

Abstract
The paradoxical role of reactive oxygen species in cell death versus cell survival establishes a delicate balance between chemotherapy efficacy and management of detrimental side effects. Normal proliferative signaling requires that cells remain inside a redox range that allows reversible protein oxidation to occur. Shifting the redox environment toward highly reducing or oxidizing states leads to cellular stress and cell death. Reactive oxygen species produced in response to Taxol and cisplatin treatment are necessary for effective cancer cell killing but the same ROS leads to damaging side effects in normal tissues. Combining antioxidants with chemotherapeutics to alleviate the unwanted side effects produces variable and often undesirable effects on cancer treatment. Here, we describe a more targeted method to improve ovarian cancer cell killing without the need for antioxidants. In ovarian cancer cells, lysophosphatidic acid (LPA) is a prominent growth factor that contributes to tumor survival and proliferation. We find that blocking LPA-dependent signaling with a specific receptor antagonist consistently increases cell death in response to both Taxol and cisplatin. We propose that inhibiting the upregulated growth factor-dependent signaling in cancer cells will target chemo-insensitivity, potentially lowering the necessary dose of the drugs and preventing harmful side effects.
Keywords: Lysophosphatidic acid (LPA), Ovarian cancer, Reactive oxygen species (ROS), Chemotherapy, Taxol, Cisplatin
Graphical abstract
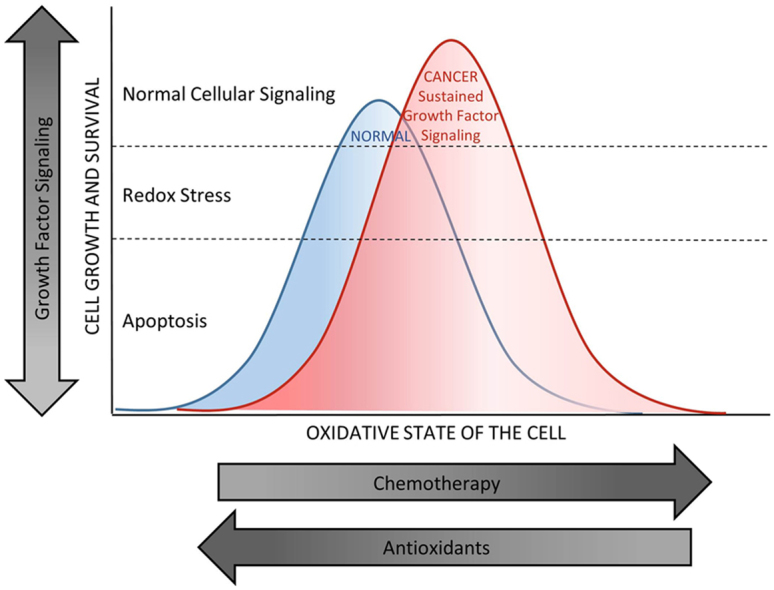
Proliferative signaling occurs inside a window that allows signaling molecules to be reversibly oxidized and reduced. Chemotherapeutic drugs push cells toward a higher oxidation state, which is necessary for effective cancer cell death. The side effect is oxidative damage to normal cells. Antioxidants have a broad range of effects on the oxidative state of normal and cancer cells. While antioxidants may help prevent oxidative damage to normal cells, the effect of the shift in redox state of a given tumor on the efficacy of chemotherapy treatment is variable. Ovarian cancer cells often make and/or respond to increased amounts of LPA, a growth factor found at high levels in ascites fluid. This sustained growth factor-dependent signaling increases cellular survival mechanisms preventing oxidative damage and promoting uncontrolled proliferation. Removing these signals dampens the peak of the survival curve allowing chemotherapeutics to more effectively kill the cancer cells and spare normal tissues.
Highlights
-
•
Chemotherapy-induced ROS are required for tumor killing, but damage normal tissue.
-
•
Antioxidants decrease side effects of ROS but have variable effects on treatment.
-
•
Blocking LPA signaling enhances chemotherapy in SKOV3 cells.
1. Introduction
The damage to normal tissues by reactive oxygen species (ROS) produced in response to chemotherapeutics is a major complication in cancer treatment. Taxol and cisplatin are two common chemotherapeutic agents often used in combination as a first line of defense to treat many cancers, including ovarian carcinomas [1], [2], [3]. Both drugs non-specifically target rapidly proliferating cells, but in mechanistically different ways. Taxol directly interacts with tubulin and reduces depolymerization of the microtubules [1], [2], [4], [5]. This blocks cells in the G2/M phase of the cell cycle and prevents proliferation [2], [4], [6]. Cisplatin crosslinks purine bases in genomic DNA which interferes with DNA repair and causes a DNA damage response resulting in apoptosis in cancer cells [7], [8], [9], [10]. Both drugs increase ROS production, not only in tumor cells where the increased oxidative stress leads to a favorable outcome, but also in surrounding tissues which leads to painful neuropathy, kidney damage, hearing loss, and gastrointestinal side effects [9], [10], [11]. The ROS-induced damage to normal tissues increases dose responsively, often causing the course of treatment to remain below a maximally effective level.
Both dietary and pharmaceutical antioxidant supplements have been used in clinical trials with modest success in preventing side effects [12], [13], [14], [15], [16], [17]. Clinically, broad range or systemic antioxidant approaches have been applied such as n-acetylcysteine (NAC), a potent ROS scavenger, or all trans retinoic acid (ATRA), the animal form of Vitamin A [5], [12], [16]. Additionally, patients often self-medicate with naturally occurring antioxidants such as green tea, Vitamin E, muscadine extract, resveratrol, and fish oil [5], [12], [13], [18], [19], [20], [21], [22]. Clinical studies are currently underway with NAC, in conjunction with chemotherapy (NIH Clinical Trial #NCT01878695) to test the effects of decreasing ROS production on tumor metabolism, as well as fatigue and post-treatment recovery in patients with breast cancer. Global inhibition of ROS has previously been shown to inhibit peripheral neuropathy in patients treated with Taxol [1], and a separate study observed that kidney damage was reduced when using antioxidants in combination with cisplatin therapy [8], [23]. However, the predictability of a cancer cell's response to combining these types of treatments with a chemotherapy or radiation regimen is complicated, with some clinical trials reporting lowered rates of survival for patients treated in combination with antioxidant therapies as opposed to those treated with chemotherapeutics alone [5], [16], [24]. The overarching conclusion is that decreasing the ROS produced in response to chemotherapy has variable, and sometimes undesirable, effects on the efficacy of the treatment.
Reactive oxygen species also play essential roles in normal cell proliferation and metabolism. They have been established as important signaling molecules in response to growth factors and cytokines allowing cells to respond to environmental changes [25], [26], [27], [28], [29]. Reversible protein oxidation plays a significant role in cell survival and proliferative pathways that protect mitochondrial membrane potential, inhibit apoptosis, and increase proliferative signaling. These pathways require the redox state of the cell to remain within a specific range, referred to as the redox window, where reversible redox-dependent proliferative signaling can occur [30]. When cellular redox homeostasis shifts outside the window to either excess oxidation or reduction, normal cellular signaling is disrupted, and apoptosis or cell death occurs [29], [31].
In cancer cells, constitutive growth factor-dependent signaling promotes sustained proliferation and resistance to cell death [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42]. Growth factor-stimulated ROS production and alterations in metabolism contribute to higher intracellular oxidative states in cancer than in normal tissues [9], [32], [43], [44]. In ovarian cancer, a predominant growth factor responsible for stimulating proliferation and survival is lysophosphatidic acid (LPA). Levels of LPA found in the ascites fluid of ovarian carcinomas reach concentrations between 2–20 µM, indicating its importance in tumorigenesis and making it a potential biomarker for disease [45], [46], [47], [48], [49]. LPA interacts with endothelial differentiation gene (Edg) family G-protein coupled receptors (GPCR) that signal through various mechanisms to increase the expression of survival signaling molecules and growth factors that promote cancer proliferation and survival [50], [51], [52]. We previously reported that LPA also stimulates NADPH oxidase-dependent generation of ROS in endosomes containing LPA receptors [53], [54]. This is essential to NF-κB, ERK, and Akt signaling and leads to increased proliferation in SKOV3 ovarian cancer cells [53]. Upregulation of these pathways in cancer also allows tumor cells to resist oxidative stress and apoptosis through the increased production of Bcl-2, Bcl-xL and other proteins that protect mitochondrial membrane potential, as well as increased resistance to ER stress-induced mitochondrial ROS production [55], [56], [57]. Treatment of these cells with a specific LPA receptor antagonist eliminates the LPA-dependent ROS production, protein oxidation, and leads to apoptosis [53], [54].
The paradoxical role of ROS in cell death versus cell survival establishes a delicate balance between chemotherapy efficacy and management of detrimental side effects. This is further complicated by the shift in the redox window necessary for optimal survival of normal tissues versus that of cancer. Presently it is not practical to measure the changing redox state of a tumor. Drug treatment, angiogenesis, hypoxia, and increased proliferation and survival signaling all play a role. Thus, using broad range ROS scavengers and systemic antioxidants to prevent unwanted side effects of chemotherapy is not widely successful. Here, we examine the role of LPA-dependent survival signaling as a source of chemo-resistance to Taxol and cisplatin in SKOV3 ovarian cancer cells. We propose that inhibiting growth factor-dependent signaling to abrogate survival and proliferative signaling in cancer cells is a more specific target to combat chemo-insensitivity and prevent harmful side effects. A better understanding of the source of ROS in response to each chemotherapeutic agent and the time and location of ROS production are needed to maximize efficacy with minimal toxicity.
2. Materials and methods
2.1. Reagents and antibodies
Cisplatin, Taxol, and NAC were purchased from Sigma Aldrich. Primary antibodies for Western blots to detect cleaved caspase-3, cleaved caspase-7, and anti-rabbit secondary antibody were from Cell Signaling Technology. Dichlorofluorescein diacetate, RPMI 1640 medium, and Opti-MEM media were from Invitrogen. MitoSOX reagent was purchased from Molecular Probes. FuGENE 6 Transfection Reagent was purchased from Promega. The plasmid for expression of HyPer was from Evrogen. Fetal bovine serum was from Lonza. Nitrocellulose membranes were from Bio-Rad and Super Signal chemiluminescence reagent was from Pierce. VPC32183 and alkyl-linked 18:1 lysophosphatidic acid (LPA) [1-(9Z-octadecenyl)-2-hydroxy-sn-glycero-3-phosphate (ammonium salt)] was from Avanti Polar Lipids, Inc.
2.2. Cell culture and treatments
SKOV3 cells (from ATCC stocks) were grown, maintained, and treated at 37 °C with 5% CO2 in RPMI 1640 medium supplemented with 10% fetal bovine serum, L-glutamine, penicillin, and streptomycin. LPA, supplied in chloroform, was dried under a stream of nitrogen, resuspended to a concentration of 1 mM in phosphate buffered saline (PBS) containing 1% fatty acid-free bovine serum albumin (BSA), and then diluted into culture medium to the indicated concentrations.
2.3. Proliferation assay
SKOV3 cells were plated in 96 well plates at 1.5 × 103 cells per well and incubated overnight at 37 °C, 5% CO2. Cells were treated with Taxol, cisplatin, or vehicle control concurrently with NAC where indicated. Cellular reactions were stopped by removing the culture media and fixing the cells with 10% (w/v) trichloroacetic acid, followed by staining with sulforhodamine B (0.4% w/v in 1% acetic acid) for 10 min. The excess dye was removed by washing repeatedly with 1% (vol/vol) acetic acid. The protein-bound dye was finally dissolved in 10 mM Tris base solution (pH unadjusted) for OD determination at 564 nm using a Molecular Devices VersaMax tunable microplate reader.
2.4. Western blotting
For Western blotting, cells were plated at 5 × 105 cells per dish in 100 mm dishes, treated or not treated with pharmacological agents, washed with cold, calcium-free PBS, scraped into lysis buffer (50 mM Tris-HCl, 100 mM NaCl, 2 mM EDTA, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM PMSF, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 50 mM NaF, and 1 mM sodium vanadate), and centrifuged to remove cell debris after one freeze/thaw cycle. Protein concentration was measured (Pierce BCA protein assay) and samples (typically 40 μg protein/lane) were resolved on SDS polyacrylamide gels, then transferred to nitrocellulose membranes, probed with protein-specific antibodies and visualized using Super Signal chemiluminescence reagent.
2.5. Microscopy
Confocal Microscopy. For mitochondrial ROS production measurements using MitoSOX, 4×104 cells were plated in 0.5 mL of media in 4-well Lab-Tek II Chambered #1.5 Coverglass and incubated for 24 h. To monitor cytoplasmic ROS production, cells were transiently transfected with pHyPer-cyto, a reporter for H2O2 levels in the cytoplasm, using FuGene6 Transfection Reagent according to manufacturer's protocol, and incubated for 24 h [58]. Where indicated, cells were pretreated with LPA or receptor antagonist before labeling with 1 µM MitoSOX for 10 min [59]. MitoSOX reagent was removed and cells were washed three times with media. The field of view was located and focused before the addition of the Taxol or cisplatin. Images were collected over time using Zeiss LSM510 laser scanning confocal microscope.
Fluorescent Microscopy. For measurement of long term ROS production, SKOV3 cells (5 × 104) were plated in 2 mL RPMI supplemented with 10% FBS in 35 mm dishes. Cells were treated 24 h after plating with indicated concentrations of cisplatin for 24 h then incubated with dichlorofluorescein diacetate for 10 min, washed and visualized using an Olympus inverted epi-fluorescent microscope with FITC filters.
2.6. TEM
SKOV3 cells were plated at 5 × 105 cells per dish in 100 mm dishes, pre-treated or not treated as indicated before treating with Taxol for 24 h, then washed with cold, calcium-free PBS and fixed with 2.5% glutaraldehyde in 0.1 M Millonig's phosphate buffer pH 7.3 for a minimum of one hour. Subsequently, the samples were washed 3 times in Millonig's buffer and post-fixed with 1% osmium tetroxide in phosphate buffer for one hour. After 3 washes in buffer the samples were dehydrated with ethanol. The cultured cells were scraped to form a pellet once they were in 50% ethanol and subsequently dehydrated to 100%. The samples were prepared for resin infiltration by incubation in propylene oxide for two changes of 15 min each. Finally, the samples were gradually infiltrated with 1:1, 1:2 and pure solutions of Spurr's resin after which they were allowed to cure in a 70 °C oven overnight. Finally, 90 nm sections were obtained with a Reichert-Jung Ultracut E ultramicrotome, stained with lead citrate and uranyl acetate and viewed with a FEI Tecnai Spirit TEM operating at 80 kV. Images were obtained with an AMT 2Vu CCD camera.
3. Results
3.1. ROS scavenger treatment has a differential effect on SKOV3 cell death in response to Taxol and cisplatin
Taxol and cisplatin both stimulate apoptosis in SKOV3 ovarian cancer cells as determined by caspase cleavage at 24 and 48 h post treatment (Fig. 1A). Both drugs increase ROS production, not only in tumor cells, but also in normal tissues which leads to negative side effects [22], [23], [24]. In order to determine the effect of reducing the ROS level on cell survival during chemotherapy treatment, we challenged SKOV3 cells with Taxol or cisplatin, in combination with increasing concentrations of n-acetylcysteine (NAC), as indicated for 48 h and measured the relative cell number by sulforhodamine B staining. This method functions independently of dyes that are influenced by the reducing effects of NAC. As expected, we found that treating SKOV3 cells with Taxol or cisplatin causes a decrease in cell number (Fig. 1). Interestingly, the effect on cell death is very different in the presence of NAC (Fig. 1B). While NAC has little effect on SKOV3 cells challenged with Taxol, cells are resistant to cisplatin dependent cell death when treated with NAC and at the higher doses have similar numbers to untreated. These data indicate that the effect of combining broad range ROS scavengers with chemotherapy drugs is variable in SKOV3 cells, and may lead to undesired effects on cancer cell survival.
Fig. 1.

Treatment with NAC, an ROS scavenger, inhibits cisplatin but not Taxol-induced cell death. Panel A. SKOV3 cells treated with either cisplatin or Taxol enter caspase-dependent apoptosis as measured by caspase cleavage. The Western blots are representative of three separate experiments. Panel B. Sulforhodamine B staining was used to determine relative cell number following 48 h of treatment with chemotherapy drugs alone or in combination with NAC. Combining NAC with Taxol treatment has no effect on the response to chemotherapy. However, NAC inhibits the effect of cisplatin on SKOV3 ovarian cancer cell death. Error bars represent the standard error of the mean of three biological replicates done in triplicate. The statistical significance of data points relative to the respective dose of NAC alone was calculated using Student's two tailed paired t-test. Values are indicated by * p-value < 1 × 10−2, ** p-value < 2 × 10−4, ‡ p-value < 2 × 10−6.
3.2. ROS production is immediate and localized to mitochondria in response to Taxol, but not cisplatin
The fact that both Taxol and cisplatin cause apoptosis, but have variable responses to redox modulation during treatment in SKOV3 cells, led us to investigate the source of ROS production in response to chemotherapeutic treatment. In order to monitor the production of cytosolic or mitochondrial ROS, SKOV3 cells were transiently transfected with the gene encoding HyPer-cyto, a cytosolic H2O2 specific fluorescent reporter, and incubated for 48 h to allow reporter expression. Cells were then labeled with MitoSOX, a fluorogenic dye for the selective detection of superoxide in the mitochondria, prior to treatment with Taxol or cisplatin [58], [59]. Using confocal time-lapse microscopy we found that Taxol significantly stimulated mitochondrial production of superoxide within 30 min following treatment, but did not increase cytosolic ROS (Fig. 2A and B). This is in sharp contrast to cisplatin which does not lead to an increase in mitochondrial ROS production (images not shown). Neither drug led to an increase in cytosolic H2O2 production during the one hour time course. Cisplatin is well known to increase intracellular ROS. Therefore, to confirm that in our hands ROS production is stimulated in response to cisplatin, we monitored the oxidative state of the cells longer term using dichlorofluorescein diacetate, which fluoresces in response to global oxidant exposure. We found that cisplatin does elicit an ROS increase across the entire cell 24 h post treatment (Fig. 2C). While cisplatin-induced ROS production has been linked to mitochondria; studies suggest that it is a downstream effect of DNA damage and ER stress [60].
Fig. 2.

Localization of ROS production. Panels A and B. SKOV3 cells transiently expressing pHyPer-cyto, a reporter for cytoplasmic H2O2, were labeled with MitoSOX to measure mitochondrial superoxide production, before treatment with 100 mM Taxol. Taxol treatment leads to increased ROS production in mitochondria but not in the cytosol of the cell 30 min following treatment. Panel B. Changes in MitoSOX fluorescence were measured using a Zeiss LSM510 laser scanning confocal microscope with a 40× objective, and individual cells were analyzed for ROS production using LSM Image Browser Software from Zeiss. The data represent the average change in MitoSOX fluorescence between time zero and 30 min post treatment of 10–30 cells total from at least three separate time courses. Error bars represent the standard error of the mean. Statistical significance relative to control was calculated using Student's two tailed t-test assuming equal variances. Values are indicated by * p-value < 2 × 10−3, ** p-value < 7 × 10−5, ‡ p-value < 2 × 10–15. Panel C. SKOV3 cells were treated for 24 h with indicated concentrations of cisplatin, then labeled with DCF-DA for 10 min before imaging with an inverted epifluorescence microscope.
3.3. Modulation of the LPA signaling pathway significantly alters mitochondrial response to Taxol
LPA-dependent survival signaling is increased in ovarian cancer [45], [46], [50], [51], [52]. The NF-κB pathway which leads to the upregulation of proteins involved in maintaining mitochondrial membrane potential is shown to be stimulated by LPA [53]. We therefore examined the effect of modulating the LPA-dependent signaling on the Taxol-induced mitochondrial ROS production (Fig. 2B). We monitored mitochondrial ROS production in cells that were pretreated for 30 min or 6 h with LPA before Taxol addition. These times for LPA treatment were used because maximal LPA-dependent pERK1/2 phosphorylation is 30 min after treatment with exogenous LPA, while maximal NF-κB activation is 4–6 h post LPA treatment [53]. We found that the LPA-dependent signaling prevented the increase in mitochondrial ROS production seen with Taxol alone (Fig. 2). In an alternative experiment, we pretreated the cells for 24 h with the LPA receptor antagonist VPC32183 prior to challenge with Taxol. This caused a five-fold increase in mitochondrial superoxide production in the cells. The dose of VPC32183 treatment used is two-fold less than those that induce apoptosis in 24 h [53]. To further investigate the effect of blocking LPA-dependent signaling on the mitochondrial response to Taxol, we used TEM to image mitochondria from cells treated with Taxol, or a combination of VPC32183 and Taxol. Pretreatment with LPA receptor antagonist before Taxol challenge has a marked visual effect on mitochondrial membrane integrity beyond Taxol treatment alone (Fig. 3). Interestingly, we also observe an increased association of the mitochondria with ER in cells treated with both the LPA receptor antagonist and Taxol. This may be indicative of an ER stress response, however, further investigation is necessary.
Fig. 3.

Mitochondrial membrane potential during Taxol treatment is protected by LPA receptor-dependent signaling. Mitochondria from SKOV3 cells treated with Taxol or Taxol in combination with 5 uM VPC32183 for 24 h were imaged by TEM. Images are representative of 20 or more slices containing multiple mitochondria.
3.4. Blocking the LPA-dependent survival signaling pathway in SKOV3 cells increases apoptosis in response to both Taxol and cisplatin
Given the striking difference in cell survival between Taxol and cisplatin treatment in the presence of NAC, as well as the differences in time and location of ROS production, we investigated whether blocking the LPA-dependent survival signaling pathways would enhance apoptosis in response to both drugs. For these experiments, the VPC32183 was used at concentrations that we confirmed are non-toxic to normal NIH3T3 cells. Indeed, VPC32183 pretreatment caused a significant increase in caspase cleavage in response to both Taxol and cisplatin (Fig. 4). Conversely, pretreatment with exogenous LPA appears to slightly abrogate the effect of the two drugs. These findings suggest that blocking specific, prominent growth factor-dependent survival signaling in cancer is a more targeted approach for chemo combination therapy than broad range scavengers or systemic antioxidants [61], [62], [63], [64], [65], [66], [67], [68].
Fig. 4.

Blocking LPA-dependent signaling increases the death response to both Taxol and cisplatin. SKOV3 cells were incubated with LPA or VPC32183 at indicated concentrations for 24 h before harvesting and immunoblotting for caspase cleavage. The data shown are representative of Western blots from three independent experiments.
4. Discussion
Normal proliferative signaling occurs within a cellular redox window that allows reversible oxidation of signaling molecules. Chemotherapeutic drugs push cells toward a higher oxidation state, which is necessary for effective cancer cell death. One adverse side effect of this is oxidative damage to normal cells. Antioxidants have a broad range of effects on the oxidative state of normal and cancer cells. While antioxidants may help prevent oxidative damage to normal cells, in cancer the effect of the change in redox state of a given tumor on the efficacy of chemotherapy treatment is variable. To begin to untangle the complications of broad antioxidant approaches for mitigating the damaging side effects during chemotherapy treatment, it is important to consider the roles of ROS in normal and cancerous cellular processes, as well as the source and localization of the ROS production.
Clinically, there is evidence that the effect of antioxidant co-treatment during chemotherapy is not predictable and may lead to undesirable outcomes [12], [13], [15], [16], [17], [20], [65]. Here, we showed that Taxol and cisplatin treatment of SKOV3 ovarian cancer cells leads to apoptosis, and that the mechanisms of action of these drugs are dependent on ROS. We found that cisplatin induced cell death is reduced with treatment of NAC, a ROS scavenger, while NAC has no effect on Taxol induced cell death. We also determined that Taxol, but not cisplatin, induces an early ROS production in the mitochondria of the cell, and that higher levels of mitochondrial ROS lead to increased loss of mitochondrial membrane potential and increased apoptosis. These data show the variability of effects of using broad antioxidant treatments in conjunction with chemotherapeutic drugs on SKOV3 ovarian cancer cells. They also reveal the differences in the nature of ROS production induced by the chemicals in these cells.
ROS production is a double edged sword. While negative side effects can be attributed to ROS, they are also required for normal cell proliferation and survival signaling. Many growth factors and cytokines have been shown to signal through ROS as a second messenger [33], [44], [50], [69], [70]. These ROS are often produced in endosomes where signal specific protein oxidation occurs [54], [70]. Normal cellular processes depend on the reversible reduction and oxidation of phosphatases and kinases. Shifting this required cellular signaling to a more reducing environment with antioxidants may perpetuate the harmful side effects because a reduced redox state might lower survival signaling and increase harmful ROS release.
Cancer cells have developed the ability to easily adapt to a changing redox environment, increasing the range of their optimal redox window. It is proposed that the mechanism of adaptation stems from the upregulation of survival signaling pathways like pERK, NF-κB, and Akt that protect against ER stress, maintain mitochondrial membrane potential, and inhibit apoptosis [31], [53], [71]. NF-κB functions to upregulate proteins such as Bcl-2, Bcl-xL, and IAPs that maintain mitochondrial membrane potential and directly inhibit apoptosis. Activation of the ERK MAP kinase cascade results in transcription of important tumor suppressor genes such as EGR1. These pathways can be upregulated in cancer cells in response to increased growth factor receptor expression or growth factor production. In ovarian cancer cells, increased production of LPA serves as a major tumorigenic factor [45], [46], [47], [48], [49]. Ascites fluid surrounding ovarian tumors contains a high level of LPA [49]. Additionally, ovarian tumors often make LPA from membrane phospholipids, and respond to LPA in the ascites fluid by upregulating survival and proliferative signaling [53], [72]. Blocking LPA-dependent survival signaling with VPC32183, a specific and competitive LPA 1 and 3 receptor antagonist has been shown to decrease NF-κB signaling, decrease phosphorylation of pERK1/2, and cause SKOV3 cells to die by apoptosis [46].
Consistent with this, we see that treating SKOV3 cells with LPA prevents mitochondrial ROS production in response to Taxol. Conversely, we also show that blocking LPA receptor activation with VPC32183 significantly increases the mitochondrial ROS production. Our TEM studies confirm that the increased levels of ROS have a deleterious effect on the mitochondrial membrane. The decrease in mitochondrial membrane potential is known to lead to caspase dependent apoptosis [11], [32], [73]. Furthermore, we show treatment of SKOV3 cells with VPC32183 increases apoptosis in response to Taxol. Interestingly, we also observe an increase in apoptosis response to VPC32183 and cisplatin.
Blocking the LPA-dependent survival and proliferative signaling pathways in SKOV3 cells is an effective method to increase cell death in response to both Taxol and cisplatin, in contrast to the variable result of NAC co-treatment. Given the unpredictable and delicate redox balance during chemotherapy treatment, we propose that blocking the growth factor stimulation of cancer cells which allows them to become more chemo-resistant may be an alternative strategy to general antioxidant therapies targeting chemotherapy-induced ROS. While normal cells may respond to the same growth factors as cancer cells, identifying the specific growth factor or growth factor receptors that are upregulated in certain cancers provides a more specific target to direct the chemotherapeutic effects toward the cancer cells, potentially allowing a lower effective dose and decreasing the harmful side effects on normal cells by leaving their redox-dependent survival signaling intact. Moving forward, it will be useful to find protein targets of the proliferation and survival pathways that may be targeted to lower the survival response that allow cancers to resist chemotherapy.
Acknowledgments
We thank Ken Grant of the Wake Forest Comprehensive Cancer Center Microscopy shared resource for his assistance with electron microscopy. This work has been supported through funding from the NIH (CA142838 to LWD), the Comprehensive Cancer Center of Wake Forest University National Cancer Institute Cancer Center Support Grant P30CA012197, and pilot funding from the Wake Forest School of Medicine Center for Redox Biology in Medicine (to TH and NS).
Acknowledgments
Declarations of interest
None.
Contributor Information
Thomas Hollis, Email: thollis@wakehealth.edu.
Larry W. Daniel, Email: ldaniel@wakehealth.edu.
References
- 1.Fidanboylu M., Griffiths L.A., Flatters S.J.L. Global inhibition of reactive oxygen species (ROS) inhibits paclitaxel-induced painful peripheral neuropathy. PLoS One. 2011;6 doi: 10.1371/journal.pone.0025212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiao H., Verdier-Pinard P., Fernandez-Fuentes N., Burd B., Angeletti R., Fiser A., Horwitz S.B., Orr G.A. Insights into the mechanism of microtubule stabilization by Taxol. Proc. Natl. Acad. Sci. USA. 2006;103:10166–10173. doi: 10.1073/pnas.0603704103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrelli F., Coinu A., Riboldi V., Borgonovo K., Ghilardi M., Cabiddu M., Lonati V., Sarti E., Barni S. Concomitant platinum-based chemotherapy or cetuximab with radiotherapy for locally advanced head and neck cancer: a systematic review and meta-analysis of published studies. Oral Oncol. 2014;50:1041–1048. doi: 10.1016/j.oraloncology.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Jordan M., Toso R.J., Thrower D., Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. USA. 1993;90:9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saeidnia S., Abdollahi M. Antioxidants: friends or foe in prevention or treatment of cancer: the debate of the century. Toxicol. Appl. Pharmacol. 2013;271:49–63. doi: 10.1016/j.taap.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Rieder C.L., Cole R.W., Khodjakov A., Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 1995;130:941–948. doi: 10.1083/jcb.130.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dasari S., Tchounwou P. Bernard. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin L., Zheng J., Zhu W., Jia N. Nephroprotective effect of gelsemine against cisplatin-induced toxicity is mediated via attenuation of oxidative stress. Cell Biochem. Biophys. 2014;71:535–541. doi: 10.1007/s12013-014-0231-y. [DOI] [PubMed] [Google Scholar]
- 9.Marullo R., Werner E., Degtyareva N., Moore B., Altavilla G., Ramalingam S.S., Doetsch P.W. Cisplatin induces a mitochondrial-ros response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS One. 2013;8:1–15. doi: 10.1371/journal.pone.0081162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi Y.M., Kim H.K., Shim W., Anwar M.A., Kwon J.W., Kwon H.K., Kim H.J., Jeong H., Kim H.M., Hwang D., Kim H.S., Choi S. Mechanism of cisplatin-induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PLoS One. 2015;10:1–21. doi: 10.1371/journal.pone.0135083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varbirio G., Veres B., Gallyas F.J.R., Sumegi B. Direct effect of taxol on free radical formation and mitochondrial permeability transition. Free Radic. Biol. Med. 2001;31:548–558. doi: 10.1016/s0891-5849(01)00616-5. [DOI] [PubMed] [Google Scholar]
- 12.Goodman M., Bostick R.M., Kucuk O., Jones D.P. Clinical trials of antioxidants as cancer prevention agents: past, present, and future. Free Radic. Biol. Med. 2011;51:1068–1084. doi: 10.1016/j.freeradbiomed.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 13.Bennett L.L., Rojas S., Seefeldt T. Role of antioxidants in the prevention of cancer. J. Exp. Clin. Med. 2012;4:215–222. [Google Scholar]
- 14.Bavarsad Shahripour R., Harrigan M.R., Alexandrov A.V. N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav. 2014;4:108–122. doi: 10.1002/brb3.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sayin V.I., Ibrahim M.X., Larsson E., Nilsson J.A., Lindahl P., Bergo M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014;6:221ra15. doi: 10.1126/scitranslmed.3007653. [DOI] [PubMed] [Google Scholar]
- 16.Lawenda B.D., Kelly K.M., Ladas E.J., Sagar S.M., Vickers A., Blumberg J.B. Should supplemental antioxidant administration be avoided during chemotherapy and radiation therapy? J. Natl. Cancer Inst. 2008;100:773–783. doi: 10.1093/jnci/djn148. [DOI] [PubMed] [Google Scholar]
- 17.Le Gal K., Ibrahim M.X., Wiel C., Sayin V.I., Akula M.K., Karlsson C., Dalin M.G., Akyurek L.M., Lindahl P., Nilsson J., Bergo M.O. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015;7 doi: 10.1126/scitranslmed.aad3740. (308re8-308re8) [DOI] [PubMed] [Google Scholar]
- 18.Ford E.S., Ajani U.A., Mokdad A.H. Brief communication: the prevalence of high intake of vitamin E from the use of supplements among U.S. adults. Ann. Intern. Med. 2005;143:116–121. doi: 10.7326/0003-4819-143-2-200507190-00010. [DOI] [PubMed] [Google Scholar]
- 19.T.A.-T, B.C.C.P.S. Group The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1990;330:1029–1035. doi: 10.1056/NEJM199404143301501. [DOI] [PubMed] [Google Scholar]
- 20.Thompson I.M., Tangen C.M., Crowley J.J., Lucia M.S., Goodman P.J., Minasian L.M., Ford L.G., Parnes H.L., Gaziano J.M., Karp D.D., Lieber M.M., Walther P.J., Klotz L., Parsons J.K., Chin J.L., Darke A.K., Lippman S.M., Goodman G.E. Vitamin E and the risk of prostate cancer. JAMA. 2011;306:1549–1556. doi: 10.1001/jama.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Omenn E., Goodman G.S., Thornquist G.E., Al M.D. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N. Engl. J. Med. 1996;334:1150–1155. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- 22.Roh J., Kim E.H., Jang H., Shin D. Aspirin plus sorafenib potentiates cisplatin cytotoxicity in resistant head and neck cancer cells through xCT inhibition. Free Radic. Biol. Med. 2017;104:1–9. doi: 10.1016/j.freeradbiomed.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Sultana S., Verma K., Khan R. Nephroprotective efficacy of chrysin against cisplatin-induced toxicity via attenuation of oxidative stress. J. Pharm. Pharmacol. 2012;64:872–881. doi: 10.1111/j.2042-7158.2012.01470.x. [DOI] [PubMed] [Google Scholar]
- 24.Di Meo S., Reed T.T., Venditti P., Victor V.M. Harmful and Beneficial Role of ROS. Oxid. Med. Cell. Longev. 2016;2016:1–3. doi: 10.1155/2016/7909186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buettner G.R. Moving free radical and redox biology ahead in the next decade(s) Free Radic. Biol. Med. 2015;78:236–238. doi: 10.1016/j.freeradbiomed.2014.10.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forman H.J., Augusto O., Brigelius-Flohe R., Dennery P.A., Kalyanaraman B., Ischiropoulos H., Mann G.E., Radi R., Roberts L.J., Vina J., Davies K.J.A. Even free radicals should follow some rules: a guide to free radical research terminology and methodology. Free Radic. Biol. Med. 2015;78:233–235. doi: 10.1016/j.freeradbiomed.2014.10.504. [DOI] [PubMed] [Google Scholar]
- 27.Reczek C.R., Chandel N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015;33:8–13. doi: 10.1016/j.ceb.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Autréaux B., Toledano M.B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 30.Yun J., Rocic P., Pung Y.F., Belmadani S., Carrao A.C.R., Ohanyan V., Chilian W.M. Redox-dependent mechanisms in coronary collateral growth: the “redox window” hypothesis. Antioxid. Redox Signal. 2009;11:1961–1974. doi: 10.1089/ars.2009.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trachootham D., Lu W., Ogasawara M.A., Nilsa R.D., Huang P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 33.Schieber M., Chandel N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bae Y.S., Sung J.-Y., Kim O.-S., Kim Y.J., Hur K.C., Kazlauskas A., Rhee S.G. Platelet-derived growth factor-induced H2O2 production requires the activation of phosphatidylinositol 3-kinase. J. Biol. Chem. 2000;275:10527–10531. doi: 10.1074/jbc.275.14.10527. [DOI] [PubMed] [Google Scholar]
- 35.Burdon R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995;18:775–794. doi: 10.1016/0891-5849(94)00198-s. [DOI] [PubMed] [Google Scholar]
- 36.Goustin A.S., Leof E.B., Shipley G.D., Moses2 H.L. Growth factors and cancer 1. Cancer Res. 1986;46:1015–1029. 〈http://cancerres.aacrjournals.org/content/canres/46/3/1015.full.pdf〉 [PubMed] [Google Scholar]
- 37.Lo Y.Y., Cruz T.F. Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. J. Biol. Chem. 1995;270:11727–11730. doi: 10.1074/jbc.270.20.11727. [DOI] [PubMed] [Google Scholar]
- 38.Meier B., Radeke H.H., Selle S., Younes M., Sies H., Resch K., Habermehl G.G. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem. J. 1989;263:539–545. doi: 10.1042/bj2630539. 〈http://www.ncbi.nlm.nih.gov/pubmed/2556998〉 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohba M., Shibanuma M., Kuroki T., Nose K. Production of hydrogen peroxide by transforming growth factor-/31 and its involvement in induction of egr-1 in mouse osteoblastic cells. J. Cell Biol. 1994;126:1079–1088. doi: 10.1083/jcb.126.4.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Q., Olashaw N., Wu J. Participation of reactive oxygen species in the lysophosphatidic acid-stimulated mitogen-activated protein kinase kinase activation pathway. J. Biol. Chem. 1995;270:28499–28502. doi: 10.1074/jbc.270.48.28499. [DOI] [PubMed] [Google Scholar]
- 41.Griendling K.K., Minieri C.A., Ollerenshaw J.D., Alexander R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 42.Roy D., Sarkar S., Felty Q. Levels of IL-1 beta control stimulatory/inhibitory growth of cancer cells. Front. Biosci. 2006;11:889–898. doi: 10.2741/1845. [DOI] [PubMed] [Google Scholar]
- 43.Trachootham D., Alexandre J., Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 44.M.-Y. Liou, P. Storz, Reactive Oxygen Species in Cancer, 2010. 〈http://dx.doi.org/10.3109/10715761003667554.Reactive〉. [DOI] [PMC free article] [PubMed]
- 45.Bese T., Barbaros M., Baykara E., Guralp O., Cengiz S., Demirkiran F., Sanioglu C., Arvas M.I. Comparison of total plasma lysophosphatidic acid and serum CA-125 as a tumor marker in the diagnosis and follow-up of patients with epithelial ovarian cancer. J. Gynecol. Oncol. 2010;21:248–254. doi: 10.3802/jgo.2010.21.4.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu J., Xiao Y., Baudhuin Yj, L.M., Hong G., Xu Y. Role of ether-linked lysophosphatidic acids in ovarian cancer cells. J. Lipid Res. 2002;43:463–476. 〈http://www.ncbi.nlm.nih.gov/pubmed/11893783〉 [PubMed] [Google Scholar]
- 47.Zhang S.Y., Shi W., Cheng P., Zaworotko M.J. A mixed-crystal lanthanide zeolite-like metal-organic framework as a fluorescent indicator for lysophosphatidic acid, a cancer biomarker. J. Am. Chem. Soc. 2015;137:12203–12206. doi: 10.1021/jacs.5b06929. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y., Pei H., Jia Y., Liu J., Li Z., Ai K., Lu Z., Lu L. Synergistic tailoring of electrostatic and hydrophobic interactions for rapid and specific recognition of lysophosphatidic acid, an early-stage ovarian cancer biomarker. J. Am. Chem. Soc. 2017;139:11616–11621. doi: 10.1021/jacs.7b06885. [DOI] [PubMed] [Google Scholar]
- 49.Cao L., Zhang Y., Fu Z., Dong L., Yang S., Meng W., Li Y., Zhang W., Zhang J., Zheng C., Zhu H., Fan L. Diagnostic value of plasma lysophosphatidic acid levels in ovarian cancer patients: a case-control study and updated meta-analysis. J. Obstet. Gynaecol. Res. 2015;41:1951–1958. doi: 10.1111/jog.12806. [DOI] [PubMed] [Google Scholar]
- 50.Jesionowska A., Cecerska-Heryc E., Matoszka N., Dolegowska B. Lysophosphatidic acid signaling in ovarian cancer. J. Recept. Signal Transduct. 2015;35:578–584. doi: 10.3109/10799893.2015.1026444. [DOI] [PubMed] [Google Scholar]
- 51.Yu S., Murph M.M., Lu Y., Liu S., Hall H.S., Liu J., Stephens C., Fang X., Mills G.B. Lysophosphatidic acid receptors determine tumorigenicity and aggressiveness of ovarian cancer cells. J. Natl. Cancer Inst. 2008;100:1630–1642. doi: 10.1093/jnci/djn378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee J., Park S.Y., Lee E.K., Park C.G., Chung H.C., Rha S.Y., Kim Y.K., Bae G.U., Kim B.K., Han J.W., Lee H.Y. Activation of hypoxia-inducible factor-1a is necessary for lysophosphatidic acid-induced vascular endothelial growth factor expression. Clin. Cancer Res. 2006;12:6351–6358. doi: 10.1158/1078-0432.CCR-06-1252. [DOI] [PubMed] [Google Scholar]
- 53.Saunders J.A., Rogers L.C., Klomsiri C., Poole L.B., Daniel L.W. Reactive oxygen species mediate lysophosphatidic acid induced signaling in ovarian cancer cells. Free Radic. Biol. Med. 2010;49:2058–2067. doi: 10.1016/j.freeradbiomed.2010.10.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klomsiri C., Rogers L.C., Soito L., McCauley A.K., King S.B., Nelson K.J., Poole L.B., Daniel L.W. Endosomal H2O2 production leads to localized cysteine sulfenic acid formation on proteins during lysophosphatidic acid-mediated cell signaling. Free Radic. Biol. Med. 2014;71:49–60. doi: 10.1016/j.freeradbiomed.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaufman R.J., Malhotra J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. Biophys. Acta - Mol. Cell Res. 2014;1843:2233–2239. doi: 10.1016/j.bbamcr.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harding H.P., Zhang Y., Bertolotti A., Zeng H., Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 57.Peng T.-I., Jou M.-J. Oxidative stress caused by mitochondrial calcium overload. Ann. N.Y. Acad. Sci. 2010;1201:183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- 58.Belousov V.V., Fradkov A.F., Lukyanov K.A., Staroverov D.B., Shakhbazov K.S., Terskikh A.V., Lukyanov S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 59.Johnson-Cadwell L.I., Jekabsons M.B., Wang A., Polster B.M., Nicholls D.G. “Mild Uncoupling” does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J. Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- 60.Yang Y., Liu H., Liu F., Dong Z. Mitochondrial dysregulation and protection in cisplatin nephrotoxicity. Arch. Toxicol. 2014;88:1249–1256. doi: 10.1007/s00204-014-1239-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang F., Liu S., Shen Y., Zhuang R., Xi J., Fang H., Pan X., Sun J., Cai Z. Protective effects of N-acetylcysteine on cisplatin-induced oxidative stress and DNA damage in HepG2 cells. Exp. Ther. Med. 2014;8:1939–1945. doi: 10.3892/etm.2014.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rocha C.R.R., Garcia C.C.M., Vieira D.B., Quinet A., de Andrade-Lima L.C., Munford V., Belizário J.E., Menck C.F.M. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. 2014;5:e1505. doi: 10.1038/cddis.2014.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo J., Tsuji T., Yasuda H., Sun Y., Fujigaki Y., Hishida A. The molecular mechanisms of the attenuation of cisplatin-induced acute renal failure by N-acetylcysteine in rats. Nephrol. Dial. Transplant. 2008;23:2198–2205. doi: 10.1093/ndt/gfn090. [DOI] [PubMed] [Google Scholar]
- 64.Kolb R.J., Ghazi A.M., Barfuss D.W. Inhibition of basolateral transport and cellular accumulation of cDDP and N-acetyl- L-cysteine-cDDP by TEA and PAH in the renal proximal tubule. Cancer Chemother. Pharmacol. 2003;51:132–138. doi: 10.1007/s00280-002-0537-0. [DOI] [PubMed] [Google Scholar]
- 65.Dickey D.T., Muldoon L.L., Doolittle N.D., Peterson D.R., Kraemer D.F., Neuwelt E.A. Effect of N-acetylcysteine route of administration on chemoprotection against cisplatin-induced toxicity in rat models. Cancer Chemother. Pharmacol. 2008;62:235–241. doi: 10.1007/s00280-007-0597-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neuwelt A.J., Wu Y.J., Knap N., Losin M., Neuwelt E.A., Pagel M.A., Warmann S., Fuchs J., Czauderna P., Wozniak M. Using acetaminophen's toxicity mechanism to enhance cisplatin efficacy in hepatocarcinoma and hepatoblastoma cell lines. Neoplasia. 2009;11:1003–1011. doi: 10.1593/neo.09688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muldoon L.L., Wu Y.J., Pagel M.A., Neuwelt E.A. N-acetylcysteine chemoprotection without decreased cisplatin antitumor efficacy in pediatric tumor models. J. Neurooncol. 2015;121:433–440. doi: 10.1007/s11060-014-1657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu Y.J., Muldoon L.L., Neuwelt E.A. The chemoprotective agent N-acetylcysteine blocks cisplatin-induced apoptosis through caspase signaling pathway. J. Pharmacol. Exp. Ther. 2005;312:424–431. doi: 10.1124/jpet.104.075119. [DOI] [PubMed] [Google Scholar]
- 69.Li Q., Spencer N.Y., Oakley F.D., Buettner G.R., Engelhardt J.F. Endosomal Nox2 facilitates redox-dependent induction of NF-kappaB by TNF-alpha. Antioxid. Redox Signal. 2009;11:1249–1263. doi: 10.1089/ars.2008.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oakley F.D., Abbott D., Li Q., Engelhardt J.F. Signaling components of redox active endosomes: the redoxosomes. Antioxid. Redox Signal. 2009;11:1313–1333. doi: 10.1089/ars.2008.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aggarwal B.B. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 72.Umezu-Goto M., Tanyi J., Lahad J., Liu S., Yu S., Lapushin R., Hasegawa Y., Lu Y., Trost R., Bevers T., Jonasch E., Aldape K., Liu J., James R.D., Ferguson C.G., Xu Y., Prestwich G.D., Mills G.B. Lysophosphatidic acid production and action: validated targets in cancer? J. Cell. Biochem. 2004;92:1115–1140. doi: 10.1002/jcb.20113. [DOI] [PubMed] [Google Scholar]
- 73.Jiang Z., Fletcher N.M., Ali-Fehmi R., Diamond M.P., Abu-Soud H.M., Munkarah A.R., Saed G.M. Modulation of redox signaling promotes apoptosis in epithelial ovarian cancer cells. Gynecol. Oncol. 2011;122:418–423. doi: 10.1016/j.ygyno.2011.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]