

Abstract
AIM
To study the genes responsible for retinitis pigmentosa.
METHODS
A total of 15 Chinese families with retinitis pigmentosa, containing 94 sporadically afflicted cases, were recruited. The targeted sequences were captured using the Target_Eye_365_V3 chip and sequenced using the BGISEQ-500 sequencer, according to the manufacturer's instructions. Data were aligned to UCSC Genome Browser build hg19, using the Burroughs Wheeler Aligner MEM algorithm. Local realignment was performed with the Genome Analysis Toolkit (GATK v.3.3.0) IndelRealigner, and variants were called with the Genome Analysis Toolkit Haplotypecaller, without any use of imputation. Variants were filtered against a panel derived from 1000 Genomes Project, 1000G_ASN, ESP6500, ExAC and dbSNP138. In all members of Family ONE and Family TWO with available DNA samples, the genetic variant was validated using Sanger sequencing.
RESULTS
A novel, pathogenic variant of retinitis pigmentosa, c.357_358delAA (p.Ser119SerfsX5) was identified in PRPF31 in 2 of 15 autosomal-dominant retinitis pigmentosa (ADRP) families, as well as in one, sporadic case. Sanger sequencing was performed upon probands, as well as upon other family members. This novel, pathogenic genotype co-segregated with retinitis pigmentosa phenotype in these two families.
CONCLUSION
ADRP is a subtype of retinitis pigmentosa, defined by its genotype, which accounts for 20%-40% of the retinitis pigmentosa patients. Our study thus expands the spectrum of PRPF31 mutations known to occur in ADRP, and provides further demonstration of the applicability of the BGISEQ500 sequencer for genomics research.
Keywords: retinitis pigmentosa, PRPF31, BGISEQ-500
INTRODUCTION
Worldwide, nearly 1.5 million people suffered retinitis pigmentosa (RP). This hereditary, retinal-degenerative disease onsets with the death of photoreceptor cells, leading to stepwise visual damage[1]–[3], and has a frequency of 0.1% in China.
The inheritance pattern of RP includes three Mendelian subtypes: autosomal dominant RP (ADRP), autosomal recessive RP (ARRP), and X-linked RP (XLRP)[1]. ADRP accounts for nearly 40 percent of symptomatic RP, and more than 25 genes have been reported to be implicated in the ADRP subtype. Furthermore, a specific subtype of ADRP causative genes has been delineated in the literature: precursor mRNA processing genes[4]. Eight of these are either precursors of six ubiquitous core snRNP proteins (PRPF3, PRPF8, PRPF31, PRPF4, SNRNP200, and PRPF6), or splicing factors of two others (RP9 and DHX38). All eight of these have been implicated in RP. These eight genes are widely expressed, and are crucial to both symptomatic and asymptomatic individuals. Individuals with retina-specific phenotypes are evidenced pathogenic mutations in these specific genes[5].
Incomplete dominance and penetrance are ubiquitous in Mendelian disease, and the genetic architecture underlying many Mendelian diseases remains unclear. It is important to elucidate how polymorphisms are associated with the development of disease, and the relationship between genetic variants and clinical phenotypes. Imperfect understanding of disease severity and incomplete penetrance retards implementation of genetic counseling in the clinic. PRPF31-associated RP is a canonical example of incomplete penetrance, because it is almost universal to observe the presence of symptomatic and asymptomatic mutation carriers in affected families, while the causes of incomplete penetrance are yet to be discovered. Mutations in PRPF31 tend to occur in the major ADRP locus (Table 1), causing the disease subtype termed RP11 (defined by genetic variants of chromosome 19q13.4, OMIM#60138)[6]–[7]. Meanwhile, genetic variants of PRPF31 compose a large proportion of ADRP, and account for almost 10 percent of cases[8]–[11]. There are nonsense, missense, frameshift and large deletion variants. Previous studies have reported that the wildtype PRPF31 allele, which was inherited from the unaffected parent, promoted the generation of asymptomatic offspring[12]–[13]. Symptomatic and asymptomatic offspring inherited distinct variants of the cis-typal wild type PRPF31 alleles from the parents, despite a genetic predisposition. This suggested that disease phenotype may be prevented by inheritance of particular parental factors[12]–[13]. Compared with symptomatic relatives, the expression of a wild type PRPF31 allele has been shown to twofold higher in asymptomatic ones[14]–[15]. This suggests that disease-preventative allele may be a common one. The disease-preventative allele acted in cis to improve the expression of the wild type PRPF31. A treatment for the symptoms resulting from inheriting the pathogenic PRPF31 variant causative of RP may be contrived, if the genetic factor in 19q13.4/PRPF31 which prevents disease phenotype can be identified and understood[16]. The MSR1 element has two forms, 3 repeats and 4 repeats, in human genome. The 4 repeats allele was along with higher expression of PRPF31. This is also inferred elsewhere to affect phenotypes[17].
Table 1. List of mutations of PRPF31 correlated with RP.
| Mutations | Gene | Disease | Clinical significance (last reviewed) |
| c.400delG (p.Asp134Ilefs) | PRPF31 | Retinal dystrophy | Likely pathogenic |
| c.527+1G>T | PRPF31 | Not provided | Pathogenic (Nov. 7, 2016) |
| c.527+3A>G | PRPF31 | Retinitis pigmentosa 11 | Pathogenic (Mar. 1, 2007) |
| c.562G>T (p.Glu188Ter) | PRPF31 | Retinitis pigmentosa | Likely pathogenic |
| c.581C>A (p.Ala194Glu) | PRPF31 | Retinitis pigmentosa 11 | Pathogenic (Apr. 6, 2007) |
| c.615C>G (p.Tyr205Ter) | PRPF31 | Retinitis pigmentosa | Likely pathogenic |
| c.615C>A (p.Tyr205Ter) | PRPF31 | Not provided | Pathogenic (Jul. 30, 2013) |
| c.646G>C (p.Ala216Pro) | PRPF31 | Retinitis pigmentosa 11 | Pathogenic (Jun. 1, 2009) |
| c.764A>T (p.Gln255Leu) | PRPF31 | Retinitis pigmentosa | Likely pathogenic |
| c.950delG (p.Gly317Alafs) | PRPF31 | Not provided | Pathogenic (May 11, 2016) |
| c.967G>T (p.Glu323Ter) | PRPF31 | Retinal dystrophy | Likely pathogenic |
| c.994C>T (p.Gln332Ter) | PRPF31 | Retinal dystrophy | Likely pathogenic |
| c.1060C>T (p.Arg354Ter) | PRPF31 | Retinal dystrophy | Likely pathogenic |
| c.1073+1G>A | PRPF31 | Retinitis pigmentosa 11 | Pathogenic (Apr. 1, 2015) |
| c.1115_1125delGGAAGCAGGCC (p.Arg372Glnfs) | PRPF31 | Retinitis pigmentosa 11 | Pathogenic (Aug. 1, 2001) |
| c.1120C>T (p.Gln374Ter) | PRPF31 | Retinal dystrophy | Likely pathogenic (Jan. 30, 2015) |
| c.1273C>T (p.Gln425Ter) | PRPF31 | Retinitis pigmentosa 11 | Pathogenic (Oct. 8, 2015) |
| c.1374+654C>G | PRPF31 | Retinitis pigmentosa 11 | Pathogenic (Sep. 1, 2009) |
SUBJECTS AND METHODS
Subjects and Clinical Testing
Shenyang the Fourth People's Hospital (SFPH) agreed with this case research. This research abided by the tenets of the Helsinki Declaration. Subjects who took part in this research had signed the informed consent.
Totally 15 Chinese families with 94 sporadic RP cases participated in the study. All of the patients accepted carefully inspection which included regular eye examinations, such as the conditions of the fundus. Totally 200 normal subjects were recruited as controls. They have no relationship with the patients.
BGISEQ-500 Sequencer and DNA Sequence Analysis
In line with the manufacturer's instructions, the extraction of genomic DNA depended on the DNA extraction kits (TianGen, Beijing). A total of 5 µg genomic DNA was sequenced by a BGISEQ-500 sequencer. The targeted sequences were captured using the Target_Eye_365_V3 chip, which contained 365 genes related to eye diseases according to OMIM. First, BGISEQ-500 libraries for each sample were prepared. Second, the target areas were concentrated. Third, hybrid-capture libraries were prepared for paired-end sequencing with read lengths of 50 bp. Finally, each sample had an average coverage depth of nearly 300× on the BGISEQ-500 sequencer. The Burroughs Wheeler Aligner MEM algorithm was applied for aligning reads to hg19 from UCSC Genome Browser. The Genome Analysis Toolkit (GATK v.3.3.0) IndelRealigner was used for realignment. Sequence variants were called with the Genome Analysis Toolkit Haplotypecaller, without any use of imputation. Variants were filtered against a panel derived from One Thousand Genomes Project, ESP6500, ExAC and dbSNP138.
Sanger Sequencing and Pathogenic Analysis
The genetic variant was validated using Sanger sequencing in the two families with available DNA samples. Three steps were used to estimate whether the variant was the cause of the two families. Initially, the variant was positive and co-segregation in the two families. Thereafter, two websites (https://scholar.google.com.hk/; https://www.omim.org) were used to search previous reports about whether the variant existed in unaffected controls. Afterwards, the consistency of two families' phenotypes and the gene with genetic variant should be taken into consideration.
RESULTS
Clinical Evaluations
The pedigree and disease inheritance of Family ONE and Family TWO over three generations indicated a dominant inheritance pattern in both families (Figure 1A, 1B). The proband of Family ONE characterized by bilateral corrected visual acuity 0.6 with childhood onset of night blindness at 6 years old, the vision field concentric contraction less than 15 degrees. The proband of Family TWO characterized by bilateral corrected visual acuity 1.0 with childhood onset of night blindness at 10 years old, the vision field concentric contraction less than 20 degrees. The fundus findings of the patients in both families were similar, and both showed typical RP. The optic disc was waxy, the retinal vessel attenuating, and the osteocyte-like pigment deposited in peripheral retina. Scotopic rod responses of electroretinogram (ERG) in majority eyes was non-detectable, similarly photopic cone responses of ERG were non-detectable from all eyes. Moreover, the ellipsoid bands around the macular area, which could easily be detected with the optical coherence tomography (OCT), disappeared in both families with RP. Other families and sporadic cases were diagnosed with RP by clinicians, according to clinical descriptions of patients and relatives.
Figure 1. Pedigrees, Sanger sequences of identified disease-associated variants and fundus appearances.

A, B: Pedigrees of Family ONE and Family TWO. Probands are denoted with an arrow. Circles represent females, squares represent males. Filled symbols represent affected patients, empty symbols indicate unaffected controls; C, D: Sanger sequence of identified heterozygous PRPF31 variant c.357_358delAA in Family ONE and Family TWO; E, F: The right and left fundus appearances of the proband of Family ONE; G, H: The right and left fundus appearances of the proband of Family TWO.
Mutation Analysis
We performed Target_Eye_365_V3 chip sequencing on two families and 200 unaffected controls. The average of raw data for each sample was 3.2 Gb. The 99.94% of raw data could align to the targeted sequences. The mean sequencing depth was 300×. Sequencing coverage of the object region, sequencing depth more than 10×, was 98.94%.
Three steps were used as the method of screening variants. Firstly, variants of which frequency is less than 1% in ExAC, ESP6500, One Thousand Genomes Project and dbSNP138, were preserved. Secondly, variants which located in coding region or around splicing site were kept. Thirdly, variants which were loss of function or predicted to be damage were preferential for analysis. Finally, a heterozygous variant c.357_358delAA in PRPF31 gene was discovered in Family ONE, Family TWO, and in one sporadic case. Sanger sequencing confirmed the presence of this genetic variant (Figure 1C, 1D).
The heterozygous variant c.357_358delAA in PRPF31 gene was co-segregated in Family ONE and Family TWO. It signified that this heterozygous variant was found in all RP patients and was absent in all unaffected relatives in the two families (Figure 1A, 1B). This variant was a candidate causative variant for the two families. After analysis of other patients and their family members, we also identified another, previously reported[18], RP associated mutation, in a single RP patient: c.421-1G>A in PRPF31.
This primary variant, c.357_358delAA in PRPF31, co-segregated to the phenotype in Family ONE and Family TWO. The sequencing result of 200 normal subjects didn't find this variant. Furthermore, this variant didn't exist in the databases of implicitly benign variations. The genetic variant was absent in both the existing databases of genetic polymorphisms, and in the reported literatures. This primary variant appears to be discovered for the first time by our research, and can be considered to be novel.
DISCUSSION
ARRP accounts for nearly 55% of all RP patients. The ratio of XLRP is just about 10%. The proportion of ADRP is almost 35% of all RP patients[19]. Particular, there have been more than 25 genes reported to correlate with ADRP. The 10% of ADRP cases were related to mutations in PRPF31 gene[8]–[11].
Incomplete penetrance is an obvious and intriguing characteristic of RP11, which is related to PRPF31 gene and a subtype of RP. Incomplete penetrance has been discovered in numerous RP11 pedigrees which carry polymorphisms in PRPF31 gene. When we find a novel, likely pathogenic variant in PRPF31 in a single RP family with an autosomal, dominant mode of inheritance, the fact that the mutation co-segregates with the disease in the family can interpreted as evidence of pathogenicity of that particular variant. However, when it proves to not be perfectly pathogenic, we should find clues as to why this is the case by paying attention to the character of partial penetrance in other PRPF31 polymorphism carrying families. Although there already have been more than two theories concerning the cause of incomplete penetrance of PRPF31 variants. One is that the core promoter activity of PRPF31 gene is related to the forms of an MSR1 repeat[17]. Another is that CNOT3 could bind to the core promoter resulting in the decrease expression of PRPF31 gene[19]. More studies are needed to clarify the exact mechanism of incomplete penetrance.
It is known that mutations in six, widely expressed splicing factors (PRPF3, PRPF6, PRPF8, PRPF31, snRNP200 and RP9) are responsible for a significant fraction of ADRP cases. The causative mechanism underlying why polymorphisms in such “housekeeping” genes bring about retina-specific phenotype remains unclear. It may be that mutations in PRPF31 disturb pre-mRNA splicing of RP9 gene and FSCN2 gene transcripts which are more directly causative of ADRP[20]–[21], due to interactions between proteins and variants in the genes RP9 and FSCN2 (http://string-db.org/cgi/input.pl?input_page_show_search=on&UserId=fiYXDJqnz89n&sessionId=bF0VWE8QypBI).
Deletions in two residues such as c.357_358delAA may impair the structure and function of the protein resulting from PRPF31 transcription. PRPF31 gene encodes a 499-amino acid protein. The mutation c.357_358delAA in PRPF31 may cause a 123-amino truncated protein product. According to Table 1, there are already 18 pathogenic mutations found in PRPF31 which are all behind of the location of c.357_358delAA (p.Ser119SerfsX5). The models which emerge from the SWISS MODEL (https://swissmodel.expasy.org/) of the wild type protein and the protein transcribed by the mutation c.357_358delAA are shown in Figure 2A and 2B.
Figure 2. The SWISS MODEL of the wild type protein (A), and the variant protein translated by the mutation c.357_358delAA (B).
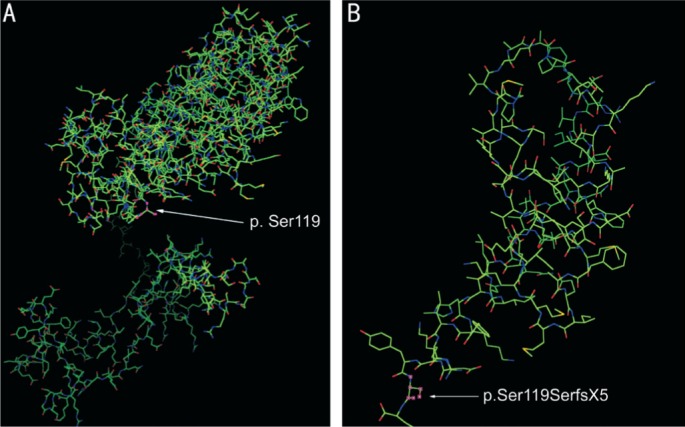
Locations of p.Ser119 polymorphism-derived protein variation are pointed out by an arrow.
In summary, one novel mutation, NM_015629 c.357_358delAA (p.Ser119SerfsX5), was identified in our study, expanding the spectrum of PRPF31 mutations known to be implicated in RP. In addition, the BGISEQ500 sequencer proves adequate for this type of genomics researches. Whole genome sequencing using the BGISEQ-500 generates an average of 100236.61 Mb raw bases in another study of this type. All whole genome sequencing data production is summarized in Table 2. After alignment, mapping rate is shown to be 99.47%, with 98.62% consisting of regions covered in excess of 4×, and an average sequencing depth of 33.02×.
Table 2. Summary of whole genome sequencing data.
| Samples | Raw bases (Mb) | Clean bases (Mb) | Clean data rate (%) | Clean read1 Q20 | Clean read2 Q20 | GC content (%) |
| NA 12878-WGSPE100 | 100236.61 | 100163.05 | 99.93 | 98.12 | 92.07 | 41.71 |
| Average | 100236.61 | 100163.05 | 99.93 | 98.12 | 92.07 | 41.71 |
For clean data where the leading sequences have been trimmed off each end of the individual reads, a Q score of 20 is obtained for almost 95% of sequence, and a Q score of 30 for almost 84% of sequence. Of the variants identified, 99.62% are represented in dbSNP, and 97.71% are annotated in the One Thousand Genomes Project database. In this study, more than 50% of sequenced genotypes proved to contain pathogenic polymorphisms in accord with diseased phenotype. Finally, the presence of those polymorphisms was verified by Sanger sequencing in all cases. This result provides further affirmation of the already high standard demonstrated elsewhere in the BGISEQ-500 sequencer for this type of genomic research.
Acknowledgments
Conflicts of Interest: Zheng Y, None; Wang HL, None; Li JK, None; Xu L, None; Tellier L, None; Li XL, None; Huang XY, None; Li W, None; Niu TT, None; Yang HM, None; Zhang JG, None; Liu DN, None.
REFERENCES
- 1.Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84(2):132–141. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu L, Hu L, Ma K, Li J, Jonas JB. Prevalence of retinitis pigmentosa in urban and rural adult Chinese: The Beijing Eye Study. Eur J Ophthalmol. 2006;16(6):865–866. doi: 10.1177/112067210601600614. [DOI] [PubMed] [Google Scholar]
- 3.Glöckle N, Kohl S, Mohr J, Scheurenbrand T, Sprecher A, Weisschuh N, Bernd A, Rudolph G, Schubach M, Poloschek C, Zrenner E, Biskup S, Berger W, Wissinger B, Neidhardt J. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet. 2014;22(1):99–104. doi: 10.1038/ejhg.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu MM, Zack DJ. Alternative splicing and retinal degeneration. Clin Genet. 2013;84(2):142–149. doi: 10.1111/cge.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Růžičková Š, Staněk D. Mutations in spliceosomal proteins and retina degeneration. RNA Biol. 2017;14(5):544–552. doi: 10.1080/15476286.2016.1191735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deery EC, Vithana EN, Newbold RJ, Gallon VA, Bhattacharya SS, Warren MJ, Hunt DM, Wilkie SE. Disease mechanism for retinitis pigmentosa (RP11) caused by mutations in the splicing factor gene PRPF31. Hum Mol Genet. 2002;11(25):3209–3219. doi: 10.1093/hmg/11.25.3209. [DOI] [PubMed] [Google Scholar]
- 7.Utz VM, Beight CD, Marino MJ, Hagstrom SA, Traboulsi EI. Autosomal dominant retinitis pigmentosa secondary to pre-mRNA splicing-factor gene PRPF31 (RP11): review of disease mechanism and report of a family with a novel 3-base pair insertion. Ophthalmic Genet. 2013;34(4):183–188. doi: 10.3109/13816810.2012.762932. [DOI] [PubMed] [Google Scholar]
- 8.Waseem NH, Vaclavik V, Webster A, Jenkins SA, Bird AC, Bhattacharya SS. Mutations in the gene coding for the pre-mRNA splicing factor, PRPF31, in patients with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2007;48(3):1330–1334. doi: 10.1167/iovs.06-0963. [DOI] [PubMed] [Google Scholar]
- 9.Van Cauwenbergh C, Coppieters F, Roels D, De Jaegere S, Flipts H, De Zaeytijd J, Walraedt S, Claes C, Fransen E, Van Camp G, Depasse F, Casteels I, de Ravel T, Leroy B P, De Baere E. Mutations in splicing factor genes are a major cause of autosomal dominant retinitis pigmentosa in Belgian families. PLoS One. 2017;12(1):e0170038. doi: 10.1371/journal.pone.0170038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Audo I, Bujakowska K, Mohand-Saïd S, Lancelot ME, Moskova-Doumanova V, Waseem NH, Antonio A, Sahel JA, Bhattacharya SS, Zeitz C. Prevalence and novelty of PRPF31 mutations in French autosomal dominant rod-cone dystrophy patients and a review of published reports. BMC Med Genet. 2010;11:145. doi: 10.1186/1471-2350-11-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu F, Sui R, Liang X, Li H, Jiang R, Dong F. Novel PRPF31 mutations associated with Chinese autosomal dominant retinitis pigmentosa patients. Mol Vis. 2012;18:3021–3028. [PMC free article] [PubMed] [Google Scholar]
- 12.McGee TL, Devoto M, Ott J, Berson EL, Dryja TP. Evidence that the penetrance of mutations at the RP11 locus causing dominant retinitis pigmentosa is influenced by a gene linked to the homologous RP11 allele. Am J Hum Genet. 1997;61(5):1059–1066. doi: 10.1086/301614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rose AM, Shah AZ, Venturini G, Rivolta C, Rose GE, Bhattacharya SS. Dominant PRPF31 mutations are hypostatic to a recessive CNOT3 polymorphism in retinitis pigmentosa: a novel phenomenon of “Linked Trans-Acting Epistasis”. Ann Hum Genet. 2014;78:62–71. doi: 10.1111/ahg.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vithana EN, Abu-Safieh L, Pelosini L, Winchester E, Hornan D, Bird AC, Hunt DM, Bustin SA, Bhattacharya SS. Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: a molecular clue for incomplete penetrance? Invest Ophthalmol Vis Sci. 2003;44(10):4204–4209. doi: 10.1167/iovs.03-0253. [DOI] [PubMed] [Google Scholar]
- 15.Rivolta C, McGee TL, Rio Frio T, Jensen RV, Berson EL, Dryja TP. Variation in retinitis pigmentosa-11 (PRPF31 or RP11) gene expression between symptomatic and asymptomatic patients with dominant RP11 mutations. Hum Mutat. 2006;27(7):644–653. doi: 10.1002/humu.20325. [DOI] [PubMed] [Google Scholar]
- 16.Rio Frio T, Civic N, Ransijn A, Beckmann JS, Rivolta C. Two trans-acting eQTLs modulate the penetrance of PRPF31 mutations. Hum Mol Genet. 2008;17(20):3154–3165. doi: 10.1093/hmg/ddn212. [DOI] [PubMed] [Google Scholar]
- 17.Rose AM, Shah AZ, Venturini G, Krishna A, Chakravarti A, Rivolta C, Bhattacharya SS. Transcriptional regulation of PRPF31 gene expression by MSR1 repeat elements causes incomplete penetrance in retinitis pigmentosa. Sci Rep. 2016;6:19450. doi: 10.1038/srep19450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu HG, Park UC, Yoon CK. Advances in Vision Research, Volume I. Japan: Springer; 2017. Retinitis Pigmentosa in Korean Patients; pp. 93–104. [Google Scholar]
- 19.Rose AM, Bhattacharya SS. Variant haploinsufficiency and phenotypic non-penetrance in PRPF31-associated retinitis pigmentosa. Clin Genet. 2016;90(2):118–126. doi: 10.1111/cge.12758. [DOI] [PubMed] [Google Scholar]
- 20.Daiger SP, Bowne SJ, Sullivan LS. Genes and mutations causing autosomal dominant retinitis pigmentosa. Cold Spring Harb Perspect Med. 2014;5(10):pii:a017129. doi: 10.1101/cshperspect.a017129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y, Tian D, Lee J, Zeng J, Zhang H, Chen S, Guo H, Xiong Z, Xia K, Hu Z, Luo J. Clinical and genetic identification of a large chinese family with autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 2015;36(1):64–69. doi: 10.3109/13816810.2013.809458. [DOI] [PubMed] [Google Scholar]