activated Tregs can directly eliminate excessive T cells by rapidly taking up IL-2, resulting in a lack of sufficient effector cells against malignant cells [25]. In addition, these inhibitory cells can produce many immunomodulatory cytokines for the suppression of T cell activity, such as TGF-β and IL-10 [26].
MDSCs
MDSCs, a major component of the immunosuppressive cells, negatively regulate immune responses against tumour progression and metastasis, impairing antitumor immunity [27, 28]. MDSCs mediate their suppression of T cell activity through a combination of major factors, such as inducible nitric oxide synthase (iNOS), arginase catalyze I (ARG1), cyclooxygenase-2 (COX-2), prostaglandin E2 (PGE2), TGF-β, IL-10, and Tregs. Additionally, the presence of MDSCs is associated with the growth of cancer cells. Therefore, inhibiting MDSCs function has been shown to improve antitumor immune responses in the TME [29, 30].
Tumor associated macrophages
M2 macrophages, termed tumor associated macrophages (TAMs), play a pivotal role during the proliferation, invasion, and metastases of tumor cells via expressing soluble proteins and cytokines, such as IL-10, matrix metalloproteinases (MMPs), urokinase-type plasminogen activator (uPA), basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), granulocyte-macrophage colony stimulating factor (GM-CSF), and migration stimulating factor (MSF) [31, 32]. Expressed IL-10 efficiently suppresses the activation of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, resulting in the rapid proliferation of tumor cells. Excessive M2 macrophages release MMP2 and MMP9 that can specifically degrade the extracellular matrix, thereby promoting the migration of tumor cells and tumor stromal cells [33]. In addition, M2 macrophages can consume L-arginine to produce ornithine and polyamine that promote tumor cell proliferation [34].
Immunosuppressive inhibitor receptor
Cytotoxic T lymphocyte-associated antigen 4
In general, T cell activity is regulated through both the activation signal pathway and the suppression signal pathway, resulting in a balanced activity of T cells in a healthy body. The activation of T cells by antigen stimulation signaling and CD28 costimulation is followed by proliferation and functional differentiation. When the activity is excessive, an inhibitory program that is mediated by cytotoxic T lymphocyte-associated antigen 4 (CTLA4) binding with B7 molecules will eventually terminate T cell function. CTLA4, a homolog of CD28, exerts an inhibitory signal to T cells, and then makes them with an inactive state [35]. Theoretically, CTLA4 recruits a phosphatase to the CD3ζ domain of T cell receptors (TCRs), thereby attenuating T cell function by the dephosphorylation of immunoreceptor tyrosine-based activation motifs (ITAM) [36]. In addition, some studies have demonstrated that TCR activation of T cells initiates the phosphorylation of the CTLA4 intracellular domain, termed the immunoreceptor tyrosine-based inhibitory motif (ITIM), and prolongs the CTLA4 expression on the activated T cell surface [37, 38]. Based on the principle of CTLA4 function, approaches that reduce CTLA4 expression or inhibit its activity have been proposed for cancer immunotherapy.
Programmed death 1
Another important immunologic target for cancer immunotherapy after CTLA4 is programmed death 1 (PD1), a cell surface receptor that belongs to the immunoglobulin superfamily expressed only on activated T lymphocytes [39]. Structurally, the mature PD1 protein comprises an extracellular recognition domain targeting its ligands (PDL1 or PDL2) presented on tumor cells and a transmembrane region followed by an intracellular tail [40]. Functionally, PD1 plays an important role in suppressing T cell activity due to the cytoplasmic tail having two phosphorylation sites, an ITIM and an immunoreceptor tyrosine-based switch motif (ITSM), that both negatively regulate T-cell receptor signals [41]. Based on CTLA4 and PD1 functions in different suppression pathways, a combination therapy of PD1 antibody and CTLA4 antibody, therefore, has emerged as an effective approach to combat cancers via checkpoint inhibition [42].
Other immune checkpoints
Although CTLA4 and PD1 pathways negatively regulate the immune response of T lymphocytes against cancers, they only represent the tip of the iceberg of potential targets that can decrease antitumor responses in the immune system. With the development of T cells for cancer immunotherapy, researches have revealed other immune checkpoints that significantly suppress the immune response, such as lymphocyte-activation gene 3 (LAG3), T-cell immunoglobulin mucin 3 (TIM3), and V-domain Ig suppressor of T cell activation (VISTA) [43–45]. The protein LAG3, an exhaustion marker, is wildly expressed on activated T cells and other immune cells, and it is an important inhibitory receptor that binds major histocompatibility complex class II (MHC II) with higher affinity than CD4, preventing antitumor immune responses [46]. TIM3, a receptor protein expressed on T cells, initiates the TIM3/Galectin-9 signaling pathway, which can inhibit lymphocyte responses and promote apoptosis [47]. VISTA, a potent negative regulator receptor expressed on hematopoietic cells and leukocytes, reduces the activity of T cell and the production of cytotoxic cytokines [48].
Intratumoral inhibitory factors
For solid tumors, a hostile microenvironment is characterized by poor oxygen (O2) and nutrient supply [49–51]. In general, cancer cells exhibit unregulated growth even with nutrient scarcity and O2 deprivation due to the constitutive activation of growth-promoting pathways dependent on unsaturated fatty acid catabolism [52]. Additionally, a lower pH of the tumor extracellular environment is frequently observed as a result of high hydrogen ions levels derived from lactic acid and carbonic acid [53]. Glycolysis produces a large amount of lactic acid under anaerobic conditions, and carbonic acid is synthesized from CO2 and H2O in the presence of a carbonic anhydrase (CA) expressed on tumor cells, which maintain an acidic microenvironment.
Lack of chemokine receptors
The primary obstacle when using CAR-modified T cells is infiltration into the solid tumor bed [54]. Some solid tumors secrete chemokines, such as CXCL12 and CXCL5, which are not conducive to T cells penetration into the intratumoral site [55]. In contrast, T cells lack some chemokine receptors that adequately match the chemokine signature secreted from solid tumors [56]. Some reports show that, although chemokines such as C-C chemokine ligand 2 (CCL2) which belongs to the C-C chemokine family, are highly secreted by solid tumors, the corresponding chemokine receptors (CCR), such as CCR2b and CCR4, that can specifically bind CCL2 are minimally expressed on T cells, resulting in a lack of homing to the tumor site [57].
Tumor antigen heterogeneity
To the best of our knowledge, CAR-T cells identify and target surface antigens presented on cancer cells through scFv structures that compose the variable domains of the light and heavy chains derived from monoclonal antibodies. The membrane protein CD19 is homogenous and widely expressed on almost all B-cell hematologic malignancies, resulting in encouraging responses from CD19-specific CAR-T cell therapy [58–61]. However, for solid tumors, the application of CAR-T cell therapy is severely limited due to a shortage of tumor-specific antigens and heterogeneity [62]. However, solid tumors are vulnerable to relapse after treatment with any therapy approach, resulting in cancer being known as a wound that does not heal.
Other immunosuppressive factors in human immune system
CD47
In the human immune system, macrophages exert phagocytosis based on a balance between phagocytic (“eat me”) and antiphagocytic (“do not eat me”) signals on target cells. The antiphagocytic signal depends on the interaction between CD47 expressed on target cells and signal regulatory protein α (SIRPα) presented on macrophages, leading to the phosphorylation of ITIMs on SIRPα. CD47, a transmembrane protein, has been found to be overexpressed in many tumor cells [63–66]. In general, the CD47-SIRPα-mediated antiphagocytic signal allows tumor cells to evade immune surveillance [67–69]. Therefore, to enhance the ability of the immune system against various cancers, CD47 can be used as a novel target for cancer immunotherapy.
CD73 and adenosine
To date, eliminating solid tumors by CAR-T cell therapy has been largely unsuccessful due to adenosine production. Adenosine is generated from extracellular adenosine monophosphate (AMP) by the ectoenzyme CD73 (ecto-5′-nucleotidase) expressed on tumor cells or immunosuppressive cells [70]. The produced adenosine significantly inhibits modified-T cell cytotoxicity through the activation of adenosine 2A receptors (A2AR), resulting in T cells that do not effectively function against solid tumors [71, 72].
Indoleamine 2,3-dioxygenase
Indoleamine 2,3-dioxygenase (IDO) is an immunosuppressive target that helps create a tolerogenic milieu in tumor sites, mainly by suppression of T cell cytotoxicity and enhancement of Treg-mediated immunosuppression [73, 74]. In addition, IDO can suppress antitumor activity of T cells through degradation of tryptophan, a key amino acid required for T cell activity [75, 76]. A series of tryptophan metabolites in the kynurenine pathway suppresses T cell activity and induces T cell apoptosis, also affecting NK cell function [77].
In summary, CAR-modified T cells are faced with numerous barriers when acting agains solid tumors with blunt immune-surveillance, achieving a poor response in some clinical trials (Fig. 1). With further research into tumor biology and immunology, some strategies have been developed to overcome these obstacles, allowing for clinical application of CAR-T cell therapy. Next, we describe these approaches to improve the TME and enhance T cell functions according to their mechanisms and their applications.
Fig. 1.
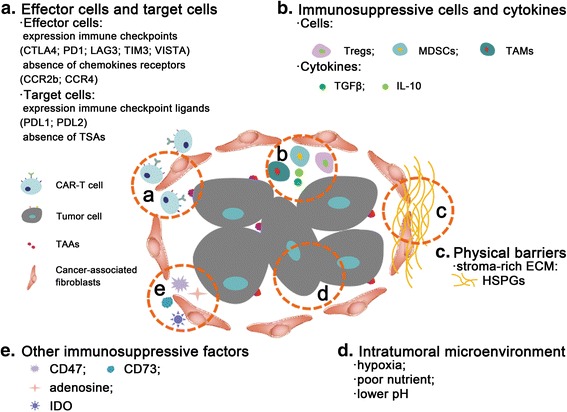
Immunosuppressive microenvironment in solid tumors. a Effector cells and target cells. Activated T cells often express some immune checkpoints that rapidly neutralize the antitumor activities, such as CTLA4, PD1, LAG3, TIM3, and VISTA, while the target cells express some immune checkpoint ligands, such as PDL1 or PDL2. In general, T cells lack some receptors, such as CCR2b or CCR4. In addition, CAR-T cell therapy is limited in solid tumors due to the paucity of tumor-specific antigens. b Immunosuppressive cells and cytokines. In the solid TEM, some immunosuppressive cells (e.g., Tregs, MDSCs, and TAMs) and released immunosuppressive cytokines (e.g., TGFβ and IL-10) significantly inhibit CAR-T cell cytotoxic function. c Physical barriers. Stroma-rich solid tumors have an abundance of ECM (e.g., HSPGs) that effectively inhibits the penetration and aggregation of T cells. d Intratumoral microenvironment. The intratumoral microenvironment is associated with hypoxia, nutrient starvation, and acidosis derived from elevated lactate generation. e Other immunosuppressive factors. CD47 allows tumor cells to evade immune surveillance mediated by CD47-SIRPα-mediated antiphagocytic signaling. Adenosine generated from extracellular AMP by the ectoenzyme CD73 results in unsuccessful application of CAR-T cell therapy. IDO contributes to tumor-induced tolerance through the degradation of tryptophan
Approaches to improve the CAR-T cell efficacy
Selecting tumor-specific antigens
The antigenic specificity of the CAR structure directly determines the precise activity and safety of modified-T cells. However, for solid tumors, the heterogeneity of tumor antigens results in invalid immune surveillance, and thereby a refractory and relapsed tumor. Some researchers have attempted to find markers of cancer stem cells, which are considered to be tumor-specific antigens in the early stage of tumor differentiation [78, 79]. The identification of a tumor-specific antigen provides a powerful tool to develop targeted therapies for solid tumors (Fig. 2a).
Fig. 2.
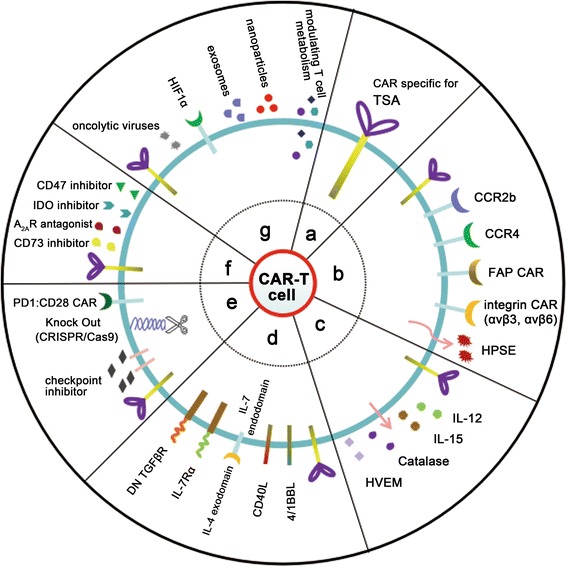
Novel strategies to enhance the efficacy of CAR-T cell therapy for solid tumors. a CAR-T cells targeting tumor specific antigens. b Infiltration and homing of CAR-T cells. CAR-modified T cells express a chemokine receptor (e.g., CCR2b or CCR4), increasing their ability to move to the tumor site. FAP-specific CAR-T cells can migrate to the tumor bed, similar to integrin αvβ3 (or αvβ6)-specific CAR-T cells. Heparanase expression enhances CAR-T cell infiltration and antitumor activity. c CAR-T cells secreting cytokines or enzymes. To resist a hostile environment, T cells have been engineered to express cytokines, such as IL-12 or IL-15. CAR-T cells are endowed with the catalase to overcome abundant ROS. CAR-T cells carrying soluble HVEM also enhance therapeutic activity against lymphomas. d CAR-T cells expressing receptors. CAR-T cells expressing costimulatory receptors (e.g., 4/1BBL or CD40L) significantly enhance T cell activity. CAR-T cells expressing a hybrid receptor comprising an IL-4 exodomain and IL-7 endodomain convert the immunosuppressive response to immune action by targeting IL-4. CAR-T cells expressing IL-7Rα specific for IL-7 have improved response in solid tumors. T cells with a dominant-negative TGFβ (DN TGF-βR) response to TGFβ. e Immune checkpoint therapy. CAR-T cell therapy can be combined with the blockage of immune checkpoints using monoclonal antibodies or the CRISPR/Cas9 system. Additionally, PD1:CD28 CAR comprising the PD1 exodomain and CD28 endodomain enhance T cell activity. f Other blockage therapies. Blockage of soluble tumor suppressive mediators in the solid tumor milieu (e.g., CD73, A2AR, IDO, or CD47) enhance CAR-T cell function. g Combination therapies. CAR-T cells can be combined with other antitumor strategies, such as oncolytic viruses, HIF-CAR, exosomes, nanoparticles, and modulating T cell metabolism
Infiltration and homing of CAR-T cells
Chemokine receptors
To enhance T cell homing to tumor cells, some chemotactic manipulations endow T cells with a strong ability to target solid tumors. A combination of chemokines secreted from tumor cells and corresponding chemokine receptors presented on genetically modified-T cells may facilitate a greater proportion of T cells to arrive at the solid tumor site (Fig. 2b). For malignant pleural mesotheliomas, T cells genetically modified to simultaneously express CCR2b and a CAR specific for mesothelin increase their ability to migrate to tumor sites and eliminate target cells. In addition, other groups have described a similar CAR methodology with augmented trafficking and antitumor efficacy, such as using CD30-specific CAR-T cells expressing CCR4 in Hodgkin’s lymphoma and GD2-specific CAR-T cells expressing CCR2b for neuroblastoma [80, 81]. In these studies, CAR T cells modified with a functional chemokine receptor have adequate tumor localization and significant antitumor efficacy in vivo.
Fibroblast activation protein
Previous reports have demonstrated that cancer-associated fibroblasts (CAFs) facilitate angiogenesis, playing a prominent role in the progression and metastasis of solid tumors [82, 83]. Therefore, CAFs are a key determinant in the malignant progression of cancer and represent an important target for cancer therapies. Schuberth and colleagues developed T cells with a CAR specific for a fibroblast activation protein (FAP), which is abundantly expressed on CAFs, and obtained an encouraging response in vivo, leading to CAF-CAR-T cells being used in an ongoing clinical trial (NCT01722149) (Fig. 2b) [84]. In summary, targeting CAFs as a therapeutic strategy is an intriguing approach to effectively eradicate cancer.
Integrin
For solid tumors, modified-T cells lack the ability to infiltrate into the tumor parenchyma, resulting in limited their effectiveness in vivo. In general, integrin αvβ3 is highly expressed on the surfaces of endothelial cells of tumor neovasculature in solid tumors. Fu and colleagues, therefore, put forward a promising strategy in which T cells are genetically engrafted with an echistatin-containing CAR (T-eCAR) specific for αvβ3 integrin (Fig. 2b) [85]. The injection of T-eCAR cells effects tumor neovasculature, leading to significant tumor shrinkage, yet has no significant off-target effect on blood vessels in normal tissues. In addition, Whilding and colleagues developed a novel CAR modality specific for αvβ6 integrin expressed on ovarian, breast, and pancreatic tumors [86].
Heparanase
Solid tumor-mediated immunosuppression may play an important role in impeding CAR-T cell activity attributed to the HSPGs-rich ECM, inhibiting the ability of modified-T cells to accumulate in tumor sites. Therefore, a strategy to degrade HSPGs around stroma-rich solid tumors is an effective way to enhance the antitumor activity of CAR-redirected T cells, such as by enzymatic degradation of heparanase (HPSE) (Fig. 2b) [87]. Caruana and colleagues reported that HPSE expression enhances CAR-redirected T cell infiltration and antitumor activity. In a neuroblastoma model, their results demonstrated that inducing the expression of HPSE in LET-T cells endowed with a GD2-specific CAR enhanced their capacity to degrade the ECM [88].
Co-expressing immune factors or enzymes
To further improve the ability of T cells to resist the evil TME, T cells have been engineered to constitutively express some cytokines, such as IL-12 or IL-15, and secrete some enzymes, such as catalase (Fig. 2c).
IL-12
The immunomodulatory cytokine IL-12 promotes the rapid reversal of immune suppression and plays an important role in the activities of T lymphocytes and other immune cells, emerging as a potent inducer of antitumor immunity [89]. For instance, Pegram and colleagues reported that CD19-targeted CAR-T cells secreting IL-12 eliminate systemic tumors and achieve optimal antitumor response [90]. In addition, CAR redirected T cells delivering IL-12, termed TRUCKs, not only enhance the activation and cytotoxicity of modified-T cells, but also result in a Th1 polarization state that attracts endogenous T cells and other innate immune cells to eradicate cancer cells [91, 92].
IL-15
Some studies have shown that the cytokine IL-15 is a potent stimulant of memory CD8+ T cells and NK cells by binding the IL-2 receptor β-common γ chain (IL2Rβγc) complex [93]. Hsu and colleagues demonstrated that IL-15 plays an important role in the trafficking of CD8+ T cells in vivo [94]. Therefore, IL-15-expressing T-cells could be used as an effective weapon to eliminate target cells within the immunosuppressive microenvironment.
Catalase
In solid tumors, infused-T cells often encounter abundant reactive oxygen species (ROS), a hallmark of the tumor milieu, which can impair the antitumor activity of modified-T cells [95]. Ligtenberg and colleagues developed a novel T cell modality (termed CAR-CAT T cell) that is endowed with a CAR specific for catalase, which catalyzes hydrogen peroxide to water and oxygen [96]. These engineered T cells could reduce the oxidative state of infiltrated tumors and preserve their antitumor activity by metabolizing hydrogen peroxide and reducing oxidative stress-mediated suppression.
HVEM
Herpesvirus entry mediator (HVEM), known as tumor necrosis factor receptor superfamily member 14 (TNFRSF14), is among the most frequently mutated genes in germinal center lymphomas (GCL) [97, 98]. Loss of HVEM usually drives the development of GCL in vivo and induces a tumor-supportive microenvironment, resulting from the disruption of the interactions between HVEM and BTLA (B and T lymphocyte attenuator) receptors [99, 100]. Boice and colleagues engineered CD19-targeted CAR-T cells carrying soluble HVEM (solHVEM) to locally and continuously enhance therapeutic activity against lymphomas by release of solHVEM [101].
Modifying some related receptors
In general, T cells expressing a CAR structure have a promising immunotherapeutic effect on B cell malignancies [102, 103]. In recent years, the design of novel CARs modified with costimulatory receptors or cytokine receptors has resulted in important effects on T cell activity, including proliferation and cytotoxicity.
4/1BBL
Some recent reports have revealed that second-generation CARs constitutively expressing ligands for CD28 or 4/1BB (CD80 or 4/1BBL, respectively) enhance T cell activity in vivo [104]. Stephan and colleagues suggested that the 4/1BB-4/1BBL signaling pathway may more effectively enhance CAR modified-T cell activation and cytotoxicity, potentially in a bidirectional manner with direct signaling within the T cells themselves and indirect triggering of other immune cells expressing the 4/1BB domain in the TME (Fig. 2d) [105].
CD40L
Cancer immunotherapy requires effective destruction of the immunosuppressive TME. Marigo and colleagues demonstrated that the CD40/CD40L axis is necessary for tumor rejection mediated by CD8+ T cells in the presence of tumor necrosis factor (TNF) derived from dendritic cells (DCs) [106]. Therefore, we envisioned that tumor elimination will be achieved by a combination of CAR-modified T cells with CD40/C40L pathway modifications (Fig. 2d).
IL-7Rα
Cytokine IL-7 is a common γ chain cytokine that is essential for the expansion of naive and memory T lymphocytes [107]. To maintain T cells with high efficacy, CTLs can be genetically modified to express IL-7Rα that can specifically bind to IL-7 (Fig. 2d). Vera and colleagues reported that the expression of IL-7Rα consistently promotes CTL activity, including activation and proliferation, in response to IL-7, and provides antitumor activity without impairing the antigen specificity or dependency of modified-CTLs [108].
DN TGF-βR
There are many immunosuppressive cytokines secreted by tumor cells in the TEM, including TGF-β, which markedly inhibits tumor-specific cellular immunity. Based on this mechanism, reducing the expression of TGF-β receptor (TGF-βR) presented on T cells is an effective strategy to surmount TGF-β-induced immune suppression [109]. Bollard and colleague demonstrated that EBV-specific T-cells endowed with a dominant-negative TGF-βR (DN TGF-βR) can resist the inhibitory effects of TGF-β, resulting in T cells with longer survival (Fig. 2d) [110].
Blockage of the immune checkpoints
Previous reports have revealed that some tumors, especially solid cancers, employ a variety of countermeasures to impair the cytotoxicity of T cells, including the expression of immune checkpoints expressed on T cells, such as PD1, CTLA4, LAG3, TIM3, or VISTA. These inhibitory receptors expressed on activated T cells have a high affinity for their ligands present on target cells, therefore the inhibitory receptor/ligand pathway is a powerful brake on T cell efficacy. Some technologies have been developed to overcome this obstacle, including monoclonal antibodies (mAb) and gene editing (Fig. 2e).
For mAb immunotherapy, clinical trials associated with the blockade of immune checkpoints have demonstrated that mAbs can reverse T cell exhaustion and restore antitumor immunity, significantly promoting tumor cell eradication, such as anti-PD1 mAb (NCT02838823, NCT02857166, NCT02836795), anti-CTLA4 mAb (NCT00610857, NCT02156804, NCT03203876), anti-LAG3 mAb (NCT02658981, NCT01968109, NCT02061761), anti-TIM3 mAb (NCT02817633), and anti-VISTA mAb (NCT02671955).
In addition, a non-viral approach to reprogram primary human T cells by disruption of immune checkpoints has been developed with the CRISPR/Cas9 technology. Su and colleagues demonstrated that gene knockout of PD1 by electroporation of plasmids encoding sgRNA and Cas9 results in significant reduction of PD1 expression, up-regulation of cytokine production, and enhanced cytotoxicity [111]. Shi and colleagues demonstrated that CTLs with knocked-out CTLA4 by CRISPR/Cas9 system significantly repress tumor growth and improve tumor eradication [112]. Therefore, a two-in-one approach including CAR-T cells and checkpoint blockade (PD1 or CTLA4) is potentially useful to improve the efficacy of T cells.
Moreover, to reverse the PDL1-mediated T cell exhaustion, Prosser and colleagues provided a novel engineering strategy in which the PD1 was converted to a T cell costimulatory receptor by exchanging its transmembrane and cytoplasmic domains with that of CD28 (Fig. 2e) [113]. In their research, tumor PDL1 significantly co-stimulated primary human T cells expressing a PD1:CD28 chimeric receptor, resulting in augmentation of cytokine secretion and increased cytotoxicity.
Other blockage therapies
CD73 inhibition and A2AR antagonist
Based on adenosine-derived immune suppression around solid tumors, there are two broad strategies to enhance CAR-T cell efficacy (Fig. 2f). One approach is to block the interaction between adenosine and A2AR by A2AR antagonists, profoundly increasing CAR-T cell cytotoxicity. For instance, Beavis and colleagues showed that human epidermal growth factor receptor 2 (HER2)-specific CAR-T cells had high efficacy against melanoma tumor cells expressing HER2 in the presence of an A2AR antagonist [114]. As a result of this preclinical work, a clinical trial (NCT02655822) is ongoing using A2AR antagonists against various solid tumors, such as non-small cell lung cancer, malignant melanoma, renal cell cancer, triple negative breast cancer, colorectal cancer, bladder cancer, and metastatic castration-resistant prostate cancer. Another approach is to generate an mAb specific for CD73 within the tumor milieu, resulting in the reduction of CD73-derived adenosine [115, 116]. In a current phase I clinical study (NCT02503774), the use of anti-CD73 mAb alone or in combination with anti-PDL1 mAb in patients with advanced solid tumors is showing promising response [117].
IDO inhibition
IDO can inhibit T cell efficacy by reducing the concentration of tryptophan and activating Tregs through kynurenine production, which contributes to immunosuppressive microenvironment [118, 119]. In a recent study, the inhibition of the IDO pathway showed significant clinical activity in patients with various cancers (Fig. 2f) [120]. Therefore, there is great interest in combining IDO inhibitors with CAR-T cell therapy for the treatment of solid tumors in future studies.
CD47 inhibition
For many malignancies, abundant CD47 expression is associated with poor patients survival. Some studies have reported that macrophage phagocytosis of tumor cells mediated by anti-CD47 antibodies is a major effector of the immune system. Additionally, Liu and colleagues demonstrated that anti-CD47-mediated tumor rejection required the activation of T cells (Fig. 2f) [121]. Hence, CAR-T cell therapy combined with CD47 inhibition could improve tumor elimination by enhancing the immune system.
Combination strategies for CAR-T therapy
T cells engineered to express CAR in adoptive transfer therapies have thus far shown significant promise in treating some hematological cancers. The clinical efficacy of CAR-T cell therapy in treating solid tumors, however, is negatively affected by a hostile TME. With advancements in tumor research, combination therapies, including T cells or modified-T cells with other antitumor strategy, should have high efficacy for solid tumor eradication (Fig. 2g).
Oncolytic viruses
Oncolytic viruses (OVs) have the capacity to selectively infect and promote tumor cell lysis and impact tumor growth via reducing tumor vasculature, while avoiding healthy tissue damage. In addition, these viruses have been shown to induce robust immune responses, thus potentiating antitumor responses to decrease the tumor burden [122, 123]. VanSeggelen and colleagues demonstrated that loading CAR-engineered T cells with low doses of virus does not impact CAR expression and activity [124]. Combining CAR-T cells with OVs represents a novel strategy to increase the efficacy of cancer treatments. In addition, Nishio and Dotti proposed to improve the migration and survival of CAR-modified T cells in solid tumors by combining them with armed OVs expressing the chemokine RANTES and the cytokine IL-15 [125]. In their experiments, the modified OVs significantly enhanced the trafficking and infiltration of CAR-T cells, thereby promoting tumor eradication.
HIF-CAR
CAR-T cell therapies raise concerns associated with “on-target, off-tumor” effects, particularly in healthy tissues expressing the targeted antigen. To effectively discriminate between healthy tissue and cancer cells, CAR-modified T cells have been endowed with other receptors specific for TME signals. As hypoxia is a hallmark of solid TME, an interesting strategy was exploited for CAR-T cell therapy by targeting hypoxia [126, 127]. For instance, Juillerat and colleagues constructed a novel CAR modality, termed HIF-CAR, that fuses an oxygen sensitive sub-domain of hypoxia-inducible factor 1-alpha (HIF1α), resulting in a CAR sensitive to a hypoxic environment [128]. HIF-CAR-T cells have specific cytotoxicity for solid tumors in the presence of both a tumor antigen and a TME signal (hypoxia), providing a basic approach for using multi-chain modified-CAR as a platform to create the next generation of CAR-T cells.
Exosomes
Exosomes play an important role in intercellular communication and mediate a cell-cell communication with a novel mechanism [129]. In addition, exosomes act as transport carriers for delivering functional proteins secreted from donor cells into the extracellular milieu, thereby improving the interaction between effector cells and target cells. Based on the properties of exosomes, CAR-T cell-derived exosomes have been used to attack tumors in place of immune cells, allowing for controlled cytotoxicity and avoiding some adverse effects of CAR-T cell-mediated immunotherapy. Additionally, exosomes derived from CAR modified-T cells pave the way for cancer immunotherapy against glioblastoma due to their nanoscaled size. Tang and colleagues created a platform, including CAR-T cells and CAR-T cell-derived exosomes, for the treatment of some tumors [130]. Kamerkar and colleagues applied exosomes to effectively facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer and significantly increase overall survival [131].
Nanoparticles
With the development of nanotechnology, nanomedicine-based immunotherapy has been used to elicit specific antitumor responses, providing a new avenue to eliminate solid tumors. Nanoparticle (NP) can be used as a delivery vehicle for releasing antitumor drugs or genes coding CAR structures, and also as immune adjuvants to stimulate the host’s immune system [132–135]. For instance, Yuan and colleagues constructed a multivalent bi-specific nanobioconjugate engager (mBiNE) derived from colloidal-NPs to promote immune-mediated tumor eradication [136]. mBiNE can excit a durable antitumor response for HER2-expressing tumors through a pro-phagocytosis signaling mediated by calreticulin. Thus, cancer immunotherapy based on mBiNE provides a new strategy to stimulate both innate and adaptive immunity. Smith and colleagues programmed T cells with tumor-recognizing capabilities in which DNA-carrying NPs facilitated T cells to express CD19-targeting CAR, thereby providing long-term tumor remission [137]. They functionalized the targeted NPs to specifically target the T cells by coupling with anti-CD3e f(ab’)2 fragments, and loaded the NPs with a vector encoding CD19-specific CAR. In summary, they established, for the first time, a practical and simplified way to modify T cells with the ability to express CD19-specific CARs by polymeric NPs, providing a new form of active immunotherapy suitable for clinical trials.
Modulating T cell metabolism
The metabolic activity of T cells is intimately linked to their efficacy against cancer. Therefore, regulating the state of T cells in the presence of exogenous active agonists would be an effective strategy to improve tumor eradication. For instance, Geiger and colleagues discovered that elevating the intracellular concentration of L-arginine modulates T cell metabolism, including a shift from glycolysis to oxidative phosphorylation, and promotes the generation of central memory-like cells, thereby enhancing the antitumor activity [138]. Yang and colleagues engineered a new mechanism of T cell cytotoxicity of by which CD8+ T cells can be potentially improved by modulating cholesterol metabolism [139]. Scheffel and colleagues reduced T cell death with the addition of the antioxidant N-acetyl cysteine (NAC), offering a potential method to improve the quality and therapeutic efficacy of adoptive T cells [140].
Taken together, the above-mentioned studies offer preclinical proof-of-concept for modulating the metabolism of therapeutic T cells for tumor eradication.
Conclusions
The great success of CAR T cell therapy in some clinical trials against hematological malignancies has provided a framework to drive this modality further to treat solid tumors with immunosuppressive factors. In recent years, the characteristics of the immunosuppressive environment have been described in detail, producing new approaches to overcoming obstacles and enhancing tumor eradication by CAR-T cell therapy. In this review, we have given a brief overview of the challenges against immunologic surveillance in the solid TME, and discussed some promising approaches to enhance CAR-mediated T cell efficacy by optimizing the CAR structure and improving tumor-induced immunosuppression. For the application of CAR-T cell therapy against solid tumors, additional strategies must be explored to modify T cells to achieve sustained cytotoxicity, and to remodel TME to further enhance tumor destruction. Finally, we envision that combination immunotherapies, including enhanced T cell activity (such as infiltration and specific targeting), and improvement of the solid intratumoral environment (such as clearing physical and biochemical barriers) will become effective means to treat solid tumors.
Acknowledgments
Not applicable.
Funding
This work was supported by the Project Program of State Key Laboratory of Natural Medicines (No. SKLNMBZ201403) and the National Science and Technology Major Projects of New Drugs (No. 2012ZX09103301-004 and No. 2014ZX09508007) in China. This project was also funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Abbreviations
- A2AR
Adenosine 2A receptor
- ALL
Acute lymphoblastic leukemia
- AMP
Adenosine monophosphate
- ARG1
Arginase catalyze I
- bFGF
Basic fibroblast growth factor
- BTLA
B and T lymphocyte attenuator
- CA
Carbonic anhydrase
- CAFs
Cancer-associated fibroblasts
- CAR
Chimeric antigen receptor
- CAR-CAT
CAR co-expression catalase
- CCL2
C-C chemokine ligand 2
- CCR
Corresponding chemokine receptors
- COX-2
Cyclooxygenase-2
- CTLA 4
Cytotoxic T lymphocyte-associated antigen 4
- CTLs
Cytotoxic T lymphocytes
- DCs
Dendritic cells
- DN TGF-βR
Dominant negative TGF-βR
- ECM
Extracellular matrix
- FAP
Fibroblast activation protein
- GCL
Germinal center lymphomas
- GM-CSF
Granulocyte-macrophage colony stimulating factor
- HER2
Human epidermal growth factor receptor 2
- HIF1α
Hypoxia-inducible factor 1-alpha
- HLA
Human leukocyte antigen
- HPSE
Heparanase
- HSPGs
Heparan sulfate proteoglycans
- HVEM
Herpesvirus entry mediator
- IDO
Indoleamine 2,3-dioxygenase
- IL
Interleukin
- IL2Rβγc
IL-2 receptor β-common γ chain
- iNOS
Inducible nitric oxide synthase
- ITAM
Immunoreceptor tyrosine-based activation motifs
- ITIM
Immunoreceptor tyrosine-based inhibition motif
- ITSM
Immunoreceptor tyrosine-based switch motif
- LAG3
Lymphocyte-activation gene 3
- mAb
Monoclonal antibody
- mBiNE
Bi-specific nanobioconjugate engager
- MDSCs
Myeloid-derived suppressor cells
- MHC II
Major histocompatibility complex class II
- MMPs
Matrix metalloproteinases
- MSF
Migrate stimulate factor
- NAC
N-acetyl cysteine
- NK
Natural killer
- NP
Nanoparticle
- OVs
Oncolytic viruses
- PD1
Programmed death 1
- PDGF
Platelet-derived growth factor
- PDL1
PD ligand 1
- PDL2
PD ligand 2
- PGE2
Prostaglandin E2
- ROS
Reactive oxygen species
- scFv
Single chain variable fragment
- SIRPα
Signal regulatory protein α
- solHVEM
Soluble HVEM
- TAMs
Tumor associated macrophages
- TCR
T cell receptor
- T-eCAR
Echistatin-containing CAR T
- TGF-β
Transforming growth factor-β
- TGF-βR
TGF-β receptor
- TIM3
T-cellimmunoglobulin mucin 3
- TME
Tumor microenvironment
- TNF
Tumor necrosis factor
- TNFRSF14
Tumor necrosis factor receptor superfamily member 14
- Tregs
Regulatory T cells
- uPA
Urokinase-type plasminogen activator
- VEGF
Vascular endothelial growth factor
- VISTA
V-domain Ig suppressor of T cell activation
Authors’ contributions
EZ designed and drafted the manuscript. JG designed and drawn the figs. HX discussed and revised the manuscript. All authors read and approved final manuscript.
Ethics approval and consent to participate
This is not applicable for this review.
Consent for publication
This is not applicable for this review.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 2.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Sci Transl Med. 2015;7:280ps7. doi: 10.1126/scitranslmed.aaa3643. [DOI] [PubMed] [Google Scholar]
- 7.Morrison C. CAR-T field booms as next-generation platforms attract big players. Nat Biotechnol. 2015;33:571–572. doi: 10.1038/nbt0615-571. [DOI] [PubMed] [Google Scholar]
- 8.Zhang E, Xu H. A new insight in chimeric antigen receptor-engineered T cells for cancer immunotherapy. J Hematol Oncol. 2017;10:1. doi: 10.1186/s13045-016-0379-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srivastava S, Riddell SR. Engineering CAR-T cells: design concepts. Trends Immunol. 2015;36:494–502. doi: 10.1016/j.it.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immun. 2017;66:1425–1436. doi: 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4/1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28:415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilham DE, Debets R, Pule M, Hawkins RE, Abken H. CAR-T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med. 2012;18:377–384. doi: 10.1016/j.molmed.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Yu S, Li A, Liu Q, Li T, Yuan X, Han X, et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol. 2017;10:78. doi: 10.1186/s13045-017-0444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beatty GL, O'Hara M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: defining the challenges and next steps. Pharmacol Ther. 2016;166:30–39. doi: 10.1016/j.pharmthera.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin C, Yu D, Essand M. Prospects to improve chimeric antigen receptor T-cell therapy for solid tumors. Immunotherapy. 2016;8:1355–1361. doi: 10.2217/imt-2016-0125. [DOI] [PubMed] [Google Scholar]
- 18.Newick K, O'Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med. 2017;68:139–152. doi: 10.1146/annurev-med-062315-120245. [DOI] [PubMed] [Google Scholar]
- 19.Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4–27. doi: 10.1016/j.addr.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 21.Digre A, Singh K, Åbrink M, Reijmers RM, Sandler S, Vlodavsky I, et al. Overexpression of heparanase enhances T lymphocyte activities and intensifies the inflammatory response in a model of murine rheumatoid arthritis. Sci Rep. 2017;7:46229. doi: 10.1038/srep46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 23.Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17:975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 26.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 27.Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5:3–8. doi: 10.1158/2326-6066.CIR-16-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125:3356–3364. doi: 10.1172/JCI80005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol. 2013;91:493–502. doi: 10.1038/icb.2013.29. [DOI] [PubMed] [Google Scholar]
- 31.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66:1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 32.Solinas G, Schiarea S, Liguori M, Fabbri M, Pesce S, Zammataro L, et al. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J Immunol. 2010;185:642–652. doi: 10.4049/jimmunol.1000413. [DOI] [PubMed] [Google Scholar]
- 33.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, et al. Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J Immunol. 2011;187:2181–2192. doi: 10.4049/jimmunol.1003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bradshaw JD, Lu P, Leytze G, Rodgers J, Schieven GL, Bennett KL, et al. Interaction of the cytoplasmic tail of CTLA-4 (CD152) with a clathrin-associated protein is negatively regulated by tyrosine phosphorylation. Biochemistry. 1997;36:15975–15982. doi: 10.1021/bi971762i. [DOI] [PubMed] [Google Scholar]
- 37.Baroja ML, Luxenberg D, Chau T, Ling V, Strathdee CA, Carreno BM, et al. The inhibitory function of CTLA-4 does not require its tyrosine phosphorylation. J Immunol. 2000;164:49–55. doi: 10.4049/jimmunol.164.1.49. [DOI] [PubMed] [Google Scholar]
- 38.Qureshi OS, Kaur S, Hou TZ, Jeffery LE, Poulter NS, Briggs Z, et al. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J Biol Chem. 2012;287:9429–9440. doi: 10.1074/jbc.M111.304329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007;56:739–745. doi: 10.1007/s00262-006-0272-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, Hercend T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171:1393–1405. doi: 10.1084/jem.171.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014;74:1924–1932. doi: 10.1158/0008-5472.CAN-13-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur J Immunol. 2005;35:2081–2088. doi: 10.1002/eji.200526090. [DOI] [PubMed] [Google Scholar]
- 47.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 48.Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577–592. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 50.Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125:5591–6. [DOI] [PubMed]
- 51.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 52.Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol. 2014;24:472–478. doi: 10.1016/j.tcb.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther. 2009;121:29–40. doi: 10.1016/j.pharmthera.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 54.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17:559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17:4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015;21:1258–1266. doi: 10.1158/1078-0432.CCR-14-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sick E, Jeanne A, Schneider C, Dedieu S, Takeda K, Martiny L. CD47 update: a multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br J Pharmacol. 2012;167:1415–1430. doi: 10.1111/j.1476-5381.2012.02099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chao MP, Weissman IL, Majeti R. The CD47–SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24:225–232. doi: 10.1016/j.coi.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2:63ra94. doi: 10.1126/scitranslmed.3001375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 67.Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25–50. doi: 10.1146/annurev-immunol-032713-120142. [DOI] [PubMed] [Google Scholar]
- 68.Yamao T, Noguchi T, Takeuchi O, Nishiyama U, Morita H, Hagiwara T, et al. Negative regulation of platelet clearance and of the macrophage phagocytic response by the transmembrane glycoprotein SHPS-1. J Biol Chem. 2002;277:39833–39839. doi: 10.1074/jbc.M203287200. [DOI] [PubMed] [Google Scholar]
- 69.Olsson M, Bruhns P, Frazier WA, Ravetch JV, Oldenborg PA. Platelet homeostasis is regulated by platelet expression of CD47 under normal conditions and in passive immune thrombocytopenia. Blood. 2005;105:3577–3582. doi: 10.1182/blood-2004-08-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol. 2012;33:231–237. doi: 10.1016/j.it.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 71.Allard B, Beavis PA, Darcy PK, Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol. 2016;29:7–16. doi: 10.1016/j.coph.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 72.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. 2006;103:13132–13137. doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9:1069–1077. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- 74.Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196:459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Terness P, Bauer TM, Röse L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weber WP, Feder-Mengus C, Chiarugi A, Rosenthal R, Reschner A, Schumacher R, et al. Differential effects of the tryptophan metabolite 3-hydroxyanthranilic acid on the proliferation of human CD8+ T cells induced by TCR triggering or homeostatic cytokines. Eur J Immunol. 2006;36:296–304. doi: 10.1002/eji.200535616. [DOI] [PubMed] [Google Scholar]
- 77.Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. 2006;108:4118–4125. doi: 10.1182/blood-2006-03-006700. [DOI] [PubMed] [Google Scholar]
- 78.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 79.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 80.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392–6402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 83.Affo S, Yu LX, Schwabe RF. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu Rev Pathol. 2017;12:153–186. doi: 10.1146/annurev-pathol-052016-100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med. 2013;11:187. doi: 10.1186/1479-5876-11-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fu X, Rivera A, Tao L, Zhang X. Genetically modified T cells targeting neovasculature efficiently destroy tumor blood vessels, shrink established solid tumors and increase nanoparticle delivery. Int J Cancer. 2013;133:2483–2492. doi: 10.1002/ijc.28269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Whilding LM, Parente-Pereira AC, Zabinski T, Davies DM, Petrovic RMG, Kao YV, et al. Targeting of aberrant αvβ6 integrin expression in solid tumors using chimeric antigen receptor-engineered T cells. Mol Ther. 2017;25:259–273. doi: 10.1016/j.ymthe.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vlodavsky I, Ilan N, Naggi A, Casu B. Heparanase: structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr Pharm Des. 2007;13:2057–2073. doi: 10.2174/138161207781039742. [DOI] [PubMed] [Google Scholar]
- 88.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524–529. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 90.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. 2011;19:751–759. doi: 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15:1145–1154. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 93.Hsu C, Hughes MS, Zheng Z, Bray RB, Rosenberg SA, Morgan RA. Primary human T lymphocytes engineered with a codon-optimized IL-15 gene resist cytokine withdrawal-induced apoptosis and persist long-term in the absence of exogenous cytokine. J Immunol. 2005;175:7226–7234. doi: 10.4049/jimmunol.175.11.7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hsu C, Jones SA, Cohen CJ, Zheng Z, Kerstann K, Zhou J, et al. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood. 2007;109:5168–5177. doi: 10.1182/blood-2006-06-029173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistent oxidative stress in cancer. FEBS Lett. 1995;358:1–3. doi: 10.1016/0014-5793(94)01368-B. [DOI] [PubMed] [Google Scholar]
- 96.Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, et al. Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol. 2016;196:759–766. doi: 10.4049/jimmunol.1401710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Launay E, Pangault C, Bertrand P, Jardin F, Lamy T, Tilly H, et al. High rate of TNFRSF14 gene alterations related to 1p36 region in de novo follicular lymphoma and impact on prognosis. Leukemia. 2012;26:559–562. doi: 10.1038/leu.2011.266. [DOI] [PubMed] [Google Scholar]
- 98.Cheung KJ, Johnson NA, Affleck JG, Severson T, Steidl C, Ben-Neriah S, et al. Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res. 2010;70:9166–9174. doi: 10.1158/0008-5472.CAN-10-2460. [DOI] [PubMed] [Google Scholar]
- 99.Pasero C, Speiser DE, Derré L, Olive D. The HVEM network: new directions in targeting novel costimulatory/co-inhibitory molecules for cancer therapy. Curr Opin Pharmacol. 2012;12:478–485. doi: 10.1016/j.coph.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 100.Steinberg MW, Cheung TC, Ware CF. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol Rev. 2011;244:169–187. doi: 10.1111/j.1600-065X.2011.01064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, et al. Loss of the HVEM tumor suppressor in lymphoma and restoration by modified CAR-T cells. Cell. 2016;167:405–418. doi: 10.1016/j.cell.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Holohan DR, Lee JC, Bluestone JA. Shifting the evolving CAR T cell platform into higher gear. Cancer Cell. 2015;28:401–402. doi: 10.1016/j.ccell.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 105.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T cell-encoded CD80 and 4/1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13:1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 106.Marigo I, Zilio S, Desantis G, Mlecnik B, Agnellini AH, Ugel S, et al. T cell cancer therapy requires CD40-CD40L activation of tumor necrosis factor and inducible nitric-oxide-Synthase-producing dendritic cells. Cancer Cell. 2016;30:377–390. doi: 10.1016/j.ccell.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Noguchi M, Nakamura Y, Russell SM, Ziegler SF, Tsang M, Cao X, et al. Interleukin-2 receptor gamma chain: a functional component of the interleukin-7 receptor. Science. 1993;262:1877–1880. doi: 10.1126/science.8266077. [DOI] [PubMed] [Google Scholar]
- 108.Vera JF, Hoyos V, Savoldo B, Quintarelli C, Giordano Attianese GM, Leen AM, et al. Genetic manipulation of tumor-specific cytotoxic T lymphocytes to restore responsiveness to IL-7. Mol Ther. 2009;17:880–888. doi: 10.1038/mt.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bollard CM, Rössig C, Calonge MJ, Huls MH, Wagner HJ, Massague J, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179–3187. doi: 10.1182/blood.V99.9.3179. [DOI] [PubMed] [Google Scholar]
- 110.Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother. 2008;31:500–505. doi: 10.1097/CJI.0b013e318177092b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Su S, Hu B, Shao J, Shen B, Du J, Du Y, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep. 2016;6:20070. doi: 10.1038/srep20070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shi L, Meng T, Zhao Z, Han J, Zhang W, Gao F, et al. CRISPR knock out CTLA-4 enhances the anti-tumor activity of cytotoxic T lymphocytes. Gene. 2017;636:36–41. doi: 10.1016/j.gene.2017.09.010. [DOI] [PubMed] [Google Scholar]
- 113.Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8+ cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol Immunol. 2012;51:263–272. doi: 10.1016/j.molimm.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 114.Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. 2017;127:929–941. doi: 10.1172/JCI89455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Antonioli L, Yegutkin GG, Pacher P, Blandizzi C, Haskó G. Anti-CD73 in cancer immunotherapy: awakening new opportunities. Trends Cancer. 2016;2:95–109. doi: 10.1016/j.trecan.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang B. CD73: a novel target for cancer immunotherapy. Cancer Res. 2010;70:6407–6411. doi: 10.1158/0008-5472.CAN-10-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A. 2010;107:1547–1552. doi: 10.1073/pnas.0908801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210:1389–1402. doi: 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015;125:3905–3916. doi: 10.1182/blood-2015-01-621474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21:1209–1215. doi: 10.1038/nm.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Buijs PR, Verhagen JH, van Eijck CH, van den Hoogen BG. Oncolytic viruses: from bench to bedside with a focus on safety. Hum Vaccin Immunother. 2015;11:1573–1584. doi: 10.1080/21645515.2015.1037058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pikor LA, Bell JC, Diallo JS. Oncolytic viruses: exploiting cancer's deal with the devil. Trends Cancer. 2015;1:266–277. doi: 10.1016/j.trecan.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 124.VanSeggelen H, Tantalo DG, Afsahi A, Hammill JA, Bramson JL. Chimeric antigen receptor-engineered T cells as oncolytic virus carriers. Mol Ther Oncolytics. 2015;2:15014. doi: 10.1038/mto.2015.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nishio N, Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology. 2015;4:e988098. doi: 10.4161/21505594.2014.988098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 127.Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 128.Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. 2017;7:39833. doi: 10.1038/srep39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- 130.Tang XJ, Sun XY, Huang KM, Zhang L, Yang ZS, Zou DD, et al. Therapeutic potential of CAR-T cell-derived exosomes: a cell-free modality for targeted cancer therapy. Oncotarget. 2015;6:44179–44190. doi: 10.18632/oncotarget.6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. doi: 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Blattman JN, Greenberg PD. Cancer immunotherapy: a treatment for the masses. Science. 2004;305:200–205. doi: 10.1126/science.1100369. [DOI] [PubMed] [Google Scholar]
- 133.Li AV, Moon JJ, Abraham W, Suh H, Elkhader J, Seidman MA, et al. Generation of effector memory T cell-based mucosal and systemic immunity with pulmonary nanoparticle vaccination. Sci Transl Med. 2013;5:204ra130. doi: 10.1126/scitranslmed.3006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cho NH, Cheong TC, Min JH, Wu JH, Lee SJ, Kim D, et al. A multifunctional core-shell nanoparticle for dendritic cell-based cancer immunotherapy. Nat Nanotechnol. 2011;6:675–682. doi: 10.1038/nnano.2011.149. [DOI] [PubMed] [Google Scholar]
- 135.Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, et al. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat Nanotechnol. 2016;11:295–303. doi: 10.1038/nnano.2015.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yuan H, Jiang W, von Roemeling CA, Qie Y, Liu X, Chen Y, et al. Multivalent bi-specific nanobioconjugate engager for targeted cancer immunotherapy. Nat Nanotechnol. 2017;12:763–769. doi: 10.1038/nnano.2017.69. [DOI] [PubMed] [Google Scholar]
- 137.Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. 2017;12:813–820. doi: 10.1038/nnano.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167:829–842. doi: 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature. 2016;531:651–655. doi: 10.1038/nature17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Scheffel MJ, Scurti G, Simms P, Garrett-Mayer E, Mehrotra S, Nishimura MI, et al. Efficacy of adoptive T-cell therapy is improved by treatment with the antioxidant N-acetyl Cysteine, which limits activation-induced T-cell death. Cancer Res. 2016;76:6006–6016. doi: 10.1158/0008-5472.CAN-16-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The material supporting the conclusion of this review has been included within the article.