

Abstract
Development of spontaneous melanoma in Xiphophorus interspecies backcross hybrid progeny, (X. hellerii × [X. maculatus Jp 163 A × X. hellerii]) is due to Mendelian segregation of a oncogene (xmrk) and a molecularly uncharacterized locus, called R(Diff), on LG5. R(Diff) is thought to suppresses the activity of xmrk in healthy X. maculatus Jp 163 A parental species that rarely develop melanoma. To better understand the molecular genetics of R(Diff), we utilized RNA-Seq to study allele-specific gene expression of spontaneous melanoma tumors and corresponding normal skin samples derived from 15 first generation backcross (BC1) hybrids and 13 fifth generation (BC5) hybrids. Allele-specific expression was determined for all genes and assigned to parental allele inheritance for each backcross hybrid individual. Results showed that genes residing in a 5.81 Mbp region on LG5 were exclusively expressed from the X. hellerii alleles in tumor-bearing BC1 hybrids. This observation indicates this region is consistently homozygous for X. hellerii alleles in tumor bearing animals, and therefore defines this region to be the R(Diff) locus. The R(Diff) locus harbors 164 gene models and includes the previously characterized R(Diff) candidate, cdkn2x. Twenty one genes in the R(Diff) region show differential expression in the tumor samples compared to normal skin tissue. These results further characterize the R(Diff) locus and suggest tumor suppression may require a multigenic region rather than a single gene variant. Differences in gene expression between tumor and normal skin tissue in this region may indicate interactions among several genes are required for backcross hybrid melanoma development.
Keywords: Bioinformatics, Gordon-Kosswig model, interspecies hybrids, Allele Specific Gene Expression, Genetic Interaction
Introduction
The Xiphophorus melanoma model was originally introduced by Myron Gordon and Kurt Kosswig in late 1920’s 1–3. This model, often termed the “Gordon-Kosswig” melanoma model, employs X. maculatus and X. hellerii interspecies hybrids to produce spontaneous melanoma in backcross hybrid progeny with certain genotypes. The X. maculatus parental line carries the spotted dorsal (Sd) macromelanophore pigmentation pattern leading to melanization of the dorsal fin. While X. hellerii does not have this pigmentation pattern 4,5, the highly inbred X. maculatus parent is homozygous for the Sd pigment pattern that is tightly linked to an oncogene, xmrk (Xiphophorus melanoma regulatory kinase) 6–9. The Sd linked xmrk oncogene is an X. maculatus-specific gene duplicate of the epidermal growth factor receptor (egfr) that has become regulated by a tumor suppressor locus, termed R(Diff), that resides on X. maculatus LG5 10,11. Neither the Sd pigment pattern, nor the xmrk oncogenic gene duplicate are present in the X. hellerii parent.
The X. maculatus gene cdkn2x has been forwarded as a candidate for the R(Diff) tumor suppressor locus. The X. maculatus cdkn2x gene is equally distant, in sequence differences, from the human CDKN2A (i.e., p16) and CDKN2B (i.e., p15) genes and maps to the R(Diff) region of LG5 12–16. X. maculatus and X. hellerii F1 interspecies hybrids express enhanced dorsal fin macromelanophore pigmentation, but do not develop invasive melanoma, presumably due to single copy regulation of xmrk by R(Diff). However, when the F1 hybrid (Sd-hellerii) is backcrossed with X. hellerii, 25% of progeny that inherit the xmrk oncogene, but do not also inherit an X. maculatus R(Diff) allele, will develop spontaneous, invasive, melanoma tumors. Other melanoma models initiated by xmrk have recently been developed, such as transgenic Japanese medaka (Oryzias latipes) in which xmrk expression is driven by the pigment cell-specific mitf promoter, leading to melanoma in medaka fry with 100% penetrance 17,18.
Although genetic linkage analysis show the candidate tumor suppressor cdkn2x mapped to the same region on LG5 as R(Diff), cdkn2x alone cannot fully account for R(Diff) function. The R(Diff) locus is expected to be homozygous for X. hellerii allele in all malignant tumor-bearing hybrids. However, ~20% of melanoma tumor-bearing fish are heterozygous for cdkn2x gene 12–15. Additionally, in these ~20% heterozygous tumor-bearing progeny, cdkn2x gene expression showed up-regulation with most of the up-regulated transcript coming from the X. maculatus allele of cdkn2x gene 13,19. This suggests that cdkn2x maps in, or very close to the actual R(Diff) region on LG5, and that the function of R(Diff) may not depend solely on cdkn2x.
To understand allele-specific expression in tumor and normal skin of animals segregating R(Diff), we sequenced mRNA from melanoma tumors and paired normal skin samples from the same hybrid animals to map parental allele genotypes to the LG5 chromosome. Results define a 5.81Mbp R(Diff) region that exhibits consistent homozygosity in all tumor-bearing fish. We incorporated transcriptome-wide genotyping information from fifth generation backcross (BC5) hybrid melanoma tumors to further restrict the R(Diff) locus to 69 gene models within the R(Diff) region. These results establish a method to employ RNA-Seq to define multigenic regions responsible for heritable tumor development.
Materials and Methods
Animal model
First generation backcross (BC1) animals used in this study were supplied by the Xiphophorus Genetic Stock Center (S1 Figure. For contact information see: http://www.xiphophorus.txstate.edu/). Specifically, a X. maculatus Jp 163 A female was artificially inseminated with sperm from a male X. hellerii (Sarabia) to produce F1 hybrids. F1 hybrid males were then backcrossed to X. hellerii females to generate the BC1 animals. Of these BC1 animals, about 25% developed melanoma tumors. At dissection, fish are anesthetized in an ice bath and upon loss of gill movement are sacrificed by cranial resection. Organ are dissected directly into TRI-Reagent (Sigma Inc. St. Louis) placed in a dry ice-ethanol bath if the RNA is isolated at the time of dissection, or into RNAlater (Ambion Inc.) and kept at −80 degree for later use. All BC1 fish were kept and samples taken in accordance with protocol approved by IACUC (IACUC2015107711).
The BC5 hybrids were produced in an independent series of crosses from F1 hybrids originating from the reciprocal cross: X. maculatus Jp 163 A males were mated to X. hellerii (Lancetilla) females. The F1 hybrid females, which had no tumors, were then successively backcrossed to X. hellerii males to produce the fifth generation of backcross hybrids (BC5). All BC5 fish used in this study were from laboratory stocks maintained in the governmentally certified animal facilities of the Biocenter. All BC5 fish were kept and samples taken in accordance with the applicable EU and national German legislation governing animal experimentation. Fish were sacrificed by over-anesthetization with MS222. We hold an authorization (568/300–1870/13) of the Veterinary Office of the District Government of Lower Franconia, Germany, in accordance with the German Animal Protection Law (TierSchG).
RNA isolation and RNA sequencing
Total RNA from 15 melanoma tumors and 15 paired normal skin samples was isolated as previously detailed 20,21. Samples were homogenized in TRI-reagent (Sigma Inc., St. Louis, MO, USA) followed by addition of 200 μl/ml chloroform, vigorously shaken, and subjected to centrifugation at 12,000 g for 5min at 4°C. Total RNA was further purified using an RNeasy mini RNA isolation kit (Qiagen, Valencia, CA, USA). Column DNase digestion at 25°C for 15 min removed residual DNA. Total RNA concentration was determined using a Qubit 2.0 fluorometer (Life Technologies, Grand Island, NY, USA). RNA quality was verified on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) to confirm that RIN scores were above 8.0 prior to sequencing.
RNA sequencing was performed upon libraries construction using the Illumina TruSeq library preparation system (Illumina, Inc., San Diego, CA, USA). RNA libraries were sequenced as 125bp or 100bp paired-end fragments using Illumina Hi-Seq 2000 system (Illumina, Inc., San Diego, CA, USA). Short sequencing reads were filtered using an in-house data processing pipeline 22. Briefly, sequencing adaptors were firstly removed from sequencing reads. Processed sequencing reads were subsequently trimmed and filtered based on quality scores by using a filtration algorithm that removed low-scoring sections of each read and preserved the longest remaining fragment. RNA-Seq statistics are summarized in S1 Table.
Genotyping
Both parental transcriptomes have been sequenced and are publically available through viewer.xgsc.txstate.edu/data/transcriptomes 23,24. To bring both parental transcriptomes to a comparable level, Reciprocal Best Hit (RBH) sequences from each parental transcriptome were retrieved to be used as species-specific transcriptome references. Processed sequencing reads were mapped to each transcriptome respectively using Bowtie2 head-to-head mode 25. The alignment files of control skin and paired-tumor samples were processed into pileup format using samtools 26 and were further processed to .vcf format using bcftools. Base call qualities were recalibrated and genotypes of both normal skin and tumor of each fish were determined based on genotype likelihood 27. Genotype was determined by the number of sequencing reads covering variant sites ≥ 40% of average sequencing depth, recalibrated sequencing read mapping quality Phred score ≥ 30, Phred genotype likelihood = 0 and alternative genotyping likelihood ≥ 20. In the BC1 data set, each gene has 4 genotype calls: GT1 (genotype call using X. maculatus as reference in skin sample), GT2 (genotype call using X. hellerii as reference in skin sample), GT3 (genotype call using X. maculatus as reference in tumor sample), and GT4 (genotype call using X. hellerii as reference in tumor sample). Genes with consistent genotype calls (GT1=GT2=GT3=GT4) were included in subsequent analysis. In BC5 data set, where only tumor data are available, genes with genotype calls that were consistent by using both parental references were included in subsequent analysis. To visualize gene models and genes of different genotypes, chromosome position for each gene model was retrieved from the X. hellerii genome annotation and was plotted on the chromosome bar graph using R (3.1.2). Heterozygous and homozygous genes were highlighted by different colors indicated in the figure legend (Fig 1, Fig 2, S4 Figure). An outline of the genotyping process is summarized in S2 Figure. One BC1 fish and one BC5 tumor showed heterozygosity genotype for the whole LG5. Previous published work shows ~20% of tumor-bearing backcross hybrids genotype as heterozygous for the cdkn2x locus. It was speculated the melanoma-bearing, cdkn2x heterozygous fish might have acquired a different mechanism from xmrk-R(Diff) interaction to initiate melanomagenesis and we therefore excluded these two fish from our study 12,28.
Figure 1. Genotyping of backcross hybrids.
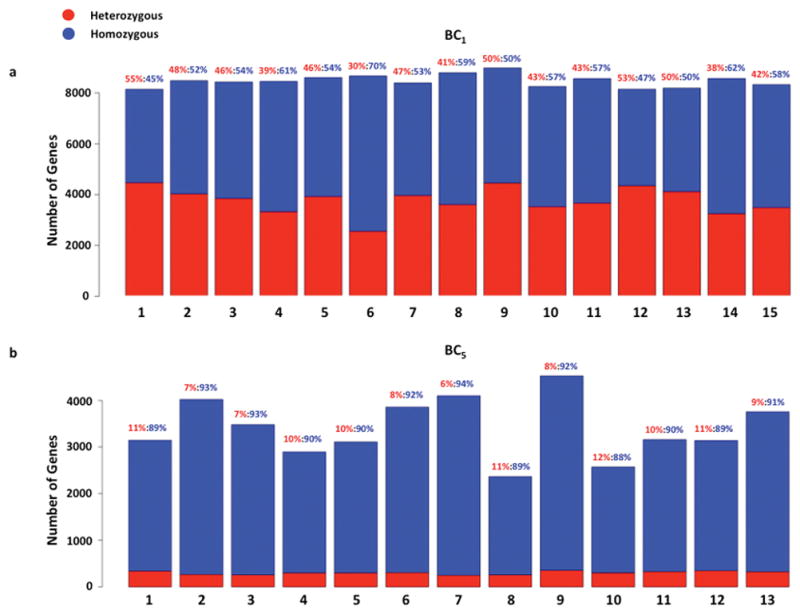
Genotypes were determined for 8,000 - 9,000 genes with high confidence for BC1 hybrids. (a) According to Mendelian inheritance, 50% of the genes in the BC1 hybrid genome should be heterozygous X. maculatus/X. hellerii, and the other 50% should be homozygous X. hellerii/X. hellerii ([heterozygous]/[homozygous]=1:1). The number of homozygous genes is 11% over-represented than expected (p=0.0086), and the number of heterozygous genes is 11% under-represented (p=0.0022). (b) Similarly, BC5 should contain 1/32 (3.13%) of their genes as heterozygous and 31/32 (96.87%) homozygous ([heterozygous]/[homozygous]=1:31). The number of heterozygous genes is over-represented by 2.87 fold (p-value=3.49E-15) and the number of homozygous genes is same as expected (p=0.25).
Figure 2. The homozygosity of X. hellerii alleles defines the R(Diff) region.

The region that only contained homozygous gene on LG5 defined the R(Diff) region. For each fish sample, heterozygous and homozygous genes were plotted on LG5 of the X. hellerii genome chromosome assembly. The genotype of each fish is represented by two LG5 genotype plots. The left one shows heterozygous genes and the right one shows homozygous genes. Genotyping information from the BC1 hybrid defined the R(Diff) region to be 5.81Mbp long (between the two dashed lines) and consists of 164 gene models. 114 genes were genotyped in our RNA-Seq experiments and were homozygous for the X. hellerii allele. Genotyping coding genes within the R(Diff) region of BC5 hybrids showed 79 genes can be genotyped. 10 of which are heterozygous and 69 of which were homozygous. Known RAD-Tag markers that were mapped to the R(Diff) region are labeled on the side of the genotype plots. Fish 9 of BC1, and Fish 13 of BC5 were excluded from this figure (See Materials and Methods for detail).
Segregation distortion
If N = backcross generation, in the backcross fish samples, a 2(N-1): 1 distribution of heterozygous: homozygous genotype is expected for each locus according to Mendelian segregation. To identify genes that experienced significant segregation bias from the expected ratio, we performed Chi-squared tests for observed vs. expected genotypes for each gene given the available sample size. Due to variation of sequencing depth and genotyping call quality, not every backcross individual received a genotype call at every loci. For each gene, numbers of heterozygous and homozygous backcross individuals were added together to determine sample size, which was subsequently used to calculate the expected number of heterozygous and homozygous individuals. The Chi-squared test was performed using Office Excel. A p < 0.05 was used to determine if a gene had a segregation distortion.
Conserved homozygous LG5 sequence analysis
To define R(Diff), where only homozygous genes for X. hellerii alleles are expected on LG5 in backcross hybrids, the genotypes of each gene on LG5 for each fish individual was plotted on the chromosome assembly of the X. hellerii genome using genome annotation as a guide 24. The LG5 chromosome plots from each individual fish were subsequently aligned to each other. The smallest region lacking heterozygosity for expressed genes for each tumor-bearing fish was defined as the R(Diff) locus. The start and end positions of the defined region were delimited by heterozygote genes. The genomic sequence of the R(Diff) region was retrieved from the X. maculatus and the X. hellerii genome chromosome assemblies, respectively 23,24.
Allele specific gene expression
To determine allele specific gene expression in the backcross hybrid genomic background, we built a hybrid reference transcriptome by combining the previously established RBH sequences of both parental alleles for each gene to allow differential mapping of sequencing reads. Short sequencing reads from each sample were mapped to this compiled transcriptome using Bowtie2 25. To quantify species-specific read counts, sequence alignment output files were filtered using custom Perl scripts. Because a short sequencing read that aligns to the common sequence of both parental alleles cannot be exclusively assigned to either allele, only sequencing reads that aligned to just one allele or the other without mismatch were kept for allele specific gene expression quantification 20.
Analysis of differentially expressed alleles
The allele-specific gene expression table was filtered based on the genotype of each gene in each fish. Only genes that were homozygous in at least two fish were kept for differential expression analysis to allow biological replicates for differential expression analysis. Any given gene might show homozygous expression in one fish, but heterozygous expression or no expression in another fish. To analyze differential expression of homozygous genes, expression counts for the homozygous samples for that gene were kept in the count table for analysis. Due to this data handling process, the alleles to be tested have different sample sizes. Therefore, to test differential expression of alleles between skin controls and melanoma tumor samples, we modeled each allele to be tested independently for differential expression test. We modeled allele count Kij as negative binomial (NB) distributed 29,30
for allele i in sample j. Mj is the library size, which equals to the total of all the X. hellerii allele counts for sample j. pip is the concentration of allele i in group p (normal skin or tumor). ϕi is the dispersion of allele i. Mean μij = Mjpip, and variance μij (1+ μijϕi). Differential expression analysis was assessed for each allele using the exact test that was implemented in edgeR to comply to over dispersed data sets 30. Alleles that change in expression levels by at least 2-fold with a False Discovery Rate (FDR) less than 0.05 (log2FC ≥ 1 or log2FC ≤ −1, FDR ≤ 0.05, log2CPM ≥ 1) were defined as Differentially Expressed Alleles.
Comparison of transcriptome gene model sequences
X. maculatus and X. hellerii allele sequences of genes in the R(Diff) region were extracted from transcriptome reference sequences from each species 24. Allelic sequence comparisons were carried out using blast (2.2.30). X. hellerii R(Diff) gene cDNA sequences were also queried against the X. maculatus protein sequence database (Xiphophorus_maculatus.Xipmac4.4.2.pep.all.fa) using blastx. Query coverage and subject coverage of both blast and blastx were calculated by blast and blastx respectively.
Data Availability
Raw sequencing data files are uploaded to GEO. The accession number will be available upon manuscript accepted for publication.
Results
Genotyping X. maculatus - X. hellerii backcross hybrids
To generate first generation backcross (BC1) hybrids, female X. maculatus Jp163 A and male X. hellerii (Sarabia) were crossed to produce F1 hybrids. These F1 hybrids exhibit enhanced pigmentation in the dorsal fin, but do not develop invasive tumors. F1 hybrid males were then backcrossed to X. hellerii females to generate the BC1 animals, about 25% of which showed very heavy pigmentation and developed invasive melanoma tumors (Figure S1).
RNA-Seq reads from normal skin and melanoma tumor, of tumor-bearing BC1 fish, were aligned to X. maculatus and X. hellerii reference transcriptomes. Sequence polymorphisms, including single nucleotide polymorphisms (SNPs), or insertion and deletions (InDels), specific to either X. maculatus or X. hellerii gene models were used as markers to trace expressed genes to each parental allele. The genotype was then inferred by alignment of allele specific sequence reads to the sequence variation sites. If a transcript showed exclusively an allele from one parent, or the other, we inferred that gene to be homozygous. In contrast, if sequencing reads with polymorphisms characteristic of both parental species were present, we inferred that gene to be heterozygous. Thus, the genotype is determined by: (a) the number of sequencing reads covering a SNP or InDel at ≥ 40% of average sequencing depth, (b) recalibrated sequence read mapping quality Phred score ≥ 30, and (c) Phred genotype likelihood = 0 with alternative genotyping likelihood ≥ 20.
Almost 9,000 genes could be genotyped in tumor and skin samples from each BC1 fish (Fig. 1). A total of 14,030 genes were genotyped based on at least one fish sample (S3 Table). The number of heterozygous genes per fish ranged from 2,559 to 4,472, while the percent of heterozygous genes ranged from 30% to 55% of the genotyped genes. Compared to an expected 50% of heterozygous genes in the BC1 hybrid, this method under-represented heterozygous genes by 5.4% (p<0.05). The number of homozygous genes per fish ranged from 3,670 to 6107, and percent of homozygous genes ranged from 45% to 70% of the genotyped genes (Fig. 1, S2 Table). Compared to an expected 50%, the homozygous genes were over-represented by 5.4% (p<0.05).
The fifth generation of backcross hybrids (BC5) were produced in an independent series of successive crosses from F1 hybrids originating from the cross: male X. maculatus Jp 163 A mated to female X. hellerii (Lancetilla) strain. F1 hybrid females, which had no tumors, were then backcrossed to X. hellerii males to produce the backcross generations leading to BC5. For each backcross generation, fish showing pigmentation enhancement (e.g., F1 phenotype), but not melanoma tumors, were used in successive backcrosses leading to the BC5 generation.
In BC5 tumor samples, a total of 10,486 genes were genotyped based on at least one tumor sample (S4 Table). The number of heterozygous genes per BC5 tumor ranged from 250 to 356 in BC5 individuals. Heterozygous genes account for 6% to 12% of genotyped genes. Heterozygous genes are expected to account for 3.13% of genotyped genes, and thus our observed heterozygous genes are over-represented (p<0.05). The number of homozygous genes per tumor ranged from 2,104 to 4,176 (Figure 1, S2 Table), with percent of homozygous genes ranging from 88% to 94% of genotyped genes. The homozygous genes are not statistically different from an expected ratio of 96.87% (p>0.05).
The homozygosity of X. hellerii alleles defines the R(Diff) region
Homozygous and heterozygous genes are expected to be randomly distributed among all BC1 individuals according to the backcross genetic background, and show no segregation distortion. We determined the majority of 14,030 genotyped loci in BC1 samples followed Mendelian expectations (S3 Table). In contrast, a total of 3,108 genes showed segregation distortion, with 2,314 genes biased toward homozygous genotypes and 794 genes biased toward heterozygous genotypes (p<0.05; S3 Table; S4 Figure a). We identified the chromosome locations for the genes showing segregation distortion, and heterozygous biased genes were found most frequently on the sex chromosome (LG21), consistent with inheritance of the xmrk oncogene. A similar result is observed in BC5 fish samples (S4 Figure c d; S4 Table). Genes with a homozygous bias in BC1 individuals were enriched on LG5, localizing around the region previously forwarded as harboring the R(Diff) locus (S4 Figure a).
To define the R(Diff) region, genomic locations of heterozygous and homozygous genes expressed in BC1 tumor bearing samples were used to set boundaries identifying regions containing only homozygous genes on LG5. This process identified a 5.81Mbp homozygous region (from 10.09Mbp to 15.89Mbp) with only X. hellerii alleles (Fig. 2). This region contains 164 gene models, including the previously forwarded R(Diff) candidate, cdkn2x (for details of all gene models, refer to S5 Table) and 44 previously reported RAD-Tag markers 31.
Extensive backcrossing of tumor bearing fish would be expected to produce relatively random recombination events in each successive backcross hybrid generation that could eliminate genes from the R(Diff) region that are not needed for melanoma development. Thus, we hypothesized the R(Diff) locus should be further restricted to genes that exhibit no heterozygosity in both BC1 and BC5 hybrids. In BC1 hybrids, 114 of the 164 gene models within R(Diff) region could be genotyped and were determined to be homozygous. 79 genes could be genotyped in BC5, with 69 showing retained homozygosity (S5 Table; Fig. 2).
Molecular characterization of the R(Diff) locus
By defining R(Diff) region, the genome sequences of the X. maculatus R(Diff) and X. hellerii R(Diff) regions can be compared. X. maculatus and X. hellerii R(Diff) share 164 gene models without inversion of gene order (Fig. 3a). To identify alleles that have primary structural differences in peptide products, amino acid sequence alignments between X. maculatus and X. hellerii alleles were compared to their nucleotide sequence alignments. If the amino acid sequence alignment is significantly shorter than transcript sequence alignment, the shorter amino acid sequence alignment is likely due to either altered amino acid sequence resulting from a frame shift, or early termination of amino acid sequence of one allele. Eleven genes show more than 20% shorter amino acid sequences when aligned to their corresponding nucleotide sequence alignment (fam184b, cnga1, zp4, atf7ip, doc2d, nuoI, zzchc7, cntf, spr40, adgrl3.1 and uncharacterized protein ENSXMAT00000017676; Fig 3b; S6 Table).
Figure 3. Sequence analysis of gene models in the R(Diff) region.

Genomic sequences of both X. maculatus and X. hellerii R(Diff) were extracted from the respective genome assemblies 24. (a) RAD-Tag markers that are mapped to the R(Diff) region are labeled with their physical location on X. maculatus R(Diff) sequences. Uncharacterized nucleotide of X. maculatus and X. hellerii R(Diff) are highlighted for both genomic sequences. X. maculatus and X. hellerii R(Diff) share the same 164 gene models (blue dots). Gene synteny is also retained between the two species. (b) Transcript and protein sequences of X. maculatus alleles and X. hellerii alleles of these 164 genes were compared to each other. Genes that showed 20% lower alignment (alignment length/X. hellerii sequence length and alignment length/X. maculatus transcript length) in protein sequence comparison (black dot) than nucleotide sequence comparison (red dot) are labeled with gene names.
Differential expression of R(Diff) genes between tumor and skin present candidates for xmrk-interacting genes
In addition to comparing gene content, we analyzed differential gene expression between tumor and normal skin samples for genes in the identified R(Diff) region to distinguish genes that may be transcriptionally relevant to melanomagenesis. Normal skin from the same melanoma-bearing hybrid progeny share the same genetic background as the melanoma tumor. In addition, the xmrk oncogene is lowly expressed in the skin while it is highly expressed in melanoma tumor. Compared to the paired normal skin, xmrk expression is 8.7 fold higher than the melanoma tumors (S7 Figure; p-adj=4.16×10−28). We identified 22 genes in the 5.81Mbp R(Diff) region that showed differential expression (log2FC≤-1 or log2FC≥1, p-adj< 0.05) in the tumor samples (eight genes were up-regulated (SPR40-like, kitb, tyrp1b, fam184b, rbm47, prf1, cdkn2a/b and eef1a1a) and thirteen genes were down-regulated in tumors (sept3, adgrl3.1, camk2d2, map9, slit2.1, slit2.2, apbb2b, suclg1, bdh2, nuol, sod3b, myoz2b, and acsl1a; Fig. 4).
Figure 4. Differential gene expression in the R(Diff) region comparing melanoma tumor and normal skin.

Differential gene expression was performed on homozygous genes in the R(Diff) region between BC1 melanoma tumors and paired normal skin samples from the same animal. Eight R(Diff) genes were over-expressed in tumors (Log2FC>2, FDR<0.05), and thirteen genes were under-expressed in tumors (Log2FC>2, FDR<0.05).
Discussion
Over the past two decades, several research groups have dedicated substantial effort in attempting to identify the molecular nature of the hypothetical R(Diff) locus that regulates melanomagenesis in Xiphophorus backcross hybrids 12–16,32–34. Genetic linkage analysis and functional interpretation directed attention towards an X. maculatus homolog of the human CDKN2A and CDKN2B gene, termed cdkn2x, to be a strong candidate for the R(Diff) 12–16. The map location and function of cdkn2x fit with the two-gene segregation model of Xiphophorus melanoma; and further, mutations in the human CDKN2A ortholog have been shown associated with human melanoma 35,36. However, these previous reports also show cdkn2x heterozygosity (for X. maculatus and X. hellerii alleles) in ~20% of tumor bearing BC1, as well as X. maculatus allele biased expression in cdkn2x heterozygous tumor bearing individuals. Therefore, inheritance of cdkn2x alone does not fully explain melanomagenesis in the Gordon-Kosswig backcross model. Of fifteen BC1 and thirteen BC5 tumor-bearing hybrids, seven of eight genotyped BC1 individuals were homozygous for cdkn2x and four of five genotyped BC5 individuals showed a homozygous genotype for this gene. These results are very similar to observations of cdkn2x genotype distributions in previous studies 12,13. Within the cdkn2x homozygous individuals, allele specific gene expression analyses showed an up-regulation of homozygous cdkn2x in melanoma tissue, yet it did not eliminate melanomagenesis, as one may expect from a tumor suppressor. Compared to previous observations that showed the X. maculatus cdkn2x allele is up-regulated in the 20% of tumor-bearing fish heterozygous for cdkn2x, our results indicate X. maculatus and X. hellerii alleles have no functional differences in the ability to repress melanomagenesis. The cdkn2x gene resides 0.42Mbp to the end of 5.81Mbp R(Diff) homozygous region defined in this study, supporting our hypothesis that cdkn2x locates close to the core R(Diff) candidate gene(s) on physical map.
Herein, we utilized newly acquired genome sequence assemblies of the parental species to define the R(Diff) region and compare the genome sequences and gene models within this region between X. maculatus and X. hellerii 23,24. We mapped RNA-Seq sequencing reads generated from tumor and normal skin of melanoma-bearing backcross individual fish to reference transcriptomes of both parental species X. maculatus and X. hellerii, and used species specific sequence variations, identified in this study, to characterize allele-specific gene expression patterns in melanoma and normal skin. Genes that we observed to express from both alleles were inferred to be heterozygous, and alternatively, homozygous if only one allele was detected. BC1 fish should show a 50% of homozygosity for X. hellerii genes based on Mendellin segregation. We observed a 5.4% over-representation of homozygous genotype (p<0.05) and a 5.4% under-representation of heterozygous genotype (p<0.05). Because the X. hellerii parent genome contributed 75% of the genome to the BC1 hybrids, and the X. maculatus genome contributed 25%, 75% of total allele specific sequence read counts were expected to be produced from X. hellerii alleles, and 25% of total allele specific sequencing read counts were expected to be derived from the X. maculatus alleles; under the hypothesis that all genes are expressed equally from both species’ alleles in the hybrid genetic background. Total sequencing read counts from the X. maculatus alleles is 15% over-represented from expected (calculated as 25% × total allele specific sequencing read count; p<0.05, S3 Figure), while total sequencing read counts form X. hellerii alleles are at expected values (75% × total allele specific sequencing read count; p=0.16, S3 Figure).
Due to dilution of X. maculatus genomic content in hybrids upon successive backcrosses to the X. hellerii parent, the X. maculatus genomic content decreased to a theoretical 1.56% in BC5 fish, and the X. hellerii genomic content increased to a theoretical 98.4% in BC5 fish (S3 Figure b). We found the total sequencing read counts from X. maculatus alleles (1.6% × total allele specific sequencing read count) and X. hellerii alleles (98.4% × total allele specific sequencing read count), and the actual total sequencing read counts from X. maculatus and X. hellerii were not statistically different from expected values (p>0.05). These observations imply allele specific gene expression reflects the genome make-up within interspecies hybrids. The decrease of X. maculatus alleles in hybrids also led to a lower probability of a gene to be heterozygous, resulting in an observed average homozygosity rate to be 91% (Fig 1b). The percent of heterozygosity is over-represented compared to expected ratio of 3.13% (or 2−5 ×100%; p<0.05) while the percent of homozygosity is not statistically different from the expected 96.87% (or 1–2−5 ×100%; p=0.25).
By SNP and InDel mapping of allele specific expression in backcross hybrids to the genome of X. hellerii, we defined R(Diff) as the smallest consistently homozygous region. We assign the R(Diff) effect to a region of 5.81Mbp containing 164 known gene models, including the previously mapped cdkn2x (S5 Table). We have observed a total of 209 recombination events in all BC1 fish. Considering the genome size of 653.69 Mb, we estimated recombination frequency is ≈0.32 recombination per Mb. Therefore, we may have expected to observe 1.86 recombination events in a randomly selected 5.81 Mbp region. However, we observed no recombination in the identified 5.81 Mbp R(Diff) region. This lack of recombination is not likely to be random (X2=1.87, Df=1, p=0.171), and future analysis of higher number of BC1 hybrids would substantiate this observation.
Our segregation distortion analysis in BC1 melanoma-bearing hybrid progeny shows genes biased to homozygosity to be highly enriched on chromosome 5 (S4 Figure b). This is consistent with the location of the R(Diff) locus. All melanoma-bearing hybrid progeny showed homozygosity for X. hellerii alleles. Genes that are biased to homozygosity were also observed to be enriched on LG 14 (138 genes) and 23 (177 genes). However, unlike the X. hellerii homozygosity for alleles in the R(Diff) locus, the genes on LG 14 and 23 were heterozygous in some of the hybrid progeny. Since these LG 14 and 23 regions were not exclusively homozygous in all melanoma bearing hybrids these regions do not define a R(Diff) locus (S6 Figure; S8 Figure).
Data from both BC1 and BC5 hybrids allowed potential recombination events to be analyzed, therefore providing more opportunity to eliminate heterozygous genes from a functional R(Diff) locus. X. hellerii genomic content in backcross progeny increases with each successive advanced backcross generation, and yet the homozygous gene markers did not further shorten from the defined 5.81Mbp R(Diff) region. In future studies, heterozygous gene markers in backcross progeny bearing enhanced melanization (xmrk +/-, X. maculatus R(Diff) +/-) may be utilized to further define the R(Diff) region.
Except for the cdkn2x gene candidate, little is known about the underlying molecular mechanism of R(Diff) function. The R(Diff) region defined in this study encodes many loci that have potentially opposing functions as candidate tumor promoting or tumor suppressing genes (i.e. kitb, cdkn2a/b), and also identifies new species-specific R(Diff) candidate genes. These observations, and the strong definition of a clearly defined 5.81 Mbp homozygous region, suggest melanomagenesis in Xiphophorus backcross hybrid models may be a consequence of gene interactions between xmrk and several genes within a multigenic R(Diff) region, rather than a simple two-gene model. However, tight clustering of several interacting genes to the same region of LG5 led to the inheritance of this tumor suppressing function behaving as a two-gene model. The replacement of X. maculatus R(Diff) with its X. hellerii counterpart results in the acquisition of X. hellerii R(Diff) unique genes and alleles, with a corresponding loss of X. maculatus R(Diff) unique genes and alleles in tumor bearing fish. We recognize that sequence differences alone in these genes could be involved in phenotypic change without transcriptional modulation of gene expression. Protein sequence alignments of 11 genes are at least 20% shorter than their cDNA sequence alignments. These shortened alignments will lead to shorter protein sequences of one allele, potentially from early termination of translation, or a different protein sequence of one allele potentially from frame shift variations. Such functional discrepancies may result in the loss-of-function or gain-of-function of X. hellerii R(Diff) genes. Further investigations will be needed to assess these many various possibilities. We notice there are seven gene models that are uniquely annotated in the X. maculatus R(Diff) region and nine gene models that are only annotated in the X. hellerii R(Diff) region. However, it is likely these differential loci in R(Diff) are due to genome annotation artifacts and/or poorly assembled genome contigs (Figure S5). Re-sequencing both X. maculatus and X. hellerii genome using long sequencing read technology (e.g., Pacific Biosciences SMRT technology) may resolve these problems and reveal true species-specific gene models in the R(Diff) locus.
Finally, we characterized gene expression changes in the R(Diff) region for tumor and paired normal skin from the same individuals to highlight genes that may interact with xmrk and lead to induction or progression of melanoma. Tumor and normal skin from the same fish share the same genetic information, yet show very distinctive gene expression patterns and phenotypes. Among the 21 differentially expressed homozygous X. hellerii alleles, down-regulated slit2 and sod3b, and up-regulated prf1, eef1a1a, kitb, tyrp1b and cdkn2x have been reported as melanoma related 12–14,28,37–55. The kitb gene and tyrp1b gene are both functionally related to the mitf gene 46. The human KIT protein activation in melanocytes leads to increased recruitment of c300 coactivator protein to MITF protein 43,50,52. MITF protein targets the TYRP1 promoter and induces the expression of TYRP1 that serves as a core enzyme for melanin synthesis 37,38,44,47,53–56. PRF1 is a T-cell effector related gene and was found to frequently co-express with memory/homing-associated genes in CD8 T-cells that are activated by melanoma cell surface marker MELAN-A 42. Thus, T-cells that up-regulated prf1 are likely to be activated in tumor-bearing fishes, and while this activation might slow tumor growth, it does not fully repress melanoma tumor formation. The gene encoding the elongation factor eEF1A is up-regulated in prostate cancer and may act as an oncogene 45. In addition to serving as an elongation factor, eEF1A plays critical roles in actin cytoskeleton organization and in functions involved in cell migration, morphology and cell death 39,40,49. Specifically inhibiting eEF1A has been shown to repress melanoma, suggesting up-regulation of eEF1A may play a central role in melanomagenesis 51. Due to a report suggesting that human SLIT2 activity reduces cellular invasion by stabilizing the interaction of N-cadherin with β-catenin, we speculate the down-regulation of slit2 could hallmark increased invasive behavior of melanoma tumor cells 41. Although the role of SOD3B gene in melanoma is unclear, it is highly expressed in melanoma tumor cells that survive chemotherapy and exhibit oxidative stress response. The down-regulation of sod3b in melanoma may indicate Xiphophorus melanoma experience less oxidative stress than normal skin, or fail to respond normally to oxidative stress 48.
Because genes in R(Diff) are functionally interrelated, and genetically linked in the Gordon-Kosswig spontaneous Xiphophorus melanoma model, we hypothesize the active R(Diff) is a cluster of genes, rather than a single gene.
In summary, we have defined the R(Diff) locus as a 5.8Mbp region, and have characterized the expression of genes within this region to expand our understanding how this interesting tumor suppression locus may function in Xiphophorus melanomagenesis. Sequences and potential functional discrepancies between both parental allelic regions, as well as altered expression of R(Diff) genes between melanoma and normal skin tissue may account for the xmrk driven melanomagenesis in the Gordon-Kosswig melanoma model.
Supplementary Material
S1 Table. Statistics of RNA-Seq
S2 Table. Statistics of genotype and allele specific expression
S3 Table. Genotyped genes in BC1 fish
S4 Table. Genotyped genes in BC5 fish
S5 Table. R(Diff) region gene models retained from X. hellerii genome
S6 Table. Sequence comparison of X. maculatus and X. hellerii genes
Xiphophorus backcross of the F1 X. maculatus Jp163 A -X. hellerii interspecies hybrid to the X. hellerii parental line. In this backcross, 25% of the BC1 progeny generate spontaneous melanoma, suggesting a two-gene model located on two separate chromosomes. BC1 progeny that shared the same pigmentation pattern with the F1 (pigmented dorsal fin, no melanoma tumor) were utilized with X. hellerii as parents to produce subsequent backcross progeny.
This study consists of 3 sections: Generating backcross hybrids and performing RNA-Seq (yellow); genotyping (blue); and quantification of allele specific gene expression (green). The software and pipelines are described in the Materials and Methods. All custom scripts are available upon request.
(a) 75% (indicated by dotted line) and 25% of the genomic content of BC1 was from X. hellerii and X. maculatus parents due to random assortment. Using these values we estimated expected sequencing read counts from BC1. Sequencing read counts from X. hellerii is same as expectation (p>0.05). Sequencing read counts form X. maculatus is 15% over-represented compared to expected read counts (p<0.05). (b) In BC5 hybrids, 98.4% (indicated by dotted line) of the hybrid genome is from X. hellerii parent, and 1.6% of the hybrid genome is the from X. maculatus. Sequencing read counts from X. maculatus and X. hellerii alleles are the same as expectation (p>0.05). S=Skin; T=tumor.
Genotypes were expected to randomly distribute among all of the BC1 progeny. Since we focused only on the tumor-bearing fish, heterozygosity of LG21 and homozygosity of LG5 were enriched (a and b). As observed in BC1, segregation distortion biased to heterozygous genotype showed enrichment on LG21, and segregation distortion biased to homozygous genotype were enriched on LG5, which was revealed by a higher density of gene models on LG21 and LG5 (a), and by normalized count of segregation distorted genes to the total number of gene models per chromosome (b). In BC5 (c and d), segregation distortion biased to heterozygous genotype showed enrichment on LG21. These observations are shown by a higher density of heterozygous genes on LG21 (c), and by normalized count of segregation distorted genes to total number of gene models per chromosome (d).
(a) There are seven genes uniquely annotated to the X. maculatus R(Diff) region. They are annotated on X. maculatus LG5 R(Diff) locus, but annotated elsewhere in the X. hellerii genome. None of the seven genes showed sequence homology between X. maculatus genome sequences and the X. hellerii R(Diff) region due to uncharacterized nucleotides in the comparable region in the X. hellerii R(Diff) region. The X. maculatus alleles of ociad1, wfs1a, drd5, egr4 and arhgef38 showed sequence alignment to the X. hellerii genomic sequences retrieved from the annotation of these genes. However, without filling the gapped sequences of X. hellerii R(Diff), we do not have enough evidence to conclude whether these genes have duplications, translocations, or are miss-annotated in X. hellerii genome. (b) There are nine genes annotated in the X. hellerii R(Diff) region. They are annotated in multiple loci, including LG5 R(Diff) region in the X. hellerii genome, but annotated in non-R(Diff) region in the X. maculatus genome. Genomic sequence for each of the nine genes was retrieved from the X. maculatus genome and was subsequently compared to sequences extracted from each annotated locus from the X. hellerii genome. For each gene, sequence comparison results showed the best alignments between X. maculatus sequence and the multiple X. hellerii sequences were not from the X. hellerii sequences from R(Diff). This indicates there are annotation errors in placing these genes on X. hellerii LG5 R(Diff) locus. Seven of the nine genes showed either no sequence alignment or only short fragment alignments between the X. hellerii and the X. maculatus alleles. ENSXMAT00000000764 (muc13a) is annotated to LG5-R(Diff) or unplaced chromosome in the X. hellerii genome, and unplaced chromosome in the X. maculatus genome. The gene muc13a genomic sequence from the X. hellerii unplaced chromosome showed 100% alignment with X. maculatus muc13a, and genomic sequence from the X. hellerii R(Diff) showed 98% alignment with X. maculatus muc13a, indicating X. hellerii has duplication of muc13a. Similarly, ENSXMAT00000004062 (alox15b) is annotated to X. hellerii R(Diff) and LG12, and the genomic sequence from X. hellerii LG12 aligns the best (100%) with X. maculatus alox15b, and sequence from R(Diff) produced 94% alignment. However, for both muc13a and alox15b, their X. hellerii LG5 sequences showed head-to-head alignments with comparable sequences of the X. maculatus LG5, therefore indicating the X. maculatus genome may have annotation errors in both alox15b and muc13a loci.
For each fish sample, heterozygous and homozygous genes were plotted on LG14 and LG23 of the X. hellerii genome/chromosome assembly. The genotype of each fish is represented by two genotype plots. The left one shows heterozygous genes and the right one shows homozygous genes.
Gene expression in xmrk was measured and normalized as Count Per Million (CPM) sequencing read counts. The xmrk gene is highly expressed in melanoma tumor showing 8.7 fold over-expression compared to expression in paired normal skin samples.
Genotypes of expressed genes were assessed (See materials and methods) and genes subsequently mapped to chromosomes.
Acknowledgments
The authors would like to thank the staff of the Xiphophorus Genetic Stock Center, Texas State University, for maintaining the pedigreed parental fish lines, performing interspecies crosses, and caring for the hybrid animals used in this study. This work was supported by the National Institutes of Health, Division of Comparative Medicine, R24-OD-011120 and R24-OD-011199.
References
- 1.GH Über Melanombildungen bei Bastarden von Xiphophorus Helleri und Platypoecilus Maculatus var. Rubra. Journal of Molecular Medicine. 1928:7. [Google Scholar]
- 2.Gordon M. The Genetics of a Viviparous Top-Minnow Platypoecilus; the Inheritance of Two Kinds of Melanophores. Genetics. 1927;12(3):253–283. doi: 10.1093/genetics/12.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.KK Über Bastarde der Teleostier Platypoecilus und Xiphophorus. Z Indukt Abstamm Vererbungsl. 1928:44. [Google Scholar]
- 4.Patton EE, Mathers ME, Schartl M. Generating and analyzing fish models of melanoma. Methods Cell Biol. 2011;105:339–366. doi: 10.1016/B978-0-12-381320-6.00014-X. [DOI] [PubMed] [Google Scholar]
- 5.Walter RB, Kazianis S. Xiphophorus interspecies hybrids as genetic models of induced neoplasia. ILAR J. 2001;42(4):299–321. doi: 10.1093/ilar.42.4.299. [DOI] [PubMed] [Google Scholar]
- 6.Adam D, Dimitrijevic N, Schartl M. Tumor suppression in Xiphophorus by an accidentally acquired promoter. Science. 1993;259(5096):816–819. doi: 10.1126/science.8430335. [DOI] [PubMed] [Google Scholar]
- 7.Adam D, Maueler W, Schartl M. Transcriptional activation of the melanoma inducing Xmrk oncogene in Xiphophorus. Oncogene. 1991;6(1):73–80. [PubMed] [Google Scholar]
- 8.Weis S, Schartl M. The macromelanophore locus and the melanoma oncogene Xmrk are separate genetic entities in the genome of Xiphophorus. Genetics. 1998;149(4):1909–1920. doi: 10.1093/genetics/149.4.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wittbrodt J, Adam D, Malitschek B, et al. Novel putative receptor tyrosine kinase encoded by the melanoma-inducing Tu locus in Xiphophorus. Nature. 1989;341(6241):415–421. doi: 10.1038/341415a0. [DOI] [PubMed] [Google Scholar]
- 10.Fornzler D, Wittbrodt J, Schartl M. Analysis of an esterase linked to a locus involved in the regulation of the melanoma oncogene and isolation of polymorphic marker sequences in Xiphophorus. Biochem Genet. 1991;29(11–12):509–524. doi: 10.1007/BF02426867. [DOI] [PubMed] [Google Scholar]
- 11.Morizot DC, Siciliano MJ. Linkage group V of platyfishes and Swordtails of the genus Xiphophorus (Poeciliidae): linkage of loci for malate dehydrogenase-2 and esterase-1 and esterase-4 with a gene controlling the severity of hybrid melanomas. J Natl Cancer Inst. 1983;71(4):809–813. [PubMed] [Google Scholar]
- 12.Kazianis S, Gutbrod H, Nairn RS, et al. Localization of a CDKN2 gene in linkage group V of Xiphophorus fishes defines it as a candidate for the DIFF tumor suppressor. Genes Chromosomes Cancer. 1998;22(3):210–220. [PubMed] [Google Scholar]
- 13.Kazianis S, Morizot DC, Coletta LD, et al. Comparative structure and characterization of a CDKN2 gene in a Xiphophorus fish melanoma model. Oncogene. 1999;18(36):5088–5099. doi: 10.1038/sj.onc.1202884. [DOI] [PubMed] [Google Scholar]
- 14.Nairn RS, Kazianis S, McEntire BB, Della Coletta L, Walter RB, Morizot DC. A CDKN2-like polymorphism in Xiphophorus LG V is associated with UV-B-induced melanoma formation in platyfish-swordtail hybrids. Proc Natl Acad Sci U S A. 1996;93(23):13042–13047. doi: 10.1073/pnas.93.23.13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nairn RS, Morizot DC, Kazianis S, Woodhead AD, Setlow RB. Nonmammalian models for sunlight carcinogenesis: genetic analysis of melanoma formation in Xiphophorus hybrid fish. Photochem Photobiol. 1996;64(3):440–448. doi: 10.1111/j.1751-1097.1996.tb03089.x. [DOI] [PubMed] [Google Scholar]
- 16.Walter RB, Rains JD, Russell JE, et al. A microsatellite genetic linkage map for Xiphophorus. Genetics. 2004;168(1):363–372. doi: 10.1534/genetics.103.019349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schartl M, Kneitz S, Wilde B, et al. Conserved expression signatures between medaka and human pigment cell tumors. PLoS One. 2012;7(5):e37880. doi: 10.1371/journal.pone.0037880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schartl M, Wilde B, Laisney JA, Taniguchi Y, Takeda S, Meierjohann S. A mutated EGFR is sufficient to induce malignant melanoma with genetic background-dependent histopathologies. J Invest Dermatol. 2010;130(1):249–258. doi: 10.1038/jid.2009.213. [DOI] [PubMed] [Google Scholar]
- 19.Butler AP, Trono D, Beard R, Fraijo R, Nairn RS. Melanoma susceptibility and cell cycle genes in Xiphophorus hybrids. Mol Carcinog. 2007;46(8):685–691. doi: 10.1002/mc.20343. [DOI] [PubMed] [Google Scholar]
- 20.Lu Y, Bowswell M, Bowswell W, Yang K, Schartl M, Walter RB. Molecular genetic response of Xiphophorus maculatus-X. couchianus interspecies hybrid skin to UVB exposure. Comp Biochem Physiol C Toxicol Pharmacol. 2015;178:86–92. doi: 10.1016/j.cbpc.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walter DJ, Boswell M, Volk de Garcia SM, et al. Characterization and differential expression of CPD and 6–4 DNA photolyases in Xiphophorus species and interspecies hybrids. Comp Biochem Physiol C Toxicol Pharmacol. 2014;163:77–85. doi: 10.1016/j.cbpc.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia TI, Shen Y, Catchen J, et al. Effects of short read quality and quantity on a de novo vertebrate transcriptome assembly. Comp Biochem Physiol C Toxicol Pharmacol. 2012;155(1):95–101. doi: 10.1016/j.cbpc.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schartl M, Walter RB, Shen Y, et al. The genome of the platyfish, Xiphophorus maculatus, provides insights into evolutionary adaptation and several complex traits. Nat Genet. 2013;45(5):567–572. doi: 10.1038/ng.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Y, Chalopin D, Garcia T, et al. X. couchianus and X. hellerii genome models provide genomic variation insight among Xiphophorus species. BMC Genomics. 2016;17:37. doi: 10.1186/s12864-015-2361-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kazianis S, Coletta LD, Morizot DC, Johnston DA, Osterndorff EA, Nairn RS. Overexpression of a fish CDKN2 gene in a hereditary melanoma model. Carcinogenesis. 2000;21(4):599–605. doi: 10.1093/carcin/21.4.599. [DOI] [PubMed] [Google Scholar]
- 29.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amores A, Catchen J, Nanda I, et al. A RAD-tag genetic map for the platyfish (Xiphophorus maculatus) reveals mechanisms of karyotype evolution among teleost fish. Genetics. 2014;197(2):625–641. doi: 10.1534/genetics.114.164293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morizot DC, Slaugenhaupt SA, Kallman KD, Chakravarti A. Genetic linkage map of fishes of the genus Xiphophorus (Teleostei: Poeciliidae) Genetics. 1991;127(2):399–410. doi: 10.1093/genetics/127.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kazianis S, Morizot DC, McEntire BB, Nairn RS, Borowsky RL. Genetic mapping in Xiphophorus hybrid fish: assignment of 43 AP-PCR/RAPD and isozyme markers to multipoint linkage groups. Genome Res. 1996;6(4):280–289. doi: 10.1101/gr.6.4.280. [DOI] [PubMed] [Google Scholar]
- 34.Kazianis S, Nairn RS, Walter RB, et al. The genetic map of Xiphophorus fishes represented by 24 multipoint linkage groups. Zebrafish. 2004;1(3):287–304. doi: 10.1089/zeb.2004.1.287. [DOI] [PubMed] [Google Scholar]
- 35.Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8(1):15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 36.Kamb A, Gruis NA, Weaver-Feldhaus J, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264(5157):436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 37.Bentley NJ, Eisen T, Goding CR. Melanocyte-specific expression of the human tyrosinase promoter: activation by the microphthalmia gene product and role of the initiator. Mol Cell Biol. 1994;14(12):7996–8006. doi: 10.1128/mcb.14.12.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertolotto C, Abbe P, Hemesath TJ, et al. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J Cell Biol. 1998;142(3):827–835. doi: 10.1083/jcb.142.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dharmawardhane S, Demma M, Yang F, Condeelis J. Compartmentalization and actin binding properties of ABP-50: the elongation factor-1 alpha of Dictyostelium. Cell Motil Cytoskeleton. 1991;20(4):279–288. doi: 10.1002/cm.970200404. [DOI] [PubMed] [Google Scholar]
- 40.Gross SR, Kinzy TG. Translation elongation factor 1A is essential for regulation of the actin cytoskeleton and cell morphology. Nat Struct Mol Biol. 2005;12(9):772–778. doi: 10.1038/nsmb979. [DOI] [PubMed] [Google Scholar]
- 41.Grossmann AH, Yoo JH, Clancy J, et al. The small GTPase ARF6 stimulates beta-catenin transcriptional activity during WNT5A-mediated melanoma invasion and metastasis. Sci Signal. 2013;6(265):ra14. doi: 10.1126/scisignal.2003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta B, Iancu EM, Gannon PO, et al. Simultaneous coexpression of memory-related and effector-related genes by individual human CD8 T cells depends on antigen specificity and differentiation. J Immunother. 2012;35(6):488–501. doi: 10.1097/CJI.0b013e31826183a7. [DOI] [PubMed] [Google Scholar]
- 43.Hemesath TJ, Price ER, Takemoto C, Badalian T, Fisher DE. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature. 1998;391(6664):298–301. doi: 10.1038/34681. [DOI] [PubMed] [Google Scholar]
- 44.Hemesath TJ, Steingrimsson E, McGill G, et al. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994;8(22):2770–2780. doi: 10.1101/gad.8.22.2770. [DOI] [PubMed] [Google Scholar]
- 45.Kido T, Hatakeyama S, Ohyama C, Lau YF. Expression of the Y-Encoded TSPY is Associated with Progression of Prostate Cancer. Genes (Basel) 2010;1(2):283–293. doi: 10.3390/genes1020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12(9):406–414. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 47.Lowings P, Yavuzer U, Goding CR. Positive and negative elements regulate a melanocyte-specific promoter. Mol Cell Biol. 1992;12(8):3653–3662. doi: 10.1128/mcb.12.8.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morvan D, Demidem A. Metabolomics and transcriptomics demonstrate severe oxidative stress in both localized chemotherapy-treated and bystander tumors. Biochim Biophys Acta. 2014;1840(3):1092–1104. doi: 10.1016/j.bbagen.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 49.Murray JW, Edmonds BT, Liu G, Condeelis J. Bundling of actin filaments by elongation factor 1 alpha inhibits polymerization at filament ends. J Cell Biol. 1996;135(5):1309–1321. doi: 10.1083/jcb.135.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price ER, Ding HF, Badalian T, et al. Lineage-specific signaling in melanocytes. C-kit stimulation recruits p300/CBP to microphthalmia. J Biol Chem. 1998;273(29):17983–17986. doi: 10.1074/jbc.273.29.17983. [DOI] [PubMed] [Google Scholar]
- 51.Van Goietsenoven G, Hutton J, Becker JP, et al. Targeting of eEF1A with Amaryllidaceae isocarbostyrils as a strategy to combat melanomas. FASEB J. 2010;24(11):4575–4584. doi: 10.1096/fj.10-162263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu M, Hemesath TJ, Takemoto CM, et al. c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev. 2000;14(3):301–312. [PMC free article] [PubMed] [Google Scholar]
- 53.Yasumoto K, Mahalingam H, Suzuki H, Yoshizawa M, Yokoyama K. Transcriptional activation of the melanocyte-specific genes by the human homolog of the mouse Microphthalmia protein. J Biochem. 1995;118(5):874–881. doi: 10.1093/jb/118.5.874. [DOI] [PubMed] [Google Scholar]
- 54.Yasumoto K, Yokoyama K, Shibata K, Tomita Y, Shibahara S. Microphthalmia-associated transcription factor as a regulator for melanocyte-specific transcription of the human tyrosinase gene. Mol Cell Biol. 1994;14(12):8058–8070. doi: 10.1128/mcb.14.12.8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yasumoto K, Yokoyama K, Takahashi K, Tomita Y, Shibahara S. Functional analysis of microphthalmia-associated transcription factor in pigment cell-specific transcription of the human tyrosinase family genes. J Biol Chem. 1997;272(1):503–509. doi: 10.1074/jbc.272.1.503. [DOI] [PubMed] [Google Scholar]
- 56.Bertolotto C, Bille K, Ortonne JP, Ballotti R. Regulation of tyrosinase gene expression by cAMP in B16 melanoma cells involves two CATGTG motifs surrounding the TATA box: implication of the microphthalmia gene product. J Cell Biol. 1996;134(3):747–755. doi: 10.1083/jcb.134.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 Table. Statistics of RNA-Seq
S2 Table. Statistics of genotype and allele specific expression
S3 Table. Genotyped genes in BC1 fish
S4 Table. Genotyped genes in BC5 fish
S5 Table. R(Diff) region gene models retained from X. hellerii genome
S6 Table. Sequence comparison of X. maculatus and X. hellerii genes
Xiphophorus backcross of the F1 X. maculatus Jp163 A -X. hellerii interspecies hybrid to the X. hellerii parental line. In this backcross, 25% of the BC1 progeny generate spontaneous melanoma, suggesting a two-gene model located on two separate chromosomes. BC1 progeny that shared the same pigmentation pattern with the F1 (pigmented dorsal fin, no melanoma tumor) were utilized with X. hellerii as parents to produce subsequent backcross progeny.
This study consists of 3 sections: Generating backcross hybrids and performing RNA-Seq (yellow); genotyping (blue); and quantification of allele specific gene expression (green). The software and pipelines are described in the Materials and Methods. All custom scripts are available upon request.
(a) 75% (indicated by dotted line) and 25% of the genomic content of BC1 was from X. hellerii and X. maculatus parents due to random assortment. Using these values we estimated expected sequencing read counts from BC1. Sequencing read counts from X. hellerii is same as expectation (p>0.05). Sequencing read counts form X. maculatus is 15% over-represented compared to expected read counts (p<0.05). (b) In BC5 hybrids, 98.4% (indicated by dotted line) of the hybrid genome is from X. hellerii parent, and 1.6% of the hybrid genome is the from X. maculatus. Sequencing read counts from X. maculatus and X. hellerii alleles are the same as expectation (p>0.05). S=Skin; T=tumor.
Genotypes were expected to randomly distribute among all of the BC1 progeny. Since we focused only on the tumor-bearing fish, heterozygosity of LG21 and homozygosity of LG5 were enriched (a and b). As observed in BC1, segregation distortion biased to heterozygous genotype showed enrichment on LG21, and segregation distortion biased to homozygous genotype were enriched on LG5, which was revealed by a higher density of gene models on LG21 and LG5 (a), and by normalized count of segregation distorted genes to the total number of gene models per chromosome (b). In BC5 (c and d), segregation distortion biased to heterozygous genotype showed enrichment on LG21. These observations are shown by a higher density of heterozygous genes on LG21 (c), and by normalized count of segregation distorted genes to total number of gene models per chromosome (d).
(a) There are seven genes uniquely annotated to the X. maculatus R(Diff) region. They are annotated on X. maculatus LG5 R(Diff) locus, but annotated elsewhere in the X. hellerii genome. None of the seven genes showed sequence homology between X. maculatus genome sequences and the X. hellerii R(Diff) region due to uncharacterized nucleotides in the comparable region in the X. hellerii R(Diff) region. The X. maculatus alleles of ociad1, wfs1a, drd5, egr4 and arhgef38 showed sequence alignment to the X. hellerii genomic sequences retrieved from the annotation of these genes. However, without filling the gapped sequences of X. hellerii R(Diff), we do not have enough evidence to conclude whether these genes have duplications, translocations, or are miss-annotated in X. hellerii genome. (b) There are nine genes annotated in the X. hellerii R(Diff) region. They are annotated in multiple loci, including LG5 R(Diff) region in the X. hellerii genome, but annotated in non-R(Diff) region in the X. maculatus genome. Genomic sequence for each of the nine genes was retrieved from the X. maculatus genome and was subsequently compared to sequences extracted from each annotated locus from the X. hellerii genome. For each gene, sequence comparison results showed the best alignments between X. maculatus sequence and the multiple X. hellerii sequences were not from the X. hellerii sequences from R(Diff). This indicates there are annotation errors in placing these genes on X. hellerii LG5 R(Diff) locus. Seven of the nine genes showed either no sequence alignment or only short fragment alignments between the X. hellerii and the X. maculatus alleles. ENSXMAT00000000764 (muc13a) is annotated to LG5-R(Diff) or unplaced chromosome in the X. hellerii genome, and unplaced chromosome in the X. maculatus genome. The gene muc13a genomic sequence from the X. hellerii unplaced chromosome showed 100% alignment with X. maculatus muc13a, and genomic sequence from the X. hellerii R(Diff) showed 98% alignment with X. maculatus muc13a, indicating X. hellerii has duplication of muc13a. Similarly, ENSXMAT00000004062 (alox15b) is annotated to X. hellerii R(Diff) and LG12, and the genomic sequence from X. hellerii LG12 aligns the best (100%) with X. maculatus alox15b, and sequence from R(Diff) produced 94% alignment. However, for both muc13a and alox15b, their X. hellerii LG5 sequences showed head-to-head alignments with comparable sequences of the X. maculatus LG5, therefore indicating the X. maculatus genome may have annotation errors in both alox15b and muc13a loci.
For each fish sample, heterozygous and homozygous genes were plotted on LG14 and LG23 of the X. hellerii genome/chromosome assembly. The genotype of each fish is represented by two genotype plots. The left one shows heterozygous genes and the right one shows homozygous genes.
Gene expression in xmrk was measured and normalized as Count Per Million (CPM) sequencing read counts. The xmrk gene is highly expressed in melanoma tumor showing 8.7 fold over-expression compared to expression in paired normal skin samples.
Genotypes of expressed genes were assessed (See materials and methods) and genes subsequently mapped to chromosomes.
Data Availability Statement
Raw sequencing data files are uploaded to GEO. The accession number will be available upon manuscript accepted for publication.