

Abstract
Recent genetic studies have provided overwhelming evidence of the involvement of microglia-related molecular networks in the pathophysiology of Alzheimer disease (AD). However, the precise mechanisms by which microglia alter the course of AD neuropathology remain poorly understood. Here we discuss current evidence of the neuroprotective functions of microglia with a focus on optical imaging studies that have revealed a role of these cells in the encapsulation of amyloid deposits (“microglia barrier”). This barrier modulates the degree of plaque compaction, amyloid fibril surface area and insulation from adjacent axons thereby reducing neurotoxicity. We discuss findings implicating genetic variants of the microglia receptor, Triggering Receptor Expressed On Myeloid Cells 2 (TREM2), in the increased risk of late onset AD. We provide evidence that increased AD risk is at least partly mediated by deficient microglia polarization towards amyloid deposits, resulting in ineffective plaque encapsulation and reduced plaque compaction, which is associated with worsened axonal pathology. Finally, we propose possible avenues for therapeutic targeting of plaque-associated microglia with the goal of enhancing the microglia barrier and potentially reducing disease progression.
Keywords: Microglia barrier, TREM2, axonal dystrophy, Alzheimer’s disease, optical imaging, neuroprotection
Introduction
Alzheimer’s disease (AD), the most prevalent neurodegenerative disorder, is characterized by slowly progressive cognitive decline that has devastating personal and socio-economic implications. Despite significant efforts to develop treatments the field has seen little success, partly due to an incomplete understanding of the mechanisms underlying disease pathogenesis. The defining neuropathological criteria for AD are extracellular deposits of aggregated β-amyloid (Aβ) peptides, and intracellular neurofibrillary tangles (NFT) composed of hyperphosphorylated microtubule associated protein (MAP) tau. A general consensus is that Aβ deposition occurs first, triggering NFT formation, synapse loss, cell death and cognitive decline(1, 2). However, the sequence of events leading to protein aggregation and the relative contributions of Aβ and tau aggregates to neural toxicity and glial reactions remain incompletely understood.
Aβ peptides are produced by enzymatic cleavage of the extracellular domain of the amyloid precursor protein (APP) (3) and are continuously released into the interstitial space. Peptides with different lengths are produced, with Aβ40 being the most abundant isoform but Aβ42 having the highest propensity to aggregate and greatest potential cytotoxicity(1). These peptides can polymerize into oligomers and fibrils, with low molecular weight species (e.g. dimers, trimers) being water soluble and diffusible(4), while protofibrils and fibrils becoming insoluble and having a tendency to coalesce into aggregates that deposit within the brain parenchyma and microvasculature (5). Once extracellular aggregation occurs, these deposits become a sink where newly formed Aβ monomers bind with high affinity, causing gradual plaque enlargement over very long intervals(6–9). Postmortem clinical-pathological correlations and Positron Emission Tomography (PET) imaging of AD patients has revealed that the build-up of Aβ precedes cognitive deficits by decades(10, 11). This suggests that a critical threshold of Aβ burden must be reached to instigate cognitive decline. However, there is a relatively weak correlation between plaque load and cognitive scores(12, 13), suggesting that additional factors contribute to modulating neural injury caused by amyloid deposits.
One such factor may be the microglial and astrocytic responses that occur around amyloid deposits. Microglia are yolk sac derived cells(14), that share functional and molecular features with tissue macrophages(15–17), and function as resident immune cells in the central nervous system (CNS). Microglia are found throughout the CNS where they are tiled into non-overlapping domains. Under homeostatic conditions, microglia are highly branched and motile, constantly extending and retracting while their cell body remains stationary(18, 19). The function of this dynamic behavior is poorly understood, but it may serve a surveillance role for detection of tissue homeostatic and pathological changes(20, 21). Recent work suggested that microglia may have additional physiological roles in the healthy brain such as synapse refinement(22, 23); monitoring synapse activity(24), and providing trophic support for neuronal plasticity(25). In response to pathogenic stimuli, cell debris and physical injury, microglia rapidly transform into activated phenotypes involving proliferation, increased phagocytosis and production of pro-inflammatory cytokines(20). In AD, microglia cluster around Aβ deposits and adopt a polarized morphology with hypertrophic processes extending towards plaques(26–28). Microglia are thought to regulate the degree of amyloid deposition by phagocytosis of amyloid aggregates with potentially protective impact on AD progression(29, 30). However, chronic microglia activation may be associated with production of neurotoxic inflammatory cytokines and reactive oxygen species(31) and microglia have been suggested to phagocytose synapses under pathological conditions(32, 33); thus, they could exert deleterious effects that contribute to disease pathogenesis. Thus, it remains unclear if microglia have a net protective or harmful effect.
The role of microglia in AD has recently gained renewed impetus due to the identification of rare coding variants associated with AD in genes highly expressed in these cells(34, 35), providing strong evidence that microglia may contribute directly to the pathogenesis of this disorder. The strongest of these associations are variants in TREM2; (Triggering Receptor Expressed on Myeloid cells 2), a gene that in the brain is virtually exclusively expressed in microglia(36). Recent evidence suggests that microglia exert neuroprotective functions that are impaired in individuals with TREM2 variants resulting in increased AD risk(34, 35, 37). Here, we review the biology of microglia neuroprotection in AD, with special emphasis on a previously unrecognized role for these cells in the encapsulation of amyloid plaques, which has marked effects on the conformation and toxicity of amyloid deposits and their insulation from adjacent neuronal processes(27, 38). We discuss studies using high-resolution optical imaging in live mice and postmortem human brain that have provided supporting evidence for these neuroprotective functions and the modulatory role by TREM2. Finally, we discuss the implications of these findings regarding therapeutic interventions and diagnostic imaging.
TREM2 variants highlight a protective microglia function in AD pathogenesis
Although microglia could play a significant role in AD pathogenesis through Aβ phagocytosis or secretion of pro-inflammatory cytokines, evidence supporting their involvement in AD has not been definitive. AD patients on chronic anti-inflammatory treatment did not show any cognitive benefits(39), suggesting that neuroinflammation was not a major disease driver. In addition, mutations in genes expressed by microglia (Complement receptor 1, HLA class II histocompatibility antigen DRB1 beta chain, CD33, Membrane-spanning 4-domains subfamily A member 6A) modified AD risk only modestly (0.9<odds ratio<1.1)(40). In contrast, single nucleotide polymorphisms in a microglia specific gene, TREM2, was found to be strongly associated with late onset AD (odds ratio ~ 3; see meta-analysis in Figure 1)(34, 35). Moreover, mutations in the TREM2-signaling partner, TYRO Protein Tyrosine Kinase Binding Protein (TYROBP; also known as DAP12) also increased AD risk(37). Although TREM2 is also expressed in peripheral monocytes (41), these cells appear to play a limited role in AD pathogenesis because they do not significantly enter the normal(42) or neurodegenerative brain(42, 43) in mice, or humans with AD (15)(43–45). Therefore, for the first-time, there is unequivocal evidence that certain microglia functions are robustly involved in AD pathogenesis.
Figure 1. Meta-analysis on the association of R47H TREM2 mutation and the risk of developing AD.
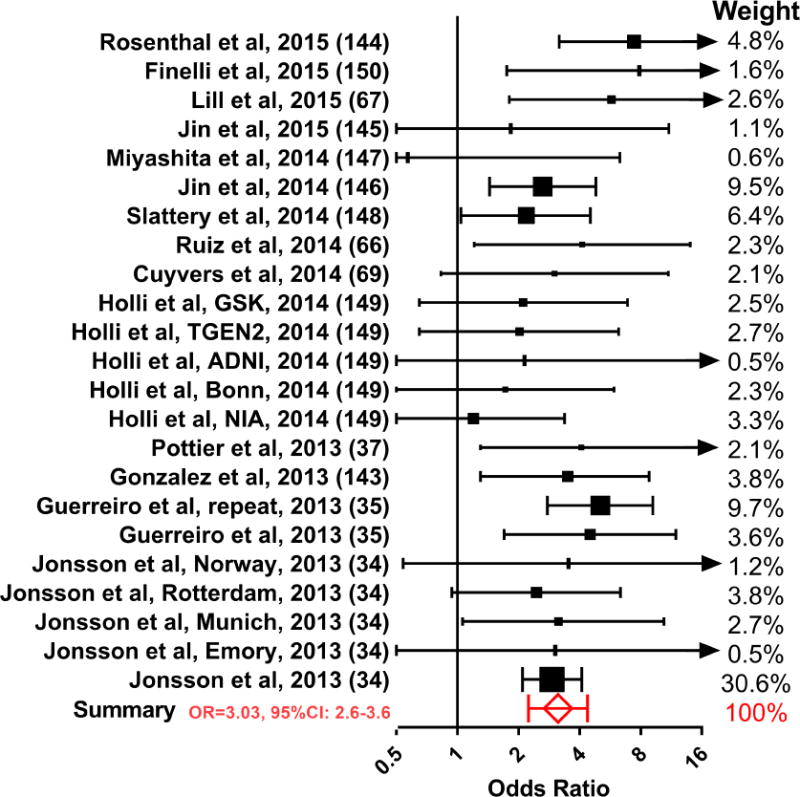
Studies (34, 35, 37, 66, 67, 69, 143–150) were selected from the combined search results of “rs75932628 Alzheimer” and “TREM2 R47H Alzheimer” on Pubmed (total of 64 search results as of September 2017), with the following criteria: 1. case-control studies examining the risk for late-onset AD associated with the single nucleotide polymorphism rs75932628 (19 studies); and 2. The studies have at least one TREM2 R47H subject in the case and control groups (14 studies). Pooled odds ratios (OR) and 95% confidence intervals (CI) were calculated by combining raw data from all studies. Arrows in the graph indicate values exceeding the axis limits. The heterogeneity among studies does not reach statistical significance with Cochran’s Q-test.
TREM2 is a single-pass transmembrane protein that was known to regulate immune responses in peripheral macrophages(41, 46) by means of lipopolysaccharide binding and bacterial phagocytosis(47). In addition, TREM2 attenuates inflammatory cytokine release from macrophages(41), promotes their differentiation and survival and stimulates phagocytosis(48). TREM2 knock-out (TREM2−/−) microglia exhibit reduced clearance of dying cells(49), or myelin debris clearance(50, 51); and loss of TREM2 increases TNF-α, IL-1β, and IL-6 release upon LPS stimulation(52). Individuals with severe loss of TREM2 function due to homozygous mutations develop polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (Nasu-Hakola disease (NHD))(53), possibly due to abnormalities in microglia-oligodendrocyte interactions (54, 55).
However, it remains unclear how TREM2 mutations contribute to increased AD risk. Given that mutations in TREM2 (i.e. R62H, R47H and D87N) are all associated with increased AD risk, it is likely that they cause loss rather than gain of function. Indeed, expression of mutant TREM2 in cultured cells led to disruption in its translocation to the cell membrane, and diminished ligand binding(56, 57). Therefore, mutations in TREM2 lead to a loss-of-function phenotype like TREM2 knockout, with apparent reduced phagocytosis and inflammation. However, there is conflicting data on whether Trem2 knockout increases or decreases overall plaque burden(58–60), even though TREM2−/− microglia have reduced phagocytic capacity in culture(56, 61). Furthermore, Trem2 deficiency does not seem to increase inflammation given that cytokines were reduced in AD mice lacking Trem2(60, 62). The extracellular domain of TREM2 can be cleaved into a soluble fragment (sTREM2), which can stimulate cytokine release(63), but the R47H mutation may reduce its capacity for inducing cytokine secretion(63). Therefore, TREM2 mutations are unlikely to increase the risk of AD through an inflammatory mechanism.
While the changes observed in phagocytosis and cytokine production are subtle, loss of TREM2 dramatically disrupts microglia engagement with amyloid plaques. Several groups have reported that TREM2 deficiency in AD mice leads to reduced microglia numbers around amyloid deposits(59, 60, 62), due to lower proliferation rates(38, 43, 64), reduced metabolic fitness(65), and increased death(60). Furthermore, the polarization of microglial processes towards the plaque surface is markedly reduced in mice with Trem2 haplodeficiency(38). A similar polarization deficiency, albeit to a lower degree, occurs in humans carrying a single allele of the R47H TREM2 mutation(38). The reduction in polarization and plaque encapsulation observed in TREM2 deficiency suggests that this microglial function may play important roles in AD pathogenesis. However, human TREM2 variants appear to also modestly increase the risk of non-β-amyloid based disorders such as amyotrophic lateral sclerosis, fronto-temporal dementia(66–70), and Nasu-Hakola disease in homozygous mutants(53, 71). This suggests that TREM2 deficiency may affect additional mechanisms independent of amyloid phagocytosis or plaque encapsulation, such as efficient corpse removal of dying cells(52, 72) or degenerating myelin(50, 51), or additional unknown TREM2 functions.
Aβ phagocytosis: what do microglia really eat in vivo?
Given that Aβ can disrupt synaptic transmission, induce oxidative stress and trigger cell death in vitro(4), microglia phagocytosis of Aβ could be a neuroprotective function. Microglia have been shown to internalize fluorescently-tagged synthetic Aβ in vitro or after infusion into the mouse brain in vivo(73, 74). Imaging of mouse or AD human tissue reveals some Aβ inside microglial phago-lysosomes(73, 75), consistent with their ability to phagocytose Aβ in vivo. Moreover, their role in Aβ clearance has been demonstrated by genetic manipulation of chemokine or pattern recognition receptors. Loss of CCR2(76), CD45(77), or TLR4(78) in microglia exacerbated amyloid load, while CX3CR1 or NLRP3 deficiency increased microglia phagocytosis and reduced amyloid burden(73, 79, 80). Moreover, passive immunization with anti-Aβ antibodies reduced fibrillar amyloid deposition in transgenic mice(81–83), and potentially in humans as assessed by PET scanning(84, 85) and postmortem histology(86, 87). Thus, under certain conditions microglia phagocytosis of Aβ can reduce the overall amyloid burden.
However, the exact Aβ species that microglia can gobble up remains controversial. Aβ exists in a variety of conformations and sizes, with nascent Aβ polymers forming dimers, oligomers and protofibrils, and plaques which are composed of β-sheet rich fibrils (3). One possibility is that microglia are not selective and phagocytose all Aβ species, including mature fibrillar plaques(29, 30). However, immunohistochemistry with conformation specific antibodies revealed that microglial lysosomes contain oligomers and protofibrils, but not β-sheet rich amyloid fibrils(73). Furthermore, time-lapse in vivo imaging of individual plaques labeled by a single pulse of a β-sheet binding dye showed no change in plaque shape over months(73), indicating no significant removal of Aβ fibrils by adjacent microglia. Consistently, in vivo studies that re-labeled plaques before each imaging time-point observed gradual growth and no disappearance of plaques(6–9),even after anti-Aβ immunization(88). Moreover, using a BACE inhibitor(89) or a regulatable transgene to turn off APP expression(82, 90) during Aβ immunotherapy remained ineffective in clearing pre-existing plaque cores; although, they did appear to reduce the diffuse protofibrillar Aβ halo surrounding them(82). In contrast, experiments tracking fluorescently tagged Aβ42 monomers infused into the subarachnoid space, which rapidly bound to the protofibrillar plaque halo did not show significant removal over intervals up to 90 days(27). Collectively, these experiments indicate that under normal circumstances microglia do not efficiently remove Aβ fibrils from compact plaques or protofibrillar halos, but may be able to phagocytose nascent Aβ polymers. Therefore, microglia phagocytosis has the potential to reduce seeding of new plaques(73, 74) but may have a limited effect once seeding has occurred. Consistent with this, ablation of microglia for 1 month did not change either soluble Aβ levels or plaque numbers in aged mice(91)(92–94). However, these studies followed animals for short intervals and in advanced stages of amyloidosis, which may have led to underestimating phagocytosis. Indeed, a recent paper demonstrated that ablation of microglia led to modest growth of the plaque halo(95). Although this could be due to a loss of ongoing microglia phagocytosis leading to plaque growth(27, 82); it is also possible that growth is due to the loss of microglia encapsulation which restricts Aβ polymerization and outward fibril extension (see discussion below).
Microglia processes form a neuroprotective barrier around plaques
Microglia processes are highly intertwined with fibrils protruding from the plaque core (26, 28). Intravital imaging in mice revealed that as amyloid deposits form, microglia concurrently cluster around and polarize their processes towards the plaque surface (96). Once enveloped around plaques, microglia processes can remain anchored to the plaque with little motility over weeks(27), in contrast to their constant motility in the normal brain(18, 19) or in microglia distant from plaques(73). Close inspection revealed that plaque regions wrapped by microglia processes appeared compact as evidenced by intense binding of Thioflavin S, while those microregions not covered by microglia appeared more diffuse(27). Interestingly, small molecule dyes like curcumin and THK-265 preferentially labeled plaque regions not covered by microglia (Figure 2)(27), possibly reflecting their affinity for protofibrillar Aβ conformation(97). These data are consistent with the possibility that the tight wrapping of microglia behaves as a physical barrier that limits the outgrowth of fibrils and compacts them into a conformation with high affinity for β-sheet binding dyes.
Figure 2. Microglia barrier and protofibrillar Aβ hotspot around amyloid plaques.
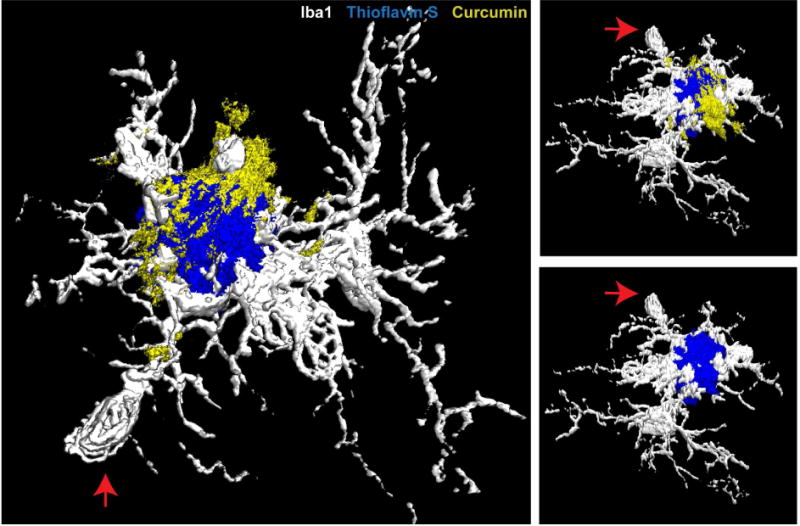
Example 3D reconstruction of a confocal image stack showing microglia coverage around an amyloid deposit in a 5XFAD mouse. Curcumin-labeled protofibrillar Aβ accumulates in the plaque regions not covered by microglia processes (27).
What are the consequences of the presence of these hotspots of protofibrillar Aβ in areas not covered by microglia? To test this, several measures of axonal dystrophy were quantified. Remarkably, plaque microregions not compacted by microglia were associated with a greater extent of dystrophic axons(27). One possible reason for this is that the freely extending fibrils not insulated by microglia protrude into the parenchyma and cause physical damage to neurites. Alternatively, the protofibrillar Aβ conformation in areas not covered by microglia may be more neurotoxic, consistent with in vitro data(98). These and other findings prompted a new hypothesis for the role of plaque-associated microglia processes. We postulated that the tight envelopment of microglia around the amyloid surface constitutes a neuroprotective barrier that limits fibril outgrowth and plaque-associated toxicity. Consistent with this, depletion of microglia in an AD mouse model showed increased plaque outgrowth and dendritic spine loss and shaft atrophy in adjacent neurons(95). Given that plaque-associated axonal dystrophy has been shown to be a good correlate of cognitive dysfunction (99, 100), this pathology may be a significant contributor to neural circuit disruption in AD. Thus, the microglia encapsulation function may play a role in preventing neural AD-associated neural dysfunction.
In AD mouse models haploinsufficient for Trem2(38, 59, 60, 62) or DAP12(38, 101), microglia clustering around plaques was found to be significantly reduced. Importantly, microglia process polarization was also dramatically diminished leading to a near complete loss of plaque encapsulation(38). As a consequence, plaques became much more diffuse, with their morphology shifting from one with compact borders to one with outward projecting fibers resembling a sea urchin(38, 43). Furthermore, super-resolution optical microscopy revealed that in Trem2-deficient mice individual Aβ filaments appeared to have a greater number of side branches(38). The increase in outwardly projecting fibers with greater number of branches would be predicted to significantly increase the total Aβ fibrils surface area that can be exposed to surrounding neural structures, with potentially damaging effects. Consistent with this view the extent of plaque-associated axonal dystrophy was exacerbated in mice lacking Trem2 or DAP12, supporting the hypothesis that the microglia barrier is a neuroprotective function. Importantly. heterozygous human carriers of the R47H TREM2 mutation also had disrupted microglia clustering and barrier formation around plaques. Similar to Trem2-haplodeficient mice, R47H mutants exhibited an increase in the number of less compact and filamentous deposits as well as a greater extent of dystrophic axons and neuronal processes with hyper-phosphorylated tau (38). The phenotypic resemblance in humans and mice suggests that the microglia barrier is a shared mechanism to limit plaque-associated neural damage. However, the conformational plaque phenotype in R47H human carriers is less dramatic than in mice. One likely explanation is that the R47H variants constitute only a partial loss of function, with less severe barrier disruption. Alternatively, the markedly faster rates of amyloid accumulation in mice may overwhelm the capacity of microglia, leading to a more severe phenotype. Interestingly, however, humans with R47H variants did have a robust disruption of axons as evidenced by the degree of axonal dystrophy and tau hyperphosphorylation. Thus, in humans the main protective function of microglia may be their ability to insulate plaques from the surrounding tissue, while their role in plaque compaction may be more limited.
Cellular mechanism involved in the microglia barrier function
Microglia sensing of amyloid deposits and their polarization towards plaques are likely important steps in the formation of an effective barrier. TREM2 may serve as both the receptor for recognizing plaque components and the trigger for downstream cytoskeleton re-organization that is required for process polarization. TREM2 does not bind to Aβ per se, but has affinity for lipids found on plaques(60). TREM2-lipid mediated signaling may be critical for barrier formation. A single point mutation in the arginine-47 (R47) residue within the TREM2 ligand-binding domain(102) leads to reduced lipid affinity (60) and disruption of microglia clustering and plaque encapsulation(38). Intriguingly, microglia do not form barrier processes around diffuse plaques. Unlike compact plaques, diffuse deposits are not decorated with lipids(103)(104), suggesting that lipidation of Aβ is a key step in TREM2-mediated microglia polarization. Upon lipid binding in vitro, the intracellular domain of TREM2 can trigger downstream DAP12-mediated tyrosine-based activation motif (ITAM)-signaling cascade, PI3K pathway activation and cytoskeletal re-organization(52). DAP12 and phosphorylated tyrosine are up-regulated and co-localized with TREM2 in the polarized microglia processes, and disrupting this signaling pathway in mice by deletion of DAP12 abolished the microglia barrier (38). However, single cell RNAseq (15) or immunohistochemistry (38, 44), have shown that TREM2 signaling appears to be activated only after microglia have fully engaged around plaques. This raises questions as to how TREM2 sensing of the plaque can occur prior to its upregulation (38) and precisely at what stage of plaque engagement TREM2 signaling becomes critical. Nevertheless, evidence argues that TREM2-DAP12 signaling mediates plaque sensing and is likely involved in the microglia process polarization necessary for barrier formation.
The exact process by which microglia induce plaque compaction remains unknown. One possibility is that microglia processes clear the diffuse protofibrillar Aβ at the plaque halo through phagocytosis and/or secretion of proteolytic enzymes, leading to the appearance of a more compact core in areas covered by microglia. However, these microglia processes are not optimally positioned to remove prefibrillar Aβ because they are closely anchored to the perimeter of the compact plaque core(27) rather than at the site of the less compact plaque halo and under normal conditions have been shown to have a limited capacity for phagocytosis of protofibrillar Aβ(73, 82). Therefore, instead of phagocytosis, we propose that microglia processes wrapping around deposits create an insulated and crowded macro-molecular environment that leads to accelerated Aβ aggregation(105), resulting in plaque compaction. Also compact plaque regions have decreased affinity for monomeric Aβ42, which may further limit the outward outgrowth of protofibrils once deposits become compact. The reduced affinity for Aβ42 could be due to microglia exerting a direct force on outwardly growing amyloid fibrils leading to their bending, which may mask their growing ends and prevent their elongation. Microglia may reduce fibirllization by physically preventing the entry of monomeric Aβ42 into the core region. However, in vivo infusion of Aβ40, which has a molecular weight similar to Aβ42, demonstrates full penetration throughout the plaque. This and other experiments(27, 73), suggest that microglia do not prevent entry of monomeric Aβ42 but rather change the affinity to Aβ42 in areas covered by microglia processes, likely by altering the conformation and compaction of accumulating protofibrils.
Failure of the microglia barrier as a general mechanism in the development of AD
While TREM2 loss-of-function mutations are found in a small percentage of AD patients (~0.5%)(34), a defective microglia barrier could also be a risk factor for the development of late onset AD. Indeed, like the defective barrier seen in TREM2 or DAP12 deficient mice(38), comparison of plaques of similar size between young and old wild type AD mice revealed that microglia coverage was significantly reduced in aging, and as predicted this was associated with enlarged protofibrillar Aβ halos as well as greater axonal dystrophy(27). Multiple mechanisms may contribute to defective microglia encapsulation given that ageing is associated with complex molecular and cellular changes (106) including reduced cell proliferation (107). Indeed, BrdU incorporation in plaque-associated microglia is significantly reduced in aging (27), suggesting that reduced proliferation limits the number of microglia available for plaque encapsulation. In addition, microglia in aging display tortuous processes and focal swellings (21, 107), suggesting cellular dysfunction that may impair polarization towards plaques. In addition, aging microglia may be less phagocytic and adopt an activated phenotype with release of pro-inflammatory cytokines (108), and a reduction in anti-inflammatory cytokines such as TGFβ (109). Thus, as protective microglial functions like phagocytosis and plaque encapsulation fail in aging, their chronic activation and changes in cytokines may increase their neurotoxic potential.
Recent studies have suggested potential modulation of microglia by mechanisms involving Apolipoprotein E (ApoE). ApoE is known for its role in brain lipid and cholesterol transport. The APOE gene has three polymorphic alleles(ε2, ε3 and ε4), and GWAS studies have shown that carriers of one ε4 allele have 2–3 times increased risk of AD, while ε4/ε4 have ~15 times increased risk(110). The APOE ε4 allele is also the most prevalent AD risk factor estimated to be in ~14% of the global population and 37% of AD patients(111). Recent studies uncovered an interaction between ApoE and TREM2 in vitro, where recombinant TREM2 exhibited high binding affinity to ApoE (112, 113). Interestingly, this affinity was reduced in mutated TREM2 (R46A, R47A, R47E and R47H), suggesting a correlation between loss of ApoE-TREM2 interaction and increased AD risk. Because ApoE can be found on the plaque surface (114), it is possible that ApoE provides a targeting signal for TREM2-expressing microglia processes. However, it remains unknown whether the different ApoE isoforms bind to TREM2 with the same affinity. Interestingly, ApoE4, but not ApoE3, was shown to activate Toll-like receptors in microglia cultures, leading to reduced TREM2 expression (115), and thus it is possible that ApoE4 could disrupt TREM2-mediated microglia polarization towards plaques in vivo. Consistent with this, in ApoE4 isoform-specific knock-in mice, microglia adopted an inflammatory phenotype and displayed abnormal processes around plaques compared to E2 and E3 knock-in mice (116, 117). Therefore, in addition to the better studied differential effects of ApoE on Aβ metabolism (117–120), it is possible that ApoE within amyloid deposits plays a role in signaling to adjacent microglia (121) for the polarization of their processes and encapsulation of plaques. However, additional microglia functions mediated through ApoE, that are independent of amyloid, may also be at play, as it has recently been shown that ApoE isoforms in mice directly modulate tau pathology and cell death (122, 123). Thus, the potential involvement of ApoE in the microglia encapsulation of plaques combined with the fact that this function diminishes with ageing, suggests that failure of the microglia barrier could constitute a general mechanism involved in AD pathogenesis.
Microglia-mediated neuroprotection as a target for AD therapies
Experimental strategies to enhance the microglia encapsulation of plaques have been demonstrated with the chemokine receptor CX3CR1 genetic deletion or by passive anti-Aβ immunization in mice (27). These manipulations led to reduced axonal dystrophy formation around plaques, indicating a possible neuroprotective effect of microglia. Importantly, in humans carrying the R47H TREM2 variant, diminished microglia encapsulation, not only worsened axonal dystrophy, but exacerbated neuronal phospho-tau accumulation around plaques (38). This suggests that boosting the microglia barrier may not only reduce axnal dystrophy but could also limit plaque-associated tau pathology. Given that the degree of tau hyperphosphorylation negatively correlates with cognitive function (124), it is plausible that enhancing microglia encapsulation of plaques could slow disease progression.
Although it is likely that most molecular manipulations will have a variety of effects on microglia function including on phagocytosis and cytokine production, ongoing research into mechanisms of microglia process polarization may suggest novel strategies to specifically manipulate their ability to encapsulate amyloid deposits. The following are potential strategies: 1) Anti-Aβ immunization: Several groups have shown that this treatment increases microglia clustering around plaques in AD mouse models(27, 82, 84). Anti-Aβ immunization increases plaque encapsulation probably by activating Fc receptors that trigger downstream signaling overlapping with that of TREM2(125). It is worth noting that microglia only express a subset of Fc receptors and different Fc receptor subtypes activate different downstream pathways(126). Particularly, while FcγRI and FcγRIII activate immunoreceptor tyrosine-based activation motif (ITAM) signaling that converges with TREM2 activation, FcγRIIB inhibits ITAM signaling. Therefore, anti-Aβ IgG antibodies with high FcγRI and low FcγRIIB affinity may be the most effective in boosting microglia barrier function due to their potential net activation of ITAM signaling. 2) Anti-ApoE immunization: Anti-ApoE immunotherapy has been shown to increase microglial recruitment around plaques in mice(127). Since ApoE can bind Aβ aggregates (114), it is likely that anti-ApoE antibodies have affinity towards plaques, similar to anti-Aβ antibodies. And likewise, anti-ApoE antibodies may be able to promote microglia process polarization by activating Fc receptors and their downstream signaling. Furthermore, given recent results showing that ApoE knock-out is neuroprotective against tau pathology(122), antibodies sequestering ApoE may have a dual beneficial effect. 3) CX3CR1 inhibition: Genetically deleting CX3CR1 in microglia leads to reduced plaque load(73, 79) and enhanced microglia barrier(27). Neutralization of CX3CR1 could thus be an approach to enhance microglia encapsulation of plaques. This may be achieved by neutralizing antibodies against CX3CR1 or its ligand Fractalkine, or by small molecule antagonist such as AZD-8797 (128). However, systemic suppression of CX3CR1 signaling may disrupt bacterial clearance by the peripheral immune system(129), while CX3CR1 deficiency may exacerbate tau hyperphosphorylation(130)(131). Thus, suppression of CX3CR1 should ideally be confined to plaque-associated microglia to increase encapsulation with minimal systemic side effects. 4) Bispecific antibodies: Antibodies with two different Fab domains can simultaneously bind to two separate epitopes. One of the two Fab domains may be against fibrillar Aβ (84), which would lead to enrichment around plaques, providing an approach to engage the second target only in the vicinity of the plaque. If the second Fab domain is anti-Fractalkine or anti-CX3CR1, then such approach may achieve simultaneous suppression of CX3CR1 signaling and activation of ITAM signaling in plaque-associated microglia, potentially leading to enhancement of the microglia barrier and/or phagocytosis.
In terms of suitability for human use of these therapeutic strategies, based on previous mouse data, it appears to be clear that the barrier function is most effective when plaques are still relatively small (27). Thus, it is likely that therapies would have to target early preclinical AD. Furthermore, as with all attempts to translate therapies based on mouse models, there is a significant uncertainty that targeting amyloid will be sufficient in the absence of direct modulation of other disease hallmarks such as tau pathology.
Potential implications for clinical human imaging
Current amyloid PET tracers are limited in their utility as predictive biomarkers due to their poor dynamic range and linearity. When patients present with mild cognitive impairment, their PET signals are near maximal and correlate poorly with the degree of cognitive impairment(10, 132, 133). Interestingly, our data shows that plaque compaction inversely correlates with the degree of axonal injury (27, 38). However, PET tracers, which are derivatives of Thioflavin T or Congo Red, have the greatest affinity for compact plaques which likely prevents detection of the potentially most neurotoxic protofibrillar species of Aβ (27, 134). In contrast, small molecule dyes such as curcumin and THK-265 preferentially bind protofibrillar Aβ (27, 97). Given that protofibrillar plaque regions are associated with more severe axonal dystrophy and neuronal process tau hyperphosphorylation (38), it is possible that novel PET probes based on compounds with affinity to protofibrillar Aβ could constitute better biomarkers of neurotoxicity. Indeed, improved brain-penetrant curcumin analogs have been developed which may have potential as amyloid PET tracers in humans(135, 136).
Current PET imaging approaches to monitor microglia activation utilize small-molecule ligands that bind the translocator protein 18kDa (TSPO) in mice(137–139) and humans(140, 141). TSPO is found in the outer mitochondrial membrane in various cell types, including neurons, astrocytes and endothelial cells (142). Thus, while TSPO is upregulated in microglia in AD (138, 139), it lacks sufficient specificity for proper quantification of microglia activation. Novel PET tracers targeting microglia receptors such as TREM2 may have greater potential because of their cell specificity and their marked upregulation in microglia surrounding amyloid deposits, which would greatly enhance the signal to noise ratios. Such probes may also provide information about the robustness of the microglia barrier and may thus be an indicator of the neuroprotective microglia that encapsulate plaques and reduce axonal dystrophy. Overall, such a strategy may offer greater resolution for tracking and interpreting the progression of AD-related neuroinflammation in parallel with existing amyloid and tau PET tracers.
Figure 3. Modulating microglia barrier alters plaque-associated protofibrillar Aβ content, axonal dystrophy and tau hyperphosphorylation.
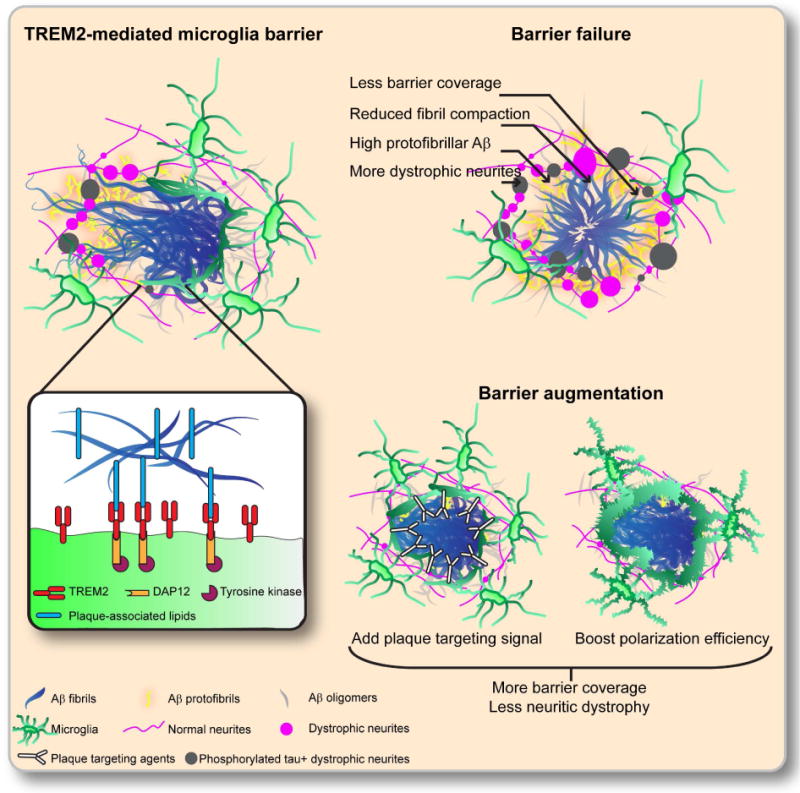
TREM2 on microglia processes binds to the lipid and lipoprotein components of amyloid plaque, triggering the downstream signaling cascade involving DAP12 and Syk tyrosine kinase. This signal leads cytoskeletal reorganization and to the polarization and expansion of the microglia processes, and the formation of microglia barrier, which increases the compaction of adjacent amyloid fibrils. Deficiency in the microglia barrier results in loosely organized amyloid fibrils, protofibrillar Aβ accumulation, increased axonal dystrophy formation and neuronal process tau hyperphosphorylation. Augmenting the microglia barrier may have therapeutic benefits in AD. Two potential strategies are: 1) plaque-targeted signaling ligands that activate TREM2 or other receptors, and 2) modulators of downstream signals that boost the microglia barrier.
Acknowledgments
This work was supported by grants from the National Institutes of Health: R01HL106815 and R21AG048181 (J.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors report no biomedical financial interests or potential conflicts of interest.
Bibliography
- 1.Selkoe DJ, Hardy J. The amyloid hypothesis of alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masters CL, Selkoe DJ. Biochemistry of amyloid β-protein and amyloid deposits in alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(6):a006262. doi: 10.1101/cshperspect.a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benilova I, Karran E, De Strooper B. The toxic aβ oligomer and alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 5.Thal DR, Griffin WST, Braak H. Parenchymal and vascular abeta-deposition and its effects on the degeneration of neurons and cognition in alzheimer’s disease. J Cell Mol Med. 2008;12(5B):1848–1862. doi: 10.1111/j.1582-4934.2008.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hefendehl JK, Wegenast-Braun BM, Liebig C, Eicke D, Milford D, Calhoun ME, et al. Long-term in vivo imaging of β-amyloid plaque appearance and growth in a mouse model of cerebral β-amyloidosis. J Neurosci. 2011;31(2):624–629. doi: 10.1523/JNEUROSCI.5147-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Condello C, Schain A, Grutzendler J. Multicolor time-stamp reveals the dynamics and toxicity of amyloid deposition. Sci Rep. 2011;1:19. doi: 10.1038/srep00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgold S, Filser S, Dorostkar MM, Schmidt B, Herms J. In vivo imaging reveals sigmoidal growth kinetic of β-amyloid plaques. Acta Neuropathol Commun. 2014;2:30. doi: 10.1186/2051-5960-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y, et al. Characterizing the appearance and growth of amyloid plaques in app/ps1 mice. J Neurosci. 2009;29(34):10706–10714. doi: 10.1523/JNEUROSCI.2637-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 11.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, et al. [11c]pib in a nondemented population: potential antecedent marker of alzheimer disease. Neurology. 2006;67(3):446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 12.Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of alzheimer’s disease. Acta Neuropathol Commun. 2014;2(1):135. doi: 10.1186/s40478-014-0135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 16.Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci. 2017;20(8):1162–1171. doi: 10.1038/nn.4597. [DOI] [PubMed] [Google Scholar]
- 17.Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, et al. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci. 2017;20(9):1300–1309. doi: 10.1038/nn.4610. [DOI] [PubMed] [Google Scholar]
- 18.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. Atp mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 19.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 20.Hanisch U-K, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 21.Hefendehl JK, Neher JJ, Sühs RB, Kohsaka S, Skodras A, Jucker M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell. 2014;13(1):60–69. doi: 10.1111/acel.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates cns synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 23.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 24.Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29(13):3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155(7):1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wegiel J, Wisniewski HM. The complex of microglial cells and amyloid star in three-dimensional reconstruction. Acta Neuropathol. 1990;81(2):116–124. doi: 10.1007/BF00334499. [DOI] [PubMed] [Google Scholar]
- 27.Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar aβ42 hotspots around plaques. Nat Commun. 2015;6:6176. doi: 10.1038/ncomms7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of alzheimer disease. J Neuroimmunol. 1989;24(3):173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 29.Lee CYD, Landreth GE. The role of microglia in amyloid clearance from the ad brain. J Neural Transm. 2010;117(8):949–960. doi: 10.1007/s00702-010-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan D. The role of microglia in antibody-mediated clearance of amyloid-beta from the brain. CNS Neurol Disord Drug Targets. 2009;8(1):7–15. doi: 10.2174/187152709787601821. [DOI] [PubMed] [Google Scholar]
- 31.Block ML, Zecca L, Hong J-S. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 32.Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, et al. Complement c3 deficiency protects against neurodegeneration in aged plaque-rich app/ps1 mice. Sci Transl Med. 2017;9(392) doi: 10.1126/scitranslmed.aaf6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in alzheimer mouse models. Science. 2016;352(6286):712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of trem2 associated with the risk of alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. Trem2 variants in alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colonna M. Trems in the immune system and beyond. Nat Rev Immunol. 2003;3(6):445–453. doi: 10.1038/nri1106. [DOI] [PubMed] [Google Scholar]
- 37.Pottier C, Ravenscroft TA, Brown PH, Finch NA, Baker M, Parsons M, et al. Tyrobp genetic variants in early-onset alzheimer’s disease. Neurobiol Aging. 2016;48:222.e9–222.e15. doi: 10.1016/j.neurobiolaging.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, et al. Trem2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016;90(4):724–739. doi: 10.1016/j.neuron.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Breitner JC, Baker LD, Montine TJ, Meinert CL, Lyketsos CG, Ashe KH, et al. Extended results of the alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement. 2011;7(4):402–411. doi: 10.1016/j.jalz.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, et al. Genome-wide association meta-analysis of neuropathologic features of alzheimer’s disease and related dementias. PLoS Genet. 2014;10(9):e1004606. doi: 10.1371/journal.pgen.1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, et al. Cutting edge: trem-2 attenuates macrophage activation. J Immunol. 2006;177(6):3520–3524. doi: 10.4049/jimmunol.177.6.3520. [DOI] [PubMed] [Google Scholar]
- 42.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain cns microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, et al. Trem2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213(5):667–675. doi: 10.1084/jem.20151948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Varvel NH, Grathwohl SA, Degenhardt K, Resch C, Bosch A, Jucker M, et al. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-β deposition in mouse models of alzheimer’s disease. J Exp Med. 2015;212(11):1803–1809. doi: 10.1084/jem.20150478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mildner A, Schlevogt B, Kierdorf K, Böttcher C, Erny D, Kummer MP, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of alzheimer’s disease. J Neurosci. 2011;31(31):11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of tlr and fcr responses in macrophages by triggering receptor expressed on myeloid cells (trem)-2 and dap12. J Immunol. 2006;177(4):2051–2055. doi: 10.4049/jimmunol.177.4.2051. [DOI] [PubMed] [Google Scholar]
- 47.N’Diaye E-N, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C, et al. Trem-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol. 2009;184(2):215–223. doi: 10.1083/jcb.200808080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu K, Byers DE, Jin X, Agapov E, Alexander-Brett J, Patel AC, et al. Trem-2 promotes macrophage survival and lung disease after respiratory viral infection. J Exp Med. 2015;212(5):681–697. doi: 10.1084/jem.20141732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. Triggering receptor expressed on myeloid cells 2 (trem2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci. 2015;35(8):3384–3396. doi: 10.1523/JNEUROSCI.2620-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, et al. Trem2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 2015;129(3):429–447. doi: 10.1007/s00401-015-1388-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, et al. Trem2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. 2015;125(5):2161–2170. doi: 10.1172/JCI77983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takahashi K, Rochford CDP, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201(4):647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71(3):656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cella M, Buonsanti C, Strader C, Kondo T, Salmaggi A, Colonna M. Impaired differentiation of osteoclasts in trem-2-deficient individuals. J Exp Med. 2003;198(4):645–651. doi: 10.1084/jem.20022220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paloneva J, Mandelin J, Kiialainen A, Bohling T, Prudlo J, Hakola P, et al. Dap12/trem2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med. 2003;198(4):669–675. doi: 10.1084/jem.20030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. Trem2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6(243):243ra86. doi: 10.1126/scitranslmed.3009093. [DOI] [PubMed] [Google Scholar]
- 57.Sirkis DW, Bonham LW, Aparicio RE, Geier EG, Ramos EM, Wang Q, et al. Rare trem2 variants associated with alzheimer’s disease display reduced cell surface expression. Acta Neuropathol Commun. 2016;4(1):98. doi: 10.1186/s40478-016-0367-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, et al. Disease progression-dependent effects of trem2 deficiency in a mouse model of alzheimer’s disease. J Neurosci. 2017;37(3):637–647. doi: 10.1523/JNEUROSCI.2110-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. Trem2 deficiency eliminates trem2+ inflammatory macrophages and ameliorates pathology in alzheimer’s disease mouse models. J Exp Med. 2015;212(3):287–295. doi: 10.1084/jem.20142322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. Trem2 lipid sensing sustains the microglial response in an alzheimer’s disease model. Cell. 2015;160(6):1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. Trem2 binds to apolipoproteins, including apoe and clu/apoj, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91(2):328–340. doi: 10.1016/j.neuron.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 62.Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, et al. Altered microglial response to aβ plaques in appps 1–21 mice heterozygous for trem2. Mol Neurodegener. 2014;9:20. doi: 10.1186/1750-1326-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhong L, Chen X-F, Wang T, Wang Z, Liao C, Wang Z, et al. Soluble trem2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017;214(3):597–607. doi: 10.1084/jem.20160844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zheng H, Jia L, Liu C-C, Rong Z, Zhong L, Yang L, et al. Trem2 promotes microglial survival by activating wnt/β-catenin pathway. J Neurosci. 2017;37(7):1772–1784. doi: 10.1523/JNEUROSCI.2459-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ulland TK, Song WM, Huang SC-C, Ulrich JD, Sergushichev A, Beatty WL, et al. Trem2 maintains microglial metabolic fitness in alzheimer’s disease. Cell. 2017;170(4):649–663.e13. doi: 10.1016/j.cell.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ruiz A, Dols-Icardo O, Bullido MJ, Pastor P, Rodríguez-Rodríguez E, López de Munain A, et al. Assessing the role of the trem2 p.r47h variant as a risk factor for alzheimer’s disease and frontotemporal dementia. Neurobiol Aging. 2014;35(2):444.e1–4. doi: 10.1016/j.neurobiolaging.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 67.Lill CM, Rengmark A, Pihlstrøm L, Fogh I, Shatunov A, Sleiman PM, et al. The role of trem2 r47h as a risk factor for alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and parkinson’s disease. Alzheimers Dement. 2015;11(12):1407–1416. doi: 10.1016/j.jalz.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guerreiro RJ, Lohmann E, Brás JM, Gibbs JR, Rohrer JD, Gurunlian N, et al. Using exome sequencing to reveal mutations in trem2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013;70(1):78–84. doi: 10.1001/jamaneurol.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, et al. Investigating the role of rare heterozygous trem2 variants in alzheimer’s disease and frontotemporal dementia. Neurobiol Aging. 2014;35(3):726.e11–9. doi: 10.1016/j.neurobiolaging.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 70.Giraldo M, Lopera F, Siniard AL, Corneveaux JJ, Schrauwen I, Carvajal J, et al. Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and alzheimer’s disease. Neurobiol Aging. 2013;34(8):2077.e11–8. doi: 10.1016/j.neurobiolaging.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klünemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, et al. The genetic causes of basal ganglia calcification, dementia, and bone cysts: dap12 and trem2. Neurology. 2005;64(9):1502–1507. doi: 10.1212/01.WNL.0000160304.00003.CA. [DOI] [PubMed] [Google Scholar]
- 72.Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, et al. A role for trem2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109(4):1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu Z, Condello C, Schain A, Harb R, Grutzendler J. Cx3cr1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J Neurosci. 2010;30(50):17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mandrekar S, Jiang Q, Lee CYD, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE. Microglia mediate the clearance of soluble abeta through fluid phase macropinocytosis. J Neurosci. 2009;29(13):4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992;84(3):225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- 76.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of alzheimer-like disease. Nat Med. 2007;13(4):432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 77.Zhu Y, Hou H, Rezai-Zadeh K, Giunta B, Ruscin A, Gemma C, et al. Cd45 deficiency drives amyloid-β peptide oligomers and neuronal loss in alzheimer’s disease mice. J Neurosci. 2011;31(4):1355–1365. doi: 10.1523/JNEUROSCI.3268-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Song M, Jin J, Lim J-E, Kou J, Pattanayak A, Rehman JA, et al. Tlr4 mutation reduces microglial activation, increases aβ deposits and exacerbates cognitive deficits in a mouse model of alzheimer’s disease. J Neuroinflammation. 2011;8:92. doi: 10.1186/1742-2094-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, et al. Cx3cr1 deficiency alters microglial activation and reduces beta-amyloid deposition in two alzheimer’s disease mouse models. Am J Pathol. 2010;177(5):2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. Nlrp3 is activated in alzheimer’s disease and contributes to pathology in app/ps1 mice. Nature. 2013;493(7434):674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Levites Y, Das P, Price RW, Rochette MJ, Kostura LA, McGowan EM, et al. Anti-abeta42 - and anti-abeta40-specific mabs attenuate amyloid deposition in an alzheimer disease mouse model. J Clin Invest. 2006;116(1):193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang A, Das P, Switzer RC, Golde TE, Jankowsky JL. Robust amyloid clearance in a mouse model of alzheimer’s disease provides novel insights into the mechanism of amyloid-beta immunotherapy. J Neurosci. 2011;31(11):4124–4136. doi: 10.1523/JNEUROSCI.5077-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, et al. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004;24(27):6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces aβ plaques in alzheimer’s disease. Nature. 2016;537(7618):50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 85.Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, et al. 11c-pib pet assessment of change in fibrillar amyloid-beta load in patients with alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9(4):363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 86.Nicoll JAR, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, et al. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65(11):1040–1048. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 87.Zotova E, Holmes C, Johnston D, Neal JW, Nicoll JAR, Boche D. Microglial alterations in human alzheimer’s disease following aβ42 immunization. Neuropathol Appl Neurobiol. 2011;37(5):513–524. doi: 10.1111/j.1365-2990.2010.01156.x. [DOI] [PubMed] [Google Scholar]
- 88.Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, et al. An effector-reduced anti-β-amyloid (aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of aβ. J Neurosci. 2012;32(28):9677–9689. doi: 10.1523/JNEUROSCI.4742-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jacobsen H, Ozmen L, Caruso A, Narquizian R, Hilpert H, Jacobsen B, et al. Combined treatment with a bace inhibitor and anti-aβ antibody gantenerumab enhances amyloid reduction in applondon mice. J Neurosci. 2014;34(35):11621–11630. doi: 10.1523/JNEUROSCI.1405-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tucker SMF, Borchelt DR, Troncoso JC. Limited clearance of pre-existing amyloid plaques after intracerebral injection of abeta antibodies in two mouse models of alzheimer disease. J Neuropathol Exp Neurol. 2008;67(1):30–40. doi: 10.1097/nen.0b013e31815f38d2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, et al. Formation and maintenance of alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12(11):1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MRP, Blurton-Jones M, et al. Eliminating microglia in alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain. 2016;139(Pt 4):1265–1281. doi: 10.1093/brain/aww016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dagher NN, Najafi AR, Kayala KMN, Elmore MRP, White TE, Medeiros R, et al. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xtg-ad mice. J Neuroinflammation. 2015;12(1):139. doi: 10.1186/s12974-015-0366-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Olmos-Alonso A, Schetters STT, Sri S, Askew K, Mancuso R, Vargas-Caballero M, et al. Pharmacological targeting of csf1r inhibits microglial proliferation and prevents the progression of alzheimer’s-like pathology. Brain. 2016;139(Pt 3):891–907. doi: 10.1093/brain/awv379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao R, Hu W, Tsai J, Li W, Gan W-B. Microglia limit the expansion of β-amyloid plaques in a mouse model of alzheimer’s disease. Mol Neurodegener. 2017;12(1):47. doi: 10.1186/s13024-017-0188-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jung CKE, Keppler K, Steinbach S, Blazquez-Llorca L, Herms J. Fibrillar amyloid plaque formation precedes microglial activation. PLoS ONE. 2015;10(3):e0119768. doi: 10.1371/journal.pone.0119768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yanagisawa D, Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992;84(3):225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- 98.O’Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, et al. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30(43):14411–14419. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Su JH, Cummings BJ, Cotman CW. Plaque biogenesis in brain aging and alzheimer’s disease. ii. progressive transformation and developmental sequence of dystrophic neurites. Acta Neuropathol. 1998;96(5):463–471. doi: 10.1007/s004010050920. [DOI] [PubMed] [Google Scholar]
- 100.Dickson TC, King CE, McCormack GH, Vickers JC. Neurochemical diversity of dystrophic neurites in the early and late stages of alzheimer’s disease. Exp Neurol. 1999;156(1):100–110. doi: 10.1006/exnr.1998.7010. [DOI] [PubMed] [Google Scholar]
- 101.Haure-Mirande J-V, Audrain M, Fanutza T, Kim SH, Klein WL, Glabe C, et al. Deficiency of tyrobp, an adapter protein for trem2 and cr3 receptors, is neuroprotective in a mouse model of early alzheimer’s pathology. Acta Neuropathol. 2017 doi: 10.1007/s00401-017-1737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kober DL, Alexander-Brett JM, Karch CM, Cruchaga C, Colonna M, Holtzman MJ, et al. Neurodegenerative disease mutations in trem2 reveal a functional surface and distinct loss-of-function mechanisms. elife. 2016;5 doi: 10.7554/eLife.20391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rak M, Del Bigio MR, Mai S, Westaway D, Gough K. Dense-core and diffuse abeta plaques in tgcrnd8 mice studied with synchrotron ftir microspectroscopy. Biopolymers. 2007;87(4):207–217. doi: 10.1002/bip.20820. [DOI] [PubMed] [Google Scholar]
- 104.Kiskis J, Fink H, Nyberg L, Thyr J, Li J-Y, Enejder A. Plaque-associated lipids in alzheimer’s diseased brain tissue visualized by nonlinear microscopy. Sci Rep. 2015;5(1):13489. doi: 10.1038/srep13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou Z, Fan J-B, Zhu H-L, Shewmaker F, Yan X, Chen X, et al. Crowded cell-like environment accelerates the nucleation step of amyloidogenic protein misfolding. J Biol Chem. 2009;284(44):30148–30158. doi: 10.1074/jbc.M109.002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. 2016;19(3):504–516. doi: 10.1038/nn.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Streit WJ. Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci. 2006;29(9):506–510. doi: 10.1016/j.tins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 108.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging alzheimer’s disease mice. J Neurosci. 2008;28(33):8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique tgf-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17(1):131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein e in alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011;10(3):241–252. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein e genotype and alzheimer disease. a meta-analysis. apoe and alzheimer disease meta analysis consortium. JAMA. 1997;278(16):1349–1356. [PubMed] [Google Scholar]
- 112.Atagi Y, Liu C-C, Painter MM, Chen X-F, Verbeeck C, Zheng H, et al. Apolipoprotein e is a ligand for triggering receptor expressed on myeloid cells 2 (trem2) J Biol Chem. 2015;290(43):26043–26050. doi: 10.1074/jbc.M115.679043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bailey CC, DeVaux LB, Farzan M. The triggering receptor expressed on myeloid cells 2 binds apolipoprotein e. J Biol Chem. 2015;290(43):26033–26042. doi: 10.1074/jbc.M115.677286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein e immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in alzheimer’s disease and kuru plaque amyloid in creutzfeldt-jakob disease. Brain Res. 1991;541(1):163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 115.Li X, Montine KS, Keene CD, Montine TJ. Different mechanisms of apolipoprotein e isoform-dependent modulation of prostaglandin e2 production and triggering receptor expressed on myeloid cells 2 (trem2) expression after innate immune activation of microglia. FASEB J. 2015;29(5):1754–1762. doi: 10.1096/fj.14-262683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rodriguez GA, Tai LM, LaDu MJ, Rebeck GW. Human apoe4 increases microglia reactivity at aβ plaques in a mouse model of aβ deposition. J Neuroinflammation. 2014;11:111. doi: 10.1186/1742-2094-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, et al. Apoe4-specific changes in aβ accumulation in a new transgenic mouse model of alzheimer disease. J Biol Chem. 2012;287(50):41774–41786. doi: 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoe isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3(89):89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hashimoto T, Serrano-Pozo A, Hori Y, Adams KW, Takeda S, Banerji AO, et al. Apolipoprotein e, especially apolipoprotein e4, increases the oligomerization of amyloid β peptide. J Neurosci. 2012;32(43):15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Huang Y-WA, Zhou B, Wernig M, Südhof TC. Apoe2, apoe3, and apoe4 differentially stimulate app transcription and aβ secretion. Cell. 2017;168(3):427–441.e21. doi: 10.1016/j.cell.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The trem2-apoe pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47(3):566–581.e9. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. Apoe4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523–527. doi: 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brecht WJ, Harris FM, Chang S, Tesseur I, Yu G-Q, Xu Q, et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24(10):2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ballatore C, Lee VM-Y, Trojanowski JQ. Tau-mediated neurodegeneration in alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8(9):663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 125.Linnartz B, Neumann H. Microglial activatory (immunoreceptor tyrosine-based activation motif) - and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia. 2013;61(1):37–46. doi: 10.1002/glia.22359. [DOI] [PubMed] [Google Scholar]
- 126.Nimmerjahn F, Ravetch JV. Fc-receptors as regulators of immunity. Vol. 96. Elsevier; 2007. pp. 179–204. 179–204. [DOI] [PubMed] [Google Scholar]
- 127.Kim J, Eltorai AEM, Jiang H, Liao F, Verghese PB, Kim J, et al. Anti-apoe immunotherapy inhibits amyloid accumulation in a transgenic mouse model of aβ amyloidosis. J Exp Med. 2012;209(12):2149–2156. doi: 10.1084/jem.20121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cederblad L, Rosengren B, Ryberg E, Hermansson N-O. Azd8797 is an allosteric non-competitive modulator of the human cx3cr1 receptor. Biochem J. 2016;473(5):641–649. doi: 10.1042/BJ20150520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, et al. Cx3cr1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307(5707):254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 130.Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.López-López A, Gelpi E, Lopategui DM, Vidal-Taboada JM. Association of the cx3cr1-v249i variant with neurofibrillary pathology progression in late-onset alzheimer’s disease. Mol Neurobiol. 2017;362:1–10. doi: 10.1007/s12035-017-0489-3. [DOI] [PubMed] [Google Scholar]
- 132.Hatashita S, Wakebe D. Amyloid-β deposition and long-term progression in mild cognitive impairment due to alzheimer’s disease defined with amyloid pet imaging. J Alzheimers Dis. 2017;57(3):765–773. doi: 10.3233/JAD-161074. [DOI] [PubMed] [Google Scholar]
- 133.Murray ME, Lowe VJ, Graff-Radford NR, Liesinger AM, Cannon A, Przybelski SA, et al. Clinicopathologic and 11c-pittsburgh compound b implications of thal amyloid phase across the alzheimer’s disease spectrum. Brain. 2015;138(Pt 5):1370–1381. doi: 10.1093/brain/awv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Matveev SV, Spielmann HP, Metts BM, Chen J, Onono F, Zhu H, et al. A distinct subfraction of aβ is responsible for the high-affinity pittsburgh compound b-binding site in alzheimer’s disease brain. J Neurochem. 2014;131(3):356–368. doi: 10.1111/jnc.12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang X, Tian Y, Yuan P, Li Y, Yaseen MA, Grutzendler J, et al. A bifunctional curcumin analogue for two-photon imaging and inhibiting crosslinking of amyloid beta in alzheimer’s disease. Chem Commun (Camb) 2014;50(78):11550–11553. doi: 10.1039/c4cc03731f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ran C, Xu X, Raymond SB, Ferrara BJ, Neal K, Bacskai BJ, et al. Design, synthesis, and testing of difluoroboron-derivatized curcumins as near-infrared probes for in vivo detection of amyloid-beta deposits. J Am Chem Soc. 2009;131(42):15257–15261. doi: 10.1021/ja9047043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu B, Le KX, Park M-A, Wang S, Belanger AP, Dubey S, et al. In vivo detection of age - and disease-related increases in neuroinflammation by 18f-ge180 tspo micropet imaging in wild-type and alzheimer’s transgenic mice. J Neurosci. 2015;35(47):15716–15730. doi: 10.1523/JNEUROSCI.0996-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.James ML, Belichenko NP, Nguyen T-VV, Andrews LE, Ding Z, Liu H, et al. Pet imaging of translocator protein (18 kda) in a mouse model of alzheimer’s disease using n-(2,5-dimethoxybenzyl)-2-18f-fluoro-n-(2-phenoxyphenyl)acetamide. J Nucl Med. 2015;56(2):311–316. doi: 10.2967/jnumed.114.141648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mirzaei N, Tang SP, Ashworth S, Coello C, Plisson C, Passchier J, et al. In vivo imaging of microglial activation by positron emission tomography with [(11)c]pbr28 in the 5xfad model of alzheimer’s disease. Glia. 2016;64(6):993–1006. doi: 10.1002/glia.22978. [DOI] [PubMed] [Google Scholar]
- 140.Kreisl WC, Lyoo CH, McGwier M, Snow J, Jenko KJ, Kimura N, et al. In vivo radioligand binding to translocator protein correlates with severity of alzheimer’s disease. Brain. 2013;136(Pt 7):2228–2238. doi: 10.1093/brain/awt145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kreisl WC, Lyoo CH, Liow J-S, Wei M, Snow J, Page E, et al. (11)c-pbr28 binding to translocator protein increases with progression of alzheimer’s disease. Neurobiol Aging. 2016;44:53–61. doi: 10.1016/j.neurobiolaging.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An rna-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gonzalez Murcia JD, Schmutz C, Munger C, Perkes A, Gustin A, Peterson M, et al. Assessment of trem2 rs75932628 association with alzheimer’s disease in a population-based sample: the cache county study. Neurobiol Aging. 2013;34(12):2889.e11–3. doi: 10.1016/j.neurobiolaging.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rosenthal SL, Bamne MN, Wang X, Berman S, Snitz BE, Klunk WE, et al. More evidence for association of a rare trem2 mutation (r47h) with alzheimer’s disease risk. Neurobiol Aging. 2015;36(8):2443.e21–6. doi: 10.1016/j.neurobiolaging.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jin SC, Carrasquillo MM, Benitez BA, Skorupa T, Carrell D, Patel D, et al. Trem2 is associated with increased risk for alzheimer’s disease in african americans. Mol Neurodegener. 2015;10(1):19. doi: 10.1186/s13024-015-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, et al. Coding variants in trem2 increase risk for alzheimer’s disease. Hum Mol Genet. 2014;23(21):5838–5846. doi: 10.1093/hmg/ddu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Miyashita A, Wen Y, Kitamura N, Matsubara E, Kawarabayashi T, Shoji M, et al. Lack of genetic association between trem2 and late-onset alzheimer’s disease in a japanese population. J Alzheimers Dis. 2014;41(4):1031–1038. doi: 10.3233/JAD-140225. [DOI] [PubMed] [Google Scholar]
- 148.Slattery CF, Beck JA, Harper L, Adamson G, Abdi Z, Uphill J, et al. R47h trem2 variant increases risk of typical early-onset alzheimer’s disease but not of prion or frontotemporal dementia. Alzheimers Dement. 2014;10(6):602–608.e4. doi: 10.1016/j.jalz.2014.05.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hooli BV, Parrado AR, Mullin K, Yip W-K, Liu T, Roehr JT, et al. The rare trem2 r47h variant exerts only a modest effect on alzheimer disease risk. Neurology. 2014;83(15):1353–1358. doi: 10.1212/WNL.0000000000000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Finelli D, Rollinson S, Harris J, Jones M, Richardson A, Gerhard A, et al. Trem2 analysis and increased risk of alzheimer’s disease. Neurobiol Aging. 2015;36(1):546.e9–13. doi: 10.1016/j.neurobiolaging.2014.08.001. [DOI] [PubMed] [Google Scholar]