

Abstract
Background
Facioscapulohumeral muscular dystrophy (FSHD) is associated with DNA hypomethylation at the 4q35 D4Z4 repeat array. Both the causal gene DUX4 and its homolog DUX4c are induced. DUX4c is immunodetected in every myonucleus of proliferative cells, while DUX4 is present in only 1/1000 of myonuclei where it initiates a gene deregulation cascade. FSHD primary myoblasts differentiate into either atrophic or disorganized myotubes. DUX4 expression induces atrophic myotubes and associated FSHD markers. Although DUX4 silencing normalizes the FSHD atrophic myotube phenotype, this is not the case for the disorganized phenotype. DUX4c overexpression increases the proliferation rate of human TE671 rhabdomyosarcoma cells and inhibits their differentiation, suggesting a normal role during muscle differentiation.
Methods
By gain- and loss-of-function experiments in primary human muscle cells, we studied the DUX4c impact on proliferation, differentiation, myotube morphology, and FSHD markers.
Results
In primary myoblasts, DUX4c overexpression increased the staining intensity of KI67 (a proliferation marker) in adjacent cells and delayed differentiation. In differentiating cells, DUX4c overexpression led to the expression of some FSHD markers including β-catenin and to the formation of disorganized myotubes presenting large clusters of nuclei and cytoskeletal defects. These were more severe when DUX4c was expressed before the cytoskeleton reorganized and myofibrils assembled. In addition, endogenous DUX4c was detected at a higher level in FSHD myotubes presenting abnormal clusters of nuclei and cytoskeletal disorganization. We found that the disorganized FSHD myotube phenotype could be rescued by silencing of DUX4c, not DUX4.
Conclusion
Excess DUX4c could disturb cytoskeletal organization and nuclear distribution in FSHD myotubes. We suggest that DUX4c up-regulation could contribute to DUX4 toxicity in the muscle fibers by favoring the clustering of myonuclei and therefore facilitating DUX4 diffusion among them. Defining DUX4c functions in the healthy skeletal muscle should help to design new targeted FSHD therapy by DUX4 or DUX4c inhibition without suppressing DUX4c normal function.
Electronic supplementary material
The online version of this article (10.1186/s13395-017-0148-4) contains supplementary material, which is available to authorized users.
Keywords: Disorganized myotubes, Myonuclear clustering, Cytoskeleton, β-catenin, Proliferation, Differentiation
Background
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common neuromuscular disorders [1]. It is characterized by muscle weakness and atrophy of the facial muscles, progressing to the upper arms, shoulder girdle, and lower limbs. FSHD is linked to the D4Z4 repeat array on 4q35 and requires both genetic and epigenetic conditions allowing the expression of DUX4 (double homeobox 4), a gene our group has identified in each D4Z4 repeat unit [2–4] (reviewed by [5]). The genetic condition is characterized by a SNP producing a polyadenylation signal in the pLAM region 3′ of the distal D4Z4 unit, leading to the expression of a stable mRNA and toxic DUX4 protein [4, 6]. The epigenetic condition is D4Z4 DNA hypomethylation, allowing DUX4 gene expression (reviewed by [7, 8]). This results either from contraction of the D4Z4 repeat array, as observed in the major FSHD1 form (OMIM#158900), or loss-of-function mutations of SMCHD1 or DNMT3B, involved in the much rarer FSHD2 form (OMIM#614982), which is clinically undistinguishable from FSHD1 (reviewed by [9]). The level of D4Z4 DNA hypomethylation generally correlates with disease variability and severity [10], as observed in severely affected individuals with inherited combined FSHD1 and 2 conditions [11, 12]. DUX4 encodes a 52-kDa transcription factor that is normally expressed in germline cells and is activated in a small proportion of FSHD muscle nuclei, from which it diffuses and initiates a transcription deregulation cascade leading to muscle atrophy, muscle differentiation defects, and oxidative stress, which are key features of FSHD [3, 4, 13–21]. The occurrence of modulators of DUX4 toxicity is suggested by the observation of two phenotypes in FSHD primary cell cultures, where myotubes are either very thin and atrophic (aFSHD) or large and disorganized with clusters of nuclei (dFSHD). The two phenotypes are found in different proportions in different myoblast primary “lines” [16, 22]. In healthy primary myotubes, DUX4 is expressed at a very low level in < 1% myonuclei, in contrast to its expression in approximately 8% (dFSHD) to 10% (aFSHD) of myonuclei in FSHD cultures. Moreover, DUX4 is induced approximately threefold in aFSHD compared to the healthy muscle cells [16, 23]. Surprisingly, DUX4 inhibition prevents the formation of atrophic but not disorganized FSHD myotubes [24]. When embryonic stem cells (hESC) with the FSHD1 genetic defect were differentiated to skeletal muscle cells, those which expressed DUX4 exhibited defects reported in FSHD cells and formed thinner myotubes, similar to aFSHD ones [25].
We have also characterized a gene, mapped 42-kb centromeric of the proximal D4Z4 element, in a truncated and inverted D4Z4 unit that encodes a DUX4 homologous protein, which we have designated DUX4c [26, 27]. DUX4c is a transcription factor that is identical to DUX4, except that it is 50 residues shorter, and its last 32 residues share 40% identity with DUX4. DUX4c is detected in primary healthy myoblasts and is induced upon their differentiation and is up-regulated together with DUX4 in FSHD muscle cells and biopsies [27, 28]. The muscles of patients with Duchenne muscular dystrophy (DMD) exhibit a high regeneration rate and increased DUX4c levels, with strong DUX4c immunolabeling being observed in some desmin-positive regenerating fibers [27, 28]. DUX4c overexpression increases the proliferation rate of human TE671 myoblasts and inhibits the differentiation of both human TE671 and mouse C2C12 myoblasts [27, 29]. This inhibition is rescued by overexpression of the myogenic factors MyoD and Myf5 in C2C12 myoblasts [29]. DUX4c induces (and interacts with) the MYF5 protein in mouse and human myoblasts [27]. DUX4c overexpression also induces myogenic microRNAs, and DUX4c knockdown causes stronger inhibition of these miRNAs in FSHD compared with healthy myotubes [30]. All of these data support a role for DUX4c during the proliferation and differentiation of healthy muscle cells and suggest that its abnormal induction could contribute to FSHD muscle pathology. A recent transcriptomic study supports the role of DUX4c in muscle development and disease [31].
In the present study, we investigated the role of DUX4c in human myotube phenotypes. Its overexpression in myotubes caused differentiation defects characterized by an abnormal troponin T distribution and large clusters of nuclei, leading to a disorganized phenotype. In contrast, DUX4c inhibition in FSHD myoblasts prevented the formation of disorganized myotubes. These gain- and loss-of-function experiments support a possible contribution of DUX4c to the FSHD pathophysiology.
Methods
Myogenic cell culture, transfections, and induction
Primary human myoblasts from an unaffected control and a patient with FSHD were isolated from muscle biopsies, purified, and established as described previously [22]. The myoblasts were grown in 35-mm collagen-coated dishes (Ywaki, Japan) in DMEM with 4.5 g/l glucose and L-glutamine (Lonza) as well as gentamicin (50 μg/ml, Sigma-Aldrich), 10% fetal bovine serum (FBS) (Invitrogen), and 1% Ultroser G (Pall BioSepra, Cergy-St-Christophe, France) at 37 °C under 5% CO2. Confluent myoblast cultures were differentiated by switching the medium to DMEM/gentamicin (50 μg/ml) with 2% FBS. Myoblasts were transfected in their culture medium 24 h after seeding, with Fugene HD (Roche Diagnostics) and DNA at a ratio of 6:2 (μl of Fugene HD:μg of DNA), according to the manufacturer’s instructions [32].
The vectors were pCIneo (Promega), pCIneo-DUX4c, pAC1M2-DUX4c, p3-kb-DUX4c, and p7.5-kb-DUX4c [27]. Doxycycline induction was performed as previously described [27].
Transfection with siRNA
Three siRNAs were designed against the DUX4c 3′UTR sequence (custom siRNA design, Ambion). Among these, siRNA1 (5′-ccagagtttcagcaaaagg-3′) was previously described for its efficiency to inhibit DUX4c overexpression and its specificity as it did not interfere with DUX4 expression [27]. For cell transfection, we used the “Silencer siRNA Starter Kit” (Ambion) with the “SiPORT NeoFX” transfection agent, as described in [32]. We used 4 μl of siPORTNeoFX and 10 nM siRNA for primary human myoblasts. All transfections were performed in duplicate wells and were repeated three times to ensure consistency.
RT-qPCR and 3′RACE
Primary myoblasts were harvested during proliferation or 1 day after the induction of differentiation. Total RNA was extracted and retro-transcribed, and quantitative PCR was performed using a specific γ-Catenin TaqMan assay (Hs00158408_m1, ThermoFisher Scientific). The quantity of γ-Catenin mRNA is provided relative to its expression level in healthy cells, which was set to 1. The means and standard errors are indicated. The 3′RACE experiments were performed as previously described [27]. The PCR products were cloned and sequenced.
Immunofluorescence
Human primary myoblasts were fixed in PBS containing 4% paraformaldehyde (Sigma-Aldrich) and treated with PBS/0.5% Triton X-100. After blocking in PBS/20% FBS, the cells were incubated with primary antibodies for 2 h at room temperature. The following antibodies and dilutions were used: mouse monoclonal (mAb) anti-troponin T 1/100 (clone JLT-12, Sigma-Aldrich), rabbit polyclonal anti-β-catenin 1/200 (ECM Biosciences), anti-KI67 1/200 (mAb, Abcam), anti-α-Tubulin mAb 1/500 (Abcam), rabbit polyclonal anti-αB-crystallin 1/50 (Enzo Life Sciences), mAb 9A12 (which we developed against DUX4) 1/50 ([4]; clone 9A12, Merck Millipore), and rabbit antiserum directed against a DUX4c peptide 1/150 [27]. After washing and blocking, cells were incubated for 1 h at room temperature with Alexa Fluor secondary antibodies at 1/100 (goat anti-mouse 488 and anti-rabbit 555, Invitrogen) then washed again and mounted with Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, Biosciences).
Images were acquired with either a Nikon Eclipse i20 microscope equipped with filters allowing the detection of proteins showing low expression and with a DS-U3 DS camera control unit at room temperature. Plan Fluor 20 X, Plan Fluor 409, and 609 Apo-VC high-resolution oil immersion objectives were used, with 350, 480, and 540 nm excitation for the 4,6-diamidino-2-phenylindole (DAPI), fluorescein isothiocyanate (FITC), and tetramethylrhodamine isothiocyanate (TRITC) channels, respectively. The Nikon Instrument Software NIS-elements Basic Research analysis software was used both for image acquisition and for measure of KI67 staining intensity per nucleus. ImageJ was used for image merging.
Immunodetection through Western blotting
Cells were lysed in hypertonic buffer containing 50 mM Tris, pH 7, 50 mM NaCl, 0.1% NP40, a protease inhibitor cocktail (Roche Diagnostics), and 1 mM DTT. Each cell lysate was separated via electrophoresis in a 10 or 12% polyacrylamide gel in the presence of SDS, followed by transfer to a nitrocellulose membrane (GE Healthcare Europe GmbH, Diegem, Belgium). Protein transfer was confirmed through Ponceau red staining. The blot was blocked with 5% non-fat dry milk diluted in PBS for 1 h at room temperature. The membranes were then incubated at 4 °C overnight with primary antibodies in PBS/2% BSA. The following antibodies and dilutions were used: mAb 9A12 1/1000, rabbit DUX4c antiserum 1/1000, anti-troponin T mAb 1/1000 (clone JLT-12, Sigma-Aldrich), rabbit polyclonal anti-β-catenin 1/1000 (ECM Biosciences, KY, USA), anti-JUP 1/1000 (ECM Biosciences, KY, USA), rabbit polyclonal anti-MYF5 1/500 (Santa Cruz Biotechnologies), rabbit polyclonal anti-Atrogin-1 1/1000 (or anti-Atrogin-1/MAFbx, ECM Biosciences), rabbit polyclonal anti-MuRF1 1/1000 (ECM Biosciences), anti-CRYM mAb 1/1000 (or anti-mu-crystallin, Abnova, Heidelberg, Germany), and anti-TP53 mAb 1/1000 (Abcam, Cambridge, UK). The membranes were subsequently washed in PBS and incubated with secondary antibodies coupled to horseradish peroxidase (HRP) 1/10000 (GE Healthcare) for 1 h at room temperature. Proteins were detected on Amersham Hyperfilm ECL (GE Healthcare) with the LiteAbLot (Euroclone, Victoria, Australia), Lumilight (Roche Diagnostics), or Super signal West Femto maximum sensitivity substrate kit (Thermo Scientific). For standardization, the membranes were stripped (200 nM glycine, 0.5 M NaCl, 0.1% SDS, pH 2.2), and immunostaining was performed with rabbit polyclonal anti-actin serum 1/1000 (Sigma-Aldrich), followed by HRP-coupled secondary antibodies 1/10,000 (GE Healthcare). Densitometry of the immunoreactive bands was performed with LabImage 1D Software (Kapelan Bio-Imaging) or the Thermo Scientific MyImageAnalysis Software v1.1. The data are normalized to control loading levels in each sample.
Statistical analyses
Statistical significance was evaluated using Student’s t test, except for KI67 immunofluorescence in gain-of-function experiment for which the Mann-Whitney test was used.
Results
DUX4c overexpression in myoblasts induces the disorganized myotube phenotype
DUX4c overexpression inhibits the differentiation of C2C12 and TE671 myoblasts into myotubes [27, 29]. We transfected primary human myoblasts with a DUX4c expression vector (pCIneo-DUX4c) and switched the cells to differentiation medium. Three days later, the cell morphology differed between the healthy myocytes (transfected with the empty pCIneo vector), which were elongated and aligned to fuse, and the DUX4c-overexpressing cells, which remained rounder and had not aligned (data not shown), as we previously observed when DUX4c was overexpressed in TE671 cells [27]. After 6 days of differentiation, healthy myotubes exhibited normally aligned nuclei, whereas DUX4c-positive myonuclei formed clusters in enlarged and deformed myotubes (Additional file 1: Figure S1A). A similar phenotype was observed in FSHD myotubes, showing cytoplasmic staining of endogenous DUX4c with patches of troponin T accumulation (Additional file 1: Figure S1B). We previously observed similar endogenous DUX4c staining in dFSHD myotubes and FSHD muscle sections in association with the clustering of myonuclei [24, 28].
DUX4c overexpression induces troponin T delocalization
We transfected primary human myoblasts as above but switched the culture to differentiation medium at an earlier time point (5 h instead of 24 h). Five days later, DUX4c-overexpressing myotubes exhibited clusters of nuclei and local dense troponin T staining, instead of striations across the entire cytoplasm as observed in healthy myotubes (Fig. 1a). However, global troponin T protein levels were not affected by DUX4c overexpression, as shown by immunodetection with a specific antibody in Western blots prepared with total cell extracts (Fig. 1b).
Fig. 1.
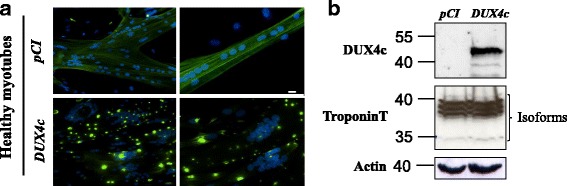
DUX4c overexpression induces misdistribution of troponin T and abnormal distribution of myonuclei. Healthy human primary myotubes were transfected with pCIneo-DUX4c or the empty pCIneo expression vector and switched to differentiation medium. a Five days later, troponin T was detected by immunofluorescence (green), and nuclei were labeled with DAPI (blue). Scale bar: 20 μM. b A 20 μg sample of total protein extracted from the same cells was separated via electrophoresis (PAGE-SDS), then transferred to a Western blot, immunodetected with the indicated primary antibodies and appropriate secondary antibodies coupled to HRP, and revealed through chemiluminescence
DUX4c overexpression in myotubes induces a disorganized phenotype
In the previous experiments, we expressed DUX4c in myoblasts before or at the beginning of differentiation. To evaluate the impact of DUX4c expression on already-formed myotubes, we transfected primary myoblasts with the doxycycline-inducible pACIM2-DUX4c expression vector [27] and switched the cells to differentiation medium. Three days later, we induced DUX4c expression with different doses of doxycycline, fixed the cells after an additional 5 days, and immunodetected DUX4c and troponin T. Without doxycycline, basal nuclear DUX4c staining was observed in cells with clusters of two to five nuclei, suggesting recent fusion or myoblasts about to fuse (Fig. 2, top panels), as previously observed [28]. With increasing amounts of doxycycline, the myotubes appeared progressively larger and more deformed. They contained clusters of DAPI-stained nuclei more frequently and included more nuclei with increasing DUX4c expression. Cytoplasmic DUX4c partially co-localizing with troponin T was also detected on one side of the myotubes (Fig. 2 bottom panels and Additional file 2: Figure S2, asterisks) or at one of their tips (Additional file 2: Figure S2 circle), as previously observed [28]. In some myotubes, clusters of DUX4c-positive nuclei formed rings; cytoplasmic troponin T immunofluorescence was observed within these structures and, more diffusely, around them (Fig. 2, Additional file 2: Figure S2). We did not observe intense spots of troponin T as in Fig. 1, but its organization was disturbed next to the large or ring-like clusters of nuclei (Fig. 2, Additional file 2: Figure S2).
Fig. 2.
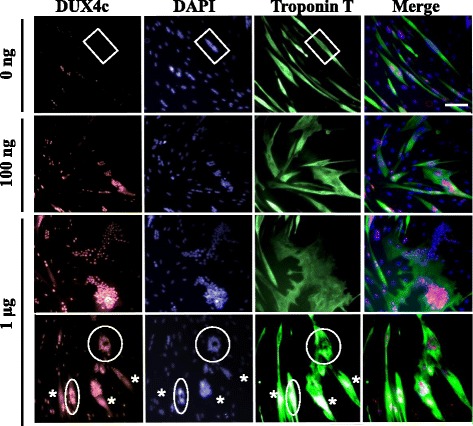
DUX4c overexpression induces the formation of disorganized myotubes with large nuclear clusters. Healthy primary myoblasts were transfected with pAC1M2-DUX4c and differentiated 48 h later. After the first myotubes formed (day 3), DUX4c expression was induced by the addition of 100 ng or 1 μg of doxycycline to the culture medium. At day 8, myotubes were fixed, and DUX4c (red) and troponin T (green) were immunodetected. Nuclei were labeled with DAPI. A cluster of nuclei (> 5 nuclei) without endogenous DUX4c labeling is boxed (0 ng Dox). Clusters of nuclei presenting a ring-like structure are circled. Stars indicate cytoplasmic DUX4c partially co-localizing with troponin T. Scale bar: 30 μm
β-catenin accumulates in disorganized FSHD myotubes and upon DUX4c overexpression
As abnormal β-catenin accumulation causes oversized and disorganized myotubes with clusters of nuclei [33], we examined the amount of β-catenin in a Western blot prepared with total extracts from myotubes in which DUX4c had been induced (a parallel culture is shown in Fig. 2). An increase in β-catenin was immunodetected (2.3-fold under the highest dose of doxycycline) (Additional file 3: Figure S3).
We then examined the intracellular location of β-catenin through immunofluorescence. In healthy primary human myotubes, the diffuse β-catenin staining observed excluded the nuclei (Fig. 3a top panels). In healthy myotubes overexpressing DUX4c and in dFSHD myotubes, β-catenin especially accumulated in the vicinity of nuclear clusters (Fig. 3a middle and bottom panels), and myotube morphology was similar in the two types of cultures (Fig. 3b). An average of four and five clusters of nuclei were counted per field in dFSHD and in DUX4c-overexpressing myotubes, respectively, while only two clusters were detected in healthy myotubes. In addition, approximately 50 and 25% of these clusters contained more than 16 nuclei in DUX4c-overexpressing and dFSHD myotubes, respectively. Healthy myotubes seldom presented such large clusters (Figs. 3c and 4).
Fig. 3.
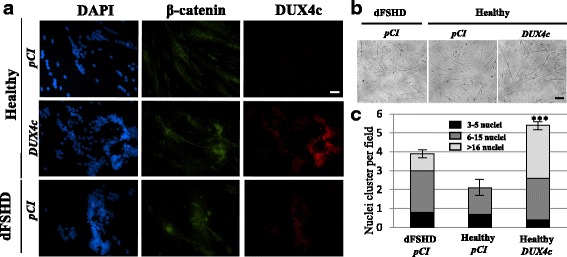
DUX4c overexpression induces abnormal clusters of myonuclei and misdistribution of β-catenin. a FSHD and healthy primary myoblasts were transfected with the empty pCIneo or pCIneo-DUX4c expression vector, and differentiation was induced 24 h later. Immunofluorescence detection with antibodies against β-catenin (green) or DUX4c (red) was performed at day 3. Nuclei were labeled with DAPI. Scale bar: 20 μm. b Representative field from each culture (white light). Scale bar: 50 μm. c The nuclei per cluster were counted in each culture (10 fields per culture) and are presented as the mean ± SD. The proportion of nuclei present per cluster is shown in grayscale. Nuclear clusters with more than 16 nuclei were seldom found in healthy myotubes; the mean is therefore close to zero. ***p < 0.001
Fig. 4.
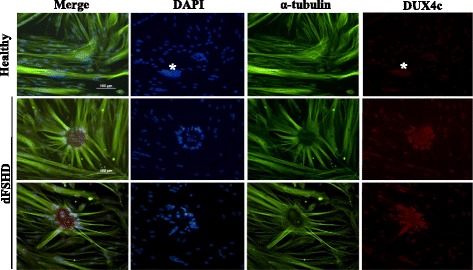
dFSHD myotubes present abnormal clusters of nuclei overexpressing DUX4c as well as cytoplasmic DUX4c and α-tubulin disorganization. Healthy and FSHD primary myoblasts were differentiated, then fixed 6 days later, and DUX4c (red) and α-tubulin (green) were detected by immunofluorescence. A large cluster of myonuclei (> 16, indicated by *) can be observed, while such clusters were very seldom found in healthy cultures. Scale bars: 100 μm
High levels of nuclear and cytoplasmic DUX4c are associated with clusters of nuclei in disorganized FSHD myotubes
After 6 days in differentiation medium, nuclei were observed in ring-shaped clusters in dFSHD myotubes, but not in a parallel healthy culture (Fig. 4). Similar ring-forming nuclei were detected in DUX4c-overexpressing myotubes after 8 days of differentiation (Fig. 2, Additional file 2: Figure S2). In dFSHD myotubes, such structures were associated with strong DUX4c immunofluorescence in nuclei surrounding a large area of cytoplasmic DUX4c staining. Several DAPI-labeled spots that were apparently smaller than nuclei were present within these nuclear rings. Intriguingly, filaments of α-tubulin seemed to converge toward these nuclear ring clusters, forming flower-like structures. As shown above, in DUX4c-overexpressing myotubes with delocalized troponin T (Figs. 1 and 2, Additional file 2: Figure S2), α-tubulin appeared disorganized (Additional file 4: Figure S4A top) or almost absent (Additional file 4: Figure S4A bottom, boxed) in the vicinity of nuclear clusters presenting strong DUX4c immunofluorescence. The nuclei in these clusters were generally smaller than the other nuclei in the same culture (Fig. 4, Additional file 4: Figure S4A). The dFSHD myotubes with delocalized α-tubulin also presented α-crystallin B chain immunofluorescence in the cytoplasm at a much higher intensity than healthy myotubes (Additional file 4: Figure S4B).
DUX4c expression induces FSHD markers
As DUX4c shares a high sequence identity with DUX4 (including the DNA binding homeodomain region), we investigated whether DUX4c could induce the expression of genes we have previously shown to be induced in FSHD muscle cells or through DUX4 expression [32]. We transfected myoblasts with DUX expression vectors and performed immunodetection on Western blots 48 h later. DUX4c overexpression induced CRYM as did DUX4, but not P53 (Additional file 5: Figure S5A). In the myotubes obtained after 8-day differentiation, we then immunodetected the E3 ubiquitin ligases MuRF1 and Atrogin-1 (also known as TRIM63 or F-box only protein 32, respectively) associated with muscle atrophy. DUX4c overexpression had not affected the level of Atrogin-1 (in contrast to DUX4) (Additional file 5: Figure S5B) but delocalized MuRF1 in the nucleus as observed following DUX4 overexpression or in FSHD myotubes (Additional file 5: Figure S5C and [32]). This is in contrast to the global MuRF1 weak staining detected in healthy myotubes (Additional file 5: Figure S5C and [32]).
Similar to the above results in myotubes (Fig. 3a), FSHD primary myoblasts presented higher levels of β-catenin, which might be attributed to DUX4 or DUX4c overexpression (Additional file 5: Figure S5B). We also observed a significant increase in mRNA expression of the β-catenin homolog γ-catenin (also known as JUP/Junction Plakoglobin). Indeed primary (aFSHD and dFSHD) as well as immortalized FSHD myoblasts presented higher JUP mRNA levels in proliferation and 1 day after the induction of differentiation as compared to healthy cells (Additional file 5: Figure S5D and data not shown). There was no regulation of JUP mRNA between proliferation and early differentiation in either cell line. At the protein level, a JUP induction in the FSHD sample or following DUX4 or DUX4c expression was less clear (stars in Additional file 5: Figure S5B), even though the gene encoding JUP has been described as a direct DUX4 target [14], and its mRNA level rapidly increases upon DUX4 induction in C2C12 cells [13].
DUX4c gain and loss of function affect myoblast proliferation.
We previously showed that DUX4c overexpression induces the proliferation of TE671 muscle cells and inhibits their differentiation [27]. In a similar experiment with healthy primary myoblasts, we detected induction of the KI67 proliferation marker (Fig. 5a). Intriguingly, the cells exhibiting the strongest nucleoplasmic KI67 labeling did not correspond to DUX4c-overexpressing cells (Fig. 5a). The number of KI67-positive nuclei did not differ between muscle cells transfected with a backbone or a DUX4c-expression vector. Nevertheless, the mean intensity of nucleoplasmic KI67 staining per nucleus was 2.5-fold higher following DUX4c overexpression (p < 0.01) (Fig. 5b). We also observed that a few DUX4c-overexpressing cells presented KI67 staining, mostly in nuclear spots that could be nucleoli (Fig. 5c).
Figure 5.
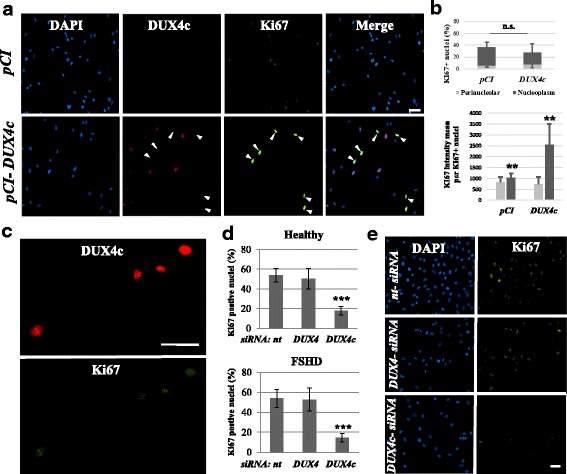
DUX4c gain and loss of function affect cell proliferation. a Representative fields of healthy myoblasts transfected with the indicated pCIneo expression vectors, fixed 24 h later, and processed for immunofluorescence detection of DUX4c (red) and KI67 (green). Arrows point to KI67-positive cells. b The percentage of perinucleolar and nucleoplasmic KI67-positive nuclei on total nuclei was counted in myoblasts showed in a. and represented as the mean ± SD. Either 10 or 15 fields were counted for myoblasts transfected either with pCIneo or pCIneo-DUX4c, respectively (total number of nuclei: 685 and 433, respectively). In addition, the perinucleolar and nucleoplasmic KI67 intensity were measured in each field. The total perinucleolar or nucleoplasmic KI67 intensity (all fields) was divided by the total number of KI67-positive nuclei (either perinucleolar or nucleoplasmic, respectively), and represented as the mean ± SD. The number of nucleoplasmic KI67-positive nuclei was 35 in the healthy culture and 31 in the FSHD culture. For the significance, a Mann-Whitney test was applied. **p < 0.01 (c). Magnification of DUX4c-expressing cells with partial co-localization of nucleolar KI67. d The number of KI67-positive nuclei was counted in 10 fields of healthy and FSHD myoblasts. The percentage of KI67-positive nuclei is represented as the mean ± SD. ***p < 0.001 was considered significant. e Representative fields of FSHD myoblasts transfected with the indicated siRNAs, then fixed 24 h later, and processed for the immunofluorescence detection of KI67. Nuclei were stained with DAPI. Scale bar: 30 μm
Three siRNAs targeting the DUX4c 3′UTR (region of lowest similarity with DUX4 sequence) were synthesized based on the DUX4c mRNA ends found either in C2C12 cells transfected with a 7.5-kb genomic vector including the DUX4c gene and natural promoter [27] or in FSHD primary myoblasts (Additional file 6: Figure S6A, B). However, DUX4c could only be knocked down using siRNA1 (Additional file 6: Figure S6C–E and [27]). We then transfected healthy and FSHD primary myoblasts with siRNAs specific to either DUX4c [27] or DUX4 [32] mRNA, or with non-targeted (nt) siRNA (Additional file 7: Figure S7). Twenty-four hours later, we evaluated myoblast proliferation by counting the number of mitotic events in the culture dishes. In the presence of the DUX4 or nt siRNA, both healthy and FSHD myoblasts exhibited five to six mitotic events per field. In contrast, myoblasts treated with the DUX4c siRNA exhibited an average of only one mitotic event per field (Additional file 8: Figure S8). In accord with a reduction of cell proliferation, a significant decrease in KI67-positive nuclei (staining observed during the late G1, S, G2, and M phases) was observed in myoblasts treated with the DUX4c siRNA compared with the DUX4 or nt siRNA (Fig. 5d, e).
DUX4c silencing suppresses the FSHD disorganized myotube phenotype
We transfected dFSHD myoblasts with the DUX4c, DUX4, or nt siRNA and switched the culture to differentiation medium. Eight days later, all of the cultures exhibited myotubes. Large deformed myotubes highlighted by troponin T staining were observed in the presence of the DUX4 or nt siRNA, which showed large clusters of myonuclei. In contrast, treatment with the DUX4c siRNA resulted in a return to a “normal” morphology, with thinner myotubes and a greater number of aligned myonuclei (Fig. 6a, b). The numbers of nuclei per cluster and clusters per field were counted in each of the three cultures: an average of four and five clusters per field were visible in the cultures of disorganized FSHD myotubes treated with the nt siRNA or the DUX4 siRNA, respectively, while an average of two clusters were counted in myotubes treated with the DUX4c siRNA (Fig. 6c).
Fig. 6.
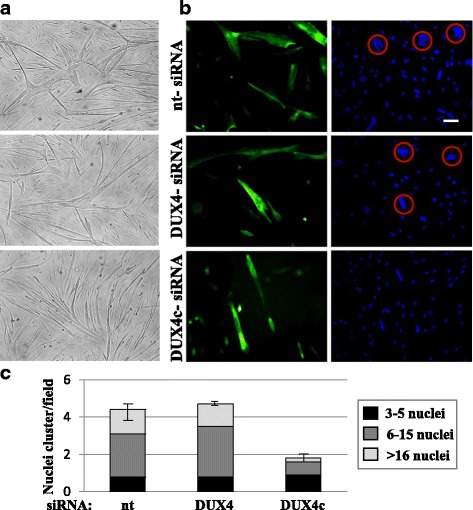
RNA interference against DUX4c suppresses the disorganized myotube phenotype. dFSHD primary myoblasts were transfected with the indicated siRNAs and differentiated. a At day 8, microscope pictures were obtained under white light. b The cells were then fixed, and troponin T (green) was detected by immunofluorescence. Nuclei were stained with DAPI. Scale bar: 30 μm. c The average (and SD) numbers of nuclei per cluster and clusters per field were counted (10 fields per culture). The proportion of nuclei per cluster is shown in grayscale. ***p < 0.001 was considered significant
Discussion
Overexpression of DUX4, the FSHD causal gene, was previously linked to muscle atrophy [32] or apoptosis [3, 18, 34–36].
Histological data on FSHD muscle sections are scarse, and these descriptions (from either highly affected or not yet/poorly affected muscles) do not cover the large known variability of affected muscles and of patient phenotypes. However, besides the expected DUX4-related atrophic fibers, some publications mention hypertrophic fibers in FSHD muscles (http://neuromuscular.wustl.edu/pathol/fsh.htm; [37]). It was speculated that these were compensatory fibers, but this was never demonstrated [37]. In addition, no pathological mechanism has been suggested to account for other histological observations made in the muscles of patients such as large numbers of splitting and branching myofibrillar bundles, myofibrillar loss and sarcomeric dysfunction, or myonuclear mispositioning, such as clustering of nuclei [20, 28, 38, 39]. Based on the present study, we suggest that these anomalies could be associated with the activation of DUX4c expression, in addition to DUX4 (see also [28]).
In the present study, we used primary cultures of myoblasts derived from FSHD muscle biopsies. In our culture and differentiation conditions (set up by [22]), FSHD myoblasts fuse to myotubes of either atrophic or disorganized (a or dFSHD) phenotypes that might reflect the atrophic and hypertrophic fiber types observed in FSHD muscles. We have previously demonstrated that DUX4 caused the aFSHD myotube phenotype [32], and we now show that it is DUX4c, not DUX4, overexpression that leads to a disorganized myotube phenotype. We could rescue dFSHD myotubes to a normal phenotype with one siRNA targeting the DUX4c mRNA. However, additional siRNAs should similarly be tested. Their design is quite difficult because the DUX4c gene is very GC rich and nearly identical to DUX4 in most of the coding sequence; a number of alternatively spliced DUX4c mRNAs were detected (Additional file 6: Figure S6B and Ansseau et al. in preparation), but not all of them have been characterized yet. Further studies of the DUX4c gene and transcripts will allow the design of additional silencing tools to help characterize DUX4c function in skeletal muscle.
DUX4c induces misdistribution of myonuclei and cytoskeletal-associated proteins
In previous studies, DUX4c overexpression was shown to result in complete inhibition of the differentiation of TE671 human rhabdomyosarcoma and mouse C2C12 muscle cells [27, 29]. Here, we showed that DUX4c gain of function at different times (before differentiation or after the induction of differentiation) in human healthy primary myoblasts did not affect their capacity to form myotubes even if their differentiation was delayed. However, myotubes with high DUX4c levels appeared hypertrophic and disorganized, with clusters of nuclei. This phenotype is similar to the giant myosacs resulting from the overexpression of skNAC [40, 41]. DUX4c and skNAC share a motif that is essential for interaction with Smyd1 [28]. Both skNAC and Smyd1 play key roles in myofibril assembly [41–43], and excess DUX4c might interfere with their association. In addition, DUX4c overexpression induced an abnormal distribution of troponin T with loss of its normal striation pattern, which could lead to sarcomere malformation and impact contraction, as previously observed following troponin T inhibition [44]. Myoblasts transfected with a backbone vector and maintained in culture more than 6 days after differentiation exhibit spontaneous contractions. However, in a preliminary experiment, we could not observe such contractions in myotubes derived from myoblasts transfected with a DUX4c-expression vector (data not shown). The absence of spontaneous contraction in myotubes with DUX4c overexpression could also partly result from delayed differentiation since myoblasts transfected with a DUX4c expression vector showed a differentiation delay. The impact on troponin T distribution was more severe when DUX4c was expressed before the cytoskeleton reorganized and myofibrils assembled during muscle differentiation. Notably, in FSHD muscles, altered splicing of troponin T mRNA has been observed, which is associated with contraction defects [45]. An abnormal localization of cytoskeletal or cytoskeleton-associated proteins, such as α-tubulin and troponin T, was observed in dFSHD primary myotubes that expressed high levels of endogenous DUX4c as well (Fig. 4, Additional file 4: Figure S4). These cells also exhibited high levels of α-crystallin B chain (CRYAB), a stress-associated chaperone that stabilizes desmin (a DUX4c partner, [28]) and thus maintains intermediate filament integrity; this chaperone is up-regulated in desminopathies as well [46, 47].
Nuclear migration and anchorage have been extensively studied in various cell types, and both the microtubule and actin cytoskeleton networks play major roles in these processes [48–50]. During the skeletal muscle development, thousands of myoblasts fuse together to form multinucleated myotubes, and nuclei progressively migrate to the periphery of the fiber [51–53]. Alterations in the microtubular network and some actin-binding proteins cause changes in the distribution of nuclei along the muscle fiber [50, 53–56]. A lack of desmin also significantly affects the positioning of nuclei [57]. DUX4c overexpression did not induce changes in the protein levels of troponin T but suppressed its normal distribution and striation pattern (Fig. 1). We identified desmin as a DUX4c partner and troponin C and specific actin, myosin, and tubulin isoforms as putative DUX4c partners [28]. We therefore hypothesize that excess DUX4c could interfere with cytoskeleton reorganization in differentiating myotubes, which may result in incorrect localization of myonuclei along the fiber and sarcomere malformations, leading to muscle contraction defects. In agreement with this hypothesis, a previous differential proteomic study showed that proteins displaying an increased relative abundance in dFSHD versus healthy myotubes were mostly associated with the cytoskeleton and particularly with the organization of the microtubule network and myofibril remodeling. This study also demonstrated that the three forms of troponin T exhibited similar abundance in dFSHD and healthy myotubes ([58], Additional file 9: Table S1), as observed in DUX4c-overexpressing myotubes in the present study. In keeping with the lack of a change in expression, no binding site for DUX4 (or, most likely, for DUX4c, as the two proteins share identical homeodomains) was identified in ChIP-seq experiments involving the three genes encoding troponin T isoforms (GEO accession: GSE33838) [14]. In the differential proteomic study, aFSHD myotubes were found to exhibit deregulated cytoskeletal proteins, with an increased abundance of proteins involved in actin cytoskeleton remodeling and a decrease in contractile muscle components ([58], Additional file 9: Table S1). We also observed abnormal troponin T localization in aFSHD myotubes [24], where DUX4 levels are higher [16]. As DUX4 shares common cytoskeletal partners with DUX4c, both proteins could similarly contribute to the observed disorganization [28].
In contrast to the major toxicity exerted by DUX4, DUX4c expression in Xenopus embryos was not observed to affect the developing musculature; however, no cytoskeletal analysis was carried out in this study [59]. In a single experiment, we observed actin disorganization in the contractile apparatus of Ciona intestinalis following the injection of zygotes with a plasmid allowing muscle-specific expression of green fluorescent protein (GFP) fused to DUX4c, but not to an unrelated protein (collaboration with A. Philips, CRBM, CNRS, Montpellier, Additional file 10: Figure S9).
Abnormal nuclear positioning in muscle diseases is generally only considered as a diagnostic marker; however, it could impact pathological development (reviewed in [50, 60]). We have shown here that DUX4c overexpression induces clusters of nuclei in muscle cell cultures and that dFSHD myotubes present nuclear clusters close to strong DUX4c immunostaining (nuclei and cytoplasm) (Fig. 4, Additional file 4: Figure S4; [28]). In a previous study, we observed DUX4c induction through immunodetection in Western blots in most FSHD muscle biopsies and also in DMD muscle biopsies (at lower levels) in contrast to healthy ones [27]. DUX4c overexpression in FSHD muscles could have an important impact on the development/progression of the pathology that should not be ignored. DUX4c was ruled out as a candidate for FSHD because an FSHD1 family presented a D4Z4 deletion removing one DUX4c allele [61, 62]. However, the other DUX4c allele was still present, and DUX4c expression might have occurred but was not analyzed in these patients. Alternatively, the loss of one DUX4c allele could be pathological ([27, 30], and see below). The generation of in vivo models will allow to investigate the impact of DUX4c overexpression in muscle fibers but not its knock-out as there is no DUX4c ortholog in rodents [63]. Other Dux genes are present in the mouse (f.i., Dux and Duxbl), but not in human [63], and Duxbl was shown to play roles in myogenesis [64]. Expression of these genes could bias the data obtained in DUX4c transgenic mice in comparison with human muscles. Finally, the study of transgenic models specifically expressing DUX4c in skeletal muscle would require an inducible expression since the highest impact of DUX4c expression in vitro was observed before myofibril formation and the induction of regeneration. We expect that DUX4c (located near the D4Z4 locus) could follow a similar kinetics of expression in patient muscle as DUX4, the FSHD causal gene; DUX4 was transiently induced in myoblasts during skeletal muscle regeneration in a mouse model carrying an FSHD D4Z4 genomic fragment [65]. This might be the case since DUX4c was detected in some DMD regenerating fibers [27], is regulated by KLF15, and could be regulated by the DUX4 myogenic enhancers (see below).
DUX4c induces β-catenin and other FSHD markers
In addition to nuclear clustering and cytoskeletal disorganization, the DUX4c expression also caused an increase in the content of β-catenin in human myotubes. In healthy proliferating myoblasts, β-catenin is stabilized by WNT signaling and locates to the nucleus, where it activates target genes. When differentiation is induced, β-catenin associates with the M-cadherin complex at the cell membrane. After myotubes have formed, the amount of membrane-associated β-catenin decreases greatly due to Ozz-E3 ubiquitin ligase and proteasome activities [66] as well as that of calpain 3, a calcium-dependent protease that degrades M-cadherin [33]. Interestingly, β-catenin accumulation due to Ozz-E3 inhibition leads to severe defects in myofibrillogenesis, accompanied by an increased frequency of myofibril branching and splitting [66]. Myofibril branching and splitting are also frequently observed in FSHD muscles and have been unexplained until now ([20, 38], Lancelot et al. in preparation). In LGMD2A, a defect in calpain 3 activity leads to hypertrophied myotubes with nuclear accumulation [33]. The clusters of nuclei present in dFSHD and DUX4c-overexpressing myotubes might be associated with simultaneous β-catenin stabilization (Fig. 3a). Canonical WNT/β-catenin was shown as the main coordinator of FSHD-associated signaling and is up-regulated by DUX4 overexpression in murine myoblasts [67]. In addition, DUX4c overexpression in murine myoblasts leads to the accumulation of nuclear beta-catenin [68]. Moreover, we showed that γ-catenin (also known as JUP), which was previously reported as a direct DUX4 target gene [14], was increased in FSHD at the RNA level, although we could not detect protein deregulation. Nuclear clusters have also been observed in FSHD muscle cell cultures selected for their high DUX4 level in a rich culture medium favoring hypertrophy [35]. We hypothesize that these myotubes were co-expressing DUX4c because 4q35 hypomethylation favors activation of both the DUX4 and DUX4c genes [69, 70]. In addition, the choice of culture medium can impact the expression of DUX4 [71] and likely that of DUX4c and might also influence myotube morphology. Accordingly, under our culture conditions, which were identical to those of Barro et al. [22] and differed from those of other studies in FSHD muscle cells, we observed higher levels of DUX4 in aFSHD than in dFSHD myotubes [16] and high DUX4c levels in dFSHD myotubes ([28] and this study).
In primary muscle cell cultures, we have shown that both DUX4c and DUX4 overexpression can induce mu-crystallin (CRYM), as frequently observed in FSHD muscle cells [32, 72]. P53 is up-regulated in FSHD and in DUX4-overexpressing muscle cells [32], but not in DUX4c-overexpressing myoblasts. This observation is in agreement with a comparative analysis of DUX4 and DUX4c following injection of their mRNAs in Xenopus embryos [59] and suggests that even if DUX4c (like DUX4) is able to transactivate PITX1 [4], it does not allow TP53 up-regulation [73]. Moreover, a recent study suggested that PITX1 transactivation could be due to an indirect effect of DUX4 [74]. DUX4 and DUX4c exhibit both shared and exclusive protein partners [28], and the choice of a given partner could lead to different activities.
DUX4 induces the muscle atrogenes MuRF1 and Atrogin-1, encoding two E3 ubiquitin ligases [32]. Atrogin-1 was not induced by DUX4c overexpression in myotubes after 8 days of differentiation. MuRF1 localizes at the M-line in healthy muscle fibers [75, 76]. However, it is translocated to the nuclei following DUX4 and DUX4c overexpression, as observed in untransfected FSHD muscle cells, and might therefore not induce cytoskeletal protein degradation in the cytoplasm. Nuclear MuRF1 localization has been reported in myocytes in association with a transcription modulator, suggesting a role in gene expression [75], which would be enhanced in FSHD and DUX4- or DUX4c-overexpressing cells.
Here and previous data [32] suggest that the induction of atrophy by DUX4 (but not DUX4c) might instead be Atrogin-1 dependent and would preferentially disrupt Z-line-associated proteins where Atrogin-1 is localized [77]. In addition to desmin, many Z-line proteins as well as proteasomal proteins are putative partners of DUX4/4c [28].
Taken together, these results suggest that DUX4 and DUX4c play a distinct role. In contrast to DUX4, DUX4c does not cause cell death [13, 23, 27, 29] and is implicated in progression to the FSHD disorganized myotube phenotype in our culture conditions, rather than to the atrophic phenotype caused by DUX4 expression [32]. Moreover, the mislocalization of nuclei and the cytoskeletal disorganization observed in FSHD myotubes suggest correlations with other myopathies, such as desminopathies or limb-girdle muscular dystrophy type 2A. In FSHD muscle sections, DUX4c was shown (as in FSHD primary myotubes) in and around abnormal nuclear clusters. Moreover, in FSHD muscle sections, desmin was abnormally localized around these nuclear clusters [28].
Altogether, these data suggest a role for DUX4c in association with the cytoskeleton and the distribution of nuclei during differentiation generating muscle cells with an aberrant number of nuclei in clusters. D.G. Miller’s group [18, 35] suggested that nuclear clustering enhances sensitivity to cell death mediated by DUX4 due to its diffusion in more nuclei and the subsequent amplification of its transcriptional activity. The DUX4 expression is necessary to develop FSHD; however, the very low DUX4 levels detected in healthy muscle cells suggest it is not sufficient [16, 23]. We speculate that DUX4c could be one factor favoring the formation of large nuclear clusters and therefore facilitating the toxic action of DUX4.
On the other hand, DUX4c expression increases during healthy and FSHD myogenic differentiation [27] (as DUX4 in differentiating FSHD myoblasts [78]). During healthy myogenic differentiation, the up-regulation of KLF15 (shown as overexpressed in FSHD muscle cells and biopsies) induces DUX4c, following its binding to and activation of the D4Z4 enhancer that comes in direct contact with the DUX4c promoter in healthy and FSHD myoblasts [69, 70]. KLF15 has been proposed to link myogenic factors (such as MYOD) with D4Z4 enhancer activity, and their association contributes to the activation of both DUX4c and nearby FRG2 genes during myogenic differentiation and in FSHD muscle cells [70]. In addition, the DUX4 myogenic enhancers DME1 and DME2 might also regulate DUX4c expression. Indeed, it was shown that DME1 and DME2 enhancers physically interact with the DUX4 and FRG2 genes in both healthy and FSHD muscle cells [79]. However, an analysis of the specific primer used to amplify the DUX4 gene in these 3C chromatin capture experiments indicates that it might also amplify the DUX4c gene (Additional file 11: Figure S10). These data reinforce a putative role for DUX4c during myogenic differentiation that might be linked to altered myofibrillogenesis in FSHD primary (atrophic [24] and disorganized) myotubes. We previously reported that DUX4c was expressed (as MYF5) in almost all myoblasts [27, 28] and up-regulated about 1.5- to 2-fold during their differentiation. We also showed that DUX4c localized in healthy myotube cytoplasm around myoblast fusion time. It was also the time when stronger DUX4c staining was observed in and around large clusters of nuclei in FSHD myotubes [28]. We speculate that DUX4c up-regulation and correct intracellular location in healthy myotubes could help myogenesis as suggested by the nature of the DUX4c-induced genes [31]. However further increased DUX4c levels and an incorrect location would perturb myofibrillogenesis as observed in FSHD myotubes. We also previously demonstrated interactions between the DUX4c and MYF5 proteins and suggested DUX4c stabilized MYF5 protein but did not induce its mRNA [27]. If these protein interactions were important during myogenesis, increased MYF5 might rescue the myogenesis defects caused by excess DUX4c as previously reported in mouse myoblast cells [29]. However, if abnormal DUX4c up-regulation occurred after differentiation, MYF5 not being expressed anymore could not counteract DUX4c activity.
DUX4c gain or loss of function interferes with proliferation
During muscle regeneration, canonical Wnt/β-catenin signaling induces satellite cell proliferation [80], and this pathway is also activated by DUX4c overexpression in myoblasts (see above). We confirmed that DUX4c overexpression induces MYF5 in human primary myoblasts, as previously shown in TE671 and C2C12 cells [27], in agreement with MYF5 induction via the canonical WNT pathway [81, 82]. We previously detected Cyclin A/DUX4c co-immunostaining (S-G2 phases) in DUX4c-overexpressing human TE671 cells as well as induction of PCNA [27]. These cancer cells harbor an activating mutation of N-RAS [83], in contrast to primary myoblasts. In the latter, we only observed a few cells presenting DUX4c staining concomitant with a particular KI67 staining (corresponding either to late G1 or G2 phase, [84]). Other cells with nucleoplasmic KI67 (S or M phases, [84]) did generally not present DUX4c labeling, which suggests incompatibility of DUX4c expression with complete cell cycle progression in primary cells. Alternatively, DUX4c overexpression in transfected cells could induce proliferation of untransfected cells in the vicinity through paracrine regulation. However, our results need to be confirmed in other primary cells.
A lack of Myf5 reduces the myoblast proliferation rate [85, 86]. We have now shown DUX4c inhibition induced myoblast proliferation defects associated with a decrease in KI67 abundance and mitotic events. These results suggest that DUX4c inhibition might affect the proliferation of satellite cells or myoblasts, which are essential for muscle regeneration [87].
A transcriptional study in murine muscle cells showed no effect of DUX4c on proliferative genes in contrast to DUX4 which down-regulated them [31]. However, this could be related to variability among species as shown for DUX4 [31, 88]. Indeed, we previously observed induction of human TE671 but not mouse C2C12 cell proliferation following DUX4c overexpression [27, 29]. Alternatively, DUX4c impact on human myoblast proliferation could be independent of its transcriptional activity, such as direct protein or RNA interactions [28], as previously shown with MYF5 [27], or a paracrine effect resulting from the activation of specific signaling pathways (i.e., the canonical WNT pathway) or both effects.
Taken together, these data indicate that DUX4c could induce proliferation in a WNT-MYF5-dependent manner, which may be prevented by DUX4c inhibition. Therefore, DUX4c might contribute to the FSHD phenotype by interfering with myoblast proliferation in one of two ways: (i) in most patients, DUX4c induction in myoblasts could perturb cell cycle progression or promote it in adjacent cells or (ii) in the few families in which the FSHD deletion has extended to loss of the DUX4c gene, there might not be sufficient protein to activate myoblast proliferation.
Conclusions
In previous studies, we developed antisense oligomers (AOs) and siRNAs targeting DUX4 mRNA to suppress DUX4 protein expression as a therapeutic approach for FSHD, with a focus on the atrophic myotube phenotype observed in primary cultures [32], and found that DUX4 mRNA silencing did not correct the disorganized FSHD myotube phenotype [24]. Our gain- and loss-of-function approaches showed that this phenotype was caused by DUX4c expression and suggested that therapeutic strategies for FSHD should take DUX4c into account, in addition to DUX4. Nevertheless, complete DUX4c suppression should not be the goal because a small amount of this protein appears to be required to sustain the myoblast proliferation necessary for muscle regeneration. Other research groups are developing various strategies with the aim of reducing DUX4 expression or toxicity [89–92]. We hypothesize that a precise DUX4/DUX4c ratio might be necessary for therapeutic applications to avoid an imbalance between the atrophic and disorganized myotube morphology. Further studies may better define the interplay between the very similar DUX4/4c proteins and, more precisely, their putative competition for interactions with common partners involved in transcription [14, 93, 94], mRNA processing/regulation/translation [28, 95], or cytoskeletal and myofibril organization [28]. The D4Z4 DNA hypomethylation associated with FSHD causes activation of not only DUX4 but also DUX4c [69, 70]. In contrast to DUX4, which is only immunodetected in 1/1000 to 1/200 of FSHD myonuclei [16, 96], DUX4c is observed in almost all FSHD myoblasts [28], and the present study suggests that DUX4c could initiate muscle fiber disorganization, favor formation of large clusters of nuclei, and disturb regeneration, thereby providing a “weakened” background, facilitating DUX4 toxicity. The levels of both proteins could differ among patients or muscles and during disease progression, and their fluctuating ratios might contribute to disease heterogeneity. Defining DUX4c functions in healthy skeletal muscle should help to design new targeted FSHD therapy by DUX4 or DUX4c inhibition without suppressing DUX4c normal function.
Additional files
DUX4c overexpression affects the morphology of human primary myotubes. A. Healthy myoblasts were transfected with the empty vector pCIneo or pCIneo-DUX4c, and differentiation was induced 24 h after transfection. Six days later, myoblasts were observed under white light (left) before the immunodetection of DUX4c (red) and troponin T (green). B. FSHD myotubes were fixed after 6 days of differentiation. DUX4c and troponin T were detected by immunofluorescence, as described above. Clusters of nuclei are circled and were correlated with troponin T accumulation. Scale bars: 20 μm. (PDF 112 kb)
DUX4c overexpression induces the formation of ring-like clusters of nuclei and detection of DUX4c in nuclei and the cytoplasm. Healthy primary myoblasts were transfected with pAC1M2-DUX4c and differentiated 48 h later. After formation of the first myotubes (day 3), DUX4c expression was induced by the addition of doxycycline (0 ng, 100 ng, or 1 μg) to the culture medium. At day 8, myotubes were fixed. DUX4c (red) and troponin T (green) were detected by immunofluorescence. Nuclei were labeled with DAPI. The circle indicates the cytoplasmic detection of DUX4c at one myotube tip, where troponin T expression was highest in the elongated myotubes. Scale bar: 30 μm. (PDF 89 kb)
DUX4c overexpression induces β-catenin. Healthy primary myoblasts were transfected with pAC1M2-DUX4c and differentiated 48 h later. After the first myotubes formed (day 3), DUX4c expression was induced by the addition of 100 ng or 1 μg of doxycycline to the culture medium in parallel to the cultures showed in Fig. 2. At day 8, myotubes were fixed and proteins were extracted, separated, transferred to a Western blot, and the indicated proteins were immunodetected as described in Fig. 1b. Histograms: densitometry of the immunoreactive bands normalized to the actin levels (Ponceau red) in each sample. Dox: doxycycline. (PDF 115 kb)
dFSHD myotubes present abnormal clusters of nuclei overexpressing DUX4c, cytoplasmic DUX4c, and α-tubulin as well as α-crystallin B chain delocalization. A. Healthy and FSHD primary myoblasts were differentiated and fixed 6 days later, and DUX4c (red) and α-tubulin (green) were detected by immunofluorescence. Clusters of nuclei are surrounded by abnormal or almost an absence of α-tubulin. B. The α-crystallin B chain is highly expressed in the cytoplasm of dFSHD myotubes, in contrast to the low nuclear expression observed in healthy myotubes. (PDF 266 kb)
DUX4c overexpression induces expression of FSHD markers. FSHD and healthy primary myoblasts were transfected with the indicated pCIneo expression vectors. A. Total protein extracts were prepared 48 h after transfection. A 30 μg sample of each extract were separated by electrophoresis, transferred to a Western blot, and the indicated proteins immunodetected. This image was used in Vanderplanck et al. (2011) for FSHD, healthy, and DUX4-overexpressing myoblasts. We only added the lane corresponding to DUX4c-overexpressing myoblasts. B. Total protein extracts were prepared 48 h after transfection (top) or 8 days after the induction of differentiation (middle). A 30 μg sample of the extracts was separated via electrophoresis, transferred to a Western blot, and immunodetected. Actin was stained with Ponceau red on the same membrane before immunodetection and was used as the loading control. N.B.: In these conditions, neither endogenous DUX4 nor DUX4c could be immunodetected with MAb 9A12. C. Immunodetection of MuRF1 in healthy FSHD and in DUX4- or DUX4c-overexpressing primary myotubes fixed 7 days after the induction of differentiation. Scale bar: 20 μm. D. γ-Catenin (JUP) mRNA quantification by RT-qPCR in RNA of healthy and FSHD primary muscle cells. The quantity of γ-Catenin mRNA was expressed relative to its amount in healthy cells and set to 1. The means and standard errors are indicated. (PDF 452 kb)
DUX4c siRNA design and efficiency evaluation. A. Schematic representation of the DUX4c 3′UTR (positions from Genbank accession number AY500824). The STOP codon, two purine-rich regions which could be used as an alternative 3′end processing (as shown for some histone genes, [97]), the restriction sites EcoRI and AflIII (used for p3kb-DUX4c and 7.5-kb-DUX4c plasmid constructs, [26]) and the localization of the three designed siRNAs are indicated. Full-length DUX4c transcripts were already described in healthy and FSHD muscle cells [27]. B. Different DUX4c RNA ends found following transfection of C2C12 cells with p7.5-kb-DUX4c or in primary FSHD myoblasts (indicated by the asterisk). C. Evaluation of DUX4c knock-down using the three siRNAs shown in A. Human muscle TE671 cells were transfected or not (NT) with pCIneo-DUX4c expression vector (DUX4c) and with or without an siRNA targeting DUX4c as indicated (si1, si2, si3). Protein extracts were prepared 3 days later and analyzed by Western blot with the rabbit anti-DUX4c serum. Actin was used as a loading control. The panel with siRNA1 was previously shown in Ansseau et al. 2009 as part of Additional file 3: Figure S3 to confirm anti-DUX4c serum specificity. NT: not transfected. D, E. DUX4c expression in DUX4c-inducible stable TE671 cells [27] transfected with 20 nM DUX4c siRNA1. Four hours later, DUX4c expression was either induced or not with 1 μg doxycycline. C. Three or 5 days later, proteins were extracted, and 20 μg were separated on a 10% PAGE-SDS gel and transferred to a Western blot. DUX4c and actin were immunodetected. D. DUX4c detection by immunofluorescence (red) on parallel cultures. (PDF 90 kb)
siRNA1 specifically silences endogenous DUX4c. RNA interference as described in Fig. 6. Endogenous DUX4c was detected by immunofluorescence (red). (PDF 51 kb)
DUX4c inhibition in myoblasts decreases mitosis. DUX4c inhibition causes cell proliferation defects. FSHD and control primary myoblasts were transfected with siRNAs targeting either the DUX4 or DUX4c mRNA or a non-targeted (nt) siRNA. Top: microscope pictures in white light taken of representative fields of each culture. Bottom: The number of mitosis present per field was counted in each culture (10 fields per culture). Histograms show the mean of the mitosis number present per field in each culture. ***p < 0.001 was considered highly significant. (PDF 286 kb)
Cytoskeleton-associated protein quantification in healthy and FSHD myotubes : reorganization of the proteomic data presented separately in Tassin et al (2012: Figure 3, supplemental tables; and unpublished data). Data were subdivided in the three classes of cytoskeletal proteins : actin thin filaments, intermediate filaments and microtubules. In summary, primary healthy, aFSHD (atrophic) and dFSHD (disorganized) myotubes were harvested 4 days after induction of differentiation and protein extracts (TE: total extract; NE: nuclear extract) were analysed by post-digest ICPL coupled to LC-MS/MS. UniProt accession number; Hugo Gene symbol; Protein name; H/L: fold change (v: identified protein without quantification); SD: geometric standard deviation (n.d.: not determined when an abnormal distribution is observed); #: number of peptides used for quantification; *: statistical significance (p<0.05) determined by Student’s t-test. Proteins with an H/L ratio greater than 1.5 are highlighted in red; those with a ratio greater than 1.3 are in pink and those with a ratio greater than 1.2 are in light pink. Proteins with an H/L ratio less than 0.7 are highlighted in green and those with an H/L ratio of 0.7 - 0.8 are highlighted in light green. (PDF 788 kb)
DUX4c expression affects actin in the muscle cells of Ciona intestinalis zygotes. Injection of a plasmid expressing GFP-DUX4c or GFP fused to an unrelated protein under a muscle-specific promoter in Ciona intestinalis zygotes led to actin (phalloidin staining) disorganization in the contractile apparatus (collaboration with A. Philips, CRBM, CNRS, Montpellier). (PDF 122 kb)
DUX4 myogenic enhancer (DME1 and 2) might interact with the DUX4c gene. The DUX4 forward primer used in a 3C capture experiment [79] is highlighted in a common region in DUX4 and DUX4c ORF sharing 100% identity (Accession numbers AF AF117653 and AY500824). In Himeda et al. [79], RT-qPCR was performed using this primer in combination with either DIR1 (containing DME1) or DIR2 (containing DME2) primer to confirm DUX4-DME1 or DUX4-DME2 interactions. The BlgII site (boxed) used to digest chromatin-linked DNA regions is also shown and is located 226-bp downstream of the 5’end of the DUX4 primer. The region between DUX4 primer and the BglII site share 100% identity. (PDF 287 kb)
Acknowledgements
We acknowledge the human cell culture platform from the Institute of Myology (Paris, France) for providing the immortalized myoblasts and Dr. Baptist Leroy and Prof. Ruddy Wattiez for the previous help in proteomic data experiments and analyses published in [56]. We thank Prof. Anne-Emilie Declèves for her support during the finalization of this study and American Journal Experts (AJE) for the English language editing.
Funding
We acknowledge the funding from the Association Belge contre les Maladies Neuro-Musculaires (https://www.telethon.be/), the Association Française contre les Myopathies (www.afm-telethon.fr), and the FSH Society (USA) (https://www.fshsociety.org).
CV successively held a doctoral fellowship from the Fonds pour la Formation à la Recherche dans l’Industrie et dans l’Agriculture (FRIA Belgium) (www.fnrs.be) and the FSHD Global Foundation (Australia) (http://fshdglobal.org/). EA and AT were post-doctoral associates of the Fonds National de la Recherche Scientifique (http://www.fnrs.be) (Belgium).
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Abbreviations
- aFSHD
Atrophic FSHD
- CRYAB
Alpha-crystallin B chain
- CRYM
Ketimine reductase mu-crystallin
- dFSHD
Disorganized FSHD
- DMD
Duchenne muscular dystrophy
- DNMT3B
DNA (cytosine-5)-methyltransferase 3B
- DUX4
Double homeobox 4
- DUX4c
Double homeobox 4 centromeric
- FSHD
Facioscapulohumeral dystrophy
- GFP
Green fluorescent protein
- JUP
Junction plakoglobin
- KI67
Proliferation marker protein Ki-67
- KLF15
Krüppel-like transcription factor 15
- LGMD2A
Limb-girdle muscular dystrophy type 2A
- MAFbx
Muscle atrophy F-box protein
- mRNA
Messenger ribonucleic acid
- MuRF1
Muscle-specific RING finger protein 1
- MYF5/Myf5
Human/mouse myogenic factor 5
- nt
Non-targeted
- PCNA
Proliferating cell nuclear antigen
- PITX1
Pituitary homeobox 1
- siRNA
Silencing RNA
- SkNAC
Skeletal nascent polypeptide-associated complex alpha, a muscle-specific isoform of Naca
- SMCHD1
Structural maintenance of chromosomes flexible hinge domain-containing protein 1
- SMYD
SET and MYND domain-containing protein 1
- SNP
Single nucleotide polymorphism
- TP53/P53
Gene/protein cellular tumor antigen p53
- WNT
Wingless-related integration site
Authors’ contributions
CV, AT, EA, SC, AW, AB, and FC conceived and designed the experiments. CV, AT, SC, AW, and FC performed the experiments. CV, AT, EA, SC, AW, CL, KV, AB, and FC analyzed the data. DL-C contributed to the access and purification of primary myoblasts. CV, AB, and FC wrote the paper. All authors read and approved the final manuscript.
Authors’ information
CV, AT, SC, and AW contributed to this manuscript during their Ph.D. or post-doctoral studies in the Laboratory of Molecular Biology. The current position of CV, AT, SC, and AW are respectively at SGS (Belgium), Laboratory of Respiratory Physiology and Rehabilitation (Research Institute for Health Sciences and Technology) at the University of Mons, GSK (Belgium) and Straticell (Belgium).
Ethics approval and consent to participate
Primary human myoblasts were derived from muscle biopsies performed according to the current ethical and legislative rules of France, and written informed consent was obtained from all subjects, as directed by the ethical committee of CHU de Villeneuve (Montpellier, France) (Barro et al. 2010). In addition, the use of this material was approved by the ethics committee of the University of Mons (ref # A901) and the ethics committee of ULB-Erasme (Brussels ref. #B2011/003 and #P2015/516).
Consent for publication
Prof. Peter S. Zammit gave us permission to refer to the data his group presented at the FSHD International Research Consortium 2009, Boston, Massachusetts.
Dr. Alexandre Philips (CRBM, CNRS, Montpellier), now retired from research, gave us permission to publish our collaborative study.
Competing interests
The authors (CV, EA, FC, and AB) are inventors in a patent application for antisense agents useful in treating FSHD and targeting either DUX4 or DUX4c (EP patent # 242 62 03, published in the European Patent Bulletin (March 7 2012) and US patent # US20120225034 A, published on September 12, 2012.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13395-017-0148-4) contains supplementary material, which is available to authorized users.
Contributor Information
Céline Vanderplanck, Email: celine.vanderplanck@sgs.com.
Alexandra Tassin, Email: alexandra.tassin@umons.ac.be.
Eugénie Ansseau, Email: eugenie.ansseau@umons.ac.be.
Sébastien Charron, Email: sebastien.x.charron@gsk.com.
Armelle Wauters, Email: armelle.wauters@gmail.com.
Céline Lancelot, Email: celine.lancelot@umons.ac.be.
Kelly Vancutsem, Email: kelly.vancutsem@umons.ac.be.
Dalila Laoudj-Chenivesse, Email: dalila.laoudj-chenivesse@inserm.fr.
Alexandra Belayew, Email: alexandra.belayew@umons.ac.be.
Frédérique Coppée, Email: frederique.coppee@umons.ac.be.
References
- 1.Deenen JCW, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJGM, Bakker E, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83:1056–1059. doi: 10.1212/WNL.0000000000000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabriëls J, Beckers MC, Ding H, De Vriese A, Plaisance S, van der Maarel SM, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236:25–32. doi: 10.1016/S0378-1119(99)00267-X. [DOI] [PubMed] [Google Scholar]
- 3.Kowaljow V, Marcowycz A, Ansseau E, Conde CB, Sauvage S, Mattéotti C, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord. 2007;17:611–623. doi: 10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Dixit M, Ansseau E, Tassin A, Winokur S, Shi R, Qian H, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci USA. 2007;104:18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards M, Coppée F, Thomas N, Belayew A, Upadhyaya M. Facioscapulohumeral muscular dystrophy (FSHD): an enigma unravelled? Hum Genet. 2012;131:325–340. doi: 10.1007/s00439-011-1100-z. [DOI] [PubMed] [Google Scholar]
- 6.Lemmers RJLF, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mul K, van den Boogaard ML, van der Maarel SM, van Engelen BGM. Integrating clinical and genetic observations in facioscapulohumeral muscular dystrophy. Curr Opin Neurol. 2016;29:606–613. doi: 10.1097/WCO.0000000000000360. [DOI] [PubMed] [Google Scholar]
- 8.Gatica LV, Rosa AL. A complex interplay of genetic and epigenetic events leads to abnormal expression of the DUX4 gene in facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2016;26:844–852. doi: 10.1016/j.nmd.2016.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Wang LH, Tawil R. Facioscapulohumeral dystrophy. Curr Neurol Neurosci Rep. 2016;16:66. doi: 10.1007/s11910-016-0667-0. [DOI] [PubMed] [Google Scholar]
- 10.Lemmers RJLF, Goeman JJ, van der Vliet PJ, van Nieuwenhuizen MP, Balog J, Vos-Versteeg M, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet. 2015;24:659–669. doi: 10.1093/hmg/ddu486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sacconi S, Lemmers RJLF, Balog J, van der Vliet PJ, Lahaut P, van Nieuwenhuizen MP, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet. 2013;93:744–751. doi: 10.1016/j.ajhg.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsen M, Rost S, El Hajj N, Ferbert A, Deschauer M, Walter MC, et al. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur J Hum Genet. 2015;23:808–816. doi: 10.1038/ejhg.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bosnakovski D, Xu Z, Gang EJ, Galindo CL, Liu M, Simsek T, et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008;27:2766–2779. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell. 2012;22:38–51. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao Z, Snider L, Balog J, Lemmers RJLF, Van Der Maarel SM, Tawil R, et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum Mol Genet. 2014;23:5342–5352. doi: 10.1093/hmg/ddu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tassin A, Laoudj-Chenivesse D, Vanderplanck C, Barro M, Charron S, Ansseau E, et al. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy? J Cell Mol Med. 2013;17:76–89. doi: 10.1111/j.1582-4934.2012.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ehrlich M, Lacey M. Deciphering transcription dysregulation in FSH muscular dystrophy. J Hum Genet. 2012;57:477–484. doi: 10.1038/jhg.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rickard AM, Petek LM, Miller DG. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum Mol Genet. 2015;24:5901–5914. doi: 10.1093/hmg/ddv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snider L, Geng LN, Lemmers RJLF, Kyba M, Ware CB, Nelson AM, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turki A, Hayot M, Carnac G, Pillard F, Passerieux E, Bommart S, et al. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic Biol Med. 2012;53:1068–1079. doi: 10.1016/j.freeradbiomed.2012.06.041. [DOI] [PubMed] [Google Scholar]
- 21.Dmitriev P, Kiseleva E, Kharchenko O, Ivashkin E, Pichugin A, Dessen P, et al. Dux4 controls migration of mesenchymal stem cells through the Cxcr4-Sdf1 axis. Oncotarget. 2016;7(40):65090-65108. [DOI] [PMC free article] [PubMed]
- 22.Barro M, Carnac G, Flavier S, Mercier J, Vassetzky Y, Laoudj-Chenivesse D. Myoblasts from affected and non-affected FSHD muscles exhibit morphological differentiation defects. J Cell Mol Med. 2010;14:275–289. doi: 10.1111/j.1582-4934.2008.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones TI, Chen JCJ, Rahimov F, Homma S, Arashiro P, Beermann ML, et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum Mol Genet. 2012;21:4419–4430. doi: 10.1093/hmg/dds284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ansseau E, Vanderplanck C, Wauters A, Harper SQ, Coppée F, Belayew A. Antisense oligonucleotides used to target the DUX4 mRNA as therapeutic approaches in faciosscapulohumeral muscular dystrophy (FSHD). Genes (Basel). 2017;8(3). 10.3390/genes8030093. [DOI] [PMC free article] [PubMed]
- 25.Caron L, Kher D, Lee KL, McKernan R, Dumevska B, Hidalgo A, et al. A human pluripotent stem cell model of facioscapulohumeral muscular dystrophy-affected skeletal muscles. Stem Cells Transl Med. 2016;5:1145–1161. doi: 10.5966/sctm.2015-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coppée F, Mattéotti C, Ansseau E, Sauvage S, Leclercq I, Leroy A, et al. The DUX gene family and FSHD. FSHD facioscapulohumeral muscular dystrophy: clinical medicine and molecular cell biology. BIOS Scientific Publishers. London, New York: Meena Upadhyaya and David N. Cooper; 2004. pp. 117–131. [Google Scholar]
- 27.Ansseau E, Laoudj-Chenivesse D, Marcowycz A, Tassin A, Vanderplanck C, Sauvage S, et al. DUX4c is up-regulated in FSHD. It induces the MYF5 protein and human myoblast proliferation. PLoS One. 2009;4:e7482. doi: 10.1371/journal.pone.0007482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ansseau E, Eidahl JO, Lancelot C, Tassin A, Matteotti C, Yip C, et al. Homologous transcription factors DUX4 and DUX4c associate with cytoplasmic proteins during muscle differentiation. PLoS One. 2016;11:e0146893. doi: 10.1371/journal.pone.0146893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bosnakovski D, Lamb S, Simsek T, Xu Z, Belayew A, Perlingeiro R, et al. DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp Neurol. 2008;214:87–96. doi: 10.1016/j.expneurol.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 30.Dmitriev P, Stankevicins L, Ansseau E, Petrov A, Barat A, Dessen P, et al. Defective regulation of microRNA target genes in myoblasts from facioscapulohumeral dystrophy patients. J Biol Chem. 2013;288:34989–35002. doi: 10.1074/jbc.M113.504522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knopp P, Krom YD, Banerji CRS, Panamarova M, Moyle LA, den Hamer B, et al. DUX4 induces a transcriptome more characteristic of a less-differentiated cell state and inhibits myogenesis. J Cell Sci. 2016;129:3816–3831. doi: 10.1242/jcs.180372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanderplanck C, Ansseau E, Charron S, Stricwant N, Tassin A, Laoudj-Chenivesse D, et al. The FSHD atrophic myotube phenotype is caused by DUX4 expression. PLoS One. 2011;6:e26820. doi: 10.1371/journal.pone.0026820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kramerova I, Kudryashova E, Wu B, Spencer MJ. Regulation of the M-cadherin-beta-catenin complex by calpain 3 during terminal stages of myogenic differentiation. Mol Cell Biol. 2006;26:8437–8447. doi: 10.1128/MCB.01296-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallace LM, Garwick SE, Mei W, Belayew A, Coppee F, Ladner KJ, et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol. 2011;69:540–552. doi: 10.1002/ana.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Block GJ, Narayanan D, Amell AM, Petek LM, Davidson KC, Bird TD, et al. WNT/β-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum Mol Genet. 2013;22:4661–4672. doi: 10.1093/hmg/ddt314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shadle SC, Zhong JW, Campbell AE, Conerly ML, Jagannathan S, Wong C-J, et al. DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy. PLoS Genet. 2017;13:e1006658. doi: 10.1371/journal.pgen.1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pakula A, Schneider J, Janke J, Zacharias U, Schulz H, Hübner N, et al. Altered expression of cyclin A 1 in muscle of patients with facioscapulohumeral muscle dystrophy (FSHD-1). Beard N, editor. PLoS One. 2013;8:e73573. [DOI] [PMC free article] [PubMed]
- 38.Rogers MT, Upadhyaya M, Sewry CA. Histological, immunological, molecular and ultrastructural characterization of FSHD muscle. FSHD facioscapulohumeral muscular dystrophy: clinical medicine and molecular cell biology. BIOS Scientific Publishers. London, New York: Meena Upadhyaya and David N. Cooper; 2004. pp. 277–298. [Google Scholar]
- 39.Lassche S, Ottenheijm CAC, Voermans NC, Westeneng H-J, Janssen BH, van der Maarel SM, et al. Determining the role of sarcomeric proteins in facioscapulohumeral muscular dystrophy: a study protocol. BMC Neurol. 2013;13:144. doi: 10.1186/1471-2377-13-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yotov WV, St-Arnaud R. Differential splicing-in of a proline-rich exon converts alphaNAC into a muscle-specific transcription factor. Genes Dev. 1996;10:1763–1772. doi: 10.1101/gad.10.14.1763. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Randall WR, Du S-J. skNAC (skeletal Naca), a muscle-specific isoform of Naca (nascent polypeptide-associated complex alpha), is required for myofibril organization. FASEB J. 2009;23:1988–2000. doi: 10.1096/fj.08-125542. [DOI] [PubMed] [Google Scholar]
- 42.Tan X, Rotllant J, Li H, De Deyne P, DeDeyne P, Du SJ. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc Natl Acad Sci USA. 2006;103:2713–2718. doi: 10.1073/pnas.0509503103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao J, Li J, Li B-J, Yagil E, Zhang J, Du SJ. Expression and functional characterization of Smyd1a in myofibril organization of skeletal muscles. PLoS One. 2014;9:e86808. doi: 10.1371/journal.pone.0086808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rui Y, Bai J, Perrimon N. Sarcomere formation occurs by the assembly of multiple latent protein complexes. PLoS Genet. 2010;6:e1001208. doi: 10.1371/journal.pgen.1001208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sancisi V, Germinario E, Esposito A, Morini E, Peron S, Moggio M, et al. Altered Tnnt3 characterizes selective weakness of fast fibers in mice overexpressing FSHD region gene 1 (FRG1) Am J Physiol Regul Integr Comp Physiol. 2014;306:R124–R137. doi: 10.1152/ajpregu.00379.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Head MW, Hurwitz L, Kegel K, Goldman JE. AlphaB-crystallin regulates intermediate filament organization in situ. Neuroreport. 2000;11:361–365. doi: 10.1097/00001756-200002070-00028. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, et al. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 48.Starr DA, Han M. ANChors away: an actin based mechanism of nuclear positioning. J Cell Sci. 2003;116:211–216. doi: 10.1242/jcs.00248. [DOI] [PubMed] [Google Scholar]
- 49.Morris NR. Nuclear positioning: the means is at the ends. Curr Opin Cell Biol. 2003;15:54–59. doi: 10.1016/S0955-0674(02)00004-2. [DOI] [PubMed] [Google Scholar]
- 50.Cadot B, Gache V, Gomes ER. Moving and positioning the nucleus in skeletal muscle—one step at a time. Nucleus. 2015;6(5):373–81. [DOI] [PMC free article] [PubMed]
- 51.Englander LL, Rubin LL. Acetylcholine receptor clustering and nuclear movement in muscle fibers in culture. J Cell Biol. 1987;104:87–95. doi: 10.1083/jcb.104.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmalbruch H, Lewis DM. Dynamics of nuclei of muscle fibers and connective tissue cells in normal and denervated rat muscles. Muscle Nerve. 2000;23:617–626. doi: 10.1002/(SICI)1097-4598(200004)23:4<617::AID-MUS22>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 53.Bruusgaard JC, Liestøl K, Gundersen K. Distribution of myonuclei and microtubules in live muscle fibers of young, middle-aged, and old mice. J Appl Physiol. 2006;100:2024–2030. doi: 10.1152/japplphysiol.00913.2005. [DOI] [PubMed] [Google Scholar]
- 54.Apel ED, Lewis RM, Grady RM, Sanes JR. Syne-1, a dystrophin- and Klarsicht-related protein associated with synaptic nuclei at the neuromuscular junction. J Biol Chem. 2000;275:31986–31995. doi: 10.1074/jbc.M004775200. [DOI] [PubMed] [Google Scholar]
- 55.Grady RM, Starr DA, Ackerman GL, Sanes JR, Han M. Syne proteins anchor muscle nuclei at the neuromuscular junction. Proc Natl Acad Sci USA. 2005;102:4359–4364. doi: 10.1073/pnas.0500711102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang X, Xu R, Zhu B, Yang X, Ding X, Duan S, et al. Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development. 2007;134:901–908. doi: 10.1242/dev.02783. [DOI] [PubMed] [Google Scholar]
- 57.Ralston E, Lu Z, Biscocho N, Soumaka E, Mavroidis M, Prats C, et al. Blood vessels and desmin control the positioning of nuclei in skeletal muscle fibers. J Cell Physiol. 2006;209:874–882. doi: 10.1002/jcp.20780. [DOI] [PubMed] [Google Scholar]
- 58.Tassin A, Leroy B, Laoudj-Chenivesse D, Wauters A, Vanderplanck C, Le Bihan M-C, et al. FSHD myotubes with different phenotypes exhibit distinct proteomes. PLoS One. 2012;7:e51865. doi: 10.1371/journal.pone.0051865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wuebbles RD, Long SW, Hanel ML, Jones PL. Testing the effects of FSHD candidate gene expression in vertebrate muscle development. Int J Clin Exp Pathol. 2010;3:386–400. [PMC free article] [PubMed] [Google Scholar]
- 60.Folker ES, Baylies MK. Nuclear positioning in muscle development and disease. Frontiers in Physiology [Internet]. 2013. [cited 2015 Dec 14];4. Available from: http://journal.frontiersin.org/article/10.3389/fphys.2013.00363/abstract [DOI] [PMC free article] [PubMed]
- 61.Tupler R, Berardinelli A, Barbierato L, Frants R, Hewitt JE, Lanzi G, et al. Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J Med Genet. 1996;33:366–370. doi: 10.1136/jmg.33.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deak KL, Lemmers RJLF, Stajich JM, Klooster R, Tawil R, Frants RR, et al. Genotype-phenotype study in an FSHD family with a proximal deletion encompassing p13E-11 and D4Z4. Neurology. 2007;68:578–582. doi: 10.1212/01.wnl.0000254991.21818.f3. [DOI] [PubMed] [Google Scholar]
- 63.Leidenroth A, Clapp J, Mitchell LM, Coneyworth D, Dearden FL, Iannuzzi L, et al. Evolution of DUX gene macrosatellites in placental mammals. Chromosoma. 2012;121:489–497. doi: 10.1007/s00412-012-0380-y. [DOI] [PubMed] [Google Scholar]
- 64.Wu S-L, Li G-Z, Chou C-Y, Tsai M-S, Chen Y-P, Li C-J, et al. Double homeobox gene, Duxbl, promotes myoblast proliferation and abolishes myoblast differentiation by blocking MyoD transactivation. Cell Tissue Res. 2014;358:551–566. doi: 10.1007/s00441-014-1974-x. [DOI] [PubMed] [Google Scholar]
- 65.Krom YD, Thijssen PE, Young JM, den Hamer B, Balog J, Yao Z, et al. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet. 2013;9:e1003415. doi: 10.1371/journal.pgen.1003415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nastasi T, Bongiovanni A, Campos Y, Mann L, Toy JN, Bostrom J, et al. Ozz-E3, a muscle-specific ubiquitin ligase, regulates beta-catenin degradation during myogenesis. Dev Cell. 2004;6:269–282. doi: 10.1016/S1534-5807(04)00020-6. [DOI] [PubMed] [Google Scholar]
- 67.Banerji CRS, Knopp P, Moyle LA, Severini S, Orrell RW, Teschendorff AE, et al. β-catenin is central to DUX4-driven network rewiring in facioscapulohumeral muscular dystrophy. J R Soc Interface. 2015;12:20140797. doi: 10.1098/rsif.2014.0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Knopp and Zammit. Assessing the effects of FRG1, DUX4 and DUX4c on muscle satellite cell function. Boston: The FSHD International Research Consortium; 2009.
- 69.Pirozhkova I, Petrov A, Dmitriev P, Laoudj D, Lipinski M, Vassetzky Y. A functional role for 4qA/B in the structural rearrangement of the 4q35 region and in the regulation of FRG1 and ANT1 in facioscapulohumeral dystrophy. PLoS One. 2008;3:e3389. doi: 10.1371/journal.pone.0003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dmitriev P, Petrov A, Ansseau E, Stankevicins L, Charron S, Kim E, et al. The Krüppel-like factor 15 as a molecular link between myogenic factors and a chromosome 4q transcriptional enhancer implicated in facioscapulohumeral dystrophy. J Biol Chem. 2011;286:44620–44631. doi: 10.1074/jbc.M111.254052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pandey SN, Khawaja H, Chen Y-W. Culture conditions affect expression of DUX4 in FSHD myoblasts. Molecules. 2015;20:8304–8315. doi: 10.3390/molecules20058304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reed PW, Corse AM, Porter NC, Flanigan KM, Bloch RJ. Abnormal expression of mu-crystallin in facioscapulohumeral muscular dystrophy. Exp Neurol. 2007;205:583–586. doi: 10.1016/j.expneurol.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 73.Liu DX, Lobie PE. Transcriptional activation of p53 by Pitx1. Cell Death Differ. 2007;14:1893–1907. doi: 10.1038/sj.cdd.4402209. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Y, Lee JK, Toso EA, Lee JS, Choi SH, Slattery M, et al. DNA-binding sequence specificity of DUX4. Skelet Muscle. 2016;6:8. doi: 10.1186/s13395-016-0080-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.AS ME, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol. 2002;157:125–136. doi: 10.1083/jcb.200108089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bogomolovas J, Gasch A, Simkovic F, Rigden DJ, Labeit S, Mayans O. Titin kinase is an inactive pseudokinase scaffold that supports MuRF1 recruitment to the sarcomeric M-line. Open Biol. 2014;4:140041. doi: 10.1098/rsob.140041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li H-H, Kedar V, Zhang C, McDonough H, Arya R, Wang D-Z, et al. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI200422220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Balog J, Thijssen PE, Shadle S, Straasheijm KR, van der Vliet PJ, Krom YD, et al. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics. 2015;10:1133–1142. doi: 10.1080/15592294.2015.1113798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Himeda CL, Debarnot C, Homma S, Beermann ML, Miller JB, Jones PL, et al. Myogenic enhancers regulate expression of the facioscapulohumeral muscular dystrophy-associated DUX4 gene. Mol Cell Biol. 2014;34:1942–1955. doi: 10.1128/MCB.00149-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Otto A, Schmidt C, Luke G, Allen S, Valasek P, Muntoni F, et al. Canonical Wnt signalling induces satellite-cell proliferation during adult skeletal muscle regeneration. J Cell Sci. 2008;121:2939–2950. doi: 10.1242/jcs.026534. [DOI] [PubMed] [Google Scholar]
- 81.Fujimaki S, Hidaka R, Asashima M, Takemasa T, Kuwabara T. Wnt protein-mediated satellite cell conversion in adult and aged mice following voluntary wheel running. J Biol Chem. 2014;289:7399–7412. doi: 10.1074/jbc.M113.539247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Han XH, Jin Y-R, Seto M, Yoon JK. A WNT/beta-catenin signaling activator, R-spondin, plays positive regulatory roles during skeletal myogenesis. J Biol Chem. 2011;286:10649–10659. doi: 10.1074/jbc.M110.169391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stratton MR, Darling J, Pilkington GJ, Lantos PL, Reeves BR, Cooper CS. Characterization of the human cell line TE671. Carcinogenesis. 1989;10:899–905. doi: 10.1093/carcin/10.5.899. [DOI] [PubMed] [Google Scholar]
- 84.du Manoir S, Guillaud P, Camus E, Seigneurin D, Brugal G. Ki-67 labeling in postmitotic cells defines different Ki-67 pathways within the 2c compartment. Cytometry. 1991;12:455–463. doi: 10.1002/cyto.990120511. [DOI] [PubMed] [Google Scholar]
- 85.Gayraud-Morel B, Chrétien F, Flamant P, Gomès D, Zammit PS, Tajbakhsh S. A role for the myogenic determination gene Myf5 in adult regenerative myogenesis. Dev Biol. 2007;312:13–28. doi: 10.1016/j.ydbio.2007.08.059. [DOI] [PubMed] [Google Scholar]
- 86.Ustanina S, Carvajal J, Rigby P, Braun T. The myogenic factor Myf5 supports efficient skeletal muscle regeneration by enabling transient myoblast amplification. Stem Cells. 2007;25:2006–2016. doi: 10.1634/stemcells.2006-0736. [DOI] [PubMed] [Google Scholar]
- 87.Chargé SBP, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 88.Sharma V, Harafuji N, Belayew A, Chen Y-W. DUX4 differentially regulates transcriptomes of human rhabdomyosarcoma and mouse C2C12 cells. PLoS One. 2013;8:e64691. doi: 10.1371/journal.pone.0064691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bosnakovski D, Choi SH, Strasser JM, Toso EA, Walters MA, Kyba M. High-throughput screening identifies inhibitors of DUX4-induced myoblast toxicity. Skelet Muscle. 2014;4:4. doi: 10.1186/2044-5040-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marsollier A-C, Ciszewski L, Mariot V, Popplewell L, Voit T, Dickson G, et al. Antisense targeting of 3′ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum Mol Genet. 2016;25:1468–1478. doi: 10.1093/hmg/ddw015. [DOI] [PubMed] [Google Scholar]
- 91.Chen JC, King OD, Zhang Y, Clayton NP, Spencer C, Wentworth BM, et al. Morpholino-mediated knockdown of DUX4 toward facioscapulohumeral muscular dystrophy therapeutics. Mol Ther. 2016;24:1405–1411. doi: 10.1038/mt.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Choi SH, Bosnakovski D, Strasser JM, Toso EA, Walters MA, Kyba M. Transcriptional inhibitors identified in a 160,000-compound small-molecule DUX4 viability screen. J Biomol Screen. 2016;21:680–688. doi: 10.1177/1087057116651868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Young JM, Whiddon JL, Yao Z, Kasinathan B, Snider L, Geng LN, et al. DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS Genet. 2013;9:e1003947. doi: 10.1371/journal.pgen.1003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Choi SH, Gearhart MD, Cui Z, Bosnakovski D, Kim M, Schennum N, et al. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016;44:5161–5173. doi: 10.1093/nar/gkw141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Feng Q, Snider L, Jagannathan S, Tawil R, van der Maarel SM, Tapscott SJ, et al. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. elife. 2015;4 [DOI] [PMC free article] [PubMed]
- 96.Geng LN, Tyler AE, Tapscott SJ. Immunodetection of human double homeobox 4. Hybridoma (Larchmt) 2011;30:125–130. doi: 10.1089/hyb.2010.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dávila López M, Samuelsson T. Early evolution of histone mRNA 3′ end processing. RNA. 2008;14:1–10. doi: 10.1261/rna.782308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DUX4c overexpression affects the morphology of human primary myotubes. A. Healthy myoblasts were transfected with the empty vector pCIneo or pCIneo-DUX4c, and differentiation was induced 24 h after transfection. Six days later, myoblasts were observed under white light (left) before the immunodetection of DUX4c (red) and troponin T (green). B. FSHD myotubes were fixed after 6 days of differentiation. DUX4c and troponin T were detected by immunofluorescence, as described above. Clusters of nuclei are circled and were correlated with troponin T accumulation. Scale bars: 20 μm. (PDF 112 kb)
DUX4c overexpression induces the formation of ring-like clusters of nuclei and detection of DUX4c in nuclei and the cytoplasm. Healthy primary myoblasts were transfected with pAC1M2-DUX4c and differentiated 48 h later. After formation of the first myotubes (day 3), DUX4c expression was induced by the addition of doxycycline (0 ng, 100 ng, or 1 μg) to the culture medium. At day 8, myotubes were fixed. DUX4c (red) and troponin T (green) were detected by immunofluorescence. Nuclei were labeled with DAPI. The circle indicates the cytoplasmic detection of DUX4c at one myotube tip, where troponin T expression was highest in the elongated myotubes. Scale bar: 30 μm. (PDF 89 kb)
DUX4c overexpression induces β-catenin. Healthy primary myoblasts were transfected with pAC1M2-DUX4c and differentiated 48 h later. After the first myotubes formed (day 3), DUX4c expression was induced by the addition of 100 ng or 1 μg of doxycycline to the culture medium in parallel to the cultures showed in Fig. 2. At day 8, myotubes were fixed and proteins were extracted, separated, transferred to a Western blot, and the indicated proteins were immunodetected as described in Fig. 1b. Histograms: densitometry of the immunoreactive bands normalized to the actin levels (Ponceau red) in each sample. Dox: doxycycline. (PDF 115 kb)
dFSHD myotubes present abnormal clusters of nuclei overexpressing DUX4c, cytoplasmic DUX4c, and α-tubulin as well as α-crystallin B chain delocalization. A. Healthy and FSHD primary myoblasts were differentiated and fixed 6 days later, and DUX4c (red) and α-tubulin (green) were detected by immunofluorescence. Clusters of nuclei are surrounded by abnormal or almost an absence of α-tubulin. B. The α-crystallin B chain is highly expressed in the cytoplasm of dFSHD myotubes, in contrast to the low nuclear expression observed in healthy myotubes. (PDF 266 kb)
DUX4c overexpression induces expression of FSHD markers. FSHD and healthy primary myoblasts were transfected with the indicated pCIneo expression vectors. A. Total protein extracts were prepared 48 h after transfection. A 30 μg sample of each extract were separated by electrophoresis, transferred to a Western blot, and the indicated proteins immunodetected. This image was used in Vanderplanck et al. (2011) for FSHD, healthy, and DUX4-overexpressing myoblasts. We only added the lane corresponding to DUX4c-overexpressing myoblasts. B. Total protein extracts were prepared 48 h after transfection (top) or 8 days after the induction of differentiation (middle). A 30 μg sample of the extracts was separated via electrophoresis, transferred to a Western blot, and immunodetected. Actin was stained with Ponceau red on the same membrane before immunodetection and was used as the loading control. N.B.: In these conditions, neither endogenous DUX4 nor DUX4c could be immunodetected with MAb 9A12. C. Immunodetection of MuRF1 in healthy FSHD and in DUX4- or DUX4c-overexpressing primary myotubes fixed 7 days after the induction of differentiation. Scale bar: 20 μm. D. γ-Catenin (JUP) mRNA quantification by RT-qPCR in RNA of healthy and FSHD primary muscle cells. The quantity of γ-Catenin mRNA was expressed relative to its amount in healthy cells and set to 1. The means and standard errors are indicated. (PDF 452 kb)
DUX4c siRNA design and efficiency evaluation. A. Schematic representation of the DUX4c 3′UTR (positions from Genbank accession number AY500824). The STOP codon, two purine-rich regions which could be used as an alternative 3′end processing (as shown for some histone genes, [97]), the restriction sites EcoRI and AflIII (used for p3kb-DUX4c and 7.5-kb-DUX4c plasmid constructs, [26]) and the localization of the three designed siRNAs are indicated. Full-length DUX4c transcripts were already described in healthy and FSHD muscle cells [27]. B. Different DUX4c RNA ends found following transfection of C2C12 cells with p7.5-kb-DUX4c or in primary FSHD myoblasts (indicated by the asterisk). C. Evaluation of DUX4c knock-down using the three siRNAs shown in A. Human muscle TE671 cells were transfected or not (NT) with pCIneo-DUX4c expression vector (DUX4c) and with or without an siRNA targeting DUX4c as indicated (si1, si2, si3). Protein extracts were prepared 3 days later and analyzed by Western blot with the rabbit anti-DUX4c serum. Actin was used as a loading control. The panel with siRNA1 was previously shown in Ansseau et al. 2009 as part of Additional file 3: Figure S3 to confirm anti-DUX4c serum specificity. NT: not transfected. D, E. DUX4c expression in DUX4c-inducible stable TE671 cells [27] transfected with 20 nM DUX4c siRNA1. Four hours later, DUX4c expression was either induced or not with 1 μg doxycycline. C. Three or 5 days later, proteins were extracted, and 20 μg were separated on a 10% PAGE-SDS gel and transferred to a Western blot. DUX4c and actin were immunodetected. D. DUX4c detection by immunofluorescence (red) on parallel cultures. (PDF 90 kb)
siRNA1 specifically silences endogenous DUX4c. RNA interference as described in Fig. 6. Endogenous DUX4c was detected by immunofluorescence (red). (PDF 51 kb)
DUX4c inhibition in myoblasts decreases mitosis. DUX4c inhibition causes cell proliferation defects. FSHD and control primary myoblasts were transfected with siRNAs targeting either the DUX4 or DUX4c mRNA or a non-targeted (nt) siRNA. Top: microscope pictures in white light taken of representative fields of each culture. Bottom: The number of mitosis present per field was counted in each culture (10 fields per culture). Histograms show the mean of the mitosis number present per field in each culture. ***p < 0.001 was considered highly significant. (PDF 286 kb)
Cytoskeleton-associated protein quantification in healthy and FSHD myotubes : reorganization of the proteomic data presented separately in Tassin et al (2012: Figure 3, supplemental tables; and unpublished data). Data were subdivided in the three classes of cytoskeletal proteins : actin thin filaments, intermediate filaments and microtubules. In summary, primary healthy, aFSHD (atrophic) and dFSHD (disorganized) myotubes were harvested 4 days after induction of differentiation and protein extracts (TE: total extract; NE: nuclear extract) were analysed by post-digest ICPL coupled to LC-MS/MS. UniProt accession number; Hugo Gene symbol; Protein name; H/L: fold change (v: identified protein without quantification); SD: geometric standard deviation (n.d.: not determined when an abnormal distribution is observed); #: number of peptides used for quantification; *: statistical significance (p<0.05) determined by Student’s t-test. Proteins with an H/L ratio greater than 1.5 are highlighted in red; those with a ratio greater than 1.3 are in pink and those with a ratio greater than 1.2 are in light pink. Proteins with an H/L ratio less than 0.7 are highlighted in green and those with an H/L ratio of 0.7 - 0.8 are highlighted in light green. (PDF 788 kb)
DUX4c expression affects actin in the muscle cells of Ciona intestinalis zygotes. Injection of a plasmid expressing GFP-DUX4c or GFP fused to an unrelated protein under a muscle-specific promoter in Ciona intestinalis zygotes led to actin (phalloidin staining) disorganization in the contractile apparatus (collaboration with A. Philips, CRBM, CNRS, Montpellier). (PDF 122 kb)
DUX4 myogenic enhancer (DME1 and 2) might interact with the DUX4c gene. The DUX4 forward primer used in a 3C capture experiment [79] is highlighted in a common region in DUX4 and DUX4c ORF sharing 100% identity (Accession numbers AF AF117653 and AY500824). In Himeda et al. [79], RT-qPCR was performed using this primer in combination with either DIR1 (containing DME1) or DIR2 (containing DME2) primer to confirm DUX4-DME1 or DUX4-DME2 interactions. The BlgII site (boxed) used to digest chromatin-linked DNA regions is also shown and is located 226-bp downstream of the 5’end of the DUX4 primer. The region between DUX4 primer and the BglII site share 100% identity. (PDF 287 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article [and its supplementary information files].