

Abstract
It is widely accepted that free radicals in tobacco smoke lead to oxidative stress and generate the popular lipid peroxidation biomarker 8-iso-prostaglandin F2α (8-iso-PGF2α). However, 8-iso-PGF2α can simultaneously be produced in vivo by the prostaglandin-endoperoxide synthases (PGHS) induced by inflammation. This inflammation-dependent mechanism has never been considered as a source of elevated 8-iso-PGF2α in tobacco smokers.
The goal of this study is to quantify the distribution of chemical- and PGHS-dependent 8-iso-PGF2α formation in the plasma of tobacco smokers and non-smokers. The influences of gender and hormonal contraceptive use were accounted for. The distribution was determined by measuring the 8-iso-PGF2α/prostaglandin F2α (PGF2α) ratio.
When comparing smokers (n=28) against non-smokers (n=30), there was a statistically significant increase in the 8-iso-PGF2α concentration. The source of this increased 8-iso-PGF2α was primarily from PGHS. When stratifying for gender, the increase in 8-iso-PGF2α in male smokers (n=9) was primarily from PGHS. Interestingly, female smokers on hormonal contraceptives had increased 8-iso-PGF2α in both pathways, whereas those not on hormonal contraceptives did not have increased 8-iso-PGF2α.
In conclusion, increased plasma 8-iso-PGF2α in tobacco smokers has complex origins, with PGHS-dependent formation as the primary source. Accounting for both pathways provides a definitive measurement of both oxidative stress and inflammation.
Keywords: 8-iso-PGF2α / PGF2α ratio, F2-isoprostanes, oxidative stress, inflammation, biomarkers, lipid peroxidation
Graphical abstract
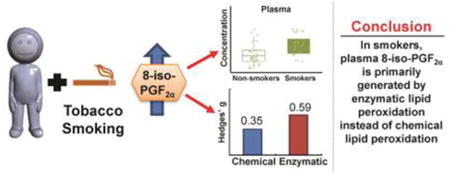
Introduction
The inhalation of combustion products from tobacco and other substances has long been hailed as an exposure synonymous with oxidative stress. Free radicals in tobacco smoke are thought to play a prominent role in oxidative stress, especially considering that a large quantity of free radicals (5×1014 per puff) is detectable within tobacco smoke [1, 2]. This detection of free radicals has led to over 3,000 publications measuring oxidative damage markers in cells, animal models and humans exposed to tobacco smoke, attempting to link oxidative stress to the myriad of adverse health conditions associated with smoking. In specimens from tobacco smokers, numerous oxidative damage markers such as F2-isoprostanes, malondialdehyde (MDA), 8-hydroxy-2′-deoxyguanosine (8-OH-dG), and protein carbonyls are all elevated [3]. Increased 8-iso-PGF2α levels among smokers have been reported numerous times across study populations and specimens, [4] and in other references in the supplemental material. However, this increase has always been attributed to increased lipid peroxidation (oxidative stress) due to the free radicals present in inhaled smoke.
Research investigations of the biomarker F2-isoprostane, 8-iso-prostaglandin F2α (8-iso-PGF2α), have shown that there is an alternate source besides oxidative stress that can generate this marker in vivo [5-15]. This source was identified as the prostaglandin endoperoxide-synthase (PGHS) enzymes, which are significantly induced in inflammation. Exposure to tobacco and other smoke induces PGHS in the lungs through non-free radical compounds as well as through tissue damage from free radicals [16-18]. Given the demonstrated induction of PGHS in chronic smokers, we hypothesized that the elevated level of 8-iso-PGF2α detected in the plasma of chronic smokers is not generated solely by non-enzymatic lipid peroxidation.
The method to distinguish chemical lipid peroxidation-dependent formation of 8-iso-PGF2α from PGHS-mediated formation has been described previously by van 't Erve et al. [14, 15] and is based on measuring the 8-iso-PGF2α / prostaglandin F2α (PGF2α) ratio.
Identifying the correct source of 8-iso-PGF2α is very important not only to establish the proper mechanism of toxicity, but also because conclusions based on oversimplified or misinterpreted results can lead to the implementation of improper and potentially dangerous intervention strategies having unintended consequences [19-21].
Materials and Methods
Materials
8-iso-PGF2α, PGF2α, 8-iso-PGF2α-d4, and PGF2α-d4 were purchased from Cayman Chemical (Ann Arbor, MI, USA).
Participant recruitment and sample collection
Healthy adult male and female volunteers were recruited to the National Institute of Environmental Health Sciences Clinical Research Unit between November 2015 and July 2016. Eligible participants were at least 18 years of age, and were asked to not take any anti-inflammatory medication (NSAIDs, corticosteroids, and/or glucocorticoids) for 7 days prior to their scheduled donation date. Participants were excluded if they had chronic diseases such as asthma, COPD, diabetes, cancer, renal disease, hypertension, and others. Smokers were defined as currently smoking tobacco cigarettes and having smoked at least 5 years with at least 100 products consumed in their life. Non-smokers were defined as not having smoked more than 100 products in their lifetime and none in the past 6 months. In addition, female participants were not pregnant. During the visit, trained staff conducted interviews to collect participant demographic information as well as an extensive medical history and smoking history. Participants were asked to fast before arrival (no food and drink except water for 8 hours). Blood was collected in lithium heparin-coated vacutainer tubes. All study protocols and questionnaires were approved by the NIEHS Institutional Review Board, and all participants gave written informed consent.
Plasma sample preparation
Whole blood was centrifuged no more than 30 min after collection (2,000 rpm for 10 min at 4 °C). Plasma was aliquoted and stored at -80 °C until all samples had been collected. Prior to analysis, samples were thawed, spiked with internal standards, and kept on ice during sample preparation. 1.0 mL of plasma was subjected to solid-phase extraction using 3 mL HLB-prime SPE columns (Waters Corp, Billerica, MA, USA). SPE columns were prewashed with 3 mL of butyl acetate, 3 mL acetone and 6 mL water. Plasma aliquots were acidified prior to deposition on the column with 50 % vol / vol of 1 % acetic acid and 5 % methanol in 100 × 13 mm glass tubes. Prewashed columns and acidified plasma were loaded into the Rapidtrace+ automated SPE workstation (Biotage LLC., Charlotte, NC, USA) 10 samples at a time. The Rapidtrace executed the following protocol: 1) load sample (2.0 mL); 2) rinse column with 5% methanol / 0.1 % acetic acid (1 mL); 3) dry with nitrogen stream for 30 seconds; and 4) collect with butyl acetate (1.0 mL). This process occurred in batches of 10 randomly selected samples. Samples were evaporated in a centrifugal vacuum evaporator maintained at 40 °C and reconstituted with 50 μL of a 33 % ethanol / 0.5 % acetic acid solution. After the above preparation, samples were kept at -80 °C in glass auto sampler vials for no more than 24 hours prior to LC/MS/MS analysis. Samples were prepared without knowledge of smoking status.
LC/MS/MS oxylipid analysis
Oxylipid levels were determined in extracted plasma samples by liquid chromatography / tandem mass spectroscopy (LC/MS/MS) as previously described [22, 23]. Online liquid chromatography and electrospray ionization tandem mass spectrometry of extracted plasma samples were performed on an UltiMate 3000 RS HPLC system and Quantiva mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Separations were achieved using a Halo C18 column (2.0 μm, 100 × 2.1 mm, Advanced Materials Technology, Wilmington, DE, USA), which was held at 50°C. The flow rate was 600 μL/min. Mobile phase A was 2 mM acetic acid in 97:3 water / n-propanol. Mobile phase B was acetonitrile. Gradient elution was used, and mobile phase B was varied as follows: 20% B at 0 min, ramp to 50% B from 2 to 6 min, and ramp to 60% B from 6 to 10 min. The injection volume was 10 μL. Samples were analyzed in duplicate. All analytes were monitored as parent ion-product ion, mass / charge pairs with specific retention times as negative ions in a multiple reaction monitoring experiment and quantified against standard curves of analytes purchased from Cayman Chemical (Ann Arbor, MI, USA).
MS data analysis
Peaks identified as 8-iso-PGF2α or PGF2α were integrated and normalized to their individual deuterated standards: 8-iso-PGF2α-d4 and PGF2α-d4. The normalized peak areas were converted to concentrations using quantitation curves generated together with each batch of samples. Each batch consisted of a random selection of 30 samples (male, female, smoker, non-smoker). Samples were analyzed without prior knowledge of smoking status.
Literature meta-analysis on smoking
A comprehensive literature analysis was performed as described in van 't Erve et al. [24] . Data was extracted specifically from manuscripts studying smoking. Thirty-nine publications were included comparing smokers to non-smokers. Forest plots of g values and confidence intervals were calculated with the R package “meta” (G. Scchwarzer - 2007) [25]
Statistics
Statistical testing for equality of nominal data between groups (Table 1) was performed using a chi square test. The 8-iso-PGF2α / PGF2α ratio and subsequent calculations of the contribution of both chemical lipid peroxidation and PGHS to the measured 8-iso-PGF2α level were performed using a custom interface for the R package “Constrained Linear Mixed Effects (CLME)” https://isoratioapp.shinyapps.io/Rserver/ [26, 27]. The calculations were derived from results in van 't Erve et al. [14]. Further statistical analysis was performed using R (version 3.2.2rc, R Core Team 2016). The Wilcoxon-Mann-Whitney test which is robust to non-normality and unequal variances between groups was used to compare the population of smokers to non-smokers. tatistical significance was assumed at p<0.05. Hedges' g values were used to assess the magnitude of change in mean 8-iso-PGF2α levels between smokers and non-smokers and was calculated using the R package “compute.es” (AC Del Re 2013).
Table 1. Demographics of the study population.
| Female | Male | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Smoking (n=19) $ | Non-smoking (n=18) $ | p value | Smoking (n=9) $ | Non-smoking (n=12) $ | p value | ||
| Age | 33.7 ± 8.1 | 32.2± 6.8 | 0.26 | 35.6 ± 10.4 | 32.6 ± 10.4 | 0.36 | |
|
| |||||||
| Race | White | 7 (37%) | 7 (39%) | 0.13 | 2 (22%) | 2 (17%) | 0.14 |
| Black or African American | 11 (58%) | 9 (50%) | 6 (67%) | 8 (67%) | |||
| Asian | - | 2 (11%) | - | 1 (8%) | |||
| Multiple | 1 (5%) | - | 1 (11%) | 1 (8%) | |||
|
| |||||||
| Ethnicity | Non-Latino or Hispanic | 17 (90%) | 18 (100%) | 0.28 | 9 (100%) | 12 (100%) | 1.0 |
| Latino or Hispanic | 1 (5%) | - | - | - | |||
| Unknown | 1 (5%) | - | - | - | |||
|
| |||||||
| Use hormonal contraceptives | 5 (26%) | 10 (55%) | 0.11 | - | - | - | |
|
| |||||||
| BMI | 29.8 ± 7.8 | 29.3 ± 6.0 | 0.62 | 26.7 ± 5.5 | 30.3 ± 4.8 | 0.06 | |
|
| |||||||
| Tobacco use per day | 10.2 ± 6.9 | - | - | 9.3 ± 6.0 | - | 0.48* | |
| Years smoked | 14 ± 7 | - | - | 15 ± 9 | - | 0.73* | |
Comparison male to female, all other p values are comparing smokers to non-smokers
Number (Percent of total) or mean ± standard deviation
Results
Participants
For this study, there were a total of 58 individuals, 21 males (9 smoker, 12 non-smoker) and 37 females (19 smoker and 18 non-smoker) (Table 1). All four sex-smoking groups were similar in age, BMI, and racial and ethnic diversity. In addition, smokers and non-smokers were not on any medication and were not diagnosed with other conditions such as COPD or asthma. Female smoking groups had a similar proportion of women who were post-menopausal, as well as participants who were currently on hormonal contraceptives. All visits occurred in the morning, and most smokers had consumed at least one tobacco cigarette within 1 hour prior to sampling. Between the groups, there was no difference in amount of tobacco cigarettes consumed per day or the duration of years smoked.
Effect of smoking
The median plasma level of 8-iso-PGF2α was significantly higher in smokers (median = 22.7 pg/mL) than non-smokers (median = 14.2 pg/mL) (p=0.009) (Figure 1A left panel, Table S1). For PGF2α, median levels were on average twice as high in smokers (80.9 pg/mL) compared to non-smokers (30.9 pg/mL) (Figure 1A right panel, Table S1). However, the increase in PGF2α levels between the smokers and non-smokers is not statistically significant (p=0.07). In comparison to 8-iso-PGF2α, PGF2α is more abundant with 1.4 times more in non-smokers and 2.6 times more in smokers.
Figure 1. Elevated plasma 8-iso-PGF2α levels in smokers in human plasma and comparison to literature data.
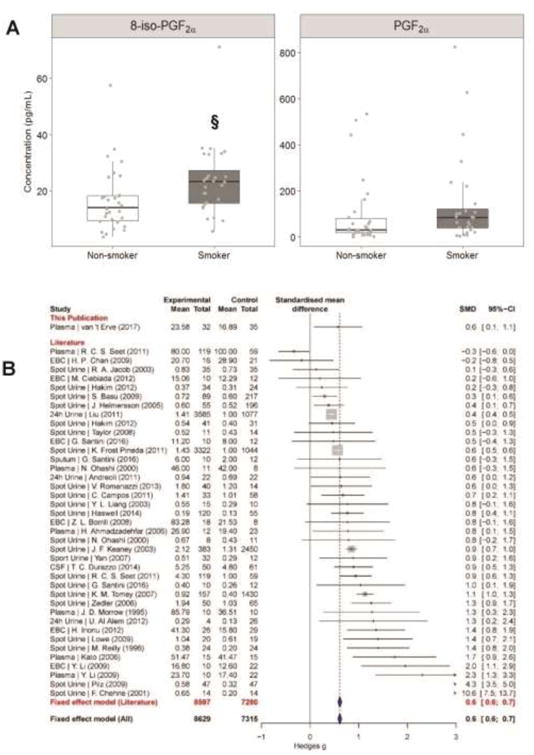
A ) Levels of 8-iso-PGF2α as well as PGF2α in box-and-whisker plots. Grey points represent each individual measurement, bars are the maximum at 1.5 IQR, the box represents the first and third quartiles, and the center band is the median value. § different from non-smokers (p<0.05). B) Forest plot of effect si e (Hedges' g) between 8-iso-PGF2α levels of non-smokers and smokers. Data obtained from supplemental references [1-37]. Fixed-effects model results are plotted in the blue diamonds. These are calculated for each subgroup with inverse variance weighting of individual studies. The mean in the experimental and control groups represents the level of 8-iso-PGF2α, and the total, the number of individuals per group. Exact median, IQR and p values are listed in the supplemental materials.
To determine a more quantitative measure of the magnitude of the effect smoking had on 8-iso-PGF2α levels, the Hedges' g value was calculated. Hedges' g is a metric of effect size calculating how much one population differs from another considering the mean, standard deviation, and number of people in each measured population. A Hedges' g value of >0.7 is considered a large effect. The magnitude of the effect smoking had on 8-iso-PGF2α in this population was modest with a Hedges' g of 0.6 (Figure 1B top line). Despite being modest, the calculated effect was statistically significantly greater than zero, thus the probability that this increase was due to chance alone was small. The measured effect in this study for increased 8-iso-PGF2αdue to smoking was comparable to those in other publications studying this relationship (Figure 1B). In addition, using an improved solid state extraction methodology for plasma, measured concentrations of 8-iso-PGF2α in non-smokers (range = 3.7 - 57.6 pg/mL) are within the range of expected values [24].
With the increase in plasma 8-iso-PGF2α levels and accompanying PGF2α levels, we can calculate the 8-iso-PGF2α/ PGF2α ratio to elucidate the source of the elevated 8-iso-PGF2αin smokers. Between non-smokers and smokers, there was a small decrease in the median 8-iso-PGF2α/ PGF2α ratio (0.38 to 0.29) (Figure 2A, Table S1); this is consistent with increased 8-iso-PGF2a produced primarily by PGHS [14, 15]. To quantitatively confirm this, the ratio for each participant was used to calculate the exact amount of 8-iso-PGF2α generated by both pathways. In this population, increased plasma 8-iso-PGF2α in smokers was shown to be generated primarily from PGHS (Figure 2B, Table S1). No statistical significance was observed for increased generation of plasma 8-iso-PGF2α by chemical lipid peroxidation. This is reflected in the calculated effect size between smokers and non-smokers for chemical lipid peroxidation and PGHS (Figure 2C, Table S2). The effect size for chemical lipid peroxidation was almost half that for PGHS (0.35 vs. 0.59), indicating that the majority of increased 8-iso-PGF2α observed in the plasma of smokers compared to non-smokers is generated by PGHS.
Figure 2. The 8-iso-PGF2α /PGF2α ratio distinguishes between 8-iso-PGF2α generated by chemical lipid peroxidation and PGHS.
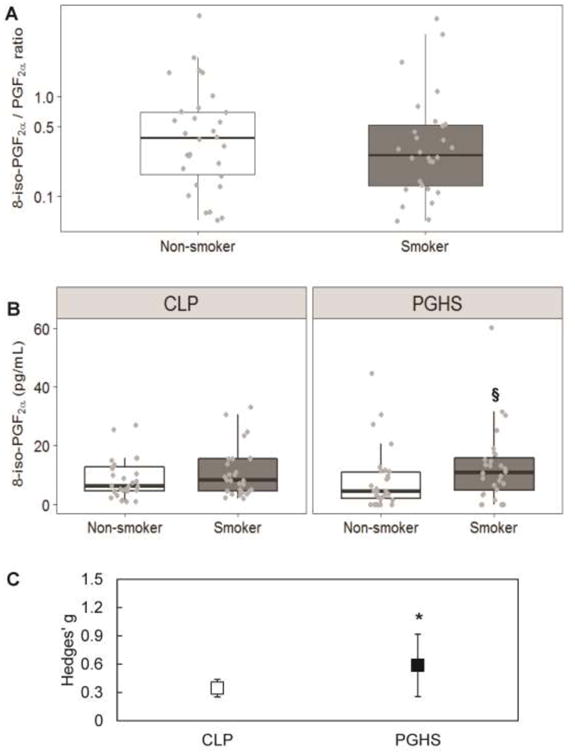
A ) The median plasma 8-iso-PGF2α /PGF2α ratio is slightly lower in smokers than non-smokers, suggesting a greater involvement of PGHS in the generation of 8-iso-PGF2α. Results are shown in box-and-whisker plots with grey points representing each individual measurement. Bars are the maximum at 1.5 IQR, the box represents the first and third quartiles, and the center band is the median value. B) Distribution of 8-iso-PGF2α generated by CLP and PGHS across smokers and non-smokers. § different from non-smokers (p<0.05). C) Mean ± standard error of the effective difference between smokers and non-smokers calculated by Hedges' g. The Hedges' g for the difference in 8-iso-PGF2α generated by CLP between smokers and non-smokers is 0.35, and the Hedges' g values from the change in PGHS-generated 8-iso-PGF2α is greater at 0.59. * Hedges' g statistically significantly greater than 0 (p<0.05). Exact median, IQR and p values are listed in the supplemental materials.
Effect of gender
We investigated the potential effect of gender on the generation of plasma 8-iso-PGF2α in smokers compared to non-smokers. Median levels of 8-iso-PGF2α were increased similarly in both male and female smokers compared to non-smokers although not statistically significant in either gender group given the smaller sample size (Figure 3A, Table S1). There was no statistical significance in the 8-iso-PGF2α levels between smoking males and females.
Figure 3. Increased plasma 8-iso-PGF2α levels upon smoking come from different sources in males and females.
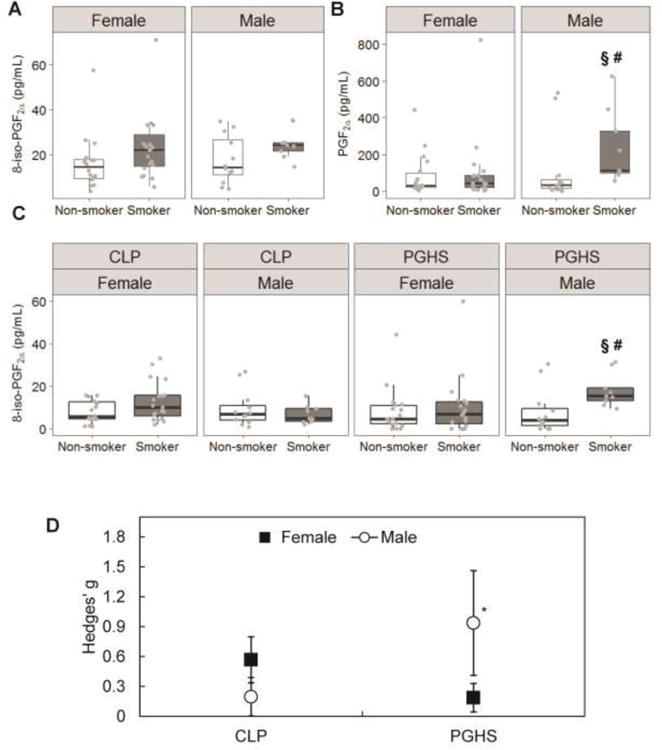
A) Levels of 8-iso-PGF2α increased in both males and females upon smoking but not statistically significantly. B) Levels of PGF2α are not changed much in females upon smoking, but statistically significantly change in males upon smoking. C) 8-iso-PGF2α formed through CLP as well as PGHS is increased upon smoking, but is statistically significant only in males. Results are shown in box-and-whisker plots with grey points representing each individual measurement. Bars are the maximum at 1.5 IQR, the box represents the first and third quartiles, and the center band is the median value. § different from non-smokers (p<0.05). # different from females (p<0.05). D) 8-iso-PGF2α is predominantly formed by CLP in females and by PGHS in males. Mean ± standard error of the effective difference between smoking and non-smoking males and females calculated by Hedges' g. *Hedges' g statistically significantly greater than 0 (p<0.05). Exact median, IQR and p values are listed in the supplemental materials.
In contrast to 8-iso-PGF2α, there was a gender difference for PGF2α, with smoking males having a far larger and statistically significant increase compared to non-smokers (Figure 3B, Table S1). No significant increase was observed in PGF2α between smoking and non-smoking females. Also, male smokers had a statistically significant increase in PGF2α compared to female smokers. No significant difference was observed in the levels of PGF2α between nonsmoking males and females.
To determine the origin of 8-iso-PGF2α in male and female smokers, the 8-iso-PGF2α / PGF2α ratio was calculated. In females, the amount of 8-iso-PGF2α generated by both chemical lipid peroxidation and PGHS was not statistically significantly increased in smokers compared to non-smokers (Figure 3C, Table S1). For males, there was similarly a non-significant increase in chemical lipid peroxidation between smokers and non-smokers. However, there was a much larger increase in PGHS-dependent 8-iso-PGF2α in smokers compared to non-smokers. The increase in PGHS-dependent 8-iso-PGF2α in smoking males was also statistically different from that in smoking females.
The trends in increased 8-iso-PGF2α generation due to smoking are best captured by the calculated effect size, Hedges' g, which gives a quantitative metric for comparison between the genders as well as pathways (Figure 3D, Table S2). For chemical lipid peroxidation, the effect of smoking on 8-iso-PGF2α generation observed in females was more than double that observed in males (Hedges' g = 0.56 compared to 0.20), but with the relatively large uncertainty in the effect size, neither estimate was statistically significantly greater than zero. Also, neither effect was particularly large. For PGHS, an opposite relationship was observed, with males having a far greater calculated effect from smoking compared to females (Hedges' g = 0.94 compared to 0.19). The calculated effect for PGHS in males was the only statistically significant effect (i.e. greater than zero).
Effect of hormonal contraceptive use in females
Almost half of the female participants, both smoking and non-smoking, were on some form of hormonal contraceptive at the time of participating in this study. There are reports in the literature that these compounds can alter susceptibility to environmental stressors and pollution. To investigate the effect of hormonal contraceptive use on our results, we stratified the female participants based on their smoking status and hormonal contraceptive use and analyzed for the sources of 8-iso-PGF2α. When comparing the measured levels of 8-iso-PGF2α between nonsmoking females on hormonal contraceptives and those who were not, a statistically significant lower level was observed, (Figure 4A, Table S1). Interestingly, this difference was not observed in the smoking groups. For PGF2α, an identical pattern was observed (Figure 4B, Table S1). For the sources of 8-iso-PGF2α, increased contributions were observed from both chemical lipid peroxidation and PGHS in females on hormonal contraceptives (Figure 4C, Table S1). Only a minimal increase in 8-iso-PGF2α by chemical lipid peroxidation was observed in females not on hormonal contraceptives, and the generation by PGHS was slightly decreased. Statistically, the only significant difference between hormonal contraceptive users and non-users was the contribution of PGHS to 8-iso-PGF2α in the plasma of non-smokers. The size of the effects of smoking was very large in the hormonal contraceptive users and basically non-existent in the hormonal contraceptive non-users (Figure 4D, Table S2).
Figure 4. Hormonal contraceptive use alters the observed contribution of PGHS and chemical lipid peroxidation.
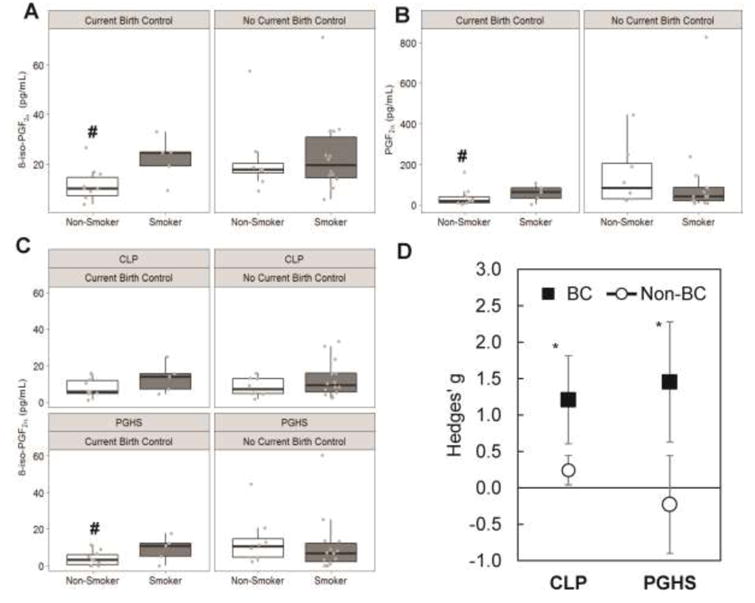
A) Levels of 8-iso-PGF2α increased in females upon smoking, this change is much greater in women using hormonal contraceptives (BC) compared to those who do not. B) Levels of PGF2α are increased in females using hormonal contraceptives upon smoking, but not in those not using hormonal contraceptives. C) Increased levels of 8-iso-PGF2α by CLP and PGHS are observed only in females currently on hormonal contraceptives. A statistically significant difference is found in the contribution of PGHS in non-smoking females. Results are shown in box-and-whisker plots with grey points representing each individual measurement. Bars are the maximum at 1.5 IQR, the box represents the first and third quartiles, and the center band is the median value. # different from hormonal contraceptive non-users (p<0.05). D) 8-iso-PGF2α is formed by both CLP and PGHS in smoking females on hormonal contraceptives. No observed effect of smoking is seen on 8-iso-PGF2α levels in females not on hormonal contraceptives. Mean ± standard error of the effective difference between smoking and non-smoking females calculated by Hedges' g. * Hedges' g statistically significantly greater than 0 (p<0.05). Exact median, IQR and p values are listed in the supplemental materials.
Discussion
Tobacco smoking is widely considered to be the gold standard model for increased oxidative stress in humans due to the large quantity of free radicals detected in the gas phase [3, 4]. This has led to at least 40 studies examining the popular biomarker of oxidative stress, the F2-isoprostanes, in various specimens of smokers. However, the conclusion of increased oxidative stress based on elevated 8-iso-PGF2α was rarely confirmed and could have arisen from induction of prostaglandin-endoperoxide synthases (PGHS) as an additional or sole source [5-15]. There is considerable literature evidence that chronic smoke inhalation induces specifically PGHS 2. Ex vivo exposure of fibroblasts to cigarette smoke shows a rapid and sustained induction of PGHS 2 [28]. Also, increased PGHS 2 mRNA levels are found in peripheral blood mononuclear cells of smokers [29]. Lastly, immune cells such as leucocytes and neutrophils are significant sources of PGHS as well as PGHS-generated eicosanoids [30]. Upon smoke inhalation, there is a rapid (>1h), over 2-fold increase in the amount of these cells in the blood stream [31]. In addition to PGHS induction in smokers, there are interesting associations between the effectiveness of PGHS inhibitors such as aspirin, NSAIDS and others to reduce the risk of developing lung cancer in chronic smokers. Several observational studies and meta-analyses consistently find a near 30 % reduction in risk of lung cancer in NSAID users [32-37]. Given the demonstrated induction of PGHS 2 and association between anti-inflammatories and smoking-related lung cancer, it is important to verify the source of elevated 8-iso-PGF2α.
PGHS contribution to 8-iso-PGF2α generation in other specimens
Reilly et al. [38] was the first to verify contribution of PGHS to 8-iso-PGF2α levels in smokers. They found significant 8-iso-PGF2α generation in serum of smokers which was inhibited by administration of the PGHS-inhibitor aspirin. However, no statistically significant inhibition of 8-iso-PGF2α was observed in urine samples with aspirin treatment. On the other hand, treatment with vitamin C or a combination of vitamin C and E led to a statistically significant reduction in urinary 8-iso-PGF2α levels. This led to the conclusion that potential PGHS generation of 8-iso-PGF2α is not a factor in urine specimens. Here we expand on this study and investigate this confounding mechanism in plasma of smokers which is another often used research specimen. The origins of 8-iso-PGF2α in plasma are potentially distinct from urine due to direct contact with peripheral blood mononuclear cells and platelets which all have high levels of PGHS. Instead of relying on pharmacological inhibitors to distinguish between enzymatic and non-enzymatic or chemical lipid peroxidation which is difficult to do consistently and without introducing bias or confounding, the 8-iso-PGF2α/ PGF2α ratio, which does not employ inhibitors was used. Confirming the pathways involved in disease etiologies is vital to further our understanding of these etiologies.
Gender dependence in 8-iso-PGF2α generation mechanism
There have been suggestions in the literature that there is a gender-dependent difference in 8-iso-PGF2α levels in chronic smokers [39-41]. These three studies combined found a near 1.5-fold increase in the mean urine 8-iso-PGF2α concentration in currently smoking females over males (Hedges' g = 0.54). In the present study, no difference in plasma 8-iso-PGF2α between male and female smokers was observed (23.2 pg/mL for smoking males and 22.1 pg/mL for smoking females). A potential reason for this discrepancy is the difference in specimens between this study and previous reports. Additionally, other factors could play a role such as the clearance rate of creatinine, which is used to normalize urinary 8-iso-PGF2α concentrations in all studies. Yan et al. [41] showed this clearly with a twofold decrease in the creatinine levels in female smokers' urine samples, which could potentially explain the gender difference.
For the formation mechanism of 8-iso-PGF2α, we observed a significantly greater than zero effect for PGHS-dependent formation in males, but not in females. After further analysis, this perceived gender-dependent difference was confounded by not accounting for hormonal contraceptive use in the female population. By stratifying the female population by current use of hormonal contraceptives, we found a response to chronic tobacco smoking. The biological rationale for this confounding effect involves the decreased expression of PGHS in endometrial tissue [42] and potentially other tissues while using hormonal contraceptives. This could explain the elevated levels of both prostaglandins and 8-iso-PGF2α and PGF2α as well as the especially large variance in PGF2α levels in females not on hormonal contraceptives. Just as reported here, other studies have found a decrease in 8-iso-PGF2α levels in females on hormonal contraceptives [43] as well as variation in the levels of 8-iso-PGF2α throughout the menstrual cycle [44].
There may have been a significant limitation to this study resulting from the oversight of not synchronizing female volunteers to participate at a specific time in their menstrual cycle. This limitation is likely also responsible for our finding of non-elevation of mean 8-iso-PGF2α and PGF2α levels upon chronic smoking in the non-hormonal contraceptive population. Future studies trying to determine the gender difference in 8-iso-PGF2α generation will need to take this into account. Ultimately, we conclude that there is likely no gender difference in the formation of 8-iso-PGF2α in smokers.
The complexity of 8-iso-PGF2α as a biomarker of oxidative stress
Despite the long-standing evidence of free radicals in cigarette smoking [1, 2], here we show that increased 8-iso-PGF2α levels observed in chronic smokers come primarily from the inflammation-induced, PGHS-dependent lipid peroxidation pathways and not directly from free radicals in the cigarette smoke. This result highlights the complex problem surrounding the exact sources of 8-iso-PGF2α in various diseases and exposures. General conclusions on increased oxidative stress when elevated level of 8-iso-PGF2α were the sole measurement can no longer be made. Careful measurement using the 8-iso-PGF2α/PGF2α ratio approach must be performed for each health condition, disease, or exposure to provide insight into the distribution of chemical and enzymatic lipid peroxidation. It is imperative that conditions which have been identified as having elevated 8-iso-PGF2α, such as those identified by van 't Erve et al. [24], need to be re-examined to identify the true sources of this biomarker. Only once the source of 8-iso-PGF2α has been identified can oxidative stress be concluded to play a role in the disease etiology and appropriate prevention strategy be designed, be it anti-oxidant, anti-inflammatory or any other strategy.
Supplementary Material
Highlights.
Elevated levels of 8-iso-PGF2α in plasma of smokers is generated primarily by PGHS
Little gender dependence is found in the origin of elevated 8-iso-PGF2α in smokers
Hormonal contraceptive use can influence the origin of elevated 8-iso-PGF2α in smokers
Acknowledgments
The authors acknowledge the invaluable contribution of the NIEHS clinical research unit (CRU) staff; Stavros Garantziotis, Christopher Lee, Brittany Mosley, Cynthia Smith, Janze Taylor, Sarah Stephenson, Catherine Wild, Jessica Luthi, Nicole Edwards, and Rebecca Church. The authors gratefully acknowledge the input of Drs. Donna D. Baird and Quaker E. Harmon on the potential confounding of hormonal contraceptive use in our study; Drs. Stephanie J. London and Kelly K. Ferguson for their technical review of this manuscript; and Jean Corbett, Dr. Ann Motten, and Mary Mason for their editorial expertise. This work was supported by the Intramural Research Program, National Institutes of Health, National Institute of Environmental Health Sciences.
Abbreviations
- 8-iso-PGF2α
8-iso-prostaglandin F2α
- PGF2α
prostaglandin F2α
- PGHS
prostaglandin-endoperoxide synthase, cyclooxygenase
- CLP
chemical lipid peroxidation
Footnotes
Conflict of Interest: The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goel R, Bitzer Z, Reilly SM, Trushin N, Foulds J, Muscat J, Liao J, Elias RJ, Richie JP., Jr Variation in Free Radical Yields from US Marketed Cigarettes. Chem Res Toxicol. 2017;30:1038–1045. doi: 10.1021/acs.chemrestox.6b00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pryor WA, Prier DG, Church DF. Electron-spin resonance study of mainstream and sidestream cigarette smoke: nature of the free radicals in gas-phase smoke and in cigarette tar. Environ Health Perspect. 1983;47:345–355. doi: 10.1289/ehp.8347345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Vaart H, Postma DS, Timens W, ten Hacken NH. Acute effects of cigarette smoke on inflammation and oxidative stress: a review. Thorax. 2004;59:713–721. doi: 10.1136/thx.2003.012468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morrow JD, Frei B, Longmire AW, Gaziano JM, Lynch SM, Shyr Y, Strauss WE, Oates JA, Roberts LJ., 2nd Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N Engl J Med. 1995;332:1198–1203. doi: 10.1056/NEJM199505043321804. [DOI] [PubMed] [Google Scholar]
- 5.Praticò D, Lawson JA, FitzGerald GA. Cyclooxygenase-dependent formation of the isoprostane, 8-epiprostaglandin F2α. J Biol Chem. 1995;270:9800–9808. doi: 10.1074/jbc.270.17.9800. [DOI] [PubMed] [Google Scholar]
- 6.Klein T, Reutter F, Schweer H, Seyberth HW, Nüsing RM. Generation of the isoprostane 8-epi-prostaglandin F2α in vitro and in vivo via the cyclooxygenases. J Pharmacol Exp Ther. 1997;282:1658–1665. [PubMed] [Google Scholar]
- 7.Praticò D, FitzGerald GA. Generation of 8-epiprostaglandin F2α by human monocytes: discriminate production by reactive oxygen species and prostaglandin endoperoxide synthase-2. J Biol Chem. 1996;271:8919–8924. doi: 10.1074/jbc.271.15.8919. [DOI] [PubMed] [Google Scholar]
- 8.Bachi A, Brambilla R, Fanelli R, Bianchi R, Zuccato E, Chiabrando C. Reduction of urinary 8-epi-prostaglandin F2α during cyclo-oxygenase inhibition in rats but not in man. Br J Pharmacol. 1997;121:1770–1774. doi: 10.1038/sj.bjp.0701321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schweer H, Watzer B, Seyberth HW, Nusing RM. Improved quantification of 8-epi-prostaglandin F2α and F2-isoprostanes by gas chromatography/triple-stage quadrupole mass spectrometry: partial cyclooxygenase-dependent formation of 8-epi-prostaglandin F2α in humans. J Mass Spectrom. 1997;32:1362–1370. doi: 10.1002/(SICI)1096-9888(199712)32:12<1362::AID-JMS606>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 10.Watkins MT, Patton GM, Soler HM, Albadawi H, Humphries DE, Evans JE, Kadowaki H. Synthesis of 8-epi-prostaglandin F2α by human endothelial cells: role of prostaglandin H2 synthase. Biochem J. 1999;344:747–754. [PMC free article] [PubMed] [Google Scholar]
- 11.Favreau F, Petit-Paris I, Hauet T, Dutheil D, Papet Y, Mauco G, Tallineau C. Cyclooxygenase 1-dependent production of F2-isoprostane and changes in redox status during warm renal ischemia-reperfusion. Free Radic Biol Med. 2004;36:1034–1042. doi: 10.1016/j.freeradbiomed.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Kadiiska MB, Gladen BC, Baird DD, Graham LB, Parker CE, Ames BN, Basu S, Fitzgerald GA, Lawson JA, Marnett LJ, Morrow JD, Murray DM, Plastaras J, Roberts LJ, II, Rokach J, Shigenaga MK, Sun J, Walter PB, Tomer KB, Barrett JC, Mason RP. Biomarkers of oxidative stress study III. Effects of the nonsteroidal anti-inflammatory agents indomethacin and meclofenamic acid on measurements of oxidative products of lipids in CCl4 poisoning. Free Radic Biol Med. 2005;38:711–718. doi: 10.1016/j.freeradbiomed.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 13.Tsikas D, Suchy MT, Niemann J, Tossios P, Schneider Y, Rothmann S, Gutzki FM, Frolich JC, Stichtenoth DO. Glutathione promotes prostaglandin H synthase (cyclooxygenase)-dependent formation of malondialdehyde and 15(S)-8-iso-prostaglandin F2alpha. FEBS Lett. 2012;586:3723–3730. doi: 10.1016/j.febslet.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 14.van 't Erve TJ, Lih FB, Kadiiska MB, Deterding LJ, Eling TE, Mason RP. Reinterpreting the best biomarker of oxidative stress: The 8-iso-PGF2α/PGF2α ratio distinguishes chemical from enzymatic lipid peroxidation. Free Radic Biol Med. 2015;83:245–251. doi: 10.1016/j.freeradbiomed.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van't Erve TJ, Lih FB, Jelsema C, Deterding LJ, Eling TE, Mason RP, Kadiiska MB. Reinterpreting the best biomarker of oxidative stress: The 8-iso-prostaglandin F2alpha/prostaglandin F2alpha ratio shows complex origins of lipid peroxidation biomarkers in animal models. Free Radic Biol Med. 2016;95:65–73. doi: 10.1016/j.freeradbiomed.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domagala-Kulawik J. Effects of cigarette smoke on the lung and systemic immunity. J Physiol Pharmacol. 2008;59 Suppl 6:19–34. [PubMed] [Google Scholar]
- 17.Bhalla DK, Hirata F, Rishi AK, Gairola CG. Cigarette smoke, inflammation, and lung injury: a mechanistic perspective. J Toxicol Environ Health B Crit Rev. 2009;12:45–64. doi: 10.1080/10937400802545094. [DOI] [PubMed] [Google Scholar]
- 18.Pappas RS. Toxic elements in tobacco and in cigarette smoke: inflammation and sensitization. Metallomics. 2011;3:1181–1198. doi: 10.1039/c1mt00066g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, Keogh JP, Meyskens FL, Jr, Valanis B, Williams JH, Jr, Barnhart S, Cherniack MG, Brodkin CA, Hammar S. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J Natl Cancer Inst. 1996;88:1550–1559. doi: 10.1093/jnci/88.21.1550. [DOI] [PubMed] [Google Scholar]
- 20.Ruano-Ravina A, Figueiras A, Freire-Garabal M, Barros-Dios JM. Antioxidant Vitamins and Risk of Lung Cancer. Current Pharmaceutical Design. 2006;12:599–613. doi: 10.2174/138161206775474396. [DOI] [PubMed] [Google Scholar]
- 21.Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants Accelerate Lung Cancer Progression in Mice. Science Translational Medicine. 2014;6 doi: 10.1126/scitranslmed.3007653. [DOI] [PubMed] [Google Scholar]
- 22.Edin ML, Wang Z, Bradbury JA, Graves JP, Lih FB, DeGraff LM, Foley JF, Torphy R, Ronnekleiv OK, Tomer KB, Lee CR, Zeldin DC. Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 2011;25:3436–3447. doi: 10.1096/fj.11-188300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CR, Imig JD, Edin ML, Foley J, DeGraff LM, Bradbury JA, Graves JP, Lih FB, Clark J, Myers P, Perrow AL, Lepp AN, Kannon MA, Ronnekleiv OK, Alkayed NJ, Falck JR, Tomer KB, Zeldin DC. Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J. 2010;24:3770–3781. doi: 10.1096/fj.10-160119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van 't Erve TJ, Kadiiska MB, London SJ, Mason RP. Classifying oxidative stress by F2-isoprostane levels across human diseases: A meta-analysis. Redox Biol. 2017;12:582–599. doi: 10.1016/j.redox.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwarzer G, Carpenter JR, Rücker G. Cham. Springer International Publishing; 2015. An Introduction to Meta-Analysis in R Meta-Analysis with R; pp. 3–17. [Google Scholar]
- 26.Jelsema CM, Peddada SD. CLME: An R Package for Linear Mixed Effects Models under Inequality Constraints. Journal of Statistical Software. 2016;75 doi: 10.18637/jss.v075.i01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farnan L, Ivanova A, Peddada SD. Linear Mixed Effects Models under Inequality Constraints with Applications. PLoS ONE. 2014;9:e84778. doi: 10.1371/journal.pone.0084778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martey CA, Pollock SJ, Turner CK, O'Reilly KM, Baglole CJ, Phipps RP, Sime PJ. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol. 2004;287:L981–991. doi: 10.1152/ajplung.00239.2003. [DOI] [PubMed] [Google Scholar]
- 29.Bonaterra GA, Hildebrandt W, Bodens A, Sauer R, Dugi KA, Deigner HP, Dröge W, Metz J, Kinscherf R. Increased cyclooxygenase-2 expression in peripheral blood mononuclear cells of smokers and hyperlipidemic subjects. Free Radical Biology and Medicine. 2005;38:235–242. doi: 10.1016/j.freeradbiomed.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 30.Fasano MB, Wells JD, McCall CE. Human neutrophils express the prostaglandin G/H synthase 2 gene when stimulated with bacterial lipopolysaccharide. Clin Immunol Immunopathol. 1998;87:304–308. doi: 10.1006/clin.1998.4545. [DOI] [PubMed] [Google Scholar]
- 31.Abboud RT, Fera T, Johal S, Richter A, Gibson N. Effect of smoking on plasma neutrophil elastase levels. J Lab Clin Med. 1986;108:294–300. [PubMed] [Google Scholar]
- 32.McCormack VA, Hung RJ, Brenner DR, Bickeboller H, Rosenberger A, Muscat JE, Lazarus P, Tjonneland A, Friis S, Christiani DC, Chun EM, Le Marchand L, Rennert G, Rennert HS, Andrew AS, Orlow I, Park B, Boffetta P, Duell EJ. Aspirin and NSAID use and lung cancer risk: a pooled analysis in the International Lung Cancer Consortium (ILCCO) Cancer Causes Control. 2011;22:1709–1720. doi: 10.1007/s10552-011-9847-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akhmedkhanov A, Toniolo P, Zeleniuch-Jacquotte A, Koenig KL, Shore RE. Aspirin and lung cancer in women. Br J Cancer. 2002;87:49–53. doi: 10.1038/sj.bjc.6600370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hernandez-Diaz S, Garcia Rodriguez LA. Nonsteroidal anti-inflammatory drugs and risk of lung cancer. Int J Cancer. 2007;120:1565–1572. doi: 10.1002/ijc.22514. [DOI] [PubMed] [Google Scholar]
- 35.Holick CN, Michaud DS, Leitzmann MF, Willett WC, Giovannucci E. Aspirin use and lung cancer in men. Br J Cancer. 2003;89:1705–1708. doi: 10.1038/sj.bjc.6601343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muscat JE, Chen SQ, Richie JP, Jr, Altorki NK, Citron M, Olson S, Neugut AI, Stellman SD. Risk of lung carcinoma among users of nonsteroidal antiinflammatory drugs. Cancer. 2003;97:1732–1736. doi: 10.1002/cncr.11242. [DOI] [PubMed] [Google Scholar]
- 37.Olsen JH, Friis S, Poulsen AH, Fryzek J, Harving H, Tjønneland A, Sørensen HT, Blot W. Use of NSAIDs, smoking and lung cancer risk. Br J Cancer. 2008;98:232–237. doi: 10.1038/sj.bjc.6604151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reilly M, Delanty N, Lawson JA, FitzGerald GA. Modulation of oxidant stress in vivo in chronic cigarette smokers. Circulation. 1996;94:19–25. doi: 10.1161/01.cir.94.1.19. [DOI] [PubMed] [Google Scholar]
- 39.Hakim IA, Harris R, Garland L, Cordova CA, Mikhael DM, Sherry Chow HH. Gender difference in systemic oxidative stress and antioxidant capacity in current and former heavy smokers. Cancer Epidemiol Biomarkers Prev. 2012;21:2193–2200. doi: 10.1158/1055-9965.EPI-12-0820. [DOI] [PubMed] [Google Scholar]
- 40.Taylor AW, Bruno RS, Traber MG. Women and smokers have elevated urinary F(2)-isoprostane metabolites: a novel extraction and LC-MS methodology. Lipids. 2008;43:925–936. doi: 10.1007/s11745-008-3222-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan W, Byrd GD, Ogden MW. Quantitation of isoprostane isomers in human urine from smokers and nonsmokers by LC-MS/MS. J Lipid Res. 2007;48:1607–1617. doi: 10.1194/jlr.M700097-JLR200. [DOI] [PubMed] [Google Scholar]
- 42.Maia H, Jr, Maltez A, Studard E, Zausner B, Athayde C, Coutinho E. Effect of the menstrual cycle and oral contraceptives on cyclooxygenase-2 expression in the endometrium. Gynecol Endocrinol. 2005;21:57–61. doi: 10.1080/09513590500099602. [DOI] [PubMed] [Google Scholar]
- 43.Smedman A, Vessby B, Samar B. Isomer-specific effects of conjugated linoleic acid on lipid peroxidation in humans: regulation by α-tocopherol and cyclo-oxygenase-2 inhibitor. Clinical science. 2004;106:67–73. doi: 10.1042/CS20030105. [DOI] [PubMed] [Google Scholar]
- 44.Schisterman EF, Gaskins AJ, Mumford SL, Browne RW, Yeung E, Trevisan M, Hediger M, Zhang C, Perkins NJ, Hovey K. Influence of endogenous reproductive hormones on F2-isoprostane levels in premenopausal women: the BioCycle Study. American Journal of Epidemiology. 2010;172:430–439. doi: 10.1093/aje/kwq131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.