

Abstract
The development of therapies for the treatment of neurological cancer faces a number of major challenges including the synthesis of small molecule agents that can penetrate the blood-brain barrier (BBB). Given the likelihood that in many cases drug exposure will be lower in the CNS than in systemic circulation, it follows that strategies should be employed that can sustain target engagement at low drug concentration. Time dependent target occupancy is a function of both the drug and target concentration as well as the thermodynamic and kinetic parameters that describe the binding reaction coordinate, and sustained target occupancy can be achieved through structural modifications that increase target (re)binding and/or that decrease the rate of drug dissociation. The discovery and deployment of compounds with optimized kinetic effects requires information on the structure–kinetic relationships that modulate the kinetics of binding, and the molecular factors that control the translation of drug–target kinetics to time-dependent drug activity in the disease state. This Review first introduces the potential benefits of drug-target kinetics, such as the ability to delineate both thermodynamic and kinetic selectivity, and then describes factors, such as target vulnerability, that impact the utility of kinetic selectivity. The Review concludes with a description of a mechanistic PK/PD model that integrates drug–target kinetics into predictions of drug activity.
Keywords: Kinetic selectivity, target vulnerability, target occupancy, drug-target residence time, PK/PD modeling, therapeutic window, blood brain barrier, CNS cancer
Introduction
The treatment of primary infiltrative and secondary metastatic CNS tumors requires the development of drugs that can penetrate the blood-brain barrier (BBB), a selectively permeable barrier composed of epithelial cells held together by tight junctions that is rich in efflux transporter proteins such as P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp).1 The BBB severely limits the ability of many drugs to penetrate into the brain and is a major impediment to the development of new CNS therapies. For instance, given the success at developing drugs that target kinases in peripheral tumors, it is salutatory that no kinase inhibitor has yet received approval for the treatment of primary CNS cancers.2 In addition, brain metastasis is a common resistance mechanism during treatment of peripheral tumors due to the inability of drugs to penetrate the BBB. Thus, since drug exposure is likely to be lower in the brain than in systemic circulation, strategies should be adopted that involve the design and synthesis of compounds that remain bound to their targets even when drug concentration is low. However, the general reliance in drug discovery programs on in vitro assays performed at constant drug concentration limits the identification and progression of compounds that display kinetic effects. This knowledge gap will also impact the development of covalent inhibitors since the benefit gained from prolonged target engagement depends on factors such as target vulnerability that can only be assessed using time-dependent assays.
Drug discovery is predicated on the identification and optimization of drug leads through a series of in vitro experiments that are usually performed at constant concentration. The quantitative metrics that result, such as the IC50 values for engagement of the purified target or for activity in a cell-based assay, are used to select and prioritize lead compounds, make assessments about the possibility for off-target effects that impact the therapeutic index, and ultimately to predict drug activity. However, equilibrium parameters are not able to fully account for time-dependent changes in target engagement in the dynamic environment of the human body where drug (and target) concentrations fluctuate. Instead, both the thermodynamics and kinetics of drug-target interactions must be utilized to fully account for time-dependent changes in target engagement. The role of drug–target kinetics in drug discovery has been discussed in a number of reviews and opinions,3−11 and key concepts include potential mechanisms that modulate the rates of drug–target complex formation (kon) and breakdown (koff),12−16 including the development of covalent inhibitors,17 and the role that drug–target residence time (1/koff), kon, and pharmacokinetics play in dictating target engagement.18−26 In addition, the role of binding kinetics has been explored in forward-thinking programs, such as the K4DD Innovative Medicines Initiative.27 The present Review reiterates some of the basic concepts that govern drug–target interactions and shows that access to the on and off rates for formation and breakdown of the drug–target complex provides an additional dimension of information that can be used to prioritize drug leads based on kinetic selectivity. Subsequently it is shown that the translation of kinetic effects to time-dependent changes in drug activity depends on target vulnerability which directly impacts kinetic selectivity. The review concludes with a discussion of pharmacokinetic/pharmacodynamic (PK/PD) models that integrate drug-target kinetics into predictions of drug activity in order to facilitate the prospective use of in vitro kinetic data.
The Thermodynamics and Kinetics of Drug–Target Interactions
Drug-target complex formation occurs because the complex is more thermodynamically stable than free unbound drug and target: thus, thermodynamics provides the driving force for drug binding. However, the value for the equilibrium constant that describes binding provides no information on the rate at which the complex forms and breaks down. Instead, the on (kon) and off (koff) rates for drug binding are controlled by the difference in free energy between the relevant ground and transition states on the binding reaction coordinate (Figure 1).
Figure 1.
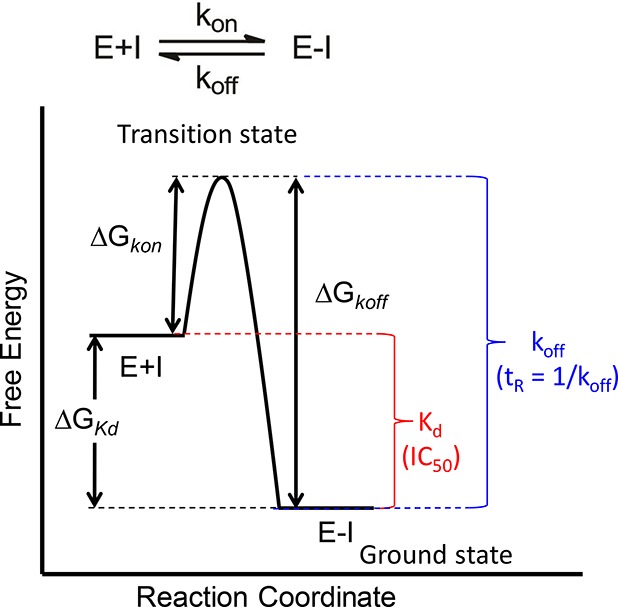
Reaction coordinate for a one-step binding event. Target (E) and drug (I) binding leads to the drug–target complex (E-I). The driving force for binding is given by the difference in free energy between E+I and E-I (ΔGKd). Experimental measurements of the equilibrium dissociation constant Kd, or parameters such as IC50 values, provide a quantitative estimate of the thermodynamics for binding. The rate at which the drug-target complex forms (kon) and dissociates (koff) is given by the difference in free energy between the respective ground states (E+I or E-I) and the rate-limiting transition state (ΔGkon and ΔGkoff). ΔGKd is related to Kd by the relationship ΔG° = −RT ln K. Assuming that parameters such as the transmission coefficient are the same for two drug molecules, then the difference in free energy for the rate of complex dissociation of the two molecules can be given by ΔΔGkoff = −RT ln(koff1/koff2). The lifetime of the drug–target complex is often quantified by the residence time, tR, where tR = 1/koff.5 The figure shows a simple one-step mechanism, although in many cases slow-binding inhibitors operate through a two-step induced-fit mechanism.13,15,28−31
Since kon and koff depend on the difference in free energy between the ground and transition states on the binding reaction coordinate, efforts to improve drug potency may have unpredictable effects on the kinetics of drug-target complex formation and breakdown. Potency is normally used as a descriptor of affinity, and thus an increase in potency is associated with an increase in the affinity of the drug-target interaction quantified by a decrease in the Kd or IC50 value. However, the impact of stabilizing the E-I ground state on the kinetics for binding depends on whether the transition state is affected.8 Several scenarios can be envisaged, the simplest of which is that stabilization of E-I has no effect on the transition state. In this case a decrease in Kd will lead to a decrease in koff with no change in kon. In other words, a more potent compound will have a slower off-rate. Alternatively, if the changes in compound structure that lead to an increase in affinity also result in equal stabilization of the transition state, then the increase in potency will have no effect on the rate at which the drug dissociates from the target. In addition, a third scenario can be envisaged where two molecules have identical affinities for the target (the same Kd or IC50 values) but different kon and koff values. Importantly, any differences in kon and koff values, either between two molecules binding to the same target or a molecule interacting with two different biological molecules (e.g., on and off-target proteins), will not be revealed by approaches that only evaluate compound affinity (potency).
Two important misconceptions abound. First, it is often assumed that an increase in potency will result in a decrease in koff. It may do, but it does not have to. Although there are many examples of long residence time compounds, kinetic data for structurally related analogs are often not available, preventing an analysis of whether or not residence time is driven by affinity. Examples where changes in structure within a compound series lead to a decrease in both IC50 and koff include inhibitors of Pseudomonas LpxC,21 human protein methyltransferase DOT1L,32 and CDK8/CycC.33 In addition, if stabilization of E-I also results in stabilization of the transition state, then once the theoretical limit for kon is reached, which is the second order rate constant for encounter of drug and target (109 M–1 s–1), any further increase in affinity must lead to a reduction in the off-rate. A simple calculation reveals that the residence time of a 1 pM drug on the target must be at least 12 min whereas a 1 fM drug will have a residence time of at least 11.5 days. However, there are also examples where changes in affinity and off-rate are disconnected, which are particularly relevant where affinities are in the micromolar to nanomolar range: a 1 nM drug may only have a residence time of 1 s on the target. For example, the quinazoline-based inhibitors gefitinib and lapatinib have Kiapp values for EGFR of 0.4 and 3 nM, respectively, but while gefitinib has a residence time of <14 min, lapatinib has a residence time of 430 min.34 Other examples include antagonists of the muscarinic M3 receptor,35 antagonists of the chemoattractant receptor-homologous molecule CRTh2/DP2,36 and inhibitors of p38α MAP kinase.37 The second major misconception is that if a compound has similar IC50 values for two different proteins, the drug-target and an off-target protein associated with unwanted side-effects, then the compound has no selectivity. Indeed, the compound has no thermodynamic selectivity, but if the kon and koff values differ between the two targets, it can still have kinetic selectivity. (Text Box 1). This is very important given the implicit relationship between selectivity and therapeutic index.
Kinetic Selectivity
The relative affinity of a compound for the target and for any known off-target proteins is commonly used as a metric for compound selectivity and the potential of the molecule for causing unwanted side-effects. As noted above, this definition of selectivity is actually thermodynamic selectivity since it is based on equilibrium binding experiments (e.g., IC50 values). Of course the ability to determine selectivity depends on knowledge and availability of known off-target proteins and, for example, kinase inhibitor discovery programs often determine compound activity toward a panel of kinases to assess the potential for off-target effects. However, thermodynamic selectivity may not map with kinetic selectivity, and indeed as noted above, kinetic selectivity can still exist even in the absence of thermodynamic selectivity.21,23,38−41
Figure 2 shows a simulation where a compound binds to four targets with the same Kd but different on and off rates, including a target in which a covalent complex is formed (koff = 0). The panels show how the occupancy of the targets change with time as a function of drug concentration (pharmacokinetics) assuming that no target turnover occurs. When compound eliminates with a half-life of 5 h (Figure 2A and C), all four targets reach >95% occupancy even at the lower initial drug dose (Figure 2C). In addition, selectivity between the three targets to which the compound binds reversibly (Targets 1–3) only occurs after more than 12 h. In contrast, if the compound half-life is only 1 h (Figure 2B and D), then there is a high degree of selectivity: for example, at 12 h for the higher drug dose (Figure 2C), Target 1 is only 5% occupied while Targets 2 and 3 are 54% and 83% occupied, respectively. Thus, despite the lack of thermodynamic selectivity, the compound demonstrates kinetic selectivity between the targets. In addition to providing an explicit example of kinetic selectivity, three additional points may be drawn from this analysis. First, the relationship between binding kinetics and drug pharmacokinetics plays a fundamental role in controlling time dependent occupancy. Second, a covalent inhibitor can in principal maximize the impact of kinetic selectivity. Third, when the compound eliminates more rapidly, Target 3 only reaches 70% occupancy at the lower dose (Figure 2D). Since Kd is kept constant, a decrease in off-rate must also lead to a reduction in the on-rate. The faster elimination results in lower peak compound concentration and, since kon is a second order rate constant, lower occupancy of the target. Thus, the level of target occupancy is a function of both kon and koff, as well as drug concentration. In addition, the analysis in Figure 2 assumes a target concentration of 1 nM which is 10-fold lower than the Kd (10 nM). However, if the Kd is similar to the target concentration, then binding to the target can modify the local drug concentration and hence the pharmacokinetics of the drug (target-mediated drug disposition),23,42,43 in a situation that is analogous to tight-binding enzyme inhibition. This effect will become more pronounced as the total drug concentration approaches the concentration of the target. This effect will become more pronounced as the total drug concentration approaches the concentration of the target. Other factors, such as the local accumulation of compounds in the plasma membrane and binding to plasma protein may also contribute to the “micropharmacokinetics” of the drug.20
Figure 2.

Time-dependent target occupancy: kinetic selectivity. A compound is assumed to bind reversibly to three targets with the same thermodynamic affinity (10 nM) but have different residence times on the three targets: Target 1, 1 s; Target 2, 10 h; and Target 3, 50 h). In addition, the compound is assumed to bind covalently to a fourth target (Target 4). Target occupancy has been simulated using Kintek,44,45 assuming either a 1.5 μM (A and B) or 0.5 μM (C and D) dose of compound that is absorbed with ka 3 h–1 but eliminated with two different rates, ke 0.139 h–1 (t1/2 5 h) (A and C) or ke 0.69 h–1 (t1/2 1 h) (B and D). Reversible binding is assumed to occur via a one-step mechanism with the following on and off-rates. Target 1: kon 100 μM–1 s–1, koff 1 s–1. Target 2: kon 2.78 × 10–3 μM–1 s–1, koff 2.78 × 10–5 s–1. Target 3: kon 5.56 × 10–4 μM–1 s–1, koff 5.56 × 10–6 s–1. For Target 4, it is assumed that the compound binds in a two-step mechanism in which the initial rapid binding of the compound to the target, defined by kon 100 μM–1 s–1 and koff 1 s–1 is followed by a second step with kinact 5.56 × 10–4 s–1 leading to the covalent drug-target complex. In each case the target concentration is fixed at 1 nM (i.e., no target turnover).
Box 1. Thermodynamic and Kinetic Selectivity.
Selectivity refers to the relative ability of a drug to engage the chosen target compared to off-target macromolecules, and provides valuable insight into the potential for unwanted side effects (i.e., the therapeutic window or therapeutic index). In many cases, selectivity is determined from affinity-based measurements, for example, by comparing IC50 values for kinase inhibition in a kinase panel. However, this is actually thermodynamic selectivity since IC50 values, as well as Kd and Ki values, are determined at constant drug concentration. Since a compound can have the same affinity for two proteins, but different on and off-rates, affinity-based assessments of selectivity provide no insight in to the possibility that a compound may show kinetic selectivity between two proteins. In other words, a drug may have the same affinity for two proteins but dramatically different binding kinetics such that the lifetimes of the two drug-target complexes differ by orders of magnitude. The contribution of kinetic selectivity to the therapeutic window is intimately related to the time-dependence of drug concentration at the target site (pharmacokinetics, PK), and drugs that eliminate rapidly relative to the lifetime of the drug-target complex will maximize the potential benefit of kinetic selectivity in situations where prolonged occupancy of the target is mitigated. The required PK for long residence time drugs will thus likely be similar to that for covalent drugs where high Cmax ensures rapid occupancy of the target and fast elimination then maximizes the therapeutic window.17 In reality the Cmax only has to be high enough to ensure that physiologically relevant levels of target engagement are achieved, and indeed the relationship between target occupancy and drug efficacy is dictated by the vulnerability of the target (see below). In addition, target turnover will also impact kinetic selectivity, since the rapid synthesis of new target will negate the effects of prolonged target occupancy at low drug concentration. Finally, the rational optimization of kinetic selectivity requires knowledge of both the ground and transition states on the binding reaction coordinate, which is important since rational drug design normally only focuses on enhancing affinity through stabilization of the drug–target ground state.
Target Vulnerability
In Figure 2 it, can be seen that target occupancy varies with time based on the kinetics of drug binding as well as drug concentration, and that in some scenarios complete occupancy of the target may not occur. The translation of target occupancy to drug pharmacodynamics depends on the relationship between occupancy and effect, which in turn depends on target vulnerability, that is, what fraction of target has to be engaged to elicit the desired response (Text Box 2). Low vulnerability targets are those where high levels of occupancy are needed to generate the desired physiological outcome. Conversely, high vulnerability targets require only low levels of occupancy to achieve the desired effect. The specific relationship between occupancy and effect can be captured using a vulnerability function, and in Figure 3 are shown examples of the functions for hypothetical low and high vulnerability targets.
Figure 3.
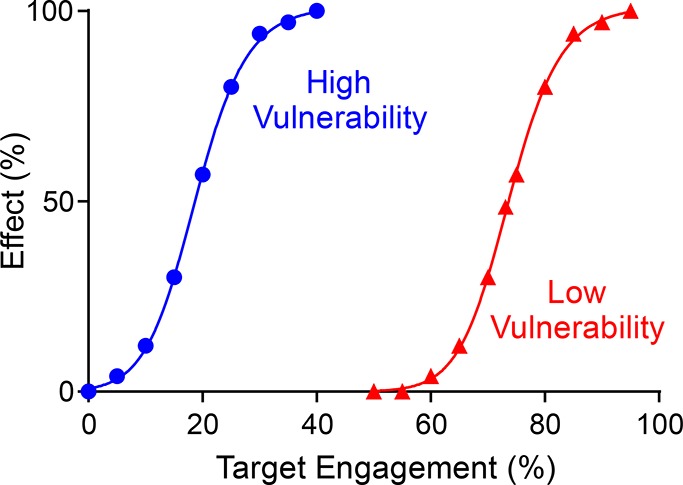
Target vulnerability plots. Vulnerability functions are shown for low (red) and high (blue) vulnerability targets. The vulnerability function is defined by the minimum level of engagement required for any effect to be observed (TOmin) and the level of engagement that leads to the maximal efficacy (TOmax). The third parameter required to define the function is the Hill coefficient or slope factor that determines the steepness of the effect response between TOmin and TOmax. For the low vulnerability target, the full physiological effect of the drug requires close to 100% target engagement, whereas only ∼35% engagement is needed for the high vulnerability target. The Hill coefficients for the two functions are 4.6 (high) and 16.4 (low).
The term vulnerability is often used in an “all-or-none” context in which targets are defined as vulnerable or not depending on whether or not target engagement results in the desired physiological effect. In Figure 3, this definition is taken one step further where we consider the degree of engagement that is required to generate the desired response. Clearly a low vulnerability target will require relatively higher levels of drug exposure to achieve pharmacologically relevant levels of engagement compared to a high vulnerability target. In addition, low vulnerability targets will be less susceptible to kinetic selectivity since it will take less time for a small fraction of active target to be generated either by drug dissociation or by the synthesis of the small percent of new target required to alleviate the impact of target engagement. Approaches that have been used to infer target vulnerability include methods that reduce the amount of target in the cell, either by genetic knockdown or by directly depleting proteins by inducible degradation.46−48 As we show below, cell-based washout experiments can also provide insight into target vulnerability.
Based on the discussion above, we would argue that the degree of target vulnerability must factor into considerations of target “druggability” since presumably it will be easier to “drug” a high vulnerability target compared one that is less vulnerable. It then follows that it is important to identify the molecular factors that influence the target vulnerability function. Under conditions that favor kinetic selectivity, for example, when the rate of drug elimination is rapid relative to the rate of drug–target complex breakdown, then the rate of target turnover will play a major role in controlling target vulnerability since rapid target resynthesis will alleviate the impact of even a covalent inhibitor once free drug has been removed. In addition, the physiological context of the target and the downstream consequences of target engagement must also play a role in vulnerability. For instance, an enzyme that catalyzes the rate limiting step in a metabolic pathway might be more vulnerable to target engagement compared to other enzymes in the pathway. Furthermore, the length of time that a target must be engaged will also be important: a more vulnerable target might be one where only transitory engagement is needed to trigger a cascade of events that result in the desired pharmacodynamic effect.
Probing Target Vulnerability: Cell Washout Experiments
Although kinetic selectivity can be defined at the level of a purified target, the role that kinetic parameters such as drug-target residence time play in drug activity will be controlled by the relationship between target engagement and time-dependent drug activity. It is thus crucial to evaluate time-dependent effects of drug treatment in more complex biological systems. As we mentioned above, compound activity is often quantified using only IC50 values obtained at constant compound concentration. This is also true for measurements of cell-based activity, which are also often only evaluated using experiments performed at constant concentration. Thus, any assessment of kinetic selectivity must include cell-based washout experiments, in which the phenotypic consequences of target engagement are evaluated once drug is “removed” from the system. Such experiments will provide direct insight into target vulnerability and the role that molecular processes such as target turnover play in controlling the coupling between drug-target residence time and time-dependent drug activity. Conceptually the approach is straightforward once a method is in place to remove compound from the media that surrounds the cells. In antibacterial space, the postantibiotic effect (PAE) of a compound is normally assessed by diluting cells exposed to drug into fresh media and monitoring the rate of regrowth.49 This approach can be employed for other types of cells that are grown in suspension. Alternative approaches are available for cells that grow on surfaces.
The cell washout experiment is very informative since it provides insight into how long it takes for the level of target engagement to fall to a point where the cell recovers from drug treatment. Intuitively we can see that target vulnerability will be a factor in controlling the length of time it takes for a cell to recover from drug removal since it will take relatively less time for processes such as target dissociation or target resynthesis to generate sufficient active target to alleviate the impact of drug binding to a low vulnerability target compared to a high vulnerability target. Thus, for a compound that has prolonged target engagement due to long residence time or covalent inhibition, the lack of long lasting effects of drug treatment following washout suggests that the target has low vulnerability, at least under the growth conditions that are employed. Conversely, a prolonged phenotypic response following washout would suggest a higher level of target vulnerability. Of course there are caveats: for example, in addition to long residence time, drug rebinding and/or accumulation of drug in the cell or membrane will result in extended target engagement. In addition, prolonged drug effects might also be due to the slow repair of essential processes that were reversibly damaged by drug treatment. Finally, it is important to reiterate a point made above: the time-dependent effects of drug treatment will depend on the growth conditions. The media used to culture cells in the lab may present a very different environment to that experienced in vivo, due to immune pressure or nutrient limitation, which could have a profound effect on target vulnerability in the same way that growth conditions can influence target essentiality. Indeed, the observation of prolonged in vivo effects after drug has been eliminated when no time-dependent effects are observed in vitro may indicate that the target is more vulnerable in vivo than in vitro.
Correlations between residence time and cellular washout have been examined in a number of systems. For antagonists of inflammatory protein complement C5a, an increase in residence time was shown to correlate with extended cellular activity following washout.50 Conversely, whereas the cellular kinetics of histone acetylation correlated with the kinetics of HDAC inhibition, washout of long residence time benzamide-based HDAC inhibitors did not result in a prolonged phenotypic response.51 In addition, we have assessed the translation of drug–target residence time to time-dependent antibacterial activity for two structurally related compound series that inhibit the FabI enoyl-ACP reductase, a target in Gram-positive and Gram-negative bacteria, and UDP-3-O-acyl-N-acetylglucosamine deacetylase (LpxC), a target in Gram-negative bacteria. Evaluation of the relationship between residence time and postantibiotic effect (PAE) revealed a steeper correlation for LpxC than FabI suggesting that LpxC is more vulnerable under the growth conditions employed (Figure 4).21,24
Figure 4.
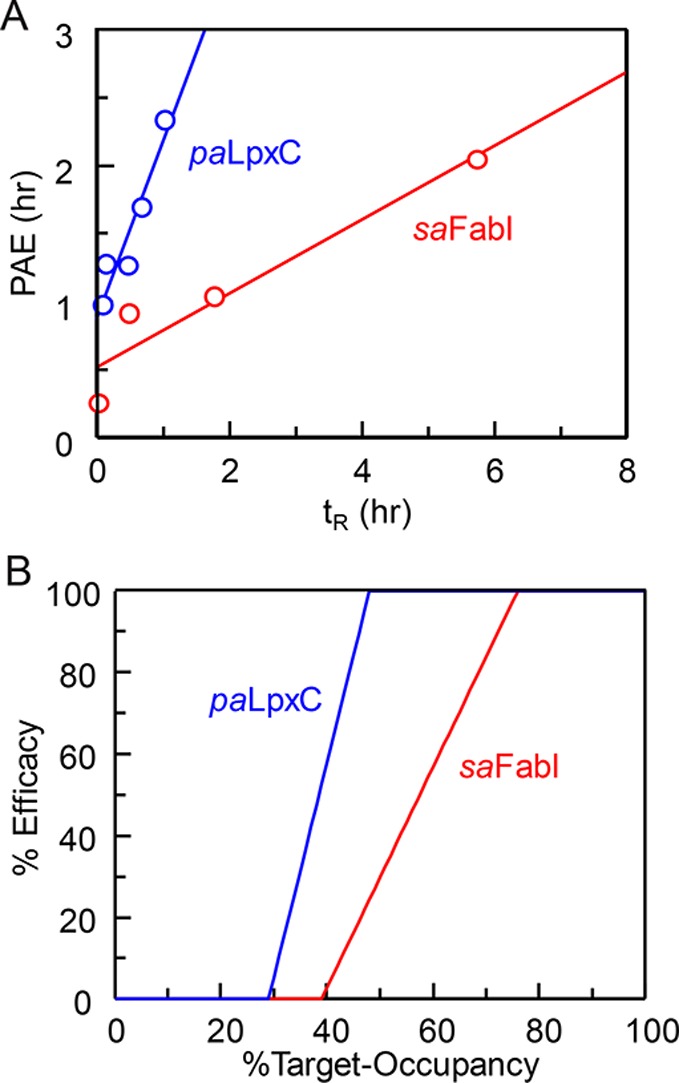
Vulnerability of two antibacterial targets: LpxC and FabI. (A) Correlation between residence time (tR) and postantibiotic effect (PAE) for two series of antibacterial compounds that target UDP-3-O-acyl-N-acetylglucosamine deacetylase from Pseudomonas aeruginosa (paLpxC) and the enoyl-ACP reductase from Staphylococcus aureus (saFabI). (B) Vulnerability functions after global fitting of the PAE data to a PK/PD model that integrates drug-target kinetics into predictions of drug activity. Both plots support the conclusion that paLpxC is more vulnerable than saFabI.21,24 Adapted from ref. (24) with permission from the Royal Society of Chemistry.
Box 2. Target Vulnerability and Cell Washout Experiments.
Target vulnerability is the fractional target occupancy required to produce the desired pharmacodynamic (PD) response and is defined by a vulnerability function given by values for TOmin, TOmax, and a Hill coefficient. Low vulnerability targets require high levels of occupancy to achieve the desired PD, whereas high vulnerability targets require lower levels of occupancy. Target vulnerability can be assessed through cell washout experiments where free drug is “removed” from the system by washing or dilution. Prolongation of the phenotypic response to drug treatment following washout can be due to several factors including continued occupancy of the target by the drug. In turn, continued target occupancy may be the result of a slow off rate, or drug rebinding. Correlations between the off rate (drug–target residence time) and a prolonged phenotypic response inform on target vulnerability and the potential for kinetic selectivity to play a role in drug pharmacology. Where possible, the drug–target kinetic data should be obtained at 37 °C since temperature will affect the rate constants for drug binding. In addition, it should be noted that target vulnerability will be affected by environmental factors such as growth conditions, and so target vulnerability in (e.g.) cell culture may be different from that found in vivo.
Translating Drug-Target Kinetics to Predictions of Drug-Activity: Mechanistic PK/PD Models
Pharmacokinetic/pharmacodynamic (PK/PD) models predict the effect time-courses resulting from administration of a drug dose. In order to fully utilize the information gained by a detailed analysis of binding kinetics, we have developed PK/PD models that integrate drug-target kinetics into predictions of drug activity.21,24,26 Essentially this involves the replacement of the Hill receptor binding equation in standard PK/PD models with the full kinetic scheme that describes the drug binding reaction coordinate (Text Box 3). In this way both the thermodynamics and kinetics of drug binding can be used to predict target engagement as a function of time and drug concentration. Importantly, the Hill receptor equation assumes that a rapid equilibrium exists between drug and target. However, this assumption can seriously underpredict target engagement in situations where a drug has a long residence time on the target relative to the rate of drug elimination. The PK/PD model was used to successfully predict the in vivo activity of a paLpxC inhibitor in an animal model of infection (Figure 5),21 and also of inhibitors of saFabI.24 Significantly, the predicted drug efficacy is much less if a rapid equilibrium is assumed between drug and target. In other words, use of a traditional PK/PD model would mandate much higher drug doses than actually needed. This analysis supports the underlying importance of the drug-target kinetic approach, and particularly the potential benefit of drugs with long residence times: compounds that dissociate slowly from their targets may have prolonged activity at low drug concentration enabling dosing frequency and/or dosing levels to be reduced thereby leading to an increase in the therapeutic window.
Figure 5.

Mechanistic PK/PD model. (A) The kinetic mechanism for two-step time-dependent inhibition replaced the Hill equation in a standard antibacterial pharmacodynamic model. (B) The model was used to successfully predict the efficacy of a paLpxC inhibitor in an animal model of infection (solid line). PK/PD modeling assuming rapid equilibrium between drug and target significantly underestimates the observed efficacy (dashed line). Figure adapted from Walkup et al.21
In Vivo Target Vulnerability: Chemical Tools to Quantify Target Engagement
The PK/PD model described above uses data from in vitro cell washout experiments to optimize the parameters that are required to predict in vivo drug activity.21,24 This approach, which involves the calculation of target engagement as a function of time and drug concentration, can be improved if in vivo target engagement can be directly quantified since this provides direct insight into the relationship between drug binding and efficacy (and indeed also evidence that the drug is actually binding to the designated target). In systems that involve covalent inhibition, such as in tyrosine kinases that have a conserved Cys at their active sites, it is possible to develop active site-directed covalent probes to quantify target engagement,52 and we have used this approach to analyze the inhibition of the Bruton’s tyrosine kinase (Btk) by the covalent inhibitor CC-292.53 Btk is a nonreceptor tyrosine kinase that is a promising target for treating diseases caused by B cell dysregulation, such as B-cell malignancies and autoimmune diseases including rheumatoid arthritis and lupus.54−58 CC-29253 as well as drugs such as ibrutinib59 contain an acrylamide electrophile that reacts with a conserved Cys (481) in the Btk active site. We synthesized a fluorescent probe based on CC-292 that was used to quantify levels of Btk engagement by CC-292 both in cell culture (Ramos cells) as well as in B lymphocytes obtained from rats dosed with CC-292. Initial values for PK/PD modeling were obtained by quantifying CC-292 binding to Btk in Ramos cells first under equilibrium conditions and then following washout of CC-292 to enable the rate of Btk turnover to be estimated. In addition, the kinetic parameters for Btk inhibition by CC-292 were calculated using the extracellular concentration of CC-292 in plasma (free fraction), thus removing the need to estimate the drug concentration across the cell membrane. Indeed, the ratio of the Ki values determined for cellular Btk compared to purified Btk was 80, suggesting that the CC-292 concentration in the cell was 80-fold lower than the concentration in the media or plasma. Fitting of time-dependent in vivo Btk engagement to a PK/PD model that explicitly included target turnover (ρ) and the ratio of [ATP] to the Km value for ATP (M = Km/[ATP]) provided a set of optimized parameters that were then used to accurately predict the efficacy of Btk in a rat model of collagen-induced arthritis (CIA). This then enabled an explicit evaluation of Btk vulnerability in vivo (Figure 6).26 The Btk vulnerability function indicates that >90% occupancy is needed to deliver the maximum efficacy in the rat CIA model, and that levels of engagement below ∼50% have no beneficial effect. This suggests that Btk is a relatively low vulnerability target.
Figure 6.

PK/PD model predicts the efficacy of CC-292, a covalent inhibitor of Btk (A) The kinetic scheme for inhibition of Btk by CC-292. Since CC-292 is an irreversible inhibitor, k6 = 0. (B) Structure of CC-292. (C) Fluorescent analogue of CC-292 used to quantify Btk engagement. (D) Predicted and observed efficacy of CC-292 in a rat model of collagen induced arthritis. (E) Vulnerability function for engagement of Btk. Adapted from ref. (26) with permission from the Royal Society of Chemistry.
Box 3. Mechanistic PK/PD Models That Include Drug-Target Kinetics.
A PK/PD model has been developed that integrates both the kinetics and thermodynamics of drug binding into predictions of drug activity. The model calculates target engagement as a function of both drug concentration and time, and then relates the time-dependence of engagement to drug pharmacodynamics. In vitro washout experiments are used to generate optimized parameters for enzyme inhibition and target turnover that are then used to predict in vivo efficacy. Quantification of target engagement using a covalent probe enables target turnover to be explicitly included in the model and provides direct insight into the vulnerability of the target in vivo.
The Btk study demonstrates that a covalent probe can be used to directly determine both TOmin and TOmax as well as the shape of the vulnerability function. For the reversible inhibitors of paLpxC and saFabI, estimates for TOmin and TOmax were obtained by calculating target engagement using the kinetic parameters for enzyme inhibition and then fitting the cellular washout (PAE) data obtained for each compound in the series to the PK/PD model. The vulnerability functions in Figure 4 were then generated by assuming a linear increase in antibacterial activity between TOmin and TOmax. While this approach can be used to determine the in vivo vulnerability functions for paLpxC and saFabI, as well as other reversible and irreversible inhibitors, approaches that correlate engagement and efficacy have traditionally relied on biomarkers, including the use of positron emission tomography (PET) radiotracers to noninvasively quantify in vivo target engagement.60−62 Examples of these approaches include the observation that the maximal effect on glucose levels was achieved with ≥80% inhibition of the type II diabetes target dipeptidyl peptidase-4 (DPP-4),60,61 and the demonstration by PET imaging that 80% occupancy of the serotonin (5-HT) transporter (5-HTT) was required for antidepressant efficacy of SSRIs,63 and >90% occupancy of the neurokinin-1 (NK1) receptor was required for the antiemetic activity of aprepitant.62
Drug–Target Kinetics and CNS Tumors
As discussed in this review, the application of drug-target kinetics to different disease states, such as in neurooncology, requires explicit knowledge of both the thermodynamics and kinetics of drug binding. In this regard, kinase inhibitor discovery programs provide a fertile ground on which to test and implement approaches based on kinetic selectivity for several reasons. First, a number of well-characterized kinases are targets for treating CNS tumors,2 and access to kinase panels enables binding to potential off-target kinases to be assessed. In addition, kinase enzymology has been heavily studied, and thus in many cases there is a detailed understanding of mechanisms that lead to long residence time inhibition, such as binding to the DFG-out conformation or the development of covalent inhibitors.33,64,65 Finally, recognition of the potential importance of kinetic selectivity has stimulated the interrogation of structure kinetic relationships for both purified kinases and for kinase inhibitors in cell-based assays.66−68 For example, as noted above, although lapatinib binds ∼10-fold less potently to EGFR than gefitinib (Kiapp values of 3 and 0.4 nM, respectively), the residence time of lapatinib on EGFR is >30-fold longer (430 and <14 min). The increase in lapatinib residence time was shown to translate to prolonged biochemical activity in a cell washout experiment (15% recovery 96 h after washout).34 In addition, drug-target kinetics have also been employed in an attempt to account for the reduced efficacy of erlotinib for treating glioblastoma based on the observation that the residence time of erlotinib on the EGFRvIII mutant found in gliomas is shorter than that for the L858R EGFR mutant associated with NSCLC.69
As mentioned at the beginning of this review, neurooncology drug discovery faces the additional hurdle of developing agents to penetrate the BBB. A recent review provides an authoritative summary of kinase inhibitors in this disease space and notes only a few examples of compounds that have been explicitly developed to be brain penetrant, such as inhibitors of EGFR and PI3K/mTOR.2 For example, in contrast to the classic NSCLC EGFR inhibitors,70 the irreversible pan-ERBB inhibitor NT113 has improved biodistribution to the CNS and has efficacy in intracranial glioblastoma xenografts, including those with high EGFRvIII expression.71 In addition, AZD3759 and GDC-0084 are reversible inhibitors of EGFR and PI3K/mTOR, respectively, both of which were designed to cross the BBB and show efficacy in preclinical models.72,73 However, other than the knowledge that NT113 is an irreversible inhibitor (which may contribute to improved efficacy of this compound compared to the classic reversible EGFR inhibitors), detailed kinetic studies on target binding and data from cell washout experiments are not available for these inhibitors limiting the conclusions that can be drawn about the relationship between target occupancy and efficacy.
Summary and Future Directions
The above discussion thus mitigates integrated efforts to generate time-dependent inhibitors of targets and evaluate the role played by parameters such as residence time in cellular washout experiments, and in more complex biological systems such as preclinical disease models. These efforts must be linked to mathematical approaches, such as PK/PD modeling, that link target engagement, drug concentration, and effect, and will be enhanced by direct measurements of target turnover, target levels, drug concentration, and target engagement. While the determination of the kinetic parameters for enzyme inhibition and receptor interactions is de rigueur for enzymologists and quantitative biochemists, and slow-binding inhibition has been recognized for more than 40 years (see Morrison and Walsh,3 and references therein), it is perhaps curious that drug-target kinetics and concepts such as kinetic selectivity are not more widely used in drug development. One reason might be that the time-dependent assays required to measure residence time are intrinsically more challenging than the straightforward dose–response relationships (IC50 values) that are derived from the end point assays used in high throughput automated compound screens and that dominate biological structure–activity relationship. Moving forward, the implementation of programs that utilize drug-target kinetics will involve the identification of compounds that display time-dependent binding and the subsequent development of structure-kinetic relationships (SKR) for target binding through combined med chem/kinetic efforts. This, coupled with structure-based design and methods to analyze and simulate protein dynamics,15,74,75 will guide the synthesis of compounds with optimized binding parameters. In turn, compound series that encompass a range of binding kinetics can then be used to explore kinetic selectivity in cells and preclinical models. We note that some CROs now offer services to generate drug-target kinetic data, and techniques such as surface plasmon resonance (SPR) are routinely used to determine binding kinetics for purified targets, supplementing approaches such as forward and reverse progress curve assays. In addition, inhibition studies often include “IC50-shift” measurements in which a reduction in IC50 following preincubation of enzyme and inhibitor is taken as evidence for slow-onset inhibition (at least for a competitive inhibitor). Future advances will involve the routine implementation of methods to measure binding kinetics in the cell, such as those derived from NanoBRET,76 approaches to quantify in vivo target engagement by reversible inhibitors,77 and the further development and parametrization of advanced mathematical models that better simulate and predict drug–target interactions in the complex environment of the human body.21,22,24,26
In summary, the availability of time-dependent target engagement data, both on purified targets and from cell washout experiments, will provide an additional dimension of information when selecting and optimizing drug leads including those that target brain cancers. In particular, prolonged target occupancy has the potential to translate into extended pharmacological activity at low drug concentration, which may be particularly important in neurooncology where drug exposure will be impacted by the BBB. However, the reliance on IC50 (or Ki, Kd) values for selecting and optimizing drug leads limits the ability to identify time-dependent inhibitors and ignores the possibility that kinetic selectivity may be present and could contribute to improvements in therapeutic window and safety.
NIH GM102864, PhRMA Foundation Fellowship, Genentech Inc.
The author declares no competing financial interest.
References
- Levin V. A.; Tonge P. J.; Gallo J. M.; Birtwistle M. R.; Dar A. C.; Iavarone A.; Paddison P. J.; Heffron T. P.; Elmquist W. F.; Lachowicz J. E.; Johnson T. W.; White F. M.; Sul J.; Smith Q. R.; Shen W.; Sarkaria J. N.; Samala R.; Wen P. Y.; Berry D. A.; Petter R. C. (2015) CNS Anticancer Drug Discovery and Development Conference White Paper. Neuro-Oncology 17 (Suppl 6), vi1–vi26. 10.1093/neuonc/nov169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffron T. P. (2016) Small Molecule Kinase Inhibitors for the Treatment of Brain Cancer. J. Med. Chem. 59, 10030–10066. 10.1021/acs.jmedchem.6b00618. [DOI] [PubMed] [Google Scholar]
- Morrison J. F.; Walsh C. T. (2006) The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 61, 201–301. 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- Swinney D. C. (2004) Biochemical mechanisms of drug action: what does it take for success?. Nat. Rev. Drug Discovery 3, 801–808. 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. (2006) Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discovery 5, 730–739. 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Swinney D. C. (2009) The role of binding kinetics in therapeutically useful drug action. Curr. Opin. Drug Discovery Dev. 12, 31–39. [PubMed] [Google Scholar]
- Zhang R.; Monsma F. (2009) The importance of drug-target residence time. Curr. Opin. Drug Discovery Dev. 12, 488–496. [PubMed] [Google Scholar]
- Lu H.; Tonge P. J. (2010) Drug-target residence time: critical information for lead optimization. Curr. Opin. Chem. Biol. 14, 467–474. 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G.; Charlton S. J. (2010) Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br. J. Pharmacol. 161, 488–508. 10.1111/j.1476-5381.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. (2010) The dynamics of drug-target interactions: drug target residence time and its impact on efficay and safety. Expert Opin. Drug Discovery 5, 305–310. 10.1517/17460441003677725. [DOI] [PubMed] [Google Scholar]
- Nunez S.; Venhorst J.; Kruse C. G. (2012) Target-drug interactions: first principles and their application to drug discovery. Drug Discovery Today 17, 10–22. 10.1016/j.drudis.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Swinney D. C. (2006) Biochemical mechanisms of New Molecular Entities (NMEs) approved by United States FDA during 2001–2004: mechanisms leading to optimal efficacy and safety. Curr. Top. Med. Chem. 6, 461–478. 10.2174/156802606776743093. [DOI] [PubMed] [Google Scholar]
- Copeland R. A. (2011) Conformational adaptation in drug-target interactions and residence time. Future Med. Chem. 3, 1491–1501. 10.4155/fmc.11.112. [DOI] [PubMed] [Google Scholar]
- Schramm V. L. (2011) Enzymatic transition states, transition-state analogs, dynamics, thermodynamics, and lifetimes. Annu. Rev. Biochem. 80, 703–732. 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. J.; Lai C. T.; Pan P.; Yu W.; Liu N.; Bommineni G. R.; Garcia-Diaz M.; Simmerling C.; Tonge P. J. (2014) A structural and energetic model for the slow-onset inhibition of the Mycobacterium tuberculosis enoyl-ACP reductase InhA. ACS Chem. Biol. 9, 986–993. 10.1021/cb400896g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G.; Van Liefde I.; Swinney D. C. (2016) On the different experimental manifestations of two-state ’induced-fit’ binding of drugs to their cellular targets. Br. J. Pharmacol. 173, 1268–1285. 10.1111/bph.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. (2011) The resurgence of covalent drugs. Nat. Rev. Drug Discovery 10, 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Lewandowicz A.; Tyler P. C.; Evans G. B.; Furneaux R. H.; Schramm V. L. (2003) Achieving the ultimate physiological goal in transition state analogue inhibitors for purine nucleoside phosphorylase. J. Biol. Chem. 278, 31465–31468. 10.1074/jbc.C300259200. [DOI] [PubMed] [Google Scholar]
- Lu H.; England K.; am Ende C.; Truglio J. J.; Luckner S.; Reddy B. G.; Marlenee N. L.; Knudson S. E.; Knudson D. L.; Bowen R. A.; Kisker C.; Slayden R. A.; Tonge P. J. (2009) Slow-onset inhibition of the FabI enoyl reductase from francisella tularensis: residence time and in vivo activity. ACS Chem. Biol. 4, 221–231. 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G. (2015) On the ’micro’-pharmacodynamic and pharmacokinetic mechanisms that contribute to long-lasting drug action. Expert Opin. Drug Discovery 10, 1085–1098. 10.1517/17460441.2015.1067196. [DOI] [PubMed] [Google Scholar]
- Walkup G. K.; You Z.; Ross P. L.; Allen E. K.; Daryaee F.; Hale M. R.; O’Donnell J.; Ehmann D. E.; Schuck V. J.; Buurman E. T.; Choy A. L.; Hajec L.; Murphy-Benenato K.; Marone V.; Patey S. A.; Grosser L. A.; Johnstone M.; Walker S. G.; Tonge P. J.; Fisher S. L. (2015) Translating slow-binding inhibition kinetics into cellular and in vivo effects. Nat. Chem. Biol. 11, 416–423. 10.1038/nchembio.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Witte W. E. A.; Wong Y. C.; Nederpelt I.; Heitman L. H.; Danhof M.; van der Graaf P. H.; Gilissen R. A.; de Lange E. C. M. (2016) Mechanistic models enable the rational use of in vitro drug-target binding kinetics for better drug effects in patients. Expert Opin. Drug Discovery 11, 45–63. 10.1517/17460441.2016.1100163. [DOI] [PubMed] [Google Scholar]
- de Witte W. E.; Danhof M.; van der Graaf P. H.; de Lange E. C. (2016) In vivo Target Residence Time and Kinetic Selectivity: The Association Rate Constant as Determinant. Trends Pharmacol. Sci. 37, 831–842. 10.1016/j.tips.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Daryaee F.; Chang A.; Schiebel J.; Lu Y.; Zhang Z.; Kapilashrami K.; Walker S. G.; Kisker C.; Sotriffer C. A.; Fisher S. L.; Tonge P. J. (2016) Correlating Drug-Target Kinetics and In vivo Pharmacodynamics: Long Residence Time Inhibitors of the FabI Enoyl-ACP Reductase. Chem. Sci. 7, 5945–5954. 10.1039/C6SC01000H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. (2016) The drug-target residence time model: a 10-year retrospective. Nat. Rev. Drug Discovery 15, 87–95. 10.1038/nrd.2015.18. [DOI] [PubMed] [Google Scholar]
- Daryaee F.; Zhang Z.; Gogarty K. R.; Li Y.; Merino J.; Fisher S. L.; Tonge P. J. (2017) A quantitative mechanistic PK/PD model directly connects Btk target engagement and in vivo efficacy. Chem. Sci. 8, 3434–3443. 10.1039/C6SC03306G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz D. A.; de Witte W. E. A.; Wong Y. C.; Knasmueller B.; Richter L.; Kokh D. B.; Sadiq S. K.; Bosma R.; Nederpelt I.; Heitman L. H.; Segala E.; Amaral M.; Guo D.; Andres D.; Georgi V.; Stoddart L. A.; Hill S.; Cooke R. M.; De Graaf C.; Leurs R.; Frech M.; Wade R. C.; de Lange E. C. M.; IJzerman A. P.; Muller-Fahrnow A.; Ecker G. F. (2017) Kinetics for Drug Discovery: an industry-driven effort to target drug residence time. Drug Discovery Today 22, 896–911. 10.1016/j.drudis.2017.02.002. [DOI] [PubMed] [Google Scholar]
- Rawat R.; Whitty A.; Tonge P. J. (2003) The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc. Natl. Acad. Sci. U. S. A. 100, 13881–13886. 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckner S. R.; Liu N.; am Ende C. W.; Tonge P. J.; Kisker C. (2010) A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J. Biol. Chem. 285, 14330–14337. 10.1074/jbc.M109.090373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney D. C.; Beavis P.; Chuang K. T.; Zheng Y.; Lee I.; Gee P.; Deval J.; Rotstein D. M.; Dioszegi M.; Ravendran P.; Zhang J.; Sankuratri S.; Kondru R.; Vauquelin G. (2014) A study of the molecular mechanism of binding kinetics and long residence times of human CCR5 receptor small molecule allosteric ligands. Br. J. Pharmacol. 171, 3364–3375. 10.1111/bph.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnuolo L. A.; Eltschkner S.; Yu W.; Daryaee F.; Davoodi S.; Knudson S. E.; Allen E. K.; Merino J.; Pschibul A.; Moree B.; Thivalapill N.; Truglio J. J.; Salafsky J.; Slayden R. A.; Kisker C.; Tonge P. J. (2017) Evaluating the Contribution of Transition-State Destabilization to Changes in the Residence Time of Triazole-Based InhA Inhibitors. J. Am. Chem. Soc. 139, 3417–3429. 10.1021/jacs.6b11148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavapathruni A.; Jin L.; Daigle S. R.; Majer C. R. a.; Therkelsen C. a.; Wigle T. J.; Kuntz K. W.; Chesworth R.; Pollock R. M.; Scott M. P.; Moyer M. P.; Richon V. M.; Copeland R. a.; Olhava E. J. (2012) Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem. Biol. Drug Des. 80, 971–980. 10.1111/cbdd.12050. [DOI] [PubMed] [Google Scholar]
- Schneider E. V.; Böttcher J.; Huber R.; Maskos K.; Neumann L. (2013) Structure-kinetic relationship study of CDK8/CycC specific compounds. Proc. Natl. Acad. Sci. U. S. A. 110, 8081–8086. 10.1073/pnas.1305378110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood E. R.; Truesdale A. T.; McDonald O. B.; Yuan D.; Hassell A.; Dickerson S. H.; Ellis B.; Pennisi C.; Horne E.; Lackey K.; Alligood K. J.; Rusnak D. W.; Gilmer T. M.; Shewchuk L. (2004) A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 64, 6652–6659. 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- Glossop P. A.; Watson C. A.; Price D. A.; Bunnage M. E.; Middleton D. S.; Wood A.; James K.; Roberts D.; Strang R. S.; Yeadon M.; Perros-Huguet C.; Clarke N. P.; Trevethick M. A.; Machin I.; Stuart E. F.; Evans S. M.; Harrison A. C.; Fairman D. A.; Agoram B.; Burrows J. L.; Feeder N.; Fulton C. K.; Dillon B. R.; Entwistle D. A.; Spence F. J. (2011) Inhalation by design: novel tertiary amine muscarinic M(3) receptor antagonists with slow off-rate binding kinetics for inhaled once-daily treatment of chronic obstructive pulmonary disease. J. Med. Chem. 54, 6888–6904. 10.1021/jm200884j. [DOI] [PubMed] [Google Scholar]
- Calbet M.; Andres M.; Armengol C.; Bravo M.; Eichhorn P.; Lopez R.; Garcia-Gonzalez V.; Roberts R.; Miralpeix M. (2016) Pharmacological characterization of CRTh2 antagonist LAS191859: Long receptor residence time translates into long-lasting in vivo efficacy. Pharmacol. Res. 111, 208–216. 10.1016/j.phrs.2016.06.014. [DOI] [PubMed] [Google Scholar]
- Wentsch H. K.; Walter N. M.; Buhrmann M.; Mayer-Wrangowski S.; Rauh D.; Zaman G. J. R.; Willemsen-Seegers N.; Buijsman R. C.; Henning M.; Dauch D.; Zender L.; Laufer S. (2017) Optimized Target Residence Time: Type I1/2 Inhibitors for p38alpha MAP Kinase with Improved Binding Kinetics through Direct Interaction with the R-Spine. Angew. Chem., Int. Ed. 56, 5363–5367. 10.1002/anie.201701185. [DOI] [PubMed] [Google Scholar]
- Guo D.; Heitman L. H.; IJzerman A. P. (2016) The Added Value of Assessing Ligand-Receptor Binding Kinetics in Drug Discovery. ACS Med. Chem. Lett. 7, 819–821. 10.1021/acsmedchemlett.6b00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes D. A.; Dowling M. R.; Leighton-Davies J.; Kent T. C.; Fawcett L.; Renard E.; Trifilieff A.; Charlton S. J. (2012) The Influence of receptor kinetics on the onset and duration of action and the therapeutic index of NVA237 and tiotropium. J. Pharmacol. Exp. Ther. 343, 520–528. 10.1124/jpet.112.194456. [DOI] [PubMed] [Google Scholar]
- Tautermann C. S.; Kiechle T.; Seeliger D.; Diehl S.; Wex E.; Banholzer R.; Gantner F.; Pieper M. P.; Casarosa P. (2013) Molecular basis for the long duration of action and kinetic selectivity of tiotropium for the muscarinic M3 receptor. J. Med. Chem. 56, 8746–8756. 10.1021/jm401219y. [DOI] [PubMed] [Google Scholar]
- Ayaz P.; Andres D.; Kwiatkowski D. A.; Kolbe C. C.; Lienau P.; Siemeister G.; Lucking U.; Stegmann C. M. (2016) Conformational Adaption May Explain the Slow Dissociation Kinetics of Roniciclib (BAY 1000394), a Type I CDK Inhibitor with Kinetic Selectivity for CDK2 and CDK9. ACS Chem. Biol. 11, 1710–1719. 10.1021/acschembio.6b00074. [DOI] [PubMed] [Google Scholar]
- Levy G. (1994) Pharmacologic target-mediated drug disposition. Clin. Pharmacol. Ther. 56, 248–252. 10.1038/clpt.1994.134. [DOI] [PubMed] [Google Scholar]
- Mager D. E. (2006) Target-mediated drug disposition and dynamics. Biochem. Pharmacol. 72, 1–10. 10.1016/j.bcp.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. (2009) FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41. 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29. 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Wei J. R.; Krishnamoorthy V.; Murphy K.; Kim J. H.; Schnappinger D.; Alber T.; Sassetti C. M.; Rhee K. Y.; Rubin E. J. (2011) Depletion of antibiotic targets has widely varying effects on growth. Proc. Natl. Acad. Sci. U. S. A. 108, 4176–4181. 10.1073/pnas.1018301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P.; Agarwal S.; Datta S. (2009) Delineating bacteriostatic and bactericidal targets in mycobacteria using IPTG inducible antisense expression. PLoS One 4, e5923. 10.1371/journal.pone.0005923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran V.; Ramachandran V.; Singh R.; Yang X.; Ragadeepthi T.; Mohapatra S.; Khandelwal S.; Patel S. (2013) Genetic and chemical knockdown: a complementary strategy for evaluating an anti-infective target. Adv. Appl. Bioinform Chem. 6, 1–13. 10.2147/AABC.S39198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundtzen R. W.; Gerber A. U.; Cohn D. L.; Craig W. A. (1981) Postantibiotic suppression of bacterial growth. Clin. Infect. Dis. 3, 28–37. 10.1093/clinids/3.1.28. [DOI] [PubMed] [Google Scholar]
- Seow V.; Lim J.; Cotterell A. J.; Yau M. K.; Xu W.; Lohman R. J.; Kok W. M.; Stoermer M. J.; Sweet M. J.; Reid R. C.; Suen J. Y.; Fairlie D. P. (2016) Receptor residence time trumps drug-likeness and oral bioavailability in determining efficacy of complement C5a antagonists. Sci. Rep. 6, 24575. 10.1038/srep24575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer B. E.; Mintzer R.; Fong R.; Mukund S.; Tam C.; Zilberleyb I.; Flicke B.; Ritscher A.; Fedorowicz G.; Vallero R.; Ortwine D. F.; Gunzner J.; Modrusan Z.; Neumann L.; Koth C. M.; Lupardus P. J.; Kaminker J. S.; Heise C. E.; Steiner P. (2013) Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 288, 26926–26943. 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair J. A.; Rauh D.; Kung C.; Yun C. H.; Fan Q. W.; Rode H.; Zhang C.; Eck M. J.; Weiss W. A.; Shokat K. M. (2007) Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat. Chem. Biol. 3, 229–238. 10.1038/nchembio866. [DOI] [PubMed] [Google Scholar]
- Evans E. K.; Tester R.; Aslanian S.; Karp R.; Sheets M.; Labenski M. T.; Witowski S. R.; Lounsbury H.; Chaturvedi P.; Mazdiyasni H.; Zhu Z.; Nacht M.; Freed M. I.; Petter R. C.; Dubrovskiy A.; Singh J.; Westlin W. F. (2013) Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J. Pharmacol. Exp. Ther. 346, 219–228. 10.1124/jpet.113.203489. [DOI] [PubMed] [Google Scholar]
- Katewa A.; Wang Y.; Hackney J. A.; Huang T.; Suto E.; Ramamoorthi N.; Austin C. D.; Bremer M.; Chen J. Z.; Crawford J. J.; Currie K. S.; Blomgren P.; DeVoss J.; DiPaolo J. A.; Hau J.; Johnson A.; Lesch J.; DeForge L. E.; Lin Z.; Liimatta M.; Lubach J. W.; McVay S.; Modrusan Z.; Nguyen A.; Poon C.; Wang J.; Liu L.; Lee W. P.; Wong H.; Young W. B.; Townsend M. J.; Reif K. (2017) Btk-specific inhibition blocks pathogenic plasma cell signatures and myeloid cell-associated damage in IFNalpha-driven lupus nephritis. JCI Insight 2, e90111. 10.1172/jci.insight.90111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo J. A.; Huang T.; Balazs M.; Barbosa J.; Barck K. H.; Bravo B. J.; Carano R. A.; Darrow J.; Davies D. R.; DeForge L. E.; Diehl L.; Ferrando R.; Gallion S. L.; Giannetti A. M.; Gribling P.; Hurez V.; Hymowitz S. G.; Jones R.; Kropf J. E.; Lee W. P.; Maciejewski P. M.; Mitchell S. A.; Rong H.; Staker B. L.; Whitney J. A.; Yeh S.; Young W. B.; Yu C.; Zhang J.; Reif K.; Currie K. S. (2011) Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat. Chem. Biol. 7, 41–50. 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- Martinez-Gamboa L.; Brezinschek H.-P.; Burmester G. R.; Dörner T. (2006) Immunopathologic role of B lymphocytes in rheumatoid arthritis: Rationale of B cell-directed therapy. Autoimmun. Rev. 5, 437–442. 10.1016/j.autrev.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Wang X.; Barbosa J.; Blomgren P.; Bremer M. C.; Chen J.; Crawford J. J.; Deng W.; Dong L.; Eigenbrot C.; Gallion S.; Hau J.; Hu H.; Johnson A. R.; Katewa A.; Kropf J. E.; Lee S. H.; Liu L.; Lubach J. W.; Macaluso J.; Maciejewski P.; Mitchell S. A.; Ortwine D. F.; DiPaolo J.; Reif K.; Scheerens H.; Schmitt A.; Wong H.; Xiong J.-M.; Xu J.; Zhao Z.; Zhou F.; Currie K. S.; Young W. B. (2017) Discovery of Potent and Selective Tricyclic Inhibitors of Bruton’s Tyrosine Kinase with Improved Drug-like Properties. ACS Med. Chem. Lett. 8, 608–613. 10.1021/acsmedchemlett.7b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak T.; Robak E. (2012) Tyrosine kinase inhibitors as potential drugs for B-cell lymphoid malignancies and autoimmune disorders. Expert Opin. Invest. Drugs 21, 921–947. 10.1517/13543784.2012.685650. [DOI] [PubMed] [Google Scholar]
- Honigberg L. A.; Smith A. M.; Sirisawad M.; Verner E.; Loury D.; Chang B.; Li S.; Pan Z.; Thamm D. H.; Miller R. A.; Buggy J. J. (2010) The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. U. S. A. 107, 13075–13080. 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J. A. (2008) Strategic approach to fit-for-purpose biomarkers in drug development. Annu. Rev. Pharmacol. Toxicol. 48, 631–651. 10.1146/annurev.pharmtox.48.113006.094611. [DOI] [PubMed] [Google Scholar]
- Durham T. B.; Blanco M. J. (2015) Target engagement in lead generation. Bioorg. Med. Chem. Lett. 25, 998–1008. 10.1016/j.bmcl.2014.12.076. [DOI] [PubMed] [Google Scholar]
- Frank R.; Hargreaves R. (2003) Clinical biomarkers in drug discovery and development. Nat. Rev. Drug Discovery 2, 566–580. 10.1038/nrd1130. [DOI] [PubMed] [Google Scholar]
- Meyer J. H.; Wilson A. A.; Sagrati S.; Hussey D.; Carella A.; Potter W. Z.; Ginovart N.; Spencer E. P.; Cheok A.; Houle S. (2004) Serotonin transporter occupancy of five selective serotonin reuptake inhibitors at different doses: an [11C]DASB positron emission tomography study. Am. J. Psychiatry 161, 826–835. 10.1176/appi.ajp.161.5.826. [DOI] [PubMed] [Google Scholar]
- Alexander L. T.; Mobitz H.; Drueckes P.; Savitsky P.; Fedorov O.; Elkins J. M.; Deane C. M.; Cowan-Jacob S. W.; Knapp S. (2015) Type II Inhibitors Targeting CDK2. ACS Chem. Biol. 10, 2116–2125. 10.1021/acschembio.5b00398. [DOI] [PubMed] [Google Scholar]
- Chaikuad A.; Tacconi E. M.; Zimmer J.; Liang Y.; Gray N. S.; Tarsounas M.; Knapp S. (2014) A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 10, 853–860. 10.1038/nchembio.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoop A.; Dey F. (2015) On-rate based optimization of structure-kinetic relationship - surfing the kinetic map. Drug Discovery Today: Technol. 17, 9–15. 10.1016/j.ddtec.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Bradshaw J. M.; McFarland J. M.; Paavilainen V. O.; Bisconte A.; Tam D.; Phan V. T.; Romanov S.; Finkle D.; Shu J.; Patel V.; Ton T.; Li X.; Loughhead D. G.; Nunn P. A.; Karr D. E.; Gerritsen M. E.; Funk J. O.; Owens T. D.; Verner E.; Brameld K. A.; Hill R. J.; Goldstein D. M.; Taunton J. (2015) Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 11, 525–531. 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. M.; Taunton J. (2014) Targeting protein kinases with selective and semipromiscuous covalent inhibitors. Methods Enzymol. 548, 93–116. 10.1016/B978-0-12-397918-6.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkovich K. J.; Hariono S.; Garske A. L.; Zhang J.; Blair J. A.; Fan Q. W.; Shokat K. M.; Nicolaides T.; Weiss W. A. (2012) Kinetics of inhibitor cycling underlie therapeutic disparities between EGFR-driven lung and brain cancers. Cancer Discovery 2, 450–457. 10.1158/2159-8290.CD-11-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broniscer A.; Panetta J. C.; O’Shaughnessy M.; Fraga C.; Bai F.; Krasin M. J.; Gajjar A.; Stewart C. F. (2007) Plasma and cerebrospinal fluid pharmacokinetics of erlotinib and its active metabolite OSI-420. Clin. Cancer Res. 13, 1511–1515. 10.1158/1078-0432.CCR-06-2372. [DOI] [PubMed] [Google Scholar]
- Yoshida Y.; Ozawa T.; Yao T. W.; Shen W.; Brown D.; Parsa A. T.; Raizer J. J.; Cheng S. Y.; Stegh A. H.; Mazar A. P.; Giles F. J.; Sarkaria J. N.; Butowski N.; Nicolaides T.; James C. D. (2014) NT113, a pan-ERBB inhibitor with high brain penetrance, inhibits the growth of glioblastoma xenografts with EGFR amplification. Mol. Cancer Ther. 13, 2919–2929. 10.1158/1535-7163.MCT-14-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q.; Wang J.; Cheng Z.; Chen K.; Johnstrom P.; Varnas K.; Li D. Y.; Yang Z. F.; Zhang X. (2015) Discovery and Evaluation of Clinical Candidate AZD3759, a Potent, Oral Active, Central Nervous System-Penetrant, Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor. J. Med. Chem. 58, 8200–8215. 10.1021/acs.jmedchem.5b01073. [DOI] [PubMed] [Google Scholar]
- Heffron T. P.; Ndubaku C. O.; Salphati L.; Alicke B.; Cheong J.; Drobnick J.; Edgar K.; Gould S. E.; Lee L. B.; Lesnick J. D.; Lewis C.; Nonomiya J.; Pang J.; Plise E. G.; Sideris S.; Wallin J.; Wang L.; Zhang X.; Olivero A. G. (2016) Discovery of Clinical Development Candidate GDC-0084, a Brain Penetrant Inhibitor of PI3K and mTOR. ACS Med. Chem. Lett. 7, 351–356. 10.1021/acsmedchemlett.6b00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph J.; Xiao Y.; Pardi A.; Ahn N. G. (2015) Slow inhibition and conformation selective properties of extracellular signal-regulated kinase 1 and 2 inhibitors. Biochemistry 54, 22–31. 10.1021/bi501101v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C. T.; Li H. J.; Yu W.; Shah S.; Bommineni G. R.; Perrone V.; Garcia-Diaz M.; Tonge P. J.; Simmerling C. (2015) Rational Modulation of the Induced-Fit Conformational Change for Slow-Onset Inhibition in Mycobacterium tuberculosis InhA. Biochemistry 54, 4683–4691. 10.1021/acs.biochem.5b00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machleidt T.; Woodroofe C. C.; Schwinn M. K.; Mendez J.; Robers M. B.; Zimmerman K.; Otto P.; Daniels D. L.; Kirkland T. A.; Wood K. V. (2015) NanoBRET--A Novel BRET Platform for the Analysis of Protein-Protein Interactions. ACS Chem. Biol. 10, 1797–1804. 10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
- Murphy D. J.; Ou Y.; Euler D. H.; Wessner K.; Adamski S.; Luo B.; Wesolowski G. A.; Vogel R.; Glantschnig H.; Lubbers L. S.; Carroll S. S.; Lai M. T. (2015) Determination of in Vivo Enzyme Occupancy Utilizing Inhibitor Dissociation Kinetics. J. Am. Chem. Soc. 137, 11230–11233. 10.1021/jacs.5b06518. [DOI] [PubMed] [Google Scholar]